DOI:
10.1039/C5QI00192G
(Research Article)
Inorg. Chem. Front., 2016,
3, 133-141
Modulating single-molecule magnet behaviour of phenoxo-O bridged lanthanide(III) dinuclear complexes by using different β-diketonate coligands†
Received
6th October 2015
, Accepted 10th November 2015
First published on 10th November 2015
Abstract
Three dinuclear Dy(III) complexes, [Dy2(tfa)4L2] (1), [Dy2(TTA)4L2] (2) and [Dy2(dbm)4L2·2CH3CN·0.5H2O] (3) (tfa = trifluoroacetylacetonate, TTA = 2-thenoyltrifluoroacetone, dbm = dibenzoylmethane and HL = 2-[((4-bromophenyl)-imino)methyl]-8-hydroxyquinoline, have been synthesized, and structurally and magnetically characterized. The Dy(III) ions are eight-coordinated with two bidentate β-diketonate and two μ2-O bridging 8-hydroxyquinoline Schiff base ligands. Magnetic studies reveal that 1–3 exhibit different magnetic relaxation behaviors with the anisotropic barriers of 9.61 K (1), 54.81 K (2) and 30.98 K (3), respectively. The different magnetic relaxation behaviors of the three Dy2 complexes originate from the different chemical environments of the central Dy3+ ions with different β-diketonate coligands.
Introduction
Single-molecule magnets (SMMs) exhibiting slow magnetic relaxation have attracted increasing interest during the past two decades because of their potential practical applications in data storage and processing.1–3 Since the discovery, in the early 1990s, of the first SMM, [Mn12] acetate,4 a large number of complexes displaying this property have been reported.5 In the early years, most of them contained only 3d ions; however, as the understanding of the magnetochemical properties of SMMs has increased, significant attention is now paid to incorporating 4f ions into SMMs.6 The DyIII ion seems to be especially useful in this respect, benefiting from both the high moment and the high anisotropy of the spin–orbit coupled DyIII Kramers doublet ground state (S = 5/2, L = 5, J = 15/2). With the aim of synthesizing new and possibly advancing the prospects of SMMs, a lot of Dy(III)-SMMs exhibiting large observed relaxation barriers, which potentially bring the goals of molecule-based information storage and processing closer to reality.7 Among these Dy(III)-SMMs, some β-diketonate-based dysprosium SMMs showed excellent magnetic properties.8 As a large number of Ln(III)-SMMs emerged, understanding the factors determining the relaxation dynamics and deeply understanding the relationship between structures and magnetic properties in such molecules have become of primary importance.
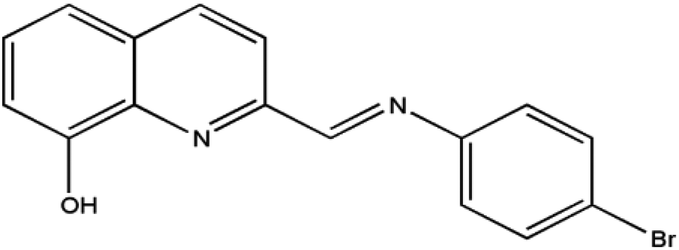
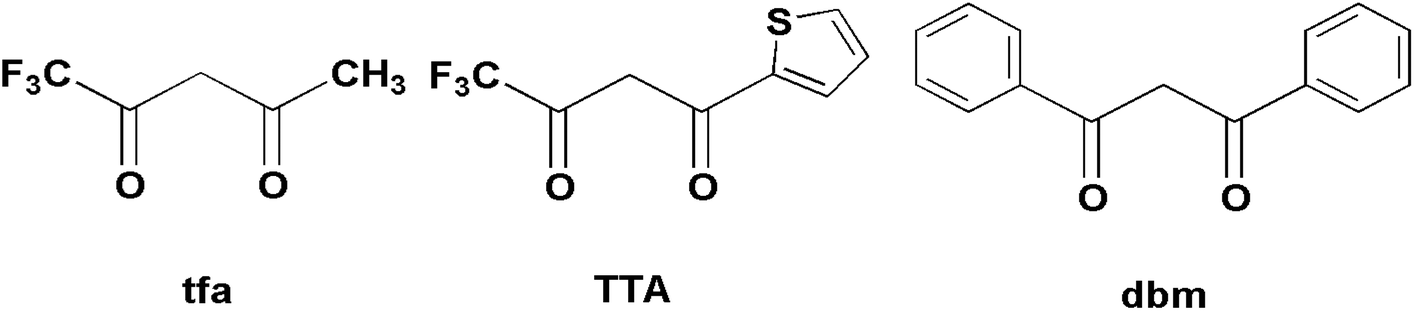
Therefore, in order to deeply understand the relationship between the structures and magnetic properties of SMMs, it is necessary and important to explore how the ligand field perturbation affects the structures of Ln(III) complexes and their magnetic relaxation behaviors.9 In view of this, we employ an attractive 8-hydroxyquinoline Schiff base ligand, HL (Scheme 1), combining with three slightly different β-diketonates (Scheme 2), trifluoroacetylacetonate (tfa), 2-thenoyltrifluoroacetone (TTA) and dibenzoylmethane (dbm), to construct Dy(III) SMMs and study their magnetic dynamics. Here, three Dy2 complexes, [Dy2(tfa)4L2] (1), [Dy2(TTA)4L2] (2) and [Dy2(dbm)4L2·2CH3CN·0.5H2O] (3) (tfa = trifluoroacetylacetonate, TTA = 2-thenoyltrifluoroacetone, dbm = dibenzoylmethane and HL = 2-[((4-bromophenyl)-imino)methyl]-8-hydroxyquinoline, have been successfully obtained and further studied by infrared spectroscopy, elemental analyses (EA), powder X-ray diffraction (PXRD) and single-crystal X-ray diffraction. Magnetic studies reveal that complexes 1–3 exhibit different magnetic relaxation behaviors with the anisotropic barriers of 9.61 K (1), 54.8 K (2) and 31.0 K (3), respectively. The different SMM behaviors of the three Dy2 complexes can be ascribed to the different chemical environments of the central Dy3+ ions. Compared with the structures and magnetic behaviors of 1–3, the β-diketonate coligand indeed plays an important role in modulating the magnetic relaxation of the three Dy2 complexes. This demonstrates that the SMM behavior of the lanthanide-8-hydroxyquinoline Schiff base derivative system can be finely tuned by changing the substituent of the β-diketonate ligand.
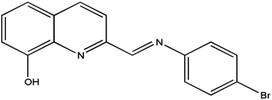 |
| | Scheme 1 The structure of HL. | |
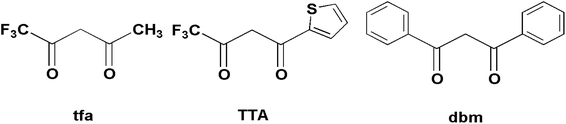 |
| | Scheme 2 Structures of three different β-diketonates. | |
Experimental section
Materials and methods
All chemicals and solvents used for the syntheses were reagent grade and used without further purification. The Dy(tfa)3·2H2O, Dy(TTA)3·2H2O and Dy(dbm)3·2H2O were synthesized according to methods reported in the literature.10 The 8-hydroxyquinoline Schiff base ligand (HL) was prepared by already reported methods.11
Elemental analyses for C, H, and N were performed on a Perkin-Elmer 240 CHN elemental analyzer. IR spectra were recorded in the range of 400–4000 cm−1 with a Bruker TENOR 27 spectrophotometer using a KBr pellet. PXRD data were examined on a Rigaku Ultima IV instrument with Cu Kα radiation (λ = 1.54056 Å), with a scan speed of 5° min−1 in the range 2θ = 5–50°. The magnetic measurements were carried out with a Quantum Design MPMS-XL7 and a PPMS-9 ACMS magnetometer. The diamagnetic corrections for the complexes were estimated using Pascal's constants, and magnetic data were corrected for diamagnetic contributions of the sample holder.12
X-ray crystallography
Single crystal X-ray diffraction data of 1–3 were collected on a computer-controlled Rigaku Saturn CCD area detector diffractometer, equipped with confocal monochromatized Mo Kα radiation with a radiation wavelength of 0.71073 Å using the ω–ϕ scan technique. The structures were solved by direct methods and refined with a full-matrix least-squares technique based on F2 using the SHELXS-97 and SHELXL-97 programs.13 Anisotropic thermal parameters were assigned to all non-hydrogen atoms. Crystallographic data and structural refinement parameters are listed in Table 1. Selected bond lengths and angles of complexes 1–3 are given in Tables S1–S3,† respectively. Crystallographic data of 1–3 are in CIF files. CCDC (1060984 – 1, 1061000 – 2 and 1425837 – 3) contain the supplementary crystallographic data for this paper.
Table 1 Crystallographic data and structure refinement summary for 1–3
| Complex |
1
|
2
|
3
|
| Empirical formula |
C52H36Br2F12N4O10Dy2 |
C64H36Br2Dy2F12N4O10S4 |
C96H71Br2Dy2N6O10.5 |
|
M
r
|
1583.60 |
1849.91 |
1961.4 |
|
T (K) |
113(2) |
113(2) |
113(2) |
| Crystal system |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
C
2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
|
a/Å |
16.741(3) |
10.769(2) |
16.748(3) |
|
b/Å |
16.621(3) |
13.002(3) |
16.799(3) |
|
c/Å |
19.953(4) |
13.289(3) |
17.363(4) |
|
β/° |
99.27(3) |
72.58(3) |
70.83(3) |
|
V/Å3 |
5479.5(19) |
1663.0(6) |
4089.8(14) |
|
Z
|
4 |
1 |
2 |
|
D
calcd/g cm−3 |
1.927 |
1.859 |
1.593 |
|
μ/mm−1 |
4.266 |
3.651 |
2.856 |
|
θ/° |
1.74 to 27.88 |
1.67 to 25.02 |
1.37 to 25.02 |
|
F(000) |
3064 |
902 |
1950 |
| Reflections collected |
24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 534 534 |
13960 |
33645 |
| Unique reflns |
6514 |
5843 |
14239 |
|
R
int
|
0.0357 |
0.0730 |
0.0453 |
| GOF (F2) |
1.061 |
0.985 |
1.071 |
|
R
1, wR2 (I > 2σ(I)) |
0.0388, 0.0875 |
0.0553, 0.1058 |
0.0409, 0.0954 |
|
R
1, wR2 (all data) |
0.0462, 0.0917 |
0.0735, 0.1142 |
0.0553, 0.1024 |
Synthesis
Synthesis of 2-[[(4-bromophenyl)imino]methyl]-8-hydroxyquinoline (HL).
To a solution of 8-hydroxyquinoline-2-carboxaldehyde (20 mmol) in methanol was added a solution of 4-bromoaniline (20 mmol) in methanol. The reaction mixture was stirred at room temperature for 4 h. Then the resulting orange precipitate was washed with methanol and dried in vacuo to give the HL ligand as a deep yellow solid. Purification by column chromatography used 1/3 dichloromethane/petroleum ether (1% triethylamine). The yield of the yellow needle product was 5.1 g, 79.2%. Elemental analysis (%) calcd for C16H11N2OBr (322): C, 59.63; H, 3.42; N, 8.70. Found: C, 59.66; H, 3.38; N, 8.75. IR (cm−1): 3400(m), 1621(m), 1566(s), 1510(s), 1317(m), 1226(m), 1197(m), 1133(w), 1068(m), 995(m), 848(s), 765(m), 608(s), 450(w).
Synthesis of [Dy2(tfa)4L2] (1).
A solution of Dy(tfa)3·2H2O (0.016 g, 0.025 mmol) in 20 mL boiling n-heptane was heated to reflux for 3 h. Then the solution was cooled to 70 °C, and a CH2Cl2 (5 mL) solution of HL (0.0082 g, 0.025 mmol) was added. The resulting mixture was stirred for 30 min at this temperature, and then cooled to room temperature. The mixture was filtered and the filtrate was kept in the dark and concentrated slowly by evaporation at 4 °C. After 7 days, block-shaped deep red-colored crystals were obtained. Yield 43% based on Dy. Elemental analysis (%) calcd for C52H36Br2F12N4O10Dy2 (1583.60): C, 39.44; H, 1.91; N, 3.54. Found: C, 39.37; H, 1.85; N, 3.59. IR (cm−1): 3418(s), 1631(s), 1594(w), 1527(m), 1453(s), 1365(w), 1288(s), 1229(s), 1018(m), 1011(w), 907(w), 842(m), 760(m), 562(m), 502(w).
Synthesis of [Dy2(TTA)4L2] (2).
The preparation of 2 was similar to that of 1, except that Dy(TTA)3·2H2O (0.0215 g, 0.025 mmol) replaced Dy(tfa)3·2H2O. Yield 38% based on Dy. Elemental analysis (%) calcd for C64H36Br2Dy2F12N4O10S4: C, 41.55; H, 1.31; N, 3.03. Found: C, 41.48; H, 1.35; N, 2.95%. IR (cm−1): 3419(m), 1632(s), 1590(w), 1460(s), 1368(w), 1289(s), 1260(w), 1227(s), 1015(w), 908(w), 793(w), 762(m), 563(m).
Synthesis of [Dy2(dbm)4L2·2CH3CN·0.5H2O] (3).
To a solution of Dy(dbm)3·2H2O (0.022 g, 0.025 mmol) in 20 mL methanol was added 5 mL dichloromethane containing 0.025 mmol (0.0082 g) of HL. The resulting mixture was stirred for about 3 h at room temperature and then the mixture was filtered and the filtrate was kept in the dark and concentrated slowly by evaporation at room temperature. After 10 days, block-shaped deep red-colored crystals were obtained. Elemental analysis (%) calcd for C96H71Br2Dy2N6O10.5: C, 64.04; H, 4.14; N, 3.11. Found: C, 64.10; H, 4.09; N, 3.18. IR (cm−1): 3420(m), 1596(s), 1460(s), 1367(w), 1291(s), 1262(w), 1170(m), 1014(w), 921(m), 792(w), 769(w), 561(m).
Results and discussion
The structures of 1–3
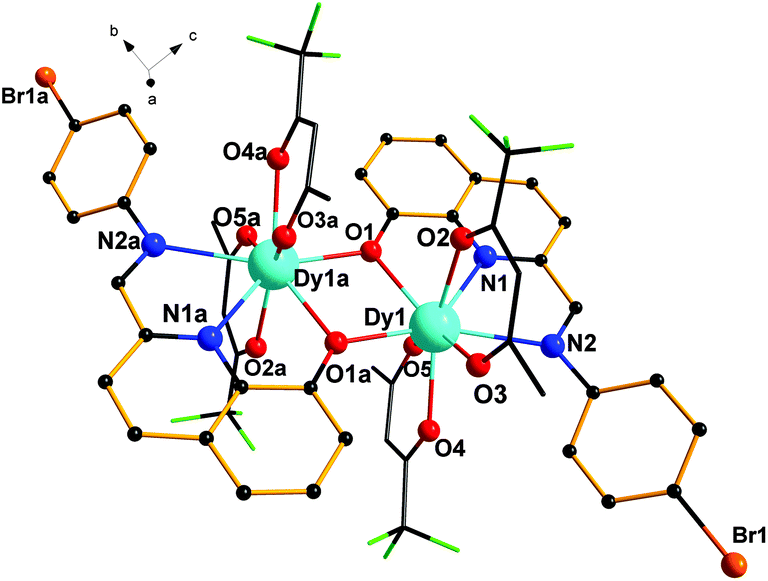
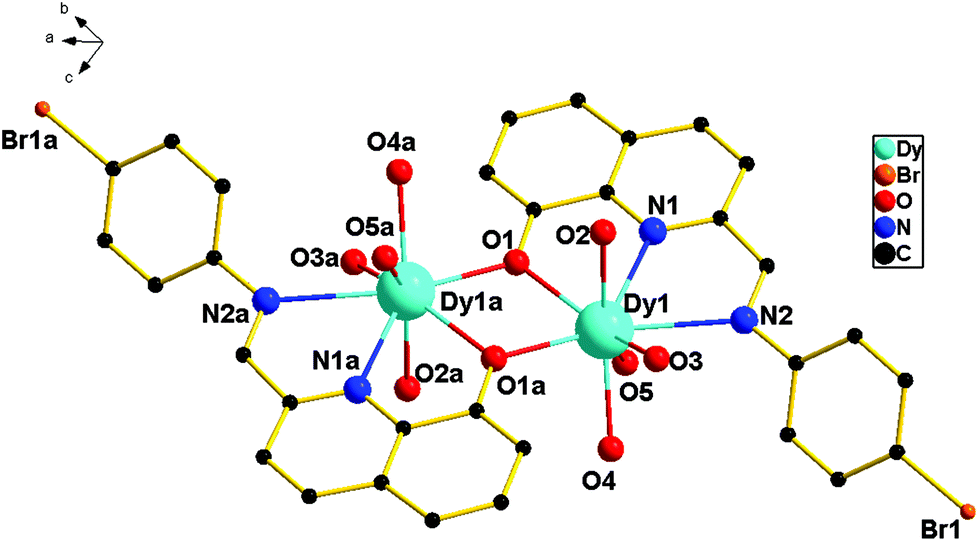
Single-crystal X-ray diffraction analyses reveal that complexes 1–3 are dinuclear complexes with very similar structures. As shown in Fig. 1, for complex 1, each molecule contains two eight-coordinated DyIII cations, two ligands (HL) and four tfa−. Each ligand serves as a tridentate ligand and chelates the Dy atom through one phenoxide oxygen atom (O1), one pyridyl ring nitrogen atom (N1) and one imine nitrogen atom (N2). Dy1 and Dy1a are bridged by two phenoxido oxygen atoms (O1 and O1a) of two ligands, leading to a four-membered Dy2O2 core with a Dy–O–Dy angle of 108.72(1)° and a Dy–Dy distance of 3.819(7) Å. The lengths of the Dy–O bonds are in the range of 2.306(3)–2.378(3) Å and the Dy–N bond distances are 2.456(4) and 2.666(3) Å. These bond distances are comparable to those reported in other phenoxo-bridged lanthanide complexes.14 Moreover, for 1, a dihedral angle of 36.89° was observed between the planes of the bromophenyl ring and the quinoline ring.
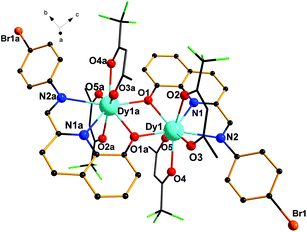 |
| | Fig. 1 Molecular structure of 1 (all hydrogen atoms are omitted for clarity). | |
Compared with 1, complex 2 is composed of two [Dy(TTA)2L] units (Fig. 2). The structures of 1 and 2 are very similar, but the subtle structural difference is in the different β-diketonate coligands. For 2, the lengths of the Dy–O bonds are in the range of 2.311(4)–2.414(4) Å and the Dy–N bond distances are 2.470(6) and 2.657(5) Å, respectively. The Dy⋯Dy distance and Dy–O–Dy angles are 3.839(2) Å and 108.68(2)°, respectively, which are slight larger than those of 1. Furthermore, the dihedral angle between the planes of the bromophenyl ring and the quinoline ring is 44.18° in 2, which is also larger than that of 1 (36.89°). These subtle but crucial structural differences of the two Dy2 complexes are mainly due to the different β-diketonate (tfa and TTA) and the introduction of the electron-donating group thiophene ring to replace the CH3 group in 2, which causes significant changes in the subtle structure of 1 and 2.
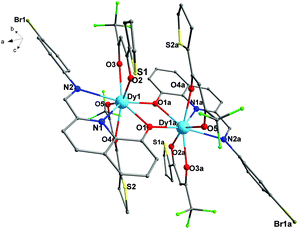 |
| | Fig. 2 Molecular structure of 2 (all hydrogen atoms are omitted for clarity). | |
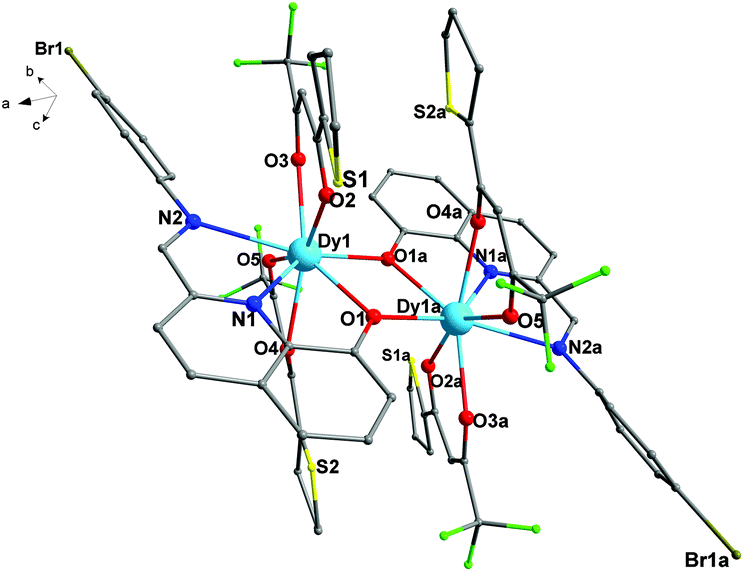
For 3, it is crystallized in the triclinic space group P. As shown in Fig. 3, the asymmetric unit is composed of two crystallographically independent [Dy2(dbm)4L2] moieties, four free CH3CN solvents and one H2O molecule. Each moiety contains two eight-coordinated DyIII cations, four dbm− and two ligands (HL). The coordination and bridging models of central DyIII ions are similar to 1 and 2. The slight difference among 1–3 is in the Dy–O–Dy bond angles and the Dy–Dy distance. The Dy1–O1–Dy1 angle of 3 is 109.93(1)° and the Dy1–Dy1a distance is 3.8836(9) Å, which are a bit larger than those of 1 and 2. The lengths of the Dy–O bonds are in the range of 2.293(3)–2.427(3) Å and the Dy–N bond distances are in the range of 2.483(4)–2.694(4) Å for 3. These bond distances are also comparable to those of already reported phenoxo-bridged lanthanide complexes.14 In addition, the dihedral angle between the planes of the bromophenyl ring and the quinoline ring of 3 is also different from 1 and 2. The dihedral angle of 3 is 28.38°, which is smaller than that of both 1 (36.89°) and 2 (44.18°). Moreover, the planes defined by four oxygen atoms of two β-diketonate coligands (O2, O3, O4, and O5) in 2 and in the Dy2 molecule of complex 3 are almost flat, but for 1, the planes defined by four oxygen atoms of two β-diketonate coligands (O2, O3, O4, and O5) are slight sloping, which are mainly caused by the electron-withdrawing groups (–CF3) in 1. Compared with 1, the introduction of the electron-donating group thiophene or benzene ring in 2 and 3 to replace the –CH3 group in 1 can greatly affect the tilt of planes which are defined by four oxygen atoms of two β-diketonate coligands (O2, O3, O4, and O5). Beyond the above, in the Dy2 molecule of complex 3, significant p–p interaction can be observed, which may be attributed to the origin of the magnetic interaction in complex 3.
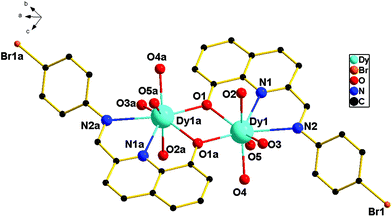 |
| | Fig. 3 The asymmetric unit for 3 (all hydrogen atoms, carbon atoms of dbm− and solvent molecule are omitted for clarity). | |
Powder X-ray diffraction
The crystalline products of 1–3 have been characterized by X-ray powder diffraction (PXRD) at room temperature (Fig. S1, ESI†). The observed XRD patterns are in good agreement with the results simulated from the single crystal data, indicating the high purity of the synthesized samples. The differences in intensity may be due to the preferred orientation of the crystalline powder samples.
Magnetic properties
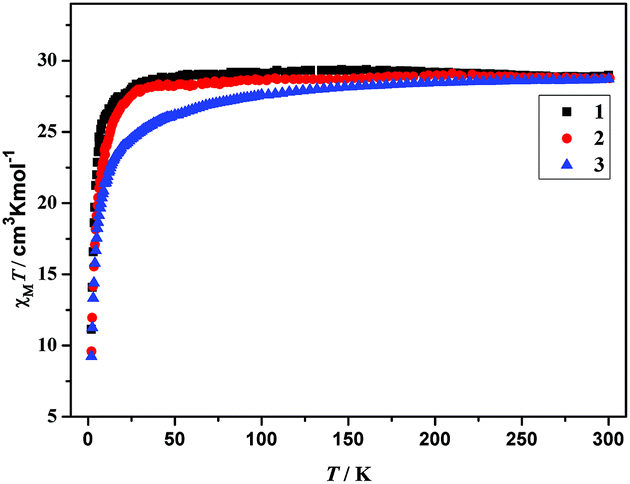
The plot of χMT versus T, where χM is the molar magnetic susceptibility, is shown in Fig. 4. For complexes 1–3, χMT values at 300 K are 28.97 (1), 28.76 (2) and 28.73 (3) cm3 K mol−1, respectively, which are slightly larger than the expected value for two isolated DyIII (6H5/2, g = 2/7), 28.34 cm3 K mol−1. Upon cooling, the χMT values of 1 and 2 remain almost unchanged between 300 and 20 K. As the temperature further decreases, a sudden decrease of χMT in 1 and 2 is observed, to reach a minimum value of 11.13 cm3 K mol−1 for 1 and 9.58 cm3 K mol−1 for 2 at 2 K. The unchanged χMT values of 1 and 2 between 300 and 20 K indicate that weak ferromagnetic interaction must operate in 1 and 2, which compensates for the drop in the χMT product caused by the depopulation of the stark sublevels of Dy3+.15a,b For 3, χMT values decrease slowly from 300 to 50 K, and then decrease distinctly to reach the minimum of 9.24 cm3 K mol−1 at 2 K. Such a behavior can be generally attributed to one or a combination of the following two phenomena: (1) the progressive depopulation of the excited stark sub-levels of the DyIII ions; (2) weak antiferromagnetic interactions between the LnIII ions within the complex.15c–g
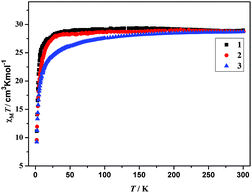 |
| | Fig. 4 Plots of χMT versus T for complexes 1–3. | |
The variation of the magnetization M with the applied magnetic field H was investigated for 1–3 in the range of 0–80 kOe at 2.0 K (Fig. S2†). M increases rapidly at low field and then grows slowly without complete saturation up to 80 kOe. The magnetization values of 1 (12.9Nβ), 2 (9.9Nβ) and 3 (11.5Nβ) at 80 kOe are much lower than the theoretical saturated value of 20Nβ anticipated for two independent Dy3+ ions, which does not reach the saturation values, indicating the presence of magnetic anisotropy and/or low-lying excited states in the system, which corresponds to the reported results.16
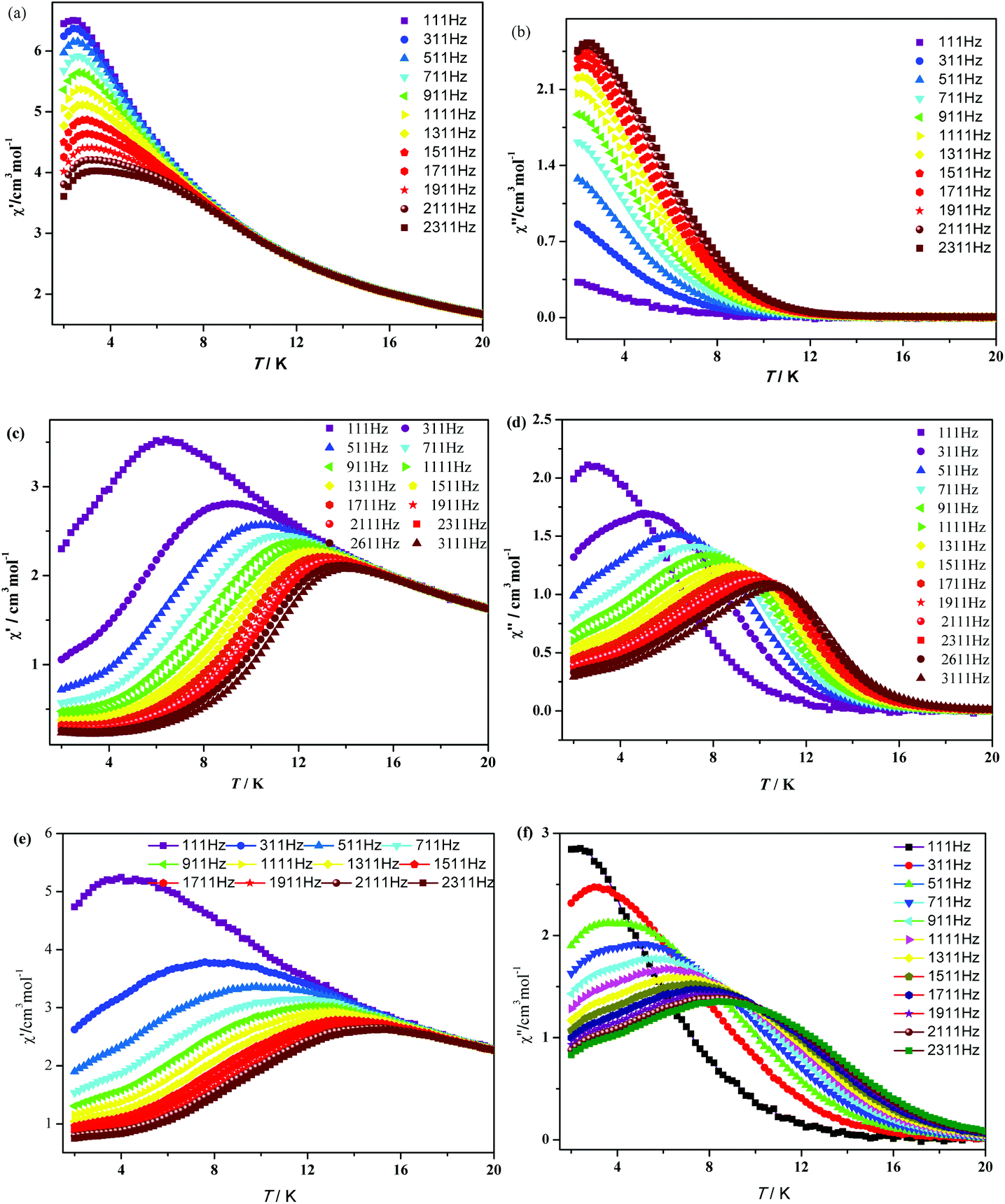
In order to investigate the dynamics of the magnetization of 1–3, the temperature dependencies of the alternating-current (ac) susceptibility were measured at 2–20 K under zero dc field with an ac field of 3.0 Oe with oscillating frequencies. As shown in Fig. 5, complexes 1–3 show strong frequency-dependent in-phase (χ′) and out-of-phase (χ′′) signals and good peak shapes are also observed, indicating the presence of slow magnetic relaxation at lower temperature, typical of SMM behavior. For 1, χ′ shows a maximum value which starts to decrease in the 2–4 K range, while the peaks of χ′′ can only be found at frequencies higher than 1111 Hz. Upon cooling, increases of χ′′ are clearly observed in the 111–1111 Hz range, indicating that blocking is not complete and thermally activated spin reversal is gradually replaced by a quantum tunneling mechanism at zero dc field.17 For 2 and 3, χ′′ defines a maximum between 2 K (111 Hz) and 11 K (2311 Hz). Unlike 1, both χ′ and χ′′ components for 2 and 3 cascade like avalanches below the blocking temperature and nearly vanish as the temperature approaches 2 K. This signals the “freezing” of the spins by the anisotropy barriers and can be taken as a clear indication of the efficient suppression of zero-field tunneling of magnetization occurring in 2 and 3.
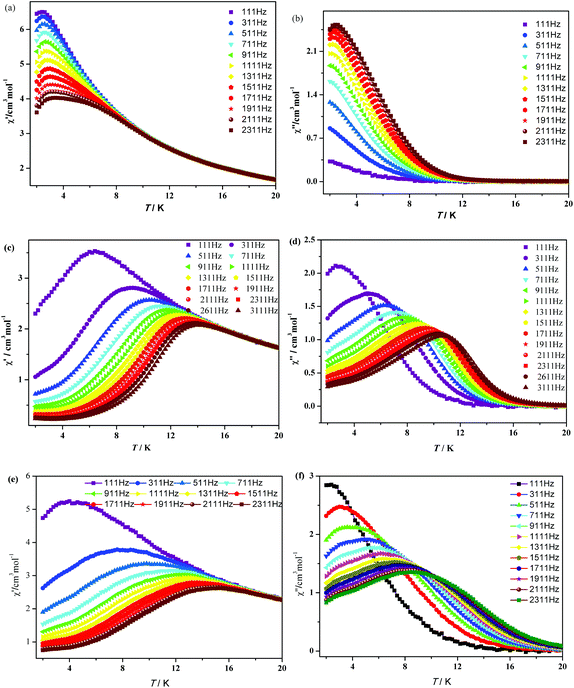 |
| | Fig. 5 Temperature dependence of the in-phase (χ′) and out-of-phase (χ′′) components of the ac magnetic susceptibility for 1 (a and b), 2 (c and d) and 3 (e and f) under zero dc field with an oscillation of 3.0 Oe. | |
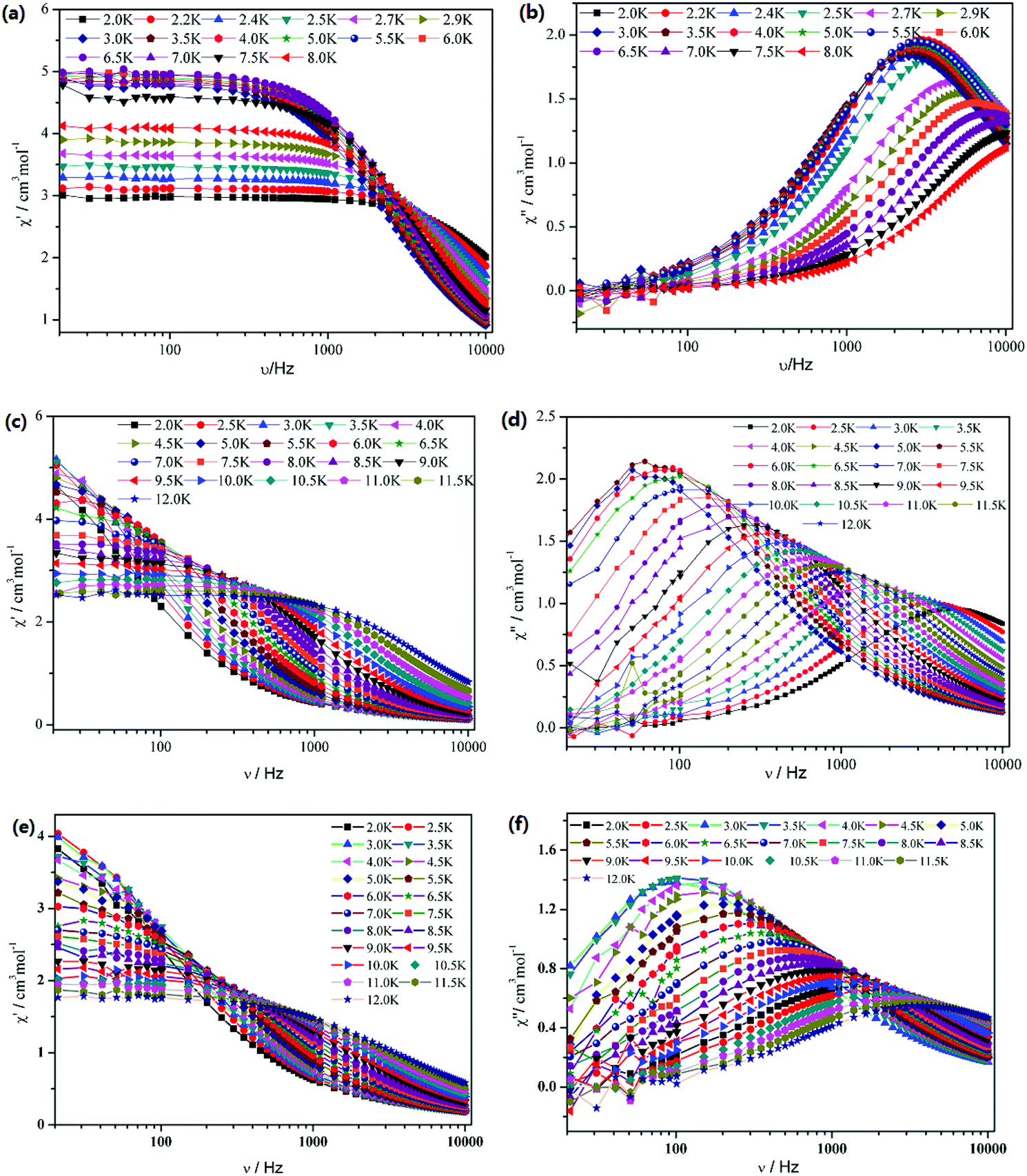
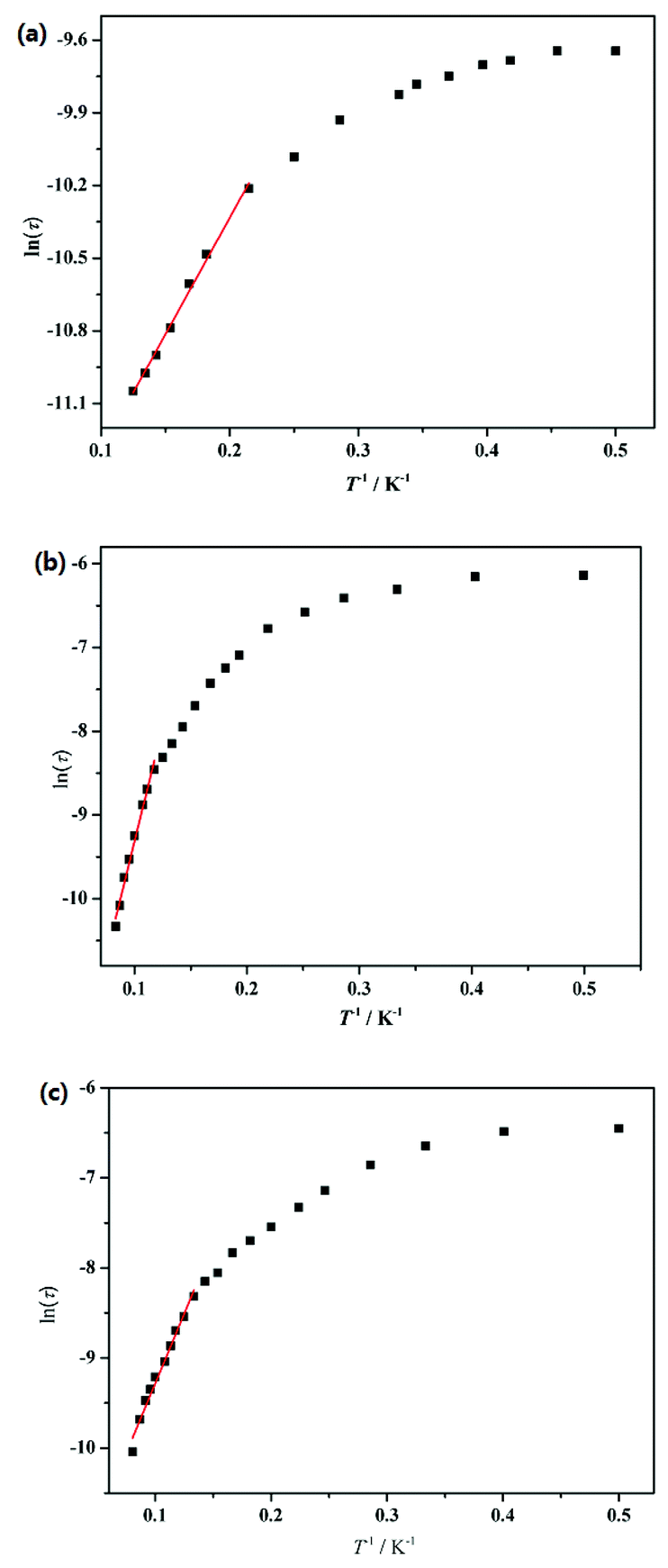
To further understand the dynamics of the magnetization of 1–3, the frequency dependencies of the alternating-current (ac) susceptibility were measured under zero dc fields. From frequency dependencies of ac susceptibility (Fig. 6), both the in-phase (χ′) and out-of-phase (χ′′) signals of 1–3 show temperature dependencies, which also reveal the presence of slow relaxation of the magnetization in 1–3. The relaxation time τ data of complexes 1–3 derived from the χ′′ peaks follow the Arrhenius law [τ = τ0exp(ΔE/kBT)] with the pre-exponential factor τ0 = 4.76 × 10−6 s and the effective barrier ΔE/kB = 9.61 K for 1, τ0 = 3.74 × 10−7 s and ΔE/kB = 54.8 K for 2 and τ0 = 4.20 × 10−6 s and ΔE/kB = 31.0 K for 3 (Fig. 7). The obtained anisotropic energy barrier is comparable to those previously reported for Dy-based compounds.18 It is worth noting that lnτ of 1–3 become weakly dependent on T as the temperature decreases. This behavior characterizes a crossover from a thermally activated Orbach mechanism that is predominant at high temperature to a direct or phonon-induced tunneling.17,19
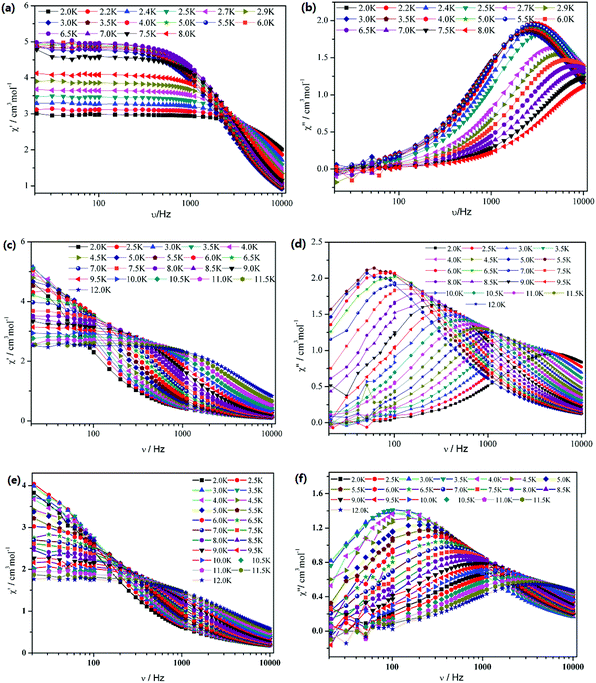 |
| | Fig. 6 Frequency dependence of the in-phase (χ′) and out-of-phase (χ′′) ac susceptibility for 1 (a and b), 2 (c and d) and 3 (e and f) under zero dc field. | |
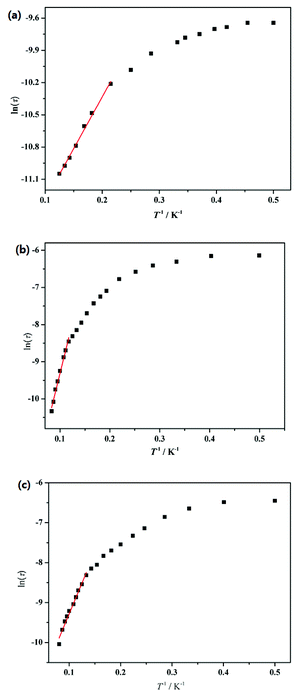 |
| | Fig. 7 Plots of ln(τ) versus T−1 fitting to the Arrhenius law for complexes 1 (a), 2 (b) and 3 (c). | |
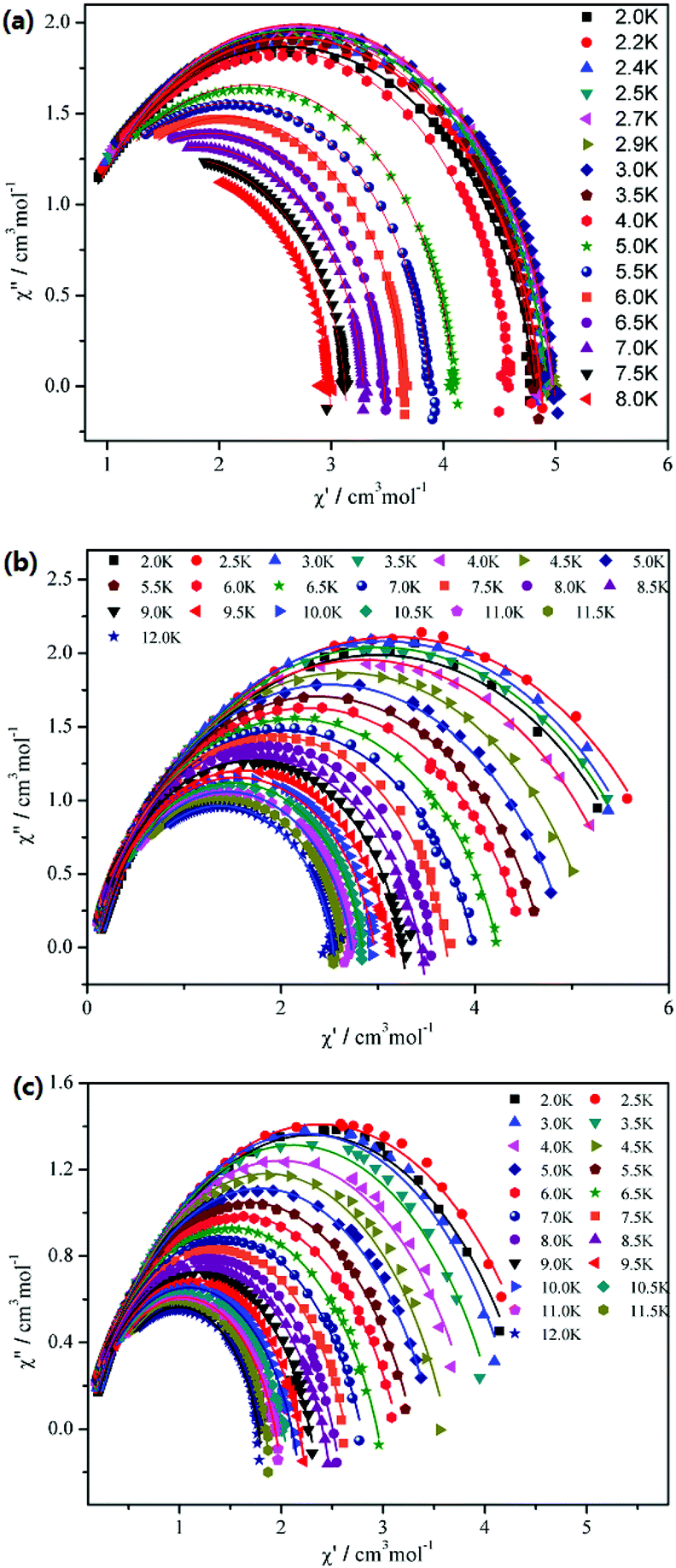
The Cole–Cole plots of compounds 1–3 are shown in Fig. 8. For 1, it shows an asymmetric shape, and the best fits of the out-of-phase (χ′′) vs. in-phase (χ′) susceptibility in the range of 2–8 K were obtained with α = 0.04–0.11 using the generalized Debye model (α = 0 corresponding to an infinitely narrow distribution of relaxation times).20 This result indicates that a single relaxation time is mainly involved in the present relaxation process independently of the temperature. However, for 2 and 3, the data plotted as Cole–Cole plots show a relatively symmetrical shape and can be fitted to the generalized Debye model, with α = 0.11–0.23 for 2 and α = 0.20–0.29 for 3, implying that thermally activated relaxation processes of both 2 and 3 have a wider distribution of relaxation time compared with 1.21
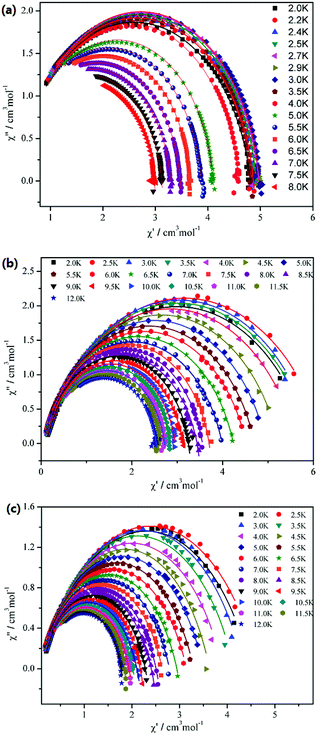 |
| | Fig. 8 Cole–Cole plots for 1 (a), 2 (b) and 3 (c) measured in zero dc field. The red solid lines are the best fit to the experimental data, obtained with the generalized Debye model with α = 0.04–0.11 for 1, α = 0.11–0.23 for 2 and α = 0.20–0.29 for 3. | |
Although complexes 1–3 show a very similar crystal structure, they exhibit different magnetic relaxation behaviors. Indeed, for 1, the χ′′ peaks only account for a component of the total susceptibility under zero field, while 2 and 3 maintain the slow relaxation of magnetization as the parent type. In addition, the energy barriers (ΔE/kB) of 2 and 3 are 54.8 K and 31.0 K, respectively, which are much higher than that of 1 (ΔE/kB = 9.61 K). These distinctive magnetic behaviors must be caused by subtle but crucial differences among the respective structures. According to the single crystal X-ray diffraction study and the semiquantitative method of polytopal analysis, for complexes 1–3, the most reasonable geometry around the Dy atom is dodecahedral (DD). However, on a closer comparison of the three Dy2 structures, the Dy⋯Dy distances and the Dy–O–Dy angles are found to be slightly different. The Dy1–O1–Dy1 angle of 3 is 109.93(1)° and the Dy1–Dy1a distance is 3.8836(9) Å, which are a bit larger than those of 1 (a Dy–O–Dy angle of 108.72(1)° and a Dy–Dy distance of 3.819(7) Å) and 2 (the Dy⋯Dy distance and Dy–O–Dy angles are 3.839(2) Å and 108.68(2)°, respectively). In addition, the dihedral angles between the planes of the bromophenyl ring and the quinoline ring of 1–3 are also discrepant. These slight differences of important bond lengths, angles and the dihedral angle may have an influence on the distorted degree of the coordination geometry of the Dy(III) ion, which can lead to the fast quantum tunneling arising from a transverse anisotropy introduced by a geometrical distortion of the coordinated sphere. These differences can also have a significant influence on the magnetic interaction between the Dy3+ ions. Hence, the slightly different chemical environment of central Dy3+ ions of the three complexes can result in different magnetic behaviors. It is evident that replacement of the CH3 group by an electron-donating group thiophene ring results in a significant change in magnetic relaxation of 2, and the introduction of the electron-donating group thiophene ring or benzene ring to replace the CH3 group in 2 and 3 causes great changes in the dynamic magnetic behavior. It seems that the electron-donating group thiophene ring or benzene ring in 2 and 3 can help to improve the energy barrier (ΔE/kB). This demonstrates that the SMM behavior of lanthanide complexes with β-diketonate as coligands can be finely tuned by changing the substituent group of the β-diketonate ligands.
Conclusions
In summary, the structures and magnetic properties of three dinuclear Dy(III) complexes based on 8-hydroxyquinoline Schiff base ligand have been reported. Complexes 1–3 are phenoxo-O bridged binuclear complexes, each central DyIII ion is eight-coordinated with two bidentate β-diketonate ligands and two μ2-O bridging two 8-hydroxyquinoline Schiff base ligands. The magnetic studies reveal that 1–3 exhibit different magnetic relaxation behaviors with the anisotropic barriers of 9.61 K (1), 54.8 K (2) and 31.0 K (3), respectively. The different magnetic relaxation behaviors of the three Dy2 complexes originated from the slightly different chemical environment around the Dy(III) ions. This demonstrates that the SMM behavior of lanthanide complexes can be finely adjusted by changing the substituent of the β-diketonate ligand.
Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (no. 21271137, 21571138 and 21561014).
Notes and references
-
(a) C. L. Hill and X. Zhang, Nature, 1995, 373, 324 CrossRef CAS;
(b) S. Hill, R. S. Edwards, N. Aliaga-Alcalde and G. Christou, Science, 2003, 302, 1015 CrossRef CAS PubMed;
(c) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS PubMed.
-
(a) H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571 CrossRef CAS;
(b) G. Li, L. Li, S. Feng, M. Wang and X. Yao, Adv. Mater., 1999, 11, 146 CrossRef CAS;
(c) K. S. Min and M. P. J. Suh, Solid State Chem., 2000, 152, 183 CrossRef CAS.
-
(a)
D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 Search PubMed;
(b) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed;
(c) F. N. Shi, L. Cunha-Silva, R. A. S. Ferreira, L. Mafra, T. Trindade, L. D. Carlos, F. A. A. Paz and J. Rocha, J. Am. Chem. Soc., 2008, 130, 150 CrossRef CAS PubMed;
(d) Y. N. Guo, G. F. Xu, W. Wernsdorfer, L. Ungur, Y. Guo, J. K. Tang, H. J. Zhang, L. F. Chibotaru and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 11948 CrossRef CAS PubMed.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS.
-
(a) H. Oshio, N. Hoshino, T. Ito and M. Nakano, J. Am. Chem. Soc., 2004, 126, 8805 CrossRef CAS PubMed;
(b) C. Boskovic, W. Wernsdorfer, K. Folting, J. C. Huffman, D. N. Hendrickson and G. Christou, Inorg. Chem., 2002, 41, 5107 CrossRef CAS PubMed;
(c) M. Murugesu, M. Habrych, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2004, 126, 4766 CrossRef CAS PubMed;
(d) A. K. Boudalis, M. Pissas, C. P. Raptopoulou, V. Psycharis, B. Abarca and R. Ballesteros, Inorg. Chem., 2008, 47, 10674 CrossRef CAS PubMed;
(e) D. Y. Wu, D. Guo, Y. Song, W. Huang, C. Y. Duan, Q. J. Meng and O. Sato, Inorg. Chem., 2009, 48, 854 CrossRef CAS PubMed;
(f) M. H. Zeng, M. X. Yao, H. Liang, W. X. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2007, 46, 1832 CrossRef CAS PubMed;
(g) M. A. Aidamen, J. M. Clemente-Juan, E. Coronado, C. Marti-Gastaldo and A. Gaita-Arino, J. Am. Chem. Soc., 2008, 130, 8874 CrossRef PubMed;
(h) P. H. Lin, T. J. Burchell, L. Ungur, L. F. Chibotaru, W. Wernsdorfer and M. Murugesu, Angew. Chem., Int. Ed., 2009, 48, 9489 CrossRef CAS PubMed;
(i) Y. N. Guo, G. F. Xu, P. Gamez, L. Zhao, S. Y. Lin, R. P. Deng, J. Tang and H. J. Zhang, J. Am. Chem. Soc., 2010, 132, 8538 CrossRef CAS PubMed;
(j) Y. Z. Zheng, Y. Lan, C. E. Anson and A. K. Powell, Inorg. Chem., 2008, 47, 10813 CrossRef CAS PubMed.
-
(a) I. J. Hewitt, J. Tang, N. T. Madhu, C. E. Anson, Y. Lan, J. Luzon, M. Etienne, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2010, 49, 6352 CrossRef CAS PubMed;
(b) R. J. Blagg, C. A. Muryn, E. J. L. McInnes, F. Tuna and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530 CrossRef CAS PubMed;
(c) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538 CrossRef CAS PubMed;
(d) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236 CrossRef CAS PubMed;
(e) J. W. Sharples, Y. Z. Zheng, F. Tuna, E. J. L. McInnes and D. Collison, Chem. Commun., 2011, 47, 7650 RSC.
-
(a) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed;
(b) Y. Z. Zheng, G. J. Zhou, Z. P. Zheng and R. E. P. Winpenny, Chem. Soc. Rev., 2014, 43, 1462 RSC;
(c) R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328 CrossRef CAS;
(d) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092 RSC.
-
(a) Y. Bi, Y. N. Guo, L. Zhao, Y. Guo, S. Y. Lin, S. D. Jiang, J. K. Tang, B. W. Wang and S. Gao, Chem. – Eur. J., 2011, 17, 12476 CrossRef CAS PubMed;
(b) G. J. Chen, Y. N. Guo, J. L. Tian, J. K. Tang, W. Gu, X. Liu, S. P. Yan, P. Cheng and D. Z. Liao, Chem. – Eur. J., 2012, 18, 2484 CrossRef CAS PubMed.
-
(a) P. Zhang, L. Zhang, C. Wang, S. F. Xue, S. Y. Lin and J. K. Tang, J. Am. Chem. Soc., 2014, 136, 4484 CrossRef CAS PubMed;
(b) P. Zhang, L. Zhang and J. K. Tang, Dalton Trans., 2015, 44, 3923 RSC;
(c) P. Zhang, Y. N. Guo and J. K. Tang, Coord. Chem. Rev., 2013, 257, 1728 CrossRef CAS;
(d) L. Ungur, S. Yan. Lin, J. K. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894 RSC.
-
(a) L. R. Melbyn, J. Rose, E. Abramson and J. C. Caris, J. Am. Chem. Soc., 1964, 86, 5117 CrossRef;
(b) S. Katagiri, Y. Tsukahara, Y. Hasegawa and Y. Wada, Bull. Chem. Soc. Jpn., 2007, 80, 1492 CrossRef CAS.
- C. Caris, P. Baret, J. L. Pierre and G. Serratrice, Tetrahedron, 1996, 52, 4659 CrossRef CAS.
-
(a)
Theory and Applications of Molecular Paramagnetism, ed. E. A. Boudreaux and L. N. Mulay, Wiley-Interscience, New York, 1976 Search PubMed;
(b) G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
-
(a)
G. M. Sheldrick, SHELXS-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(b)
G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
-
(a) S. Liu, L. Gelmini, S. J. Rettig, R. C. Thompson and C. Orvig, J. Am. Chem. Soc., 1992, 114, 6081 CrossRef CAS;
(b) L. E. Roy and T. Hughbanks, J. Am. Chem. Soc., 2006, 128, 568 CrossRef CAS PubMed;
(c) J. Long, F. Habib, P.-H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319 CrossRef CAS PubMed.
-
(a) Y. L. Hou, G. Xiong, B. Shen, B. Zhao, Z. Chen and J. Z. Cui, Dalton Trans., 2013, 42, 3587 RSC;
(b) M. Fang, J. J. Li, P. F. Shi, B. Zhao and P. Cheng, Dalton Trans., 2013, 42, 6553 RSC;
(c) I. Nemec, M. Machata, R. Herchel, R. Bočab and Z. Trávníček, Dalton Trans., 2012, 41, 14603 RSC;
(d) Y. G. Huang, F. J. Jiang, D. Q. Yuan, M. Y. Wu, Q. Gao, W. Wei and M. C. Hong, Cryst. Growth Des., 2008, 8, 166 CrossRef CAS;
(e) H. L. Gao, Y. L. Yi, Y. M. Hu, J. Qu, C. C. Hu, R. Wufu, J. Z. Cui and B. Zhai, CrystEngComm, 2012, 14, 7965 RSC;
(f) Y. G. Huang, X. T. Wang, F. J. Jiang, S. Gao, M. Y. Wu, Q. Gao, W. Wei and M. C. Hong, Chem. – Eur. J., 2008, 14, 10340 CrossRef CAS PubMed;
(g) W. T. Xu, Y. F. Zhou, D. C. Huang, W. Xiong, M. Y. Su, K. Wang, S. Han and M. C. Hong, Cryst. Growth Des., 2013, 13, 5420 CrossRef CAS.
-
(a) K. Bernot, J. Luzon, L. Bogani, M. Etienne, C. Sangregorio, M. Shanmugam, A. Caneschi, R. Sessoli and D. Gatteschi, J. Am. Chem. Soc., 2009, 131, 5573 CrossRef CAS PubMed;
(b) S. Kanegawa, M. Maeyama, M. Nakano and N. Koga, J. Am. Chem. Soc., 2008, 130, 3079 CrossRef CAS PubMed;
(c) P. H. Lin, T. J. Burchell, R. Cĺerac and M. M. Murugesu, Angew. Chem., Int. Ed., 2008, 47, 884 Search PubMed;
(d) P. Hu, M. Zhu, X. L. Mei, H. X. Tian, Y. Ma, L. C. Li and D. Z. Liao, Dalton Trans., 2012, 41, 14651 RSC.
- Y. N. Guo, X. H. Chen, S. F. Xue and J. K. Tang, Inorg. Chem., 2011, 50, 9705 CrossRef CAS PubMed.
-
(a) S. Bala, M. S. Bishwas, B. Pramanik, S. Khanra, K. M. Fromm, P. Poddar and R. Mondal, Inorg. Chem., 2015, 54, 8197 CrossRef CAS PubMed;
(b) G. Xiong, X. Y. Qin, P. F. Shi, Y. L. Hou, J. Z. Cui and B. Zhao, Chem. Commun., 2014, 50, 4255 RSC;
(c) M. Fang, H. H. Zhao, A. V. Prosvirin, D. Pinkowicz, B. Zhao, P. Cheng, W. Wernsdorfer, E. K. Brechin and K. R. Dunbar, Dalton Trans., 2013, 42, 14693 RSC;
(d) Y. L. Hou, G. Xiong, P. F. Shi, R. R. Cheng, J. Z. Cui and B. Zhao, Chem. Commun., 2013, 49, 6066 RSC;
(e) Z. L. Wu, J. Dong, W. Y. Ni, B. W. Zhang, J. Z. Cui and B. Zhao, Dalton Trans., 2014, 43, 16838 RSC.
- M. Gonidec, F. Luis, A. Vílchez, J. Esquena, D. B. Amabilino and J. Veciana, Angew. Chem., Int. Ed., 2010, 49, 1623 CrossRef CAS PubMed.
-
(a) K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341 CrossRef CAS;
(b) S. M. J. Aubin, Z. Sun, L. Pardi, J. Krzystek, K. Folting, L. C. Brunel, A. L. Rheingold, G. Christou and D. N. Hendrickson, Inorg. Chem., 1999, 38, 5329 CrossRef CAS.
-
(a) S. Xue, L. Zhao, Y. Guo, P. Zhang and J. Tang, Chem. Commun., 2012, 48, 8946 RSC;
(b) H. Wang, T. Liu, K. Wang, C. Duan and J. Jiang, Chem. – Eur. J., 2012, 18, 7691 CrossRef CAS PubMed;
(c) Y. X. Ren, X. J. Zheng, L. C. Li, D. Q. Yuan, M. An and L. P. Jin, Inorg. Chem., 2014, 53, 12234 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.