
Magnetization dynamics of a heterometallic Dy-isocarbonyl complex†
Courtney M.
Dickie
and
Michael
Nippe
*
Department of Chemistry, Texas A&M University, 3255 TAMU, College Station, TX 77843, USA. E-mail: nippe@chem.tamu.edu
First published on 7th December 2015
Abstract
The reaction of Dy(N(Si(CH3)3)2)3 with three equivalents of HW(CO)3Cp (Cp = cyclopentadienyl) furnished the first structurally characterized isocarbonyl complex of Dy3+, [{CpW(CO)2(μ-CO)}3Dy(thf)5] (1). Solid dilution via cocrystallization with the isostrucutral Y3+ analogue of 1 allowed for the observation of temperature dependent signals in the out-of-phase component of the ac magnetic susceptibility in the presence of an applied dc field. An effective barrier to magnetization reversal of 12.6 cm−1 could be extracted for compound 1 under these conditions.
 Michael Nippe | Michael Nippe was born and raised in Berlin, Germany. He received his Dipl.-Chem. in 2007 from TU-Berlin after conducting research under Andreas Grohmann and Karl Wieghardt (Max-Planck Institute for Bioinorganic Chemistry). His graduate studies at UW-Madison with John Berry focused on studying the influences of metal–metal interactions on reactivity and spectroscopy. In 2011, Michael moved as a postdoctoral researcher to Berkeley where he worked at UC-Berkeley and LBNL with Jeffrey Long, Christopher Chang, and Kenneth Raymond as part of the Joint Center for Artificial Photosynthesis and the LBNL Chemical Catalysis Program. During this time he investigated molecular electrocatalysts, molecular magnets, and supramolecular cage structures. In 2014, Michael started his independent research group at Texas A&M University. Current major research areas of the group include magnetization dynamics of heterometallic lanthanide complexes, molecular strategies towards improved activation and conversion of energy-relevant small molecules, and supramolecular approaches towards drug delivery and imaging. |
Introduction
Paramagnetic molecules that exhibit bistable high spin ground states and magnetic anisotropy can display energy barriers to reorientation of their molecular spins which can lead to slow magnetic relaxation dynamics and magnetic hysteresis.1 Such molecules are commonly referred to as single-molecule magnets or SMMs and are being investigated for their potential applications for high density data storage. Since the discovery of slow magnetic relaxation in multinuclear transition metal complexes2 research has focused on the development of mono-3 and multi-nuclear4 transition metal complexes. The large single ion anisotropy of lanthanide ions renders them attractive candidates in this regard and has led to the development of single-ion magnets with improved barriers to spin reorientation (Ueff).5 The first lanthanide-based SMMs were lanthanide phthalocyanine sandwich complexes [LnPc2]− (Ln = Tb, Dy) and a high Ueff of 230 cm−1 (331 K) was reported for diluted samples [Bu4N][Tb0.2Y0.8Pc2].6 A heteroleptic neutral [Tb3+(Pc)(Pc′)] complex (Pc′ = octa(tertbutylphenoxy)-substituted Pc ring) was shown to have an even higher Ueff of 652 cm−1 (938 K), currently a record in the field.7 Diluted yttrium-doped polymetallic Dy and Tb poly–lanthanide alkoxide cage complexes, Ln@{Y4K2}, were found to have Ueff exceeding 556 cm−1 (800 K).8 Notably, the blocking of relaxation via the first excited state led to these complexes possessing the second highest relaxation barriers reported and the slow magnetic relaxation was found to be single-ion in origin.However, the plethora of lanthanide based SMMs suffer from quantum tunnelling of the magnetization (QTM) and modern approaches circumvent this by utilizing strong exchange coupling between lanthanide ions through radical bridges.9 Magnetic coupling of lanthanide ions to transition metal ions has also been widely utilized in order to combine magnetic anisotropy and high spin states of lanthanide and transition metal ions. Most of these 4f–3d complexes feature bridging oxygen based ligands.10 These studies are further complicated as subtle changes in the ligand field of the lanthanide ion influences the energies of low lying mJ states, thereby determining Ueff.5c,11
We have started a research program that aims at the discovery of new heterometallic lanthanide–transition metal complexes and their magnetic characterization. As part of this effort we utilize anionic metal-carbonyl fragments as ligands for lanthanide ions. These efforts are motivated by the degree of tunability of ligand field strength imposed by metal-carbonyl anions as a function of the nature of the transition metal employed.12 Furthermore, utilization of paramagnetic metal carbonyl anions13 as ligands would allow to study exchange coupling through carbonyl bridges.14 Additionally, Ln-isocarbonyl complexes are potential precursors to lanthanide–transition metal bonds via photolysis: Recently, Arnold and co-workers demonstrated the formation of an uranium–cobalt bond via photolytic CO elimination of an U-isocarbonyl compound.15
We chose to start our investigations by characterizing the magnetic properties of a Dy-isocarbonyl complex, since Dy3+ is a highly anisotropic Kramers ion and its ground state is bistable, independent of ligand field.
Herein, we report the synthesis of [{CpW(CO)2(μ-CO)}3Dy(thf)5] 1via amine elimination reactions between [Dy(N(SiMe3)2)3] and [HW(CO)3Cp]. Notably, this is the first report of a structurally characterized isocarbonyl complex of Dy.16 Temperature dependent slow magnetic relaxation could be observed in dilute samples of 1·thf (Y![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Dy 12:1), with a barrier height (Ueff) of 12.6 cm−1 (18.1 K) under a 400 Oe applied dc field.
:Dy 12:1), with a barrier height (Ueff) of 12.6 cm−1 (18.1 K) under a 400 Oe applied dc field.
Experimental section
General considerations
Unless otherwise noted, all reactions and manipulations were carried out under anaerobic and anhydrous conditions in an argon-filled glovebox (Vigor). Tetrahydrofuran (thf) and toluene were dried and deoxygenated using a solvent purification system (JC Meyer Solvent System) and stored over molecular sieves in an argon-filled glovebox. [Dy(N(SiMe3)2)3] and [Y(N(SiMe3)2)3] were prepared as previously described17 and recrystallized from toluene. HW(CO)3Cp was purchased from Sigma-Aldrich and used as received. Elemental analyses were carried out by ALS Environmental (Tucson, AZ). IR spectra were recorded on a Mattson ATI Genesis FT-IR spectrometer fitted with a PIKE Technologies MIRacle ATR sampling accessory.X-ray crystallography
Single crystal X-ray diffraction measurements were carried out using a Bruker-AXS APEX-II CCD area detector diffractometer (Mo Kα radiation, λ = 0.71069 Å) diffractometer (NSF-CHE-9807975, NSF-CHE-0079822 and NSF-CHE-0215838). Crystals were mounted on nylon loops and cooled in a nitrogen stream (110(2) K). Bruker AXS APEX II software was used for data collection and reduction.19 Absorption corrections were applied using SADABS.20 Space group assignments were determined by examination of systematic absences, E-statistics, and successive refinement of the structures. Structures were solved using direct methods and refined by least-squares refinement on F2 followed by difference Fourier synthesis.21 All hydrogen atoms were included in the final structure factor calculation at idealized positions and were allowed to ride on the neighbouring atoms with relative isotropic displacement coefficients. Thermal parameters were refined anisotropically for most non-hydrogen atoms. There is partial disorder of some of the carbon atoms of one of the Cp ligands. Crystallographic data for 1·thf is given in Table S1.† The structure of isostructural Y3+ analogue 2 was confirmed by unit cell determination to match the previously reported unit cell.18Magnetic measurements
Magnetic samples were prepared by adding crushed crystalline sample to a high-purity 7 mm NMR tube and covering it with a layer of eicosane. The NMR tube was flame sealed under vacuum and the eicosane was melted in a water bath at 35 °C, evenly dispersing it throughout the sample. The sample was loaded into a straw affixed to the end of the sample rod. Magnetic measurements were carried out using a Quantum Design MPMS XL SQUID magnetometer (NSF-9974899) (compound 1·thf) and a Quantum Design MPMS 3 SQUID magnetometer (TAMU Vice President of Research) (dilution sample). Dc measurements were carried out over a temperature range of 1.8 to 300 K and an applied field of 1000 Oe. Ac measurements were carried out over a temperature range of 1.8 to 8 K, using a 2 to 5 Oe alternating field and applied dc fields of 0 to 2000 Oe. Data was corrected for sample holder and diamagnetic corrections were applied to the dc magnetic data of 1·thf, accounting for contributions from eicosane and the core diamagnetism with the diamagnetic contribution from the complex estimated from Pascal's constants.22 Cole–Cole plots (χ′Mvs. χ′′M) were fitted using least-squares regression to formulae describing χ′ and χ′′ in terms of the variables constant temperature susceptibility (χT), adiabatic susceptibility (χS), relaxation time (τ) and the distribution of relaxation times (α).Results and discussion
Synthesis
The preparation of molecular and polymeric lanthanide isocarbonyl species has previously been reported via salt elimination reactions between lanthanide halide complexes and alkali metal salts of anionic transition metal carbonyl complexes. However, structural information for the early examples is not available.23 More recently, molecular structures of lanthanide isocarbonyl complexes have been reported and it could be shown that alkane elimination reactions between lanthanide–alkyl complexes and transition metal hydrides are viable alternative synthetic routes.18 Interestingly, we find that HW(CO)3Cp is sufficiently acidic to react smoothly with [Dy(N(SiMe3)2)3] in thf to form [{CpW(CO)2(μ-CO)}3Dy(thf)5] 1via an amine elimination reaction (Scheme 1). Large block-shaped crystals of 1·thf are obtained from concentrated thf solutions at −30 °C over 24 h. Attempts to carry out the reaction and crystallizations in non-coordinating solvents were unsuccessful due to the limited solubility of HW(CO)3Cp. The isostructural yttrium analogue 2·thf has been reported; previously prepared via alkane elimination. We found that 2·thf can also be conveniently prepared from [Y(N(SiMe3)2)3] instead. | ||
| Scheme 1 Synthesis of [{CpW(CO)2(μ-CO)}3RE(thf)5], RE = Dy (1) and Y (2). | ||
Crystal structure
Compound 1·thf crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table S1†). X-ray diffraction studies revealed a distorted square antiprismatic geometry around the central Dy3+ ion. Three [CpW(CO)3]− ligand moieties are coordinated to the Dy3+ centre through the carbonyl oxygen (d(Dy–O) = 2.320(5) Å, 2.270(5) Å, 2.267(5) Å) and five thf ligands (d(Dy–Othf) = 2.384(5)–2.462(5) Å) fill the rest of the coordination sphere (Fig. 1 and Table S2†). The O–Dy–O angles concerning O donors of the same distorted square are in the range of 72°–75°. There is one additional non-coordinated thf molecule per molecule of 1 in the crystal lattice. Intermolecular Dy3+⋯Dy3+ distances in crystals of 1·thf are in the range of 10.45 to 13.66 Å, with the shortest interaction depicted in the unit cell packing diagram in Fig. S1.† Notably, this is the first structurally characterized example of a dysprosium isocarbonyl compound.
(Table S1†). X-ray diffraction studies revealed a distorted square antiprismatic geometry around the central Dy3+ ion. Three [CpW(CO)3]− ligand moieties are coordinated to the Dy3+ centre through the carbonyl oxygen (d(Dy–O) = 2.320(5) Å, 2.270(5) Å, 2.267(5) Å) and five thf ligands (d(Dy–Othf) = 2.384(5)–2.462(5) Å) fill the rest of the coordination sphere (Fig. 1 and Table S2†). The O–Dy–O angles concerning O donors of the same distorted square are in the range of 72°–75°. There is one additional non-coordinated thf molecule per molecule of 1 in the crystal lattice. Intermolecular Dy3+⋯Dy3+ distances in crystals of 1·thf are in the range of 10.45 to 13.66 Å, with the shortest interaction depicted in the unit cell packing diagram in Fig. S1.† Notably, this is the first structurally characterized example of a dysprosium isocarbonyl compound.
 | ||
| Fig. 1 Molecular structure of 1 in 1·thf. Green = Dy, purple = W, red = O, grey = C. Hydrogen atoms omitted for clarity. | ||
The W–C bond lengths to the bridging isocarbonyl moieties are with 1.875(8)–1.894(8) Å shorter than those to the terminal carbonyl W–C bonds (1.939(7)–1.955(8) Å). The C–O bond lengths of the bridging isocarbonyl groups are with 1.21(1)–1.22(1) Å longer than those of the terminal carbonyl ligands (1.16(1)–1.18(1) Å). The W–C–O (bridging) bond angles are nearly linear, with values of 177.4(7)°, 177.7(7)° and 177.9(6)°. The W–C (bridging) and C–O (bridging) bond distances and angles are in agreement with those observed in a tungsten isocarbonyl complex of cerium, [Cp′′2Ce(μ-OC)W(CO)(Cp)(μ-CO)]2,16q and of yttrium, [{CpW(CO)2(μ-CO)}3Y(thf)5].18 We note that similar shortening and elongation of M–C and C–O distances, respectively, has also been well established for simple contact ion pairs of alkali metal salts of transition metal carbonyl anions; the presence of the cationic charge is hereby expected to lower LUMO energies of the respective CO ligand and results in increased π-backbonding from the transition metal.24
Compound 1·thf is isostructural to the yttrium analogue 2·thf first prepared by Kempe et al. through an alkane elimination reaction between [Y(CH2SiMe3)3(thf)2] and 3 equivalents of [HW(CO)3Cp].18
Magnetic studies
Dc magnetic susceptibility data were collected for 1·thf over the temperature range of 2 to 300 K under an applied field of 1000 Oe (Fig. 2). The room temperature χMT value of 14.22 emu K mol−1 is in agreement with the expected value (14.17 emu K mol−1) for a single Dy3+ ion (6H15/2, S = 5/2, L = 5, g = 4/3). Upon cooling, a gradual decrease of χMT at around 100 K and a more rapid decrease below 50 K is observed, reaching a minimum value of 10.51 emu K mol−1 at 2 K. This decrease at low temperature is attributed to thermal depopulation of the Stark sublevels of Dy3+ and possible antiferromagnetic dipole–dipole interactions between Dy3+ centres.25 | ||
| Fig. 2 Temperature dependence of the molar magnetic susceptibility times temperature (χMT) product for 1·thf under a 1000 Oe applied dc field at temperatures 2 to 300 K. | ||
To investigate magnetization dynamics of 1, variable frequency ac susceptibility measurements were carried out on 1·thf. In the absence of an applied dc field, no signal was observed in the out-of-phase (χ′′M) component of the ac magnetic susceptibility at frequencies up to 1000 Hz (Fig. S2†). The absence of a signal in χ′′M under zero dc field could be due to quantum tunnelling, as this phenomenon is frequently observed in lanthanide-based SMMs.26 One commonly used method to suppress quantum tunnelling is the application of a small dc field which lifts the degeneracy of energy levels, thereby preventing this relaxation path.25,27 Application of a dc field of 1000 or 2000 Oe revealed a signal in the χ′′M component. However the maxima of these signals lie outside the accessible frequency range of the instrument (1000 Hz) (Fig. S2†).
A common cause of quantum tunnelling in SMMs is intermolecular dipole–dipole interactions between paramagnetic metal centres, allowing for mixing of the ground states.28 Dilution of a paramagnetic compound with an isostructural diamagnetic compound by co-crystallization is frequently carried out to suppress dipole–dipole interactions in Ln-SMMs.5e,6 To probe the effect of Dy3+ dipole–dipole interactions on quantum tunnelling in 1·thf, a solid magnetic dilution sample was prepared. The isostructural diamagnetic Y3+ compound, 2·thf, was prepared and crystals of a 12:1 dilution of Y:Dy were isolated.
Ac studies of the dilution sample under zero applied dc field between 1.8 and 4 K revealed a signal in the χ′′M component. However the maxima lie outside of the frequency range of the instrument (Fig. 3). Application of variable strength dc fields at 1.8 K revealed slow relaxation of the magnetization, an increase in χ′′M and a shift in the maxima of χ′′M to lower frequencies (Fig. S3†). The largest signal in the out-of-phase component (χ′′M) was obtained by applying a 400 Oe dc field. As a result, the remainder of the ac studies were carried out under a 400 Oe dc field.
 | ||
| Fig. 3 Frequency dependence of the in-phase (χ′M) (top) and out-of-phase (χ′′M) (bottom) ac susceptibility measurements of a 12:1 (Y:Dy) magnetic dilution of 1·thf under zero applied dc field and a 5 Oe switching field. Lines are a guide for the eye. | ||
The temperature dependence of χ′M and χ′′M were investigated under an applied dc field of 400 Oe in the temperature range 1.8 to 2.4 K (Fig. 4) and up to 4 K (Fig. S4†). The maxima shifted to higher frequencies with increasing temperature, moving out of the frequency limit of the instrument at temperatures above 2.4 K. The quantitative relaxation times, τ, were found by fitting the Cole–Cole plots (χ′′Mvs. χ′M) for each temperature with the generalized Debye model (solid lines) (Fig. 5, left). From the temperature dependence of τ, the barrier height Ueff can be determined from the Arrhenius plot of lnτ vs. 1/T (Fig. 5, right). The linear portion of the Arrhenius plot is indicative of an Orbach relaxation mechanism,5i with a calculated thermal relaxation barrier of Ueff = 12.6 cm−1 (18.1 K) and a pre-exponential factor of τ0 = 9.3 × 10−8. The obtained value of Ueff falls into the lower end of values determined for other mononuclear Dy3+ complexes.5e,g The non-linear dependence at low temperatures of the Arrhenius plot is indicative of the presence of other relaxation pathways such as Raman and quantum tunnelling processes.5i,28
 | ||
| Fig. 4 Frequency dependence as a function of the in-phase (χ′M) (top) and out-of-phase (χ′′M) (bottom) ac susceptibility measurements of a 12:1 (Y:Dy) magnetic dilution of 1·thf under a 400 Oe applied dc field and a 2 Oe switching field. Lines are a guide for the eye. | ||
 | ||
| Fig. 5 Left: Cole–Cole (Argand) plots for ac susceptibility collected under an applied field of 400 Oe for the 12:1 (Y:Dy) magnetic dilution of 1·thf, open circles represent experimental data, solid lines represent fits. Right: Arrhenius plot of the 12:1 (Y:Dy) magnetic dilution of 1·thf. Relaxation times, τ, were determined from non-linear least squares fits to the Cole–Cole plots. Open black circles correspond to experimental data, solid red line corresponds to a fit of the linear portion of the data (T = 2–2.4 K) and dashed red line to a fit of all the data (T = 1.8–2.4 K) to the Arrhenius expression τ = τ0exp(Ueff/kBT), affording Ueff = 12.6 cm−1 (18.1 K) and 10.7 cm−1 (15.4 K) respectively. | ||
Finally, we were interested in calculating the orientation of the easy axis of the magnetization of the Dy3+ ion in 1. For this purpose, we utilized the quantitative electrostatic model described by Chilton et al. (Fig. 6) which uses point charges on the ligand donor atoms.29 While the anionic charge in an isolated [CpW(CO)3]− anion is symmetrically delocalized, the differences in geometric parameters between bridging and end-on carbonyl ligands (see above) strongly suggest that anionic charge is (at least partially) localized on the bridging carbonyl ligands in 1.
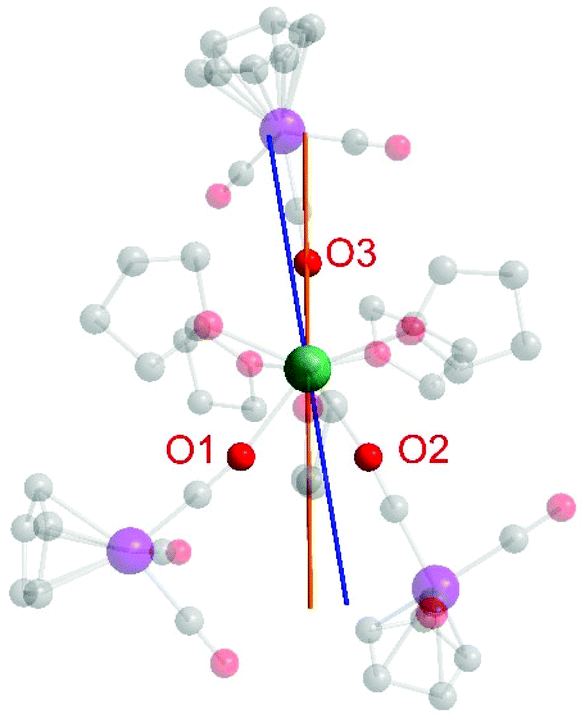 | ||
| Fig. 6 Calculated magnetic anisotropy axes in 1 assuming equivalent negative charges on O1–O3 (orange line) and inequivalent charges for O1 (−0.5), O2 (−1), and O3 (−1) (blue line). The electrostatic calculations were carried out using the FORTRAN program MAGELLAN, developed by Chilton et al.29 | ||
We considered two different scenarios: first, assuming equivalent binding and charge localization of the three [CpW(CO)3]− moieties to Dy3+ we calculated the magnetic easy axis shown in orange in Fig. 6, which is insensitive to the magnitude of the anionic charge employed for the calculation. Notably, the anionic charges of the isocarbonyl-O atoms apparently enforce the ground state electron density of Dy to be distributed perpendicular to the Dy-isocarbonyl plane. Accordingly, the easy axis is calculated to lie within that very plane.30
However, given that the Dy–O1 bond length is roughly 0.05 Å longer than the Dy–O2 and Dy–O3 distances, we also calculated the magnetic easy axis for the Dy3+ ion assuming less negative charge on O1 (−0.5) than on O2 (−1) and O3 (−1) (blue axis in Fig. 6). The change in orientation is as expected from an electrostatic model.
Overall, we prefer a formulation of equal charge distribution for O1, O2, and O3 (orange line) in 1.
Conclusions
The first structurally characterized dysprosium isocarbonyl complex, [{CpW(CO)2(μ-CO)}3Dy(thf)5] 1, has been synthesized from the amine elimination reaction between [Dy(N(SiMe3)2)3] and [HW(CO)3Cp]. Slow magnetic relaxation is observed in a dilute sample of 1·thf (Y:Dy 12:1) under a 400 Oe applied dc field, with a barrier height (Ueff) of 12.6 cm−1 (18.1 K). The utilization of lanthanide complexes stabilized by metal-carbonyl fragments may lead to new classes of heterometallic SMMs. Investigations of the electrochemical and photochemical properties of compounds such as 1 are currently ongoing and may allow for the generation of species that feature exchange coupling between lanthanide and paramagnetic transition metal ions through carbonyl bridges.
Acknowledgements
MN is grateful to the TAMU Chemistry Department for generous start-up funds and financial support by the Welch Foundation (A-1880). We thank Toby Woods and Dr Joseph Zadrozny for helpful discussions.Notes and references
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, 2006 Search PubMed.
- R. Sessoli, T. H. Lien, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Cristou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1805–1816 CrossRef CAS; R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef.
- S. Gómez-Coca, D. Aravena, R. Morales and E. Ruiz, Coord. Chem. Rev., 2015, 289–290, 379–392 CrossRef CAS; G. A. Craig and M. Murrie, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC; G. Christou, Polyhedron, 2005, 24, 2065–2075 CrossRef CAS.
- Ln SMMs reviews: (a) R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS; (b) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC; (c) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC; (d) J. Luzon and R. Sessoli, Dalton Trans., 2012, 41, 13556–13567 RSC; (e) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (f) P. Zhang, Y.-N. Guo and J. Tang, Coord. Chem. Rev., 2013, 257, 1728–1763 CrossRef CAS; (g) H. L. C. Feltham and S. Brooker, Coord. Chem. Rev., 2014, 276, 1–33 CrossRef CAS; (h) K. Lui, W. Shi and P. Cheng, Coord. Chem. Rev., 2015, 289–290, 74–122 Search PubMed; (i) S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655–6669 RSC; (j) R. A. Layfield and M. Murugesu, Lanthanides and Actinides in Molecular Magnetism, Wiley-VCH, Weinham, 2015 Search PubMed; (k) C. Benelli and D. Gatteschi, Introduction to Molecular Magnetism: From Transition Metals to Lanthanides, Wiley-VCH, Weinham, 2015 Search PubMed; (l) J. Dreiser, J. Phys.: Condens. Matter, 2015, 27, 183203 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- C. R. Ganivet, B. Ballesteros, G. de la Torre, J. M. Clemente-Juan, E. Coronado and T. Torres, Chem. – Eur. J., 2013, 19, 1457–1465 CrossRef CAS PubMed.
- R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed.
- S. Demir, I.-R. Jeon, J. R. Long and T. D. Harris, Coord. Chem. Rev., 2015, 289–290, 149–176 CrossRef CAS; J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef PubMed.
- L. R. Piquer and E. C. Sañudo, Dalton Trans., 2015, 44, 8771–8780 RSC.
- (a) L. Ungur, J. J. Le Roy, I. Korobkov, M. Murugesu and L. F. Chibotaru, Angew. Chem., Int. Ed., 2014, 53, 4413–4417 CrossRef CAS PubMed; (b) T. Pugh, F. Tuna, L. Ungur, D. Collison, E. J. L. McInnes, L. F. Chibotaru and R. A. Layfield, Nat. Commun., 2015, 6, 7492 CrossRef PubMed.
- I. P. Beletskaya, A. Z. Voskoboynikov, E. B. Chuklanova, N. I. Kirillova, A. K. Shestakova, I. N. Parshina, A. I. Gusev and G. K.-I. Magomedov, J. Am. Chem. Soc., 1993, 115, 3156–3166 CrossRef CAS; B. E. Bursten and M. G. Gatter, J. Am. Chem. Soc., 1984, 106, 2554–2558 CrossRef.
- C. A. Ghilardi, S. Mindollini and L. Sacconi, J. Organomet. Chem., 1980, 186, 279–287 CrossRef CAS.
- J. H. Osborne, A. L. Rheingold and W. C. Trogler, J. Am. Chem. Soc., 1985, 107, 6292–6297 CrossRef CAS.
- A. L. Ward, W. W. Lukens, C. C. Lu and J. Arnold, J. Am. Chem. Soc., 2014, 136, 3647–3654 CrossRef CAS PubMed.
- For examples of other lanthanide isocarbonyl compounds see Yb: (a) T. D. Tilley and R. A. Anderson, J. Am. Chem. Soc., 1982, 104, 1772–1774 CrossRef CAS; (b) M. P. Blake, N. Kaltsoyannis and P. Mountford, Chem. Commun., 2013, 49, 3315–3317 RSC; (c) M. P. Blake, N. Kaltsoyannis and P. Mountford, J. Am. Chem. Soc., 2011, 133, 15358–15361 CrossRef CAS PubMed; (d) C. Doring, A.-M. Dietel, M. V. Butovskii, V. Bezugly, F. R. Wagner and R. Kempe, Chem. – Eur. J., 2010, 16, 10679–10683 CrossRef PubMed; (e) (and Sm) S. N. Konchenko, T. Sanden, N. A. Pushkarevsky, R. Koppe and P. W. Roesky, Chem. – Eur. J., 2010, 16, 14278–14280 CrossRef CAS PubMed; (f) J. M. Boncella and R. A. Anderson, Inorg. Chem., 1984, 23, 432–437 CrossRef CAS; (g) P. V. Poplaukhin, X. Chen, E. A. Meyers and S. G. Shore, Inorg. Chem., 2006, 45, 10115–10125 CrossRef CAS PubMed; (h) (and Sm) A. C. Hiller, A. Sella and M. R. J. Elsegood, J. Chem. Soc., Dalton Trans., 1998, 3871–3874 RSC; (i) Z. Hou, K. Aida, Y. Takagi and Y. Wakatsuki, J. Organomet. Chem., 1994, 473, 101–104 CrossRef CAS; (j) C. E. Plecnik, S. Liu, J. Liu, X. Chen, E. A. Meyers and S. G. Shore, Inorg. Chem., 2002, 41, 4936–4943 CrossRef CAS PubMed; (k) K. Muller-Buschbaum, G. B. Deacon and C. M. Forsyth, Eur. J. Inorg. Chem., 2002, 3172–3177 CrossRef CAS. La: (l) A. A. Pasynskii, I. L. Eremenko, G. Z. Suleimanov, Y. A. Nuriev, I. P. Beletskaya, V. E. Shklover and Y. T. Struchkov, J. Organomet. Chem., 1984, 266, 45–52 CrossRef CAS. Sm: (m) G. Li and W.-T. Wong, J. Organomet. Chem., 1996, 522, 271–275 CrossRef; (n) A. C. Hillier, S. Y. Liu, A. Sella, O. Zekria and M. R. J. Elsegood, J. Organomet. Chem., 1997, 528, 209–215 CrossRef CAS; (o) A. Recknagel, A. Steiner, S. Brooker, D. Stalke and F. T. Edelmann, Chem. Ber., 1991, 124, 1373–1375 CrossRef CAS. Eu: (p) C. E. Plecnik, S. Liu, X. Chen, E. A. Meyers and S. G. Shore, J. Am. Chem. Soc., 2004, 126, 204–213 CrossRef CAS PubMed. Ce: (q) P. N. Hazin, J. C. Huffman and J. W. Bruno, J. Chem. Soc., Chem. Commun., 1988, 1473–1474 RSC.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 1021–1023 RSC; P. Zheng, L. Zhang, C. Wang, S. Xue, S.-Y. Lin and J. Tang, J. Am. Chem. Soc., 2014, 136, 4484–4487 CrossRef CAS PubMed.
- A. P. Sobaczynski, J. Obenauf and R. Kempe, Eur. J. Inorg. Chem., 2014, 1211–1217 CrossRef CAS.
- SAINT and APEX 2 Software for CCD Diffractometers, Bruker AXS Inc., Madison, WI, USA, 2014 Search PubMed.
- G. M. Sheldrick, SADABS, version 2.03, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2000 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2007, 64, 112–122 CrossRef PubMed; (b) G. M. Sheldrick, SHELXT, University of Göttingen, Germany, 2014 Search PubMed; (c) G. M. Sheldrick, SHELXL, University of Göttingen, Germany, 2014 Search PubMed; (d) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS; (e) OLEX2: a complete structure solution, refinement and analysis program O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- (a) A. E. Crease and P. Legzdins, J. Chem. Soc., Dalton Trans., 1973, 1501–1507 RSC; (b) G. Z. Suleimanov, L. F. Rybakova, A. B. Sigalov, Y. A. Nuriev, N. S. Kochetkova and I. P. Beletskaya, Zh. Org. Khim., 1982, 18, 2482–2485 CAS; (c) G. Z. Suleimanov and I. P. Beletskaya, Dokl. Akad. Nauk, 1981, 261, 381–383 CAS; (d) X. Wang, X. Zhou and Y. Wang, Guangpuxue Yu Guangpu Fenxi, 1987, 7, 28–30 CAS; (e) X. Wang, X. Zhou, J. Zhang, Y. Xia, R. Lui and S. Wang, Kexue Tongbao, 1985, 30, 351–355 CAS; (f) S. Li, X. Yang, Y. Sun, P. Li, X. Wang and X. Zhou, Gaodeng Xuexiao Huaxue Xuebao, 1988, 9, 1242–1245 CAS; (g) P.-F. Yan, J.-S. Gao, Z.-Y. Le and G.-J. Mao, Gaodeng Xuexiao Huaxue Xuebao, 1998, 19, 21–23 CAS.
- M. Y. Darensbourg and H. L. C. Barros, Inorg. Chem., 1979, 18, 3286–3288 CrossRef CAS; M. Y. Darensbourg and J. M. Hanckel, Organometallics, 1982, 1, 82–87 CrossRef.
- S.-D. Jiang, B.-W. Wang, G. Su, Z.-M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448 CrossRef CAS PubMed.
- Y. Bi, Y.-N. Guo, L. Zhao, Y. Guo, S.-Y. Lin, S.-D. Jiang, J. Tang, B.-W. Wang and S. Gao, Chem. – Eur. J., 2011, 17, 12476 CrossRef CAS PubMed.
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS PubMed.
- S. Demir, J. M. Zadrozny and J. R. Long, Chem. – Eur. J., 2014, 20, 9524–9529 CrossRef CAS PubMed and references therein.
- N. F. Chilton, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551 Search PubMed.
- S.-S. Liu, X. Ling, S.-D. Jiang, Y.-Q. Zhang, Y.-S. Meng, Z. Wang, B.-W. Wang, W.-X. Zhang, Z. Xi and S. Gao, Inorg. Chem., 2015, 54, 5162–5168 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structure tables, unit cell packing diagram, and additional magnetic data. CCDC 1432149. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00224a |
| This journal is © the Partner Organisations 2016 |