DOI:
10.1039/C6QI00197A
(Research Article)
Inorg. Chem. Front., 2016,
3, 1245-1255
Ruthenium(II) arene complexes containing benzhydrazone ligands: synthesis, structure and antiproliferative activity†
Received
21st June 2016
, Accepted 5th August 2016
First published on 23rd August 2016
Abstract
A suitable method for the synthesis of ruthenium(II) arene benzhydrazone complexes (1–6) of the general formula [(η6-arene)Ru(L)Cl] (arene-benzene or p-cymene; L-monobasic bidentate substituted 9-anthraldehyde benzhydrazone derivatives) has been described. The composition of the complexes has been established by elemental analysis, IR, UV-Vis, emission, NMR and ESI-MS spectral methods. The solid state molecular structure of a representative complex was determined by a single-crystal X-ray diffraction study and indicates the presence of pseudo-octahedral (piano stool) geometry. All the complexes have been thoroughly screened for their cytotoxicity against human cervical cancer cells (HeLa), human breast cancer cell line (MDA-MB-231) and human liver carcinoma cells (Hep G2) under in vitro conditions. Interestingly, the cytotoxic activity of complex 6 is much more potent than cisplatin with low IC50 values against all the cancer cell lines tested. Furthermore, the results of AO–EB, Hoechst 33258 and flow cytometry analyses reveal that these complexes induce cell death only through apoptosis. The comet assay has been employed to determine the extent of DNA fragmentation in cancer cells. A hemolysis assay with human erythrocytes demonstrated good blood biocompatibility of all the ruthenium(II) arene benzhydrazone complexes. These results highlight the strong possibility to develop highly active ruthenium complexes as anticancer agents.
Introduction
Transition metal complexes remain an important resource for the generation of chemical diversity in the search for novel therapeutic and diagnostic agents, especially in the field of anticancer drug development.1 Cisplatin represents one of the most active and clinically useful agents used in the treatment of cancer, achieving cures in testicular cancer and high response rates in ovarian and small cell lung cancer.2 Evidence from both pre-clinical studies and clinical investigations has strongly confirmed DNA as the biological target for cisplatin through the formation of irreversible adducts via the process of ligand exchange. However, in common with many other cytotoxic drugs, cisplatin induces normal tissue toxicity, particularly to the kidney, and the development of acquired drug resistance can occur in initially responsive disease types.3 Hence, there is a need for new approaches that are purposefully planned to circumvent these drawbacks. In this regard, ruthenium is considered a promising metal center for new anticancer agents, with NAMI-A4 imidazolium trans-[tetrachloro(dimethylsulfoxide)(imidazole)ruthenium(III)] and KP10195 indazolium trans-[tetrachlorobis(1H-indazole)ruthenium(III)] being the most promising ruthenium complexes reaching clinical trials.6 However, more recently, organometallic Ru arene complexes have attracted increasing attention, which are effective against resistant tumors, and have completed phase I and II clinical trials. RAPTA7 and RAED8 compounds are the most intensively investigated organoruthenium complexes and have shown promise in drug development.
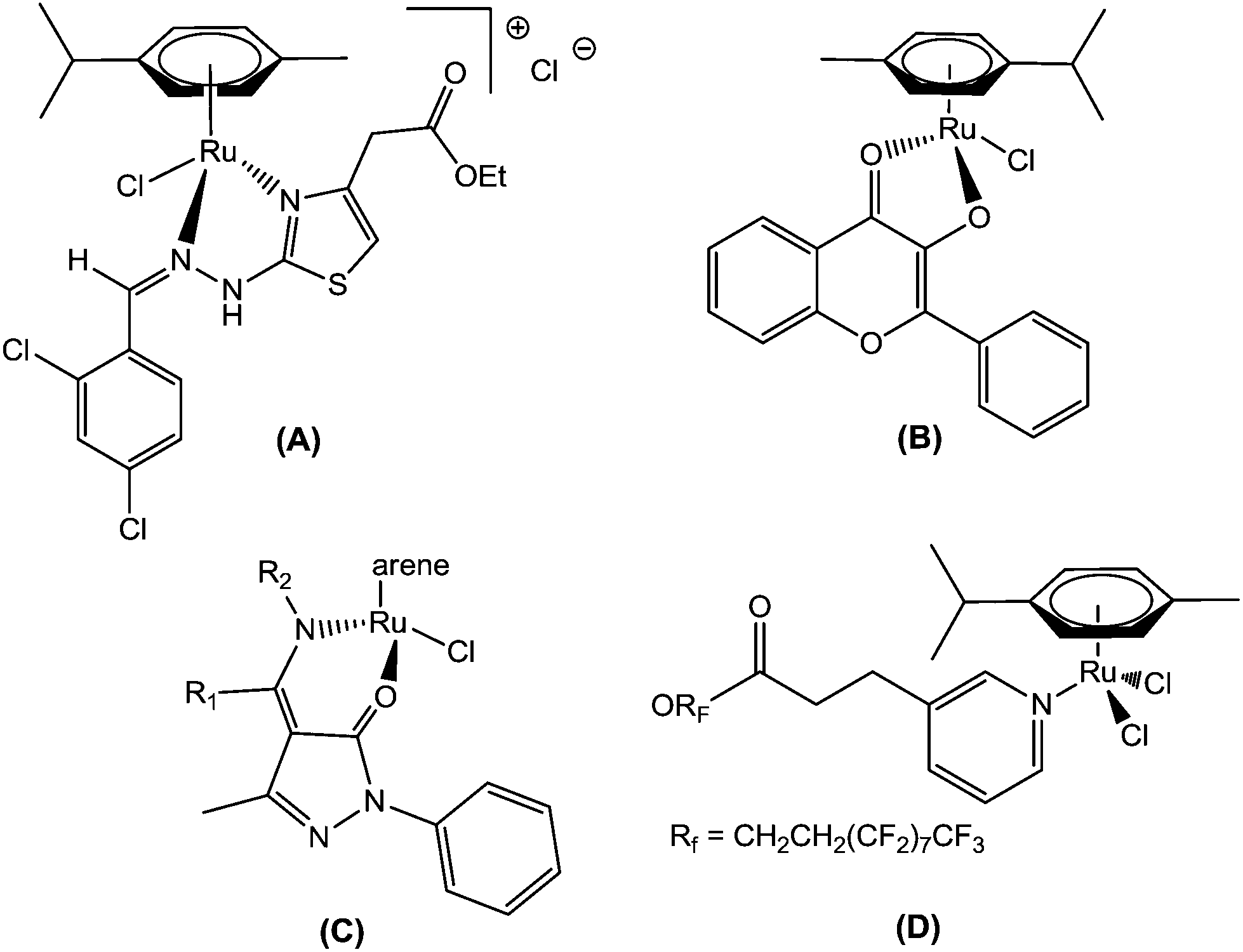
In this context, recently, attention has been focused on organometallic Ru(II)–arene complexes, which have emerged as an approach to show potential Ru-based therapeutic agents. Thus, Sadler et al. found that the Ru(II)–arene-en (en, ethylenediamine) complex exhibits efficient cytotoxic activity and also shows activity against cisplatin-resistant cell lines.9 Adriana Grozav et al. have described hydrazinyl-thiazolo arene ruthenium complexes with antiproliferative activity on three tumor cell lines (HeLa, A2780, and A2780cisR) and a noncancerous cell line (HFL-1) (A).10 Very recently, Sheldrick and coworkers have prepared half-sandwich Ru(II) complexes with methyl-substituted polypyridyl ligands, which strongly bind to DNA and also regulate apoptosis.11 The synthesis and antiproliferative activity of RuII(η6-arene) compounds carrying bioactive flavonol ligands have been reported by Hartinger et al. (B).12 Wei Su et al. have described the DNA binding properties and anticancer activity of ketone-N4-substituted thiosemicarbazones and their ruthenium(II) arene complexes.13 A series of ruthenium(II) arene complexes with the 4-(biphenyl-4-carbonyl)-3-methyl-1-phenyl-5-pyrazolonate ligand, and related 1,3,5-triaza-7-phosphaadamantane (PTA) derivatives, has been reported along with their anticancer activity with low IC50 values (C).14 Furthermore, Dyson and his co-workers have reported the synthesis of novel ruthenium half-sandwich complexes containing (N,O)-bound pyrazolone-based β-ketoamine ligands with moderate anticancer activity (D) (Fig. 1).15 P. J. Sadler and his co-workers have reported that ruthenium-arene complexes with curcuminoid analogues possess antiproliferative activity.16In vitro and in vivo evaluations of water-soluble iminophosphorane ruthenium(II) arene compounds have been reported by Isabel Marzo et al. Furthermore, these complexes were found to be cytotoxic towards cancerous cell lines (Jurkat, A549, DU-145, MiaPaca2, MDA-MB-231 and HEK-293 T) to a comparable extent to cisplatin.17 The synthesis and antiproliferative activity against SKOV-3, PC-3, MDA-MB-231 and EC109 cancer cell lines of ruthenium(II) arene N-heterocyclic carbene complexes have been described.18
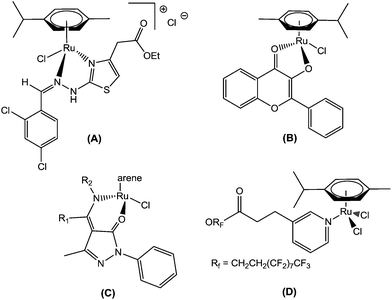 |
| | Fig. 1 Recently reported ruthenium(II) arene anticancer drugs. | |
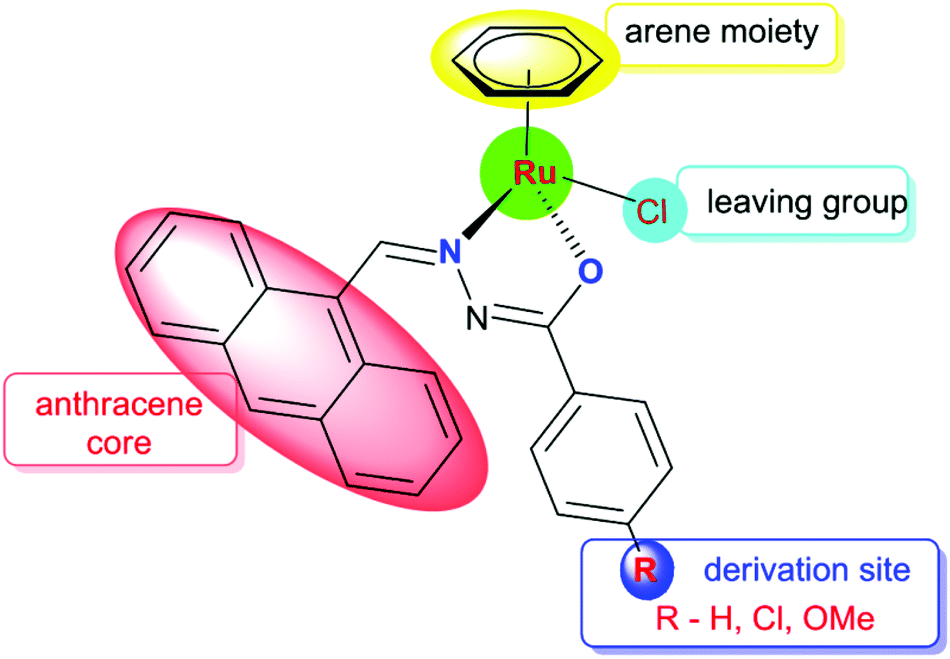
Anthracene and its derivatives are one of the most important classes of ligands with high intrinsic fluorescence and have been investigated as promising chemotherapeutic agents.19 Anthracene itself has been reported to be effective against psoriasis. The antitumor activities of anthracyclines can be attributed to their significant inhibition of the topoisomerase II activity and DNA damage. Bisantrene is a newly developed anthracycline derivative via organic synthesis for cancer treatment.20 In this background, we have focused on organic compounds used as biologically active ligands, which are derived from pharmacophore anthraquinone compounds with hydrazone moieties due to the identification of several hydrazone lead compounds showing antiproliferative activity21 and antitumor activity.22 It has been found from the literature that only a few reports are available on the synthesis, characterisation and cytotoxicity of ruthenium(II) complexes containing hydrazone ligands.23 Nevertheless, it should be pointed out that, as far as we know, the biological properties of arene ruthenium complexes bearing aroylhydrazones have not been explored until now. Therefore, in this study, we have combined the ruthenium unit with an anthraquinone moiety and the benzhydrazone ligand to generate a series of organometallic compounds with significant anticancer activity, taking advantage of the synthetic versatility of hydrazone derivatives and their promising biological activity (Fig. 2).
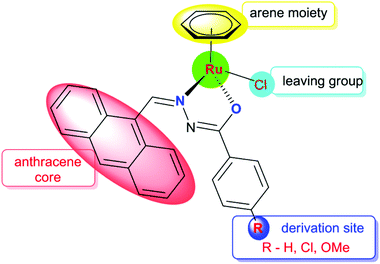 |
| | Fig. 2 Design of Ru(II) arene 9-anthraldehyde benzhydrazone complexes. | |
In the present study, the synthesis and characterization of Ru(II) arene complexes containing bidentate 9-anthraldehyde benzhydrazone ligands and chlorine were performed. All the synthesized complexes have been characterized by elemental analysis, IR, UV-Vis and NMR and ESI-MS spectroscopy techniques. The molecular structure of complex 5 was confirmed through single crystal X-ray diffraction. The in vitro cytotoxicity of complexes 1–6 against HeLa, MDA-MB-231, Hep G2 and NIH 3T3 was screened by MTT assay. The morphological changes were investigated using various biochemical apoptosis assays (AO–EB staining, Hoechst staining, flow cytometry technique and comet assay). All the complexes have exhibited negligible red hemoglobin release, implying that they are negligibly toxic or safe to normal cells.
Experimental section
Methods and instrumentation
The microanalyses of carbon, hydrogen, nitrogen and sulphur were recorded by using an analytical function testing Vario EL III CHNS elemental analyser at the Sophisticated Test and Instrumentation Centre (STIC), Cochin University, Kochi. Melting points were recorded with a Boetius micro-heating table and are corrected. FT-IR spectra were recorded in KBr pellets with a JASCO 400 plus spectrometer. Electronic spectra in chloroform solution were recorded with a CARY 300 Bio UV-visible Varian spectrometer. Emission intensity measurements were carried out by using a Jasco FP-6500 spectrofluorimeter with a 5 nm exit slit. 1H NMR spectra were recorded on a Bruker 400 MHz instrument using tetramethylsilane (TMS) as an internal reference. A Micromass Quattro II triple quadrupole mass spectrometer was employed for electrospray ionization mass spectrometry (ESI-MS).
Materials
The starting materials [(η6-p-cymene)RuCl2]2 and [(η6-benzene)RuCl2]2 were prepared according to literature methods.24
Procedure for the preparation of 9-anthraldehyde benzhydrazone ligands
A mixture of 4-substituted benzhydrazide (1 mmol) and 9-anthraldehyde (1 mmol) in ethanol (10 mL) containing a drop of glacial acetic acid was refluxed for 30 min. The separated solid was filtered and dried in air. Ligands were further purified by recrystallisation from methanol.25 Yield: 67–92%.
Procedure for the synthesis of ruthenium(II) arene benzhydrazone complexes
A mixture containing the starting material [(η6-arene)RuCl2]2 (arene = benzene or p-cymene) (0.05 mmol), 9-anthraldehyde benzhydrazone ligand (0.1 mmol) and triethylamine (0.3 mL) in benzene (20 ml) was taken in a clean 50 ml round bottom flask. The resulting mixture was allowed to react under stirring at room temperature for 2 h. A color change of the solution from dark red to orange brown was observed. The solution was concentrated to 2 mL, and hexane was added to initiate the precipitation of the complex. The reaction progress was monitored through thin layer chromatography.
[Ru(η6-C6H6)(Cl)(L1)] (1).
Brown solid. Yield = 0.160 g (68%); m.p.: 183 °C (with decomposition); calculated: C28H21ClN2ORu: C, 62.51; H, 3.93; N, 5.21%. Found: C, 62.57; H, 3.94; N, 5.25%. IR (KBr, cm−1): 1531 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NC), 1486 ν(NC–O), 1375 ν(C–O). UV-Vis (CH3CN, λmax/nm) (εmax/dm3 mol−1 cm−1): 410 (1143), 278 (6371), 232 (14
N–NC), 1486 ν(NC–O), 1375 ν(C–O). UV-Vis (CH3CN, λmax/nm) (εmax/dm3 mol−1 cm−1): 410 (1143), 278 (6371), 232 (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 757). 1H NMR (400 MHz, CDCl3) (δ ppm): 9.71 (s, 1H, HCN), 7.43–8.54 (m, 14H, aromatic), 4.64 (s, 6H, CH-benzene). ESI-MS: displays a peak at m/z 502.46 (M − Cl)+ (calcd m/z 502.48).
757). 1H NMR (400 MHz, CDCl3) (δ ppm): 9.71 (s, 1H, HCN), 7.43–8.54 (m, 14H, aromatic), 4.64 (s, 6H, CH-benzene). ESI-MS: displays a peak at m/z 502.46 (M − Cl)+ (calcd m/z 502.48).
[Ru(η6-C6H6)(Cl)(L2)] (2).
Brown solid. Yield = 0.0933 g (69%); m.p.: 196 °C (with decomposition); calculated: C28H20Cl2N2ORu: C, 58.75; H, 3.52; N, 4.89%. Found: C, 58.73; H, 3.51; N, 4.86%. IR (KBr, cm−1): 1533 ν(CN–NC), 1471 ν(NC–O), 1383 ν(C–O). UV-Vis (CH3CN, λmax/nm) (εmax/dm3 mol−1 cm−1): 419 (1044), 267 (4977), 236 (10051). 1H NMR (400 MHz, CDCl3) (δ ppm): 9.64 (s, 1H, HCN), 7.34–8.68 (m, 13H, aromatic), 4.63 (s, 6H, CH-benzene). ESI-MS: displays a peak at m/z 537.02 (M − Cl)+ (calcd m/z 536.99).
[Ru(η6-C6H6)(Cl)(L3)] (3).
Orange brown solid. Yield = 0.268 g (92%); m.p.: 174 °C (with decomposition); calculated: C29H23ClN2O2Ru: C, 61.32; H, 4.08; N, 4.93%. Found: C, 61.34; H, 4.04; N, 4.95%. IR (KBr, cm−1): 1524 ν(CN–NC), 1479 ν(NC–O), 1352 ν(C–O). UV-Vis (CH3CN, λmax/nm) (εmax/dm3 mol−1 cm−1): 421 (1496), 262 (4904), 236 (10242). 1H NMR (400 MHz, CDCl3) (δ ppm): 9.61 (s, 1H, HCN), 6.87–8.57 (m, 13H, aromatic), 4.62 (s, 6H, CH-benzene), 3.84 (s, 3H, OCH3). ESI-MS: displays a peak at m/z 532.58 (M − Cl)+ (calcd m/z 532.50).
[Ru(η6-p-cymene)(Cl)(L1)] (4).
Orange solid. Yield = 0.240 g (80%); m.p.: 189 °C (with decomposition); calculated: C32H31ClN2ORu: C, 64.47; H, 5.24; N, 4.70%. Found: C, 64.49; H, 5.26; N, 4.68%. IR (KBr, cm−1):1528 ν(CN–NC), 1486 ν(NC–O), 1371 ν(C–O). UV-Vis (CH3CN, λmax/nm) (εmax/dm3 mol−1 cm−1): 409 (1044), 259 (4941), 229 (11908). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.62 (s, 1H, HCN), 7.27–8.55 (m, 14H, aromatic), 5.37 (d, 1H, p-cym-H), 5.02 (d, 1H, p-cym-H), 4.77 (d, 1H, p-cym-H), 4.36 (d, 1H, p-cym-H), 3.11 (m, 1H, p-cym CH(CH3)2), 2.48 (s, 3H, p-cym CCH3), 1.41 (d, 3H, p-cym CH(CH3)2), 1.02 (d, 3H, p-cym CH(CH3)2). ESI-MS: displays a peak at m/z 559.05 (M − Cl)+ (calcd m/z 559.00).
[Ru(η6-p-cymene)(Cl)(L2)] (5).
Orange solid. Yield = 0.269 g (82%); m.p.: 202 °C (with decomposition); calculated: C32H28Cl2N2ORu: C, 61.15; H, 4.49; N, 4.46%. Found: C, 61.13; H, 4.53; N, 4.46%. IR (KBr, cm−1):1538 ν(CN–NC), 1489 ν(NC–O), 1369 ν(C–O). UV-Vis (CH3CN, λmax/nm) (εmax/dm3 mol−1 cm−1): 412 (1237), 267 (6908), 231 (15482). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.60 (s, 1H, HCN), 7.49–8.68 (m, 13H, aromatic), 5.35 (d, 1H, p-cym-H), 5.01 (d, 1H, p-cym-H), 5.00 (d, 1H, p-cym-H), 4.36 (d, 1H, p-cym-H), 2.48 (m, 1H, p-cym CH(CH3)2), 2.18 (s, 3H, p-cym CCH3), 1.41 (d, 3H, p-cym CH(CH3)2), 1.00 (d, 3H, p-cym CH(CH3)2). ESI-MS: displays a peak at m/z 591.75 (M − HCl)+ (calcd m/z 593.10). Single crystals suitable for X-ray diffraction were obtained by recrystallisation in DCM and methanol solution.
[Ru(η6-p-cymene)(Cl)(L3)] (6).
Orange solid. Yield = 0.180 g (78%); m.p.: 196 °C (with decomposition); calculated: C33H31ClN2O2Ru: C, 63.50; H, 5.01; N, 4.49%. Found: C, 63.48; H, 5.01; N, 4.48%. IR (KBr, cm−1):1527 ν(CN–NC), 1474 ν(NC–O), 1393 ν(C–O). UV-Vis (CH3CN, λmax/nm) (εmax/dm3 mol−1 cm−1): 418 (1576), 271 (7294), 238 (13110). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.59 (s, 1H, HCN), 6.87–8.68 (m, 13H, aromatic), 5.35 (d, 1H, p-cym-H), 5.34 (d, 1H, p-cym-H), 5.00 (d, 1H, p-cym-H), 4.36 (d, 1H, p-cym-H), 3.85 (s, 3H, OCH3), 2.48 (m, 1H, p-cym CH(CH3)2), 2.19 (s, 3H, p-cym CCH3), 1.15 (d, 3H, p-cym CH(CH3)2), 0.99 (d, 3H, p-cym CH(CH3)2). ESI-MS: displays a peak at m/z 586.59 (M − HCl)+ (calcd m/z 588.38).
X-ray crystallography
Single crystals of [Ru(η6-p-cymene)(Cl)(L2)] (5) were grown by slow evaporation of a dichloromethane–methanol solution at room temperature. A single crystal of suitable size was covered with Paratone oil, mounted on the top of a glass fiber, and transferred to a Bruker AXS Kappa APEX II single crystal X-ray diffractometer using monochromated MoKα radiation (λ = 0.71073). Data were collected at 293 K. The structure was solved by direct methods using SIR-97 and was refined by the full matrix least-squares method on F2 with SHELXL-97.26 Non-hydrogen atoms were refined with anisotropy thermal parameters. All hydrogen atoms were geometrically fixed and collected to refine using a riding model. Frame integration and data reduction were performed using the Bruker SAINT Plus (Version 7.06a) software. The multiscan absorption corrections were applied to the data using SADABS software. Fig. 1 was drawn with ORTEP27 and the structural data have been deposited at the Cambridge Crystallographic Data Centre: CCDC 1477541.
Lipophilicity
The hydrophobicity values of complexes 1–6 were measured by the “shake flask” method in octanol–water phase partitions. Complexes 1–6 were dissolved in a mixture of water and n-octanol followed by shaking for 1 hour. The mixture was allowed to settle over a period of 30 minutes and the resulting two phases were collected separately without cross contamination of one solvent layer into another. The concentration of the complexes in each phase was determined by UV-Vis absorption spectroscopy at room temperature. The results are given as the mean values obtained from three independent experiments. The sample solution concentration was used to calculate logP. Partition coefficients for 1–6 were calculated using the equation: logP = log[(1–6)oct/(1–6)aq].
Cell culture and inhibition of cell growth
Cell culture.
HeLa (human cervical cancer cell line), MDA-MB-231 (triple negative breast carcinoma), Hep G2 (human liver carcinoma cell line) and NIH 3T3 (noncancerous cell, mouse embryonic fibroblast) were obtained from the National Centre for Cell Science (NCCS), Pune. These cell lines were cultured as a monolayer in RPMI-1640 medium (Biochrom AG, Berlin, Germany), supplemented with 10% fetal bovine serum (Sigma-Aldrich, St Louis, MO, USA) and with 100 U mL−1 penicillin and 100 μg mL−1 streptomycin as antibiotics (Himedia, Mumbai, India), at 37 °C under a humidified atmosphere of 5% CO2 in a CO2 incubator (Heraeus, Hanau, Germany).
Inhibition of cell growth
The IC50 values, which are the concentrations of the tested compounds that inhibit 50% of cell growth, were determined using a 3-(4,5-dimethyl thiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Cells were plated in their growth medium at a density of 5000 cells per well in 96 flat bottomed well plates. After 24 h plating, the Ru(II) arene benzhydrazone complexes 1–6 were added at different concentrations (1–100 μM for 24 h, with a final volume in the well of 250 μL) for 24 h to study the dose dependent cytotoxic effect. To each well, 20 μL of 5 mg mL−1 MTT in phosphate buffered saline (PBS) was added. The plates were wrapped with aluminium foil and incubated for 4 h at 37 °C. The purple formazan product was dissolved by addition of 100 μL of 100% DMSO to each well. The quantity of formazan formed gave a measure of the number of viable cells. HeLa, MDA-MB-231 and Hep G2 were used for the MTT assay. The absorbance was monitored at 570 nm (measurement) and 630 nm (reference) using a 96-well plate reader (Bio-Rad, Hercules, CA, USA). Data were collected for four replicates each and used to calculate the respective means. The percentage of inhibition was calculated from this data using the formula: percentage inhibition = 100 × {Mean OD of untreated cells (control) − Mean OD of treated cells}/{Mean OD of untreated cells (control)}. The IC50 value was determined as the complex concentration that is required to reduce the absorbance to half that of the control.
Acridine orange and ethidium bromide staining experiments
The changes in chromatin organization in MDA-MB-231 cells after treatment with IC50 concentrations of complexes 4 and 6 were investigated by using acridine orange (AO) and ethidium bromide (EB). 5 × 105 cells were allowed to adhere overnight on a coverslip placed in each well of a 12-well plate. The cells were allowed to recover for 1 h, washed thrice with DPBS, stained with an AO and EB mixture (1:1, 10 μM) for 15 min, and observed with an epifluorescence microscope (Carl Zeiss, Germany).
Hoechst 33258 staining method
Hoechst 33258 staining was done using a method described earlier but with slight modifications. 5 × 105 MDA-MB-231 cells were treated with IC50 concentrations of complexes 4 and 6 for 24 h in a 6-well culture plate and fixed with 4% paraformaldehyde followed by permeabilization with 0.1% Triton X-100. Cells were then stained with 50 μg mL−1 Hoechst 33258 for 30 min at room temperature. The cells undergoing apoptosis, represented by the morphological changes of apoptotic nuclei, were observed and imaged using an epifluorescence microscope (Carl Zeiss, Germany).
Apoptosis evaluation – flow cytometry
The MDA-MB-231 cells were grown in a 6-well culture plate and exposed to IC50 concentrations of complexes 4 and 6 for 24 h. The Annexin V-FITC kit uses annexin V conjugated with fluorescein isothiocyanate (FITC) to label phosphatidylserine sites on the membrane surface of apoptotic cells. Briefly the cells were trypsinised and washed with Annexin binding buffer and incubated with Annexin V-FITC and PI for 30 minutes and immediately analysed using a flow cytometer FACS Aria-II. The results were analysed using DIVA software and the percentage of positive cells was calculated.
Cellular DNA damage by the comet assay
DNA damage was quantified by means of the comet assay as described. Assays were performed under red light at 4 °C. Cells used for the comet assay were sampled from a monolayer during the growing phase, 24 h after seeding. MDA-MB-231 cells were treated with complexes 4 and 6 at IC50 concentration, and the cells were harvested by a trypsinization process at 24 h. A total of 200 μL of 1% normal agarose in PBS at 65 °C was dropped gently onto a fully frosted microslide, covered immediately with a coverslip, and placed over a frozen ice pack for about 5 min. The coverslip was removed after the gel had set. The cell suspension from one fraction was mixed with 1% low-melting agarose at 37 °C in a 1:3 ratio. A total of 100 μL of this mixture was applied quickly on top of the gel, coated over the microslide, and allowed to set as before. A third coating of 100 μL of 1% low-melting agarose was placed on the gel containing the cell suspension and allowed to set. Similarly, slides were prepared (in duplicate) for each cell fraction. After solidification of the agarose, the coverslips were removed, and the slides were immersed in an ice-cold lysis solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris, NaOH, pH 10, 0.1% Triton X-100) and placed in a refrigerator at 4 °C for 16 h. All of the above operations were performed under low-lighting conditions in order to avoid additional DNA damage. Slides, after removal from the lysis solution, were placed horizontally in an electrophoresis tank. The reservoirs were filled with an electrophoresis buffer (300 mM NaOH and 1 mM Na2EDTA, pH > 13) until the slides were just immersed in it. The slides were allowed to stand in the buffer for about 20 min (to allow DNA unwinding) after which electrophoresis was carried out at 0.8 V cm−1 for 15 min. After electrophoresis, the slides were removed, washed thrice in a neutralization buffer (0.4 M Tris, pH 7.5), and gently dabbed to dry. Nuclear DNA was stained with 20 μL of EB (50 μg mL−1). Photographs were taken using an epifluorescence microscope (Carl Zeiss).
Hemocompatibility assay
Fresh blood was collected from healthy volunteers in sterile lithium heparin vacutainers. Further, red blood cells (RBCs) were separated by centrifugation (1500 rpm for 10 min at 4 °C) and a Ficoll density gradient. After discarding the supernatant containing plasma and platelets, the RBCs were washed thrice with sterile phosphate buffered saline (PBS). Then, the pellets (1 ml) were resuspended in 3 ml of PBS. Then, 0.1 ml of the diluted RBC suspension was added to complexes 1–6 mixed in a 0.5 ml PBS suspension at their respective IC50 concentrations (24.12, 27.04, 14.23, 18.94, 23.23, and 13.78 μM) and incubated at 37 °C for 4 h. After incubation, all the samples were centrifuged at 12000 rpm at 4 °C and supernatants were transferred to a 96-well plate. The hemolytic activity was determined by measuring the absorbance at 570 nm (Bio-Rad microplate reader model 550, Japan). Control samples of 0% lysis (PBS buffer) and 100% lysis (in 1% Triton X-100) were employed in the experiment. The percentage of hemolysis was calculated as follows:
| % Hemolysis = (As − An)/(Ap − An) × 100% |
where As, An, and Ap are the absorbances of the sample, the negative control and the positive control respectively.
Statistical analysis
Values are given as the means ± SD. Data are presented as averages of independent experiments, performed in duplicate or triplicate. Statistical analyses were done using Student's t-test. P < 0.05 was considered statistically significant.
Results and discussion
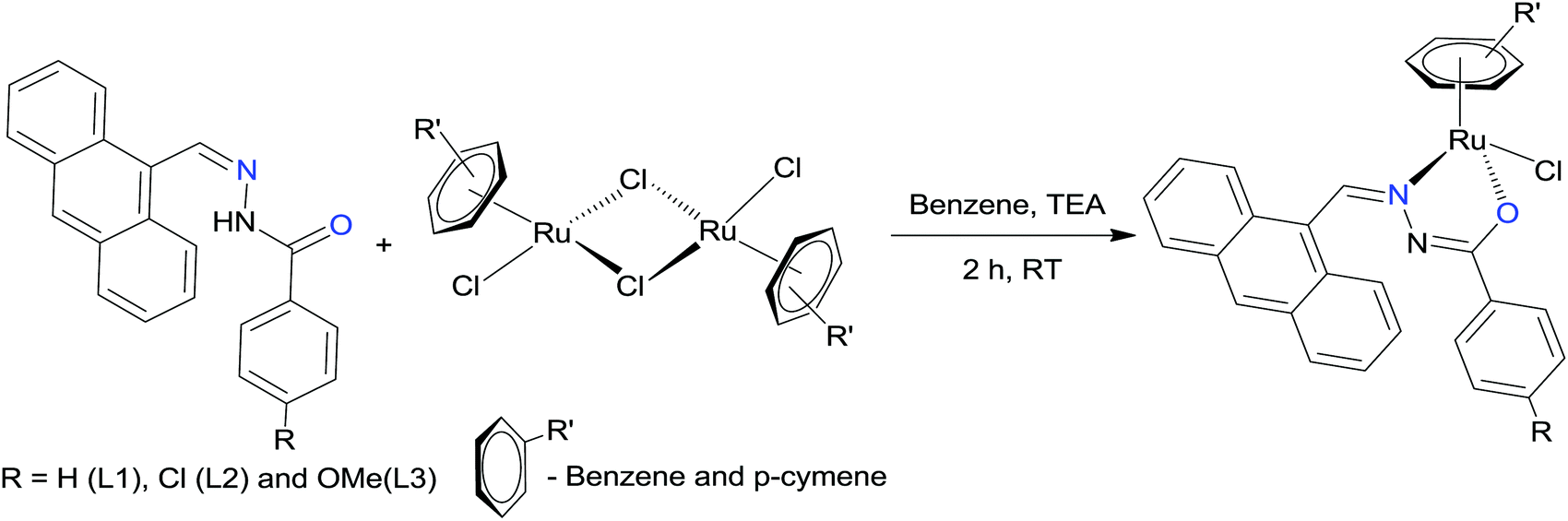
The hydrazone ligand derivatives were conveniently prepared in excellent yield by the condensation of 9-anthraldehyde with substituted benzhydrazides in an equimolar ratio. These ligands were allowed to react with the ruthenium(II) arene precursor [(η6-arene)RuCl2]2 (arene-benzene or p-cymene) in a 2:1 molar ratio in the presence of triethylamine as the base and new complexes of the general formula [(η6-arene)Ru(L)Cl] (arene-benzene or p-cymene; L-substituted 9-anthraldehyde benzhydrazone derivatives) (Scheme 1) were obtained in high yields. The addition of triethylamine to the reaction mixture was used to remove a proton from the imidol oxygen and to facilitate the coordination of the imidolate oxygen to the ruthenium(II) ion. All the complexes are air-stable and highly soluble in most organic solvents. The analytical data of all the ruthenium(II) arene benzhydrazone complexes are in good agreement with the molecular formula proposed.
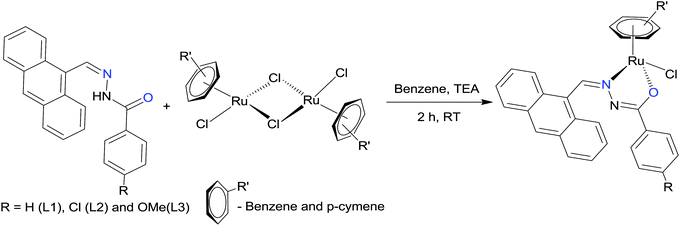 |
| | Scheme 1 Synthesis of Ru(II) arene benzhydrazone complexes. | |
Characterization of the complexes
The IR spectra of the free ligands displayed a medium to strong band in the region of 3180–3196 cm−1 which is characteristic of the N–H functional group. The free ligands also displayed νCN and νCO absorptions in the region of 1548–1576 cm−1 and 1610–1653 cm−1, respectively, which indicate that the ligands exist in the amide form in the solid state. Bands that are due to νN–H and νCO stretching vibrations were not observed with the complexes, which indicates that the ligands underwent tautomerization and subsequent coordination of the imidolate enolate form during complexation. Coordination of the ligand to the ruthenium(II) ion through an azomethine nitrogen is expected to reduce the electron density in the azomethine link and thus lower the absorption frequency upon complexation at 1527–1538 cm−1, which indicates the coordination of azomethine nitrogen to the ruthenium(II) ion. The band in the region of 1352–1393 cm−1 is due to the imidolate oxygen, which is coordinated to the metal. The IR spectra of all the complexes therefore confirm the mode of coordination of the benzhydrazone ligand to the ruthenium(II) ion via the azomethine nitrogen and imidolate oxygen.28
The absorption spectra of the ruthenium(II) arene benzhydrazone complexes in chloroform exhibited bands in the ultraviolet region below 278 nm that are very similar and are attributable to the transitions within the ligand orbitals (n–π*, π–π*) taking place in the anthracene benzhydrazone ligands. The lowest energy absorption bands in the electronic spectra of the complexes in the visible region 409–421 nm are ascribed to MLCT (metal to ligand charge transfer) transitions. Based on the pattern of the electronic spectra of all the complexes an octahedral environment around the ruthenium(II) ion has been proposed similar to that of the other octahedral ruthenium(II) arene complexes.29 The light emitting properties of all the complexes were investigated in DMSO at ambient temperature (298 K). The excitation was made at the charge transfer band for all the complexes. The emission maxima of all the complexes have experienced a positive shift of the order of 90–98 nm. The emission maximum falls in the range 490–498 nm. It is likely that the emission originates from the lowest energy metal to ligand charge transfer (MLCT) state, probably derived from the excitation involving dπ(Ru)–π* (ligand), MLCT transitions, similar to the MLCT observed in other reported Ru(II) arene complexes.29,30
The 1H NMR spectra of all the complexes were recorded in CDCl3 to confirm the bonding of the benzoylhydrazone ligand to the ruthenium(II) ion. Multiplets observed in the region δ 6.87–8.68 ppm in the complexes have been assigned to the aromatic protons of benzhydrazone ligands. The signal due to the azomethine proton appears in the region δ 9.60–9.71 ppm. The position of the azomethine signal in the complexes is slightly downfield in comparison with that of the free ligand, suggesting deshielding of the azomethine proton due to its coordination to ruthenium. The singlet due to the –NH proton of the free ligand in the region δ 11.22–11.60 ppm is absent in the complex, further supporting enolisation and coordination of the imidolate oxygen to the Ru(II) ion. Therefore, the 1H NMR spectra of the complexes confirm the bidentate coordination mode of the benzhydrazone ligands to ruthenium(II) ions. In all the complexes, the cymene protons appear in the region of δ 4.36–5.36 ppm.31 In addition, the two isopropyl methyl protons of the p-cymene appeared as two doublets in the region of δ 0.98–1.51 ppm and the methine protons come in the region of δ 2.43–2.48 ppm as septet. Furthermore, the methyl group of the p-cymene comes as a singlet around the region of δ 2.15–2.19 ppm. Additionally methoxy protons are observed as singlets for complexes 3 and 6 at δ 3.13–3.84 ppm. On the other hand, benzene arene protons displayed an upfield shift relative to complexes 4–6 in the region δ 4.62–4.64 ppm (Fig. S1, ESI†).
The ESI-MS spectra of the complexes have been acquired to explain the relative composition and stability of the complexes. Thus, we have recorded mass spectra for all the complexes which confirm the formation of the complexes. Mass spectrometric measurements were carried out under positive ion ESI mode using acetonitrile as the solvent. In their positive ESI mass spectra, 1–6 showed major peaks due to the cationic fragment [(η6-arene)Ru(L)Cl]+ generated by loss of the Cl−. The ESI spectra of complexes 1–6 display peaks at m/z 502.46 (1, M − Cl)+, 537.02 (2, M − Cl)+, 532.58 (3, M − Cl)+, 559.05 (4, M − Cl)+, 591.75 (5, M − Cl − H)+ and 586.59 (6, M − Cl − H)+ respectively. The mass spectrometry results are in good agreement with the proposed molecular formulae of the complexes and suggest that the chloro (Cl−) group is labile and possibly replaced by targeted biomolecules.
X-ray crystallographic studies
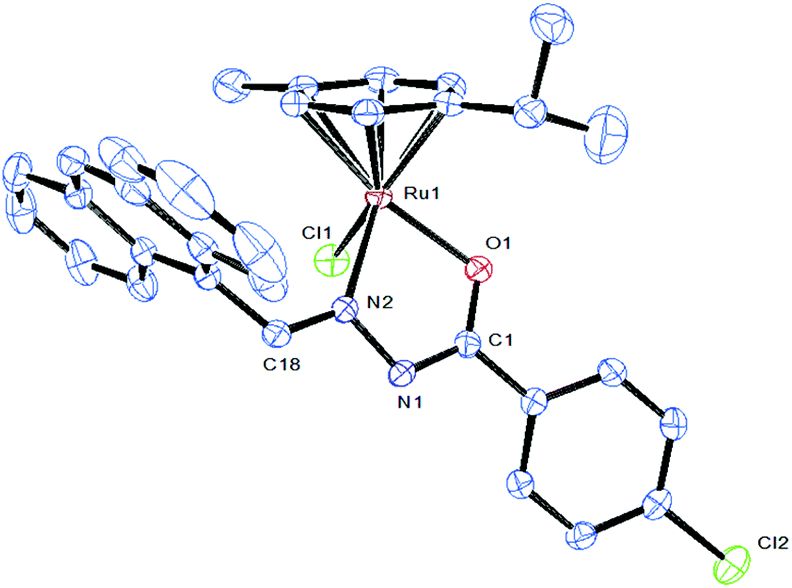
Attempts were made to grow single crystals for all the complexes to confirm the coordination mode of the ligand to metal and the geometry of the complex. Molecular structures of [Ru(η6-p-cymene)(Cl)(L5)] (5) have been determined by single-crystal X-ray diffraction analyses. Crystals of 5 grew from slow diffusion of dichloromethane into methanol solutions and crystallized in the triclinic system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The selected bond lengths and bond angles are given in Table 2 whereas crystallographic data and structural refinement parameters are gathered in Table 1. The ORTEP views of the molecules with the atom numbering are shown in Fig. 3. The molecular structure of complex 5 shows clearly that the benzhydrazone ligand coordinates in a bidentate manner to ruthenium ions via the azomethine nitrogen and imidolate oxygen in addition to one chlorine and one arene group. The complex adopts the commonly observed piano-stool geometry as reported in many half-sandwich arene ruthenium(II) complexes.32 In this case, the arene ring forms the seat of the piano-stool, while the bidentate benzhydrazone N, O and Cl ligands form the three legs of the stool. Therefore, the ruthenium(II) ion is sitting in a NOCl (η6-arene) coordination environment. The benzhydrazone ligand binds to the metal centre at N and O forming the five membered chelate ring with bite angles 75.94(9)° O(1)–Ru(1)–N(2) and 83.25(8)° N(2)–Ru(1)–Cl(1). The bond lengths of Ru(1)–N(2) and Ru(1)–O(1) are 2.105(3) Å and 2.056(2) Å respectively. The Ru–Cl bond length is found to be 2.4105(13) Å and the bond length is in agreement with other structurally characterized p-cymene ruthenium complexes.33 The ruthenium atom is π bonded to the arene ring with an average Ru–C distance of 2.173(3) Å, whereas the average C–C bond length in the arene ring is 1.482(4) Å with alternating short and long bonds. As all the complexes display similar spectral properties, the other five complexes are assumed to have a similar structure to that of [Ru(η6-p-cymene)(Cl)(L2)] (5).
space group. The selected bond lengths and bond angles are given in Table 2 whereas crystallographic data and structural refinement parameters are gathered in Table 1. The ORTEP views of the molecules with the atom numbering are shown in Fig. 3. The molecular structure of complex 5 shows clearly that the benzhydrazone ligand coordinates in a bidentate manner to ruthenium ions via the azomethine nitrogen and imidolate oxygen in addition to one chlorine and one arene group. The complex adopts the commonly observed piano-stool geometry as reported in many half-sandwich arene ruthenium(II) complexes.32 In this case, the arene ring forms the seat of the piano-stool, while the bidentate benzhydrazone N, O and Cl ligands form the three legs of the stool. Therefore, the ruthenium(II) ion is sitting in a NOCl (η6-arene) coordination environment. The benzhydrazone ligand binds to the metal centre at N and O forming the five membered chelate ring with bite angles 75.94(9)° O(1)–Ru(1)–N(2) and 83.25(8)° N(2)–Ru(1)–Cl(1). The bond lengths of Ru(1)–N(2) and Ru(1)–O(1) are 2.105(3) Å and 2.056(2) Å respectively. The Ru–Cl bond length is found to be 2.4105(13) Å and the bond length is in agreement with other structurally characterized p-cymene ruthenium complexes.33 The ruthenium atom is π bonded to the arene ring with an average Ru–C distance of 2.173(3) Å, whereas the average C–C bond length in the arene ring is 1.482(4) Å with alternating short and long bonds. As all the complexes display similar spectral properties, the other five complexes are assumed to have a similar structure to that of [Ru(η6-p-cymene)(Cl)(L2)] (5).
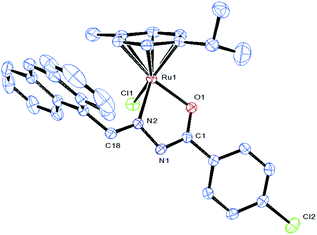 |
| | Fig. 3 ORTEP drawing of complex 5 at the 30% probability level, with hydrogen atoms being omitted for clarity. The solvent molecule has been omitted for clarity. | |
Table 1 Selected crystal data and structure refinement summary of 5
| Complex |
5
|
| Chemical formula |
C32H28Cl2N2ORu |
| Formula weight |
628.53 |
| Temperature |
296(2) K |
| Crystal system |
Triclinic |
| Space group |
P |
|
a (Å) |
9.951(2) |
|
b (Å) |
11.437(2) |
|
c (Å) |
13.867(3) |
|
α (°) |
69.55(3) |
|
β (°) |
71.55(3) |
|
γ (°) |
73.82(3) |
| Volume (Å3) |
1377.4(5) |
|
Z
|
2 |
|
ρ (Mg m−3) |
1.515 |
|
μ (mm−1) |
0.792 |
| Reflections collected |
21062 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0319, wR2 = 0.0911 |
|
R indices (all data) |
R
1 = 0.0370, wR2 = 0.1035 |
| Goodness-of-fit on F2 |
1.152 |
Table 2 Selected bond lengths (Å) and angles (°) in 5
| Distances/angles |
5
|
| Ru(1)–N(2) |
2.105(3) |
| Ru(1)–O(1) |
2.056(2) |
| Ru(1)–Cl(1) |
2.4105(13) |
| Ru(1)–C(10) |
2.173(3) |
| N(1)–N(2) |
1.400(3) |
| N(2)–C(18) |
1.283(4) |
| O(1)–C(1) |
1.292(4) |
| O(1)–Ru(1)–N(2) |
75.94(9) |
| N(2)–Ru(1)–Cl(1) |
83.25(8) |
| N(2)–N(1)–Ru(1) |
114.48(18) |
| C(1)–O(1)–Ru(1) |
112.89(18) |
| C(1)–N(2)–N(1) |
110.5(2) |
| C(13)–Ru(1)–Cl(1) |
92.62(11) |
| O(1)–Ru(1)–Cl(1) |
84.36(7) |
Lipophilicity
The hydrophobicity of metal complexes is an important parameter to determine the penetration behaviour across the cell membrane, and is investigated in terms of the partition coefficient (logP). Here, the complexes are likely to differ in their hydrophobicity due to the variation in the different substitutions present in the complexes. These measurements are based on the solubility of a given compound in an aqueous vs. organic medium, based on the concentration of a given compound distributed in the biphasic system (n-octanol–water).34 The calculated logP values for complexes 1–6 are 2.49, 2.55, 2.36, 2.18, 2.23 and 1.82 respectively (Table 3). Among all the complexes, complexes having p-cymene with a methoxy substituent (6) show higher hydrophobicity than the rest of the complexes (1–5).
Table 3 Cytotoxicity (IC50, μM) of the ligand and complexes 1–6 (n.e.: no effect) and calculated partition coefficients (logP)
| Complex |
IC50 values (μM) |
logP |
| HeLa |
MDA-MB-231 |
Hep G2 |
NIH 3T3 |
| Complex 1 |
24.12 ± 0.1 |
19.41 ± 0.3 |
21.32 ± 0.2 |
232.13 ± 0.5 |
2.49 ± 0.3 |
| Complex 2 |
27.04 ± 0.2 |
23.01 ± 0.1 |
26.49 ± 0.3 |
241.47 ± 0.6 |
2.55 ± 0.5 |
| Complex 3 |
14.23 ± 0.4 |
11.29 ± 0.4 |
13.57 ± 0.1 |
248.51 ± 0.3 |
2.36 ± 0.4 |
| Complex 4 |
18.94 ± 0.3 |
9.97 ± 0.2 |
11.86 ± 0.2 |
239.12 ± 0.4 |
2.18 ± 0.4 |
| Complex 5 |
23.23 ± 0.1 |
18.03 ± 0.3 |
23.06 ± 0.2 |
229.79 ± 0.5 |
2.23 ± 0.5 |
| Complex 6 |
13.78 ± 0.1 |
5.03 ± 0.2 |
10.18 ± 0.3 |
243.81 ± 0.5 |
1.82 ± 0.4 |
| L1 |
n.e. |
n.e. |
n.e. |
n.e. |
|
| L2 |
n.e. |
n.e. |
n.e. |
n.e. |
|
| L3 |
n.e. |
97.10 ± 0.3 |
99.12 ± 0.4 |
n.e. |
|
| Cisplatin |
21.32 ± 0.2 |
11.91 ± 0.6 |
19.16 ± 1.2 |
245.13 ± 0.4 |
|
In vitro antiproliferative activity
All the ruthenium complexes 1–6 and the free benzhydrazone ligands were evaluated for their cytotoxic activity against HeLa, MDA-MB-231 and Hep-G2 along with NIH 3T3 cell lines by using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (colorimetric assay) that measures mitochondrial dehydrogenase activity as an indication of cell viability. Because these complexes might undergo ligand substitution reactions with water molecules when dissolved in aqueous solutions, freshly made stock solutions of each compound were used. The widely used clinical drug, cisplatin, was included as a positive control. The effects of the ruthenium(II) arene complexes to arrest the proliferation of cancer cells were investigated after an exposure of 24 h. It is to be noted that the ligands did not show any inhibition of the cell growth even up to 100 μM and clearly indicate that chelation of the benzoylhydrazone ligand with metal ions is responsible for the observed cytotoxicity properties of the complexes. The results of the MTT assays revealed that complexes showed notable activity against the cell lines HeLa, MDA-MB-231 and Hep-G2 with respect to IC50 values (Table 3). From the IC50 values obtained it was inferred that complex 6 is highly active against all the cell lines with very low IC50 values compared to the well-known anticancer drug cisplatin. In addition, in vitro cytotoxic activity studies of the complexes against the mouse embryonic fibroblast cell line NIH 3T3 (normal cells) were undertaken and the IC50 values are above 229 μM, which confirmed that the complexes are very specific to cancer cells. The fact that these ruthenium(II) arene benzhydrazone complexes 1–6 possess significant cytotoxicity over the ligands may be due to the presence of extended π conjugation resulting from the chelation of Ru(II) ions with the ligand. Furthermore, the observed higher activity of complexes 4 and 6 is correlated to the nature of the chelating benzoylhydrazone ligand and arene moiety.
Furthermore, the observed cytotoxic activities of complexes 3 and 6 were found to be superior when compared with other complexes. The observed higher efficiencies of complexes 3 and 6 are related to the nature of the substitution of the benzhydrazone ligand that is coordinated to the ruthenium ion. Higher cytotoxicity is observed for complexes 3 and 6, which contain an electron-donating methoxy group that consequently increases the lipophilic character of the metal complex, which favours its permeation through the lipid layer of a cell membrane. Apart from the three different cell lines, the proliferation of the MDA-MB-231 cell line was arrested to a greater extent than that of HeLa and Hep-G2 cells by the complexes.35
On the other hand, the arene moiety group also imparts hydrophobic character to the molecule, which facilitates the passive diffusion through the cell membrane, enhancing the cellular accumulation on the antitumor activity of these ruthenium complexes. It has been observed that complexes 4–6 with a p-cymene group show higher potency than those with a benzene group in complex 3, which may be attributed to the stronger hydrophobic interactions between the Ru(II)–cymene complex and the biomolecular targets.36 Complex 6 shows high cytotoxic activity with very low IC50 values of 11.40 ± 0.7, 4.13 ± 0.4 and 9.19 ± 0.3 μM toward HeLa, MDA-MB-231 and Hep-G2. Furthermore, the IC50 values are much lower than those previously reported for other Ru(II) arene arylazo, 2-thiosalicylic acid, phenanthroimidazole or polypyridyl complexes.37
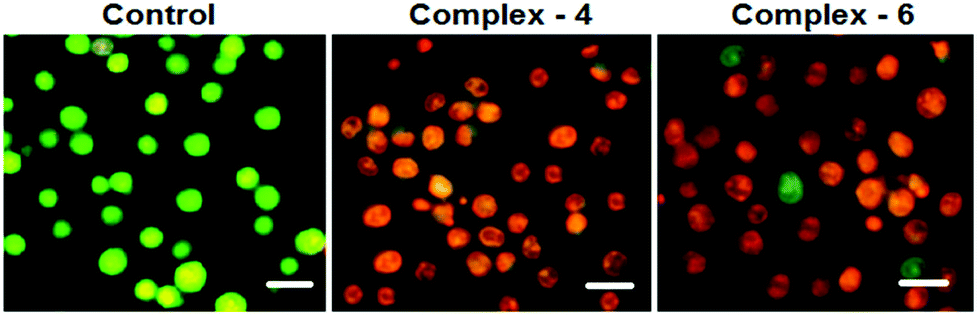
AO–EB and Hoechst staining assays
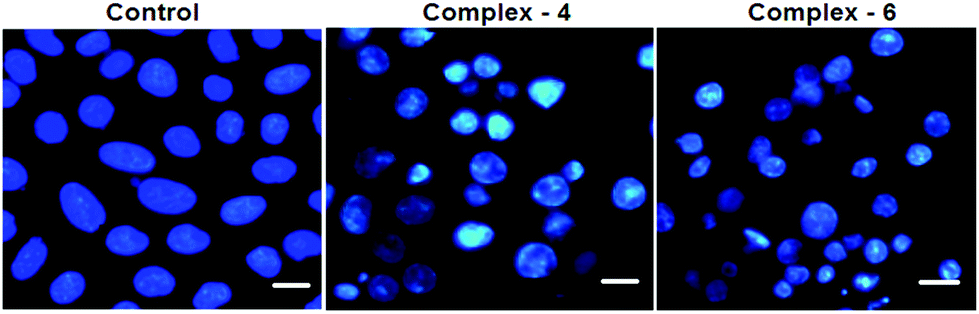
In order to observe the morphological changes, MDA-MB-231 cells were stained with acridine orange (AO) and ethidium bromide (EB). AO is a cell permeable fluorescent dye that stains nuclear DNA across an intact cell membrane and EB only stains cells that have lost membrane integrity. The living cells will be uniformly stained green, apoptotic cells are stained green and contain apoptotic characteristics such as cell blebbing, nuclear shrinkage and chromatin condensation, and necrotic cells are stained red and can be found by AO–EB double staining. After MDA-MB-231 cells were exposed to complexes 4 and 6 for 24 h at IC50 concentration, morphological changes were observed, which are shown in Fig. 4. In the control, the living cells of MDA-MB-231 were stained bright green in spots. On the treatment of MDA-MB-231 cells with complexes 4 and 6, green apoptotic cells with apoptotic characteristics such as nuclear shrinkage and chromatin condensation, as well as red necrotic cells, were observed. These observations indicate that complexes 4 and 6 can induce apoptosis in MDA-MB-231 cells.38 Additionally, when complex 4 and 6 treated MDA-MB-231 cells were stained with Hoechst 33258, apoptotic features such as nuclear shrinkage and chromatin condensation were also observed (Fig. 5). Hence the results of AO–EB and Hoechst staining assays suggest that complexes 4 and 6 induce apoptosis in MDA-MB-231 cells.39
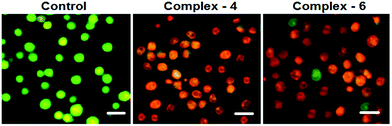 |
| | Fig. 4 Morphological assessment of the AO and EB of MDA-MB-231 cells treated with complexes 4 and 6 (IC50 concentration) for 24 h. The scale bar is 20 μm. | |
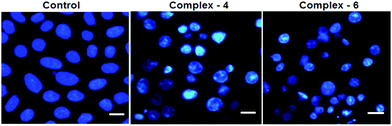 |
| | Fig. 5 Morphological assessment of complexes 4 and 6 (IC50 concentration) and MDA-MB-231 cells for 24 h. The scale bar is 20 μm. | |
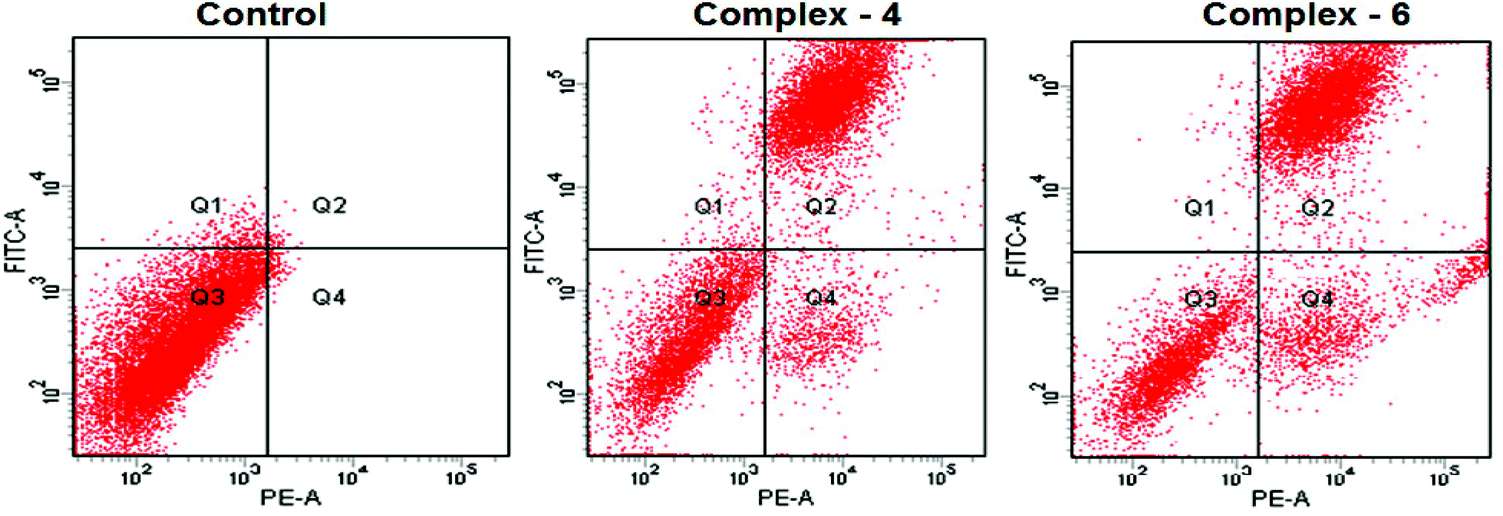
Evaluation of apoptosis – flow cytometry
The potential to induce apoptosis in cancer cells by the addition of synthesized complexes can be quantitatively investigated by flow cytometry analysis using an Annexin V protocol, with the help of an Annexin V-FITC apoptosis detection kit to perform double-staining with propidium iodide and Annexin V-FITC. Annexin V, a Ca2+ dependent phospholipid-binding protein with a high affinity for the membrane phospholipid phosphatidylserine (PS), is quite helpful for identifying apoptotic cells with exposed PS. Propidium iodide is a standard flow cytometric viability probe used to distinguish viable from non-viable cells (Fig. 6). The MDA-MB-231 cells were treated with complexes 4 and 6 at IC50 concentrations for 24 h. The cell death induced by the complexes follows a pathway from the lower left quadrant to the upper right quadrant (Annexin V+/PI+), which represents cells undergoing apoptosis.40
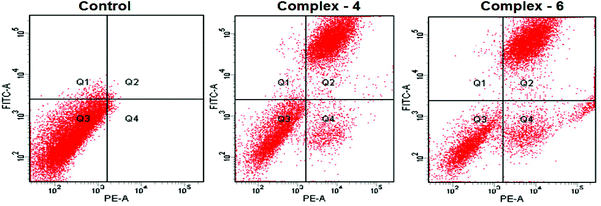 |
| | Fig. 6 AnnexinV/propidium iodide assay of MDA-MB-231 cells treated with complexes 4 and 6 (IC50 concentration) measured by flow cytometry. | |
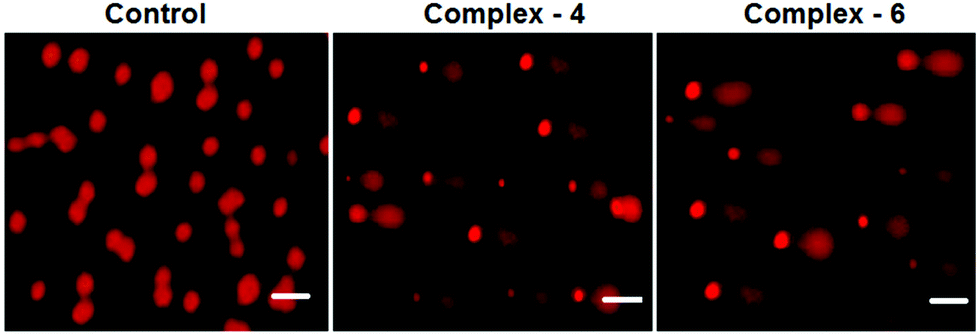
Comet assay
As is well known, DNA is considered to be the target of most of the current antitumor drugs to conquer tumors. DNA fragmentation is the hallmark of apoptosis. The single cell gel electrophoresis assay (comet assay) is commonly utilized to assess DNA integrity. As shown in Fig. 7, in the control, MDA-MB-231 cells fail to show a comet like appearance. When MDA-MB-231 cells were treated with IC50 concentrations of complexes 4 and 6 for 24 h, a statistically significant and well-formed comet was observed. The length of the comet tail is a sign of the extent of DNA damage. With the IC50 concentration of the complexes, comet tails become more and more obvious. The results show that ruthenium(II) arene complexes are capable of eliciting DNA damaging effects, as evidenced by the comet assays on MDA-MB-231 cells. It is well known that DNA fragmentation is a hallmark of apoptosis.41
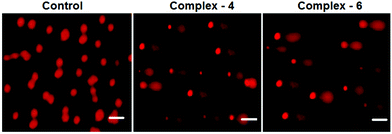 |
| | Fig. 7 Comet assay of staining of the EB control (untreated) MDA-MB-231 cells treated with complexes 4 and 6 (IC50 concentration) for 24 h. The scale bar is 40 μm. | |
Hemocompatibility assay
Interaction of the drug with the blood components, particularly human RBCs, is an important and inevitable phenomenon; thus assessing the haemolysis becomes crucial in evaluating the blood compatibility of drugs. The results show that complexes 1–6 show good compatibility with human RBCs. The mechanism of direct hemolytic activity for different toxic agents was found to be nonspecific. According to the International Organization for Standardization/Technical Report 7406, the admissible level of hemolysis of biological materials is 5%. As shown in Fig. 8, compared to the positive control (Triton X-100), all the complexes have exhibited negligible red hemoglobin release, implying that they are negligibly toxic or safe to normal cells.42
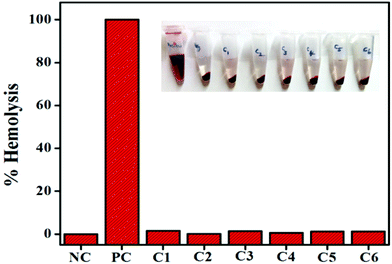 |
| | Fig. 8 Human blood compatibility analysis of complexes 1–6. | |
Conclusions
We report here the synthesis of six new ruthenium(II) arene complexes containing bidentate O and N chelating 9-anthraldehyde benzhydrazone ligands. All the complexes (1–6) have been fully characterized by analytical and spectral methods (IR, UV-vis, emission, 1H NMR and ESI-MS). Molecular structure by a X-ray diffraction study reveals that the benzhydrazone ligand coordinates to ruthenium via azomethine nitrogen and imidolate oxygen and adopts the familiar pseudo-octahedral “piano-stool” geometry. Interestingly, the cytotoxic activities of complex 6 against the tested cancer cell lines were significantly superior to those of the well-known anticancer drug cisplatin. Furthermore, fluorescence staining techniques and flow cytometry using the annexin-V assay revealed that complexes 4 and 6 induce apoptosis in MDA-MB-231 cancer cells. Alkaline comet assay confirms the single-strand break of DNA. Hemolysis assays revealed that all the complexes 1–6 were less toxic to human RBCs. On the basis of the results, we suggest that ruthenium-arene based benzhydrazone complexes are attractive agents for the development of future anticancer therapies relying on the combination of chemopreventive and chemotherapeutic agents for human cancers.
Acknowledgements
M. K. M. S. Khan thanks the University Grants Commission (UGC), New Delhi for financial assistance through a UGC-BSR fellowship (Ref. No. F.7-22/2007(BSR)). We thank Dr Yu Liu for the crystallographic discussion. We express our sincere thanks to DST-FIST, India for the use of a Bruker 400 MHz spectrometer at the School of Chemistry, Bharathidasan University, Tiruchirappalli-24.
References
-
(a) L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed
 ;
(b) J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed ;
(c) S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2014, 33, 201–214 CrossRef PubMed ;
(d) A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed .
;
(b) J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed ;
(c) S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2014, 33, 201–214 CrossRef PubMed ;
(d) A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed .
-
(a) M. Ohmichi, J. Hayakawa, K. Tasaka, H. Kurachi and Y. Murata, Trends Pharmacol. Sci., 2005, 26, 113–116 CrossRef CAS PubMed ;
(b) S. L. Cooke and J. D. Brenton, Lancet Oncol., 2011, 12, 1169–1174 CrossRef CAS PubMed ;
(c) D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed .
- G. Giaccone, Drugs, 2000, 59, 9–17 CrossRef CAS PubMed .
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed .
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS .
- M. Kubanik, H. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658–5668 CrossRef CAS .
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306(Part 1), 86–114 CrossRef CAS .
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC .
-
(a) C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS ;
(b) A. F. A. Peacock and P. J. Sadler, Chem. – Asian J., 2008, 3, 1890–1899 CrossRef CAS PubMed .
- A. Grozav, O. Balacescu, L. Balacescu, T. Cheminel, I. Berindan-Neagoe and B. Therrien, J. Med. Chem., 2015, 58, 8475–8490 CrossRef CAS PubMed .
- Y. Geldmacher, R. Rubbiani, P. Wefelmeier, A. Prokop, I. Ott and W. S. Sheldrick, J. Organomet. Chem., 2011, 696, 1023–1031 CrossRef CAS .
- A. Kurzwernhart, W. Kandioller, S. Bächler, C. Bartel, S. Martic, M. Buczkowska, G. Mühlgassner, M. A. Jakupec, H.-B. Kraatz, P. J. Bednarski, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, J. Med. Chem., 2012, 55, 10512–10522 CrossRef CAS PubMed .
- W. Su, Q. Qian, P. Li, X. Lei, Q. Xiao, S. Huang, C. Huang and J. Cui, Inorg. Chem., 2013, 52, 12440–12449 CrossRef CAS PubMed .
- R. Pettinari, C. Pettinari, F. Marchetti, B. W. Skelton, A. H. White, L. Bonfili, M. Cuccioloni, M. Mozzicafreddo, V. Cecarini, M. Angeletti, M. Nabissi and A. M. Eleuteri, J. Med. Chem., 2014, 57, 4532–4542 CrossRef CAS PubMed .
- C. M. Clavel, E. Păunescu, P. Nowak-Sliwinska, A. W. Griffioen, R. Scopelliti and P. J. Dyson, J. Med. Chem., 2015, 58, 3356–3365 CrossRef CAS PubMed .
- M. Melchart, A. Habtemariam, O. Novakova, S. A. Moggach, F. P. A. Fabbiani, S. Parsons, V. Brabec and P. J. Sadler, Inorg. Chem., 2007, 46, 8950–8962 CrossRef CAS PubMed .
- M. Frik, A. Martínez, B. T. Elie, O. Gonzalo, D. Ramírez de Mingo, M. Sanaú, R. S. Delgado, T. Sadhukha, S. Prabha, J. W. Ramos, I. Marzo and M. Contel, J. Med. Chem., 2014, 57, 9995 CrossRef CAS PubMed .
- G. Lv, L. Guo, L. Qiu, H. Yang, T. Wang, H. Liu and J. Lin, Dalton Trans., 2015, 44, 7324–7331 RSC .
-
(a) T. P. Wunz, R. T. Dorr, D. S. Alberts, C. L. Tunget, J. Einspahr, S. Milton and W. A. Remers, J. Med. Chem., 1987, 30, 1313–1321 CrossRef CAS PubMed ;
(b) H. N. Kim, J. Lim, H. N. Lee, J.-W. Ryu, M. J. Kim, J. Lee, D.-U. Lee, Y. Kim, S.-J. Kim, K. D. Lee, H.-S. Lee and J. Yoon, Org. Lett., 2011, 13, 1314–1317 CrossRef CAS PubMed ;
(c) R. F. Pittillo and C. Woolley, Appl. Environ. Microbiol., 1969, 18, 519–521 CAS .
- J. Yellol, S. A. Pérez, A. Buceta, G. Yellol, A. Donaire, P. Szumlas, P. J. Bednarski, G. Makhloufi, C. Janiak, A. Espinosa and J. Ruiz, J. Med. Chem., 2015, 58, 7310 CrossRef CAS PubMed .
- A. Romero, T. Caldés, E. Díaz-Rubio and M. Martín, Clin. Transl. Oncol., 2012, 14, 163–168 CrossRef CAS PubMed .
-
(a) M. Varache-Lembège, S. Moreau, S. Larrouture, D. Montaudon, J. Robert and A. Nuhrich, Eur. J. Med. Chem., 2008, 43, 1336–1343 CrossRef PubMed ;
(b) H. A. Abdel-Aziz, T. Aboul-Fadl, A.-R. M. Al-Obaid, M. Ghazzali, A. Al-Dhfyan and A. Contini, Arch. Pharmacal Res., 2012, 35, 1543–1552 CrossRef CAS PubMed .
-
(a) M. Alagesan, P. Sathyadevi, P. Krishnamoorthy, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2014, 43, 15829–15840 RSC ;
(b) E. Singleton and H. E. Swanepoel, Inorg. Chim. Acta, 1982, 57, 217–221 CrossRef CAS .
-
(a) M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC ;
(b) M. A. Bennett, T. N. Huang, T. W. Matheson, A. K. Smith, S. Ittel and W. Nickerson, Inorg. Synth., 1982, 74–78 CrossRef CAS .
- S. Mondal, C. Das, B. Ghosh, B. Pakhira, A. J. Blake, M. G. B. Drew and S. K. Chattopadhyay, Polyhedron, 2014, 80, 272–281 CrossRef CAS .
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed .
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS .
-
(a) R. J. Butcher, J. Jasinski, G. M. Mockler and E. Sinn, J. Chem. Soc., Dalton Trans., 1976, 1099–1102 RSC ;
(b) R. N. Prabhu and R. Ramesh, RSC Adv., 2012, 2, 4515–4524 RSC .
- K. N. Kumar, G. Venkatachalam, R. Ramesh and Y. Liu, Polyhedron, 2008, 27, 157–166 CrossRef CAS .
-
(a) A. Juris, L. Prodi, A. Harriman, R. Ziessel, M. Hissler, A. El-ghayoury, F. Wu, E. C. Riesgo and R. P. Thummel, Inorg. Chem., 2000, 39, 3590–3598 CrossRef CAS PubMed ;
(b) J. V. Ortega, K. Khin, W. E. van der Veer, J. Ziller and B. Hong, Inorg. Chem., 2000, 39, 6038–6050 CrossRef CAS PubMed ;
(c) R. N. Prabhu, D. Pandiarajan and R. Ramesh, J. Organomet. Chem., 2009, 694, 4170–4177 CrossRef CAS .
-
(a) M. U. Raja and R. Ramesh, J. Organomet. Chem., 2012, 699, 5–11 CrossRef CAS ;
(b) M. Kalidasan, R. Nagarajaprakash, S. Forbes, Y. Mozharivskyj and K. M. Rao, Z. Anorg. Allg. Chem., 2015, 641, 715–723 CrossRef CAS .
-
(a) F. Marchetti, C. Pettinari, R. Pettinari, A. Cerquetella, C. Di Nicola, A. Macchioni, D. Zuccaccia, M. Monari and F. Piccinelli, Inorg. Chem., 2008, 47, 11593–11603 CrossRef CAS PubMed ;
(b) D. Pandiarajan and R. Ramesh, J. Organomet. Chem., 2013, 723, 26–35 CrossRef CAS .
-
(a) J. Valladolid, C. Hortiguela, N. Busto, G. Espino, A. M. Rodriguez, J. M. Leal, F. A. Jalon, B. R. Manzano, A. Carbayo and B. Garcia, Dalton Trans., 2014, 43, 2629–2645 RSC ;
(b) R. Pettinari, C. Pettinari, F. Marchetti, B. W. Skelton, A. H. White, L. Bonfili, M. Cuccioloni, M. Mozzicafreddo, V. Cecarini, M. Angeletti, M. Nabissi and A. M. Eleuteri, J. Med. Chem., 2014, 57, 4532–4542 CrossRef CAS PubMed .
-
(a) V. Vajpayee, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Chem. Commun., 2011, 47, 5184–5186 RSC ;
(b) M. Kubanik, H. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658–5668 CrossRef CAS ;
(c) R. K. Gupta, G. Sharma, R. Pandey, A. Kumar, B. Koch, P.-Z. Li, Q. Xu and D. S. Pandey, Inorg. Chem., 2013, 52, 13984–13996 CrossRef CAS PubMed ;
(d) S. Mukhopadhyay, R. K. Gupta, R. P. Paitandi, N. K. Rana, G. Sharma, B. Koch, L. K. Rana, M. S. Hundal and D. S. Pandey, Organometallics, 2015, 34, 4491–4506 CrossRef CAS .
-
(a) A. Habtemariam, M. Melchart, R. Fernández, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef CAS PubMed ;
(b) R. E. Morris, R. E. Aird, P. del Socorro Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed .
-
(a) É. A. Enyedy, G. M. Bognár, T. Kiss, M. Hanif and C. G. Hartinger, J. Organomet. Chem., 2013, 734, 38–44 CrossRef ;
(b) J. Ruiz, V. Rodríguez, N. Cutillas, A. Espinosa and M. J. Hannon, Inorg. Chem., 2011, 50, 9164–9171 CrossRef CAS PubMed .
-
(a) L. He, S.-Y. Liao, C.-P. Tan, R.-R. Ye, Y.-W. Xu, M. Zhao, L.-N. Ji and Z.-W. Mao, Chem. – Eur. J., 2013, 19, 12152–12160 CrossRef CAS PubMed ;
(b) Q. Wu, C. Fan, T. Chen, C. Liu, W. Mei, S. Chen, B. Wang, Y. Chen and W. Zheng, Eur. J. Med. Chem., 2013, 63, 57–63 CrossRef CAS PubMed ;
(c) R. Pettinari, C. Pettinari, F. Marchetti, B. W. Skelton, A. H. White, L. Bonfili, M. Cuccioloni, M. Mozzicafreddo, V. Cecarini, M. Angeletti, M. Nabissi and A. M. Eleuteri, J. Med. Chem., 2014, 57, 4532–4542 CrossRef CAS PubMed ;
(d) Q. Wu, K. Zheng, S. Liao, Y. Ding, Y. Li and W. Mei, Organometallics, 2016, 35, 317–326 CrossRef CAS ;
(e) M. Hanif, S. Meier, A. Nazarov, J. Risse, A. Legin, A. Casini, M. A. Jakupec, B. K. Keppler and C. G. Hartinger, Front. Chem., 2013, 1–7 CAS .
- R. K. Gupta, R. Pandey, G. Sharma, R. Prasad, B. Koch, S. Srikrishna, P.-Z. Li, Q. Xu and D. S. Pandey, Inorg. Chem., 2013, 52, 3687–3698 CrossRef CAS PubMed .
-
(a) Z.-F. Chen, Q.-P. Qin, J.-L. Qin, Y.-C. Liu, K.-B. Huang, Y.-L. Li, T. Meng, G.-H. Zhang, Y. Peng, X.-J. Luo and H. Liang, J. Med. Chem., 2015, 58, 2159–2179 CrossRef CAS PubMed .
-
(a) J. P. Johnpeter, G. Gupta, J. M. Kumar, G. Srinivas, N. Nagesh and B. Therrien, Inorg. Chem., 2013, 52, 13663–13673 CrossRef CAS PubMed ;
(b) B. Banik, K. Somyajit, G. Nagaraju and A. R. Chakravarty, Dalton Trans., 2014, 43, 13358–13369 RSC .
- R. M. Lord, A. J. Hebden, C. M. Pask, I. R. Henderson, S. J. Allison, S. L. Shepherd, R. M. Phillips and P. C. McGowan, J. Med. Chem., 2015, 58, 4940–4953 CrossRef CAS PubMed .
-
(a) H.-W. An, S.-L. Qiao, C.-Y. Hou, Y.-X. Lin, L.-L. Li, H.-Y. Xie, Y. Wang, L. Wang and H. Wang, Chem. Commun., 2015, 51, 13488–13491 RSC ;
(b) B. Naeye, H. Deschout, M. Roding, M. Rudemo, J. Delanghe, K. Devreese, J. Demeester, K. Braeckmans, S. C. De Smedt and K. Raemdonck, Biomaterials, 2011, 32, 9120–9127 CrossRef CAS PubMed ;
(c) K. Purkait, S. Chatterjee, S. Karmakar and A. Mukherjee, Dalton Trans., 2016, 45, 8541–8555 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1477541. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00197a |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.