
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine uptake and enhanced electrical conductivity in a porous coordination polymer based on cucurbit[6]uril†
Jing-Xiang
Lin
ab,
Jun
Liang
a,
Ji-Fei
Feng
a,
Bahar
Karadeniz
c,
Jian
Lü
*ac and
Rong
Cao
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China. E-mail: lujian05@fjirsm.ac.cn; rcao@fjirsm.ac.cn
bThe School of Ocean Science and Biochemistry Engineering, Fuqing Branch of Fujian Normal University, Fuqing, 350300, The People's Republic of China
cSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK
First published on 7th September 2016
Abstract
A porous coordination polymer [(Na2I2CB[6])·8H2O]n (complex 1; CB[6] = cucurbit[6]uril), which absorbs gaseous iodine molecules and forms a polyiodide containing material I2@1, has been prepared and used as an absorbent for iodine uptake. The formation of halogen bonds between discrete iodide ions and iodine molecules has been demonstrated as predominant driving forces for iodine adsorption. Moreover, complex 1 demonstrates enhanced electrical conductivity upon iodine uptake, thanks to the formation of the polyiodide structure. This observation indicates that the discrete iodide matrix in the crystal lattice is an ideal and preferential molecular dock for iodine accommodation.
Introduction to the international collaborationThe collaboration between the Fujian Institute of Research on the Structure of Matter (FJIRSM) Chinese Academy of Sciences (CAS), and the University of Nottingham (UoN) has been initiated through an International Exchange Scheme (2011 China Costshare project) cofunded by the National Science Foundation of China (NSFC) and the Royal Society (RS). A following research grant from the RS and Sino-British Fellowship Trust (SBFT) has been offered to further consolidate the collaborative research between the two parties (2012 and 2013). More recently, the active and fruitful collaboration of FJIRSM (CAS) and UoN has been rewarded by a successful Key International Collaboration Scheme from NSFC (2014–2018). This contribution dedicated to “Sino-European” collaboration themed collection has been a snapshot of the collaborative research projects based on crystalline porous molecular materials and their applications in molecular storage & separation and beyond. |
Introduction
Iodine encapsulation has been widely studied with porous coordination polymers (PCPs)1–4 for many applications, such as nuclear waste management,5,6 luminescence,2 magnetism7 and electrical conductivity.1 It has been demonstrated that both molecular porosity and preferential binding sites in porous materials play crucial roles in determining the efficiency and capacity for iodine adsorption. Volkringer and co-workers have reported a systematic study on iodine adsorption by using metal–organic frameworks (MOFs),6 in which the pore functionality of MOF materials, that is decorating groups with various polarity and electron-donating ability, has been widely studied. Consequently, MOF materials with pore functionality of electron-donating groups, for example –OH and –NH2, typically exhibit high adsorption efficiency for iodine molecules. In this context, PCP materials with discrete iodide moieties could be an excellent choice to realize high iodine adsorption efficiency, as it is well-known that electron-rich iodide moieties can serve as electron-donating groups to bind iodine molecules via strong halogen bonds to form polyiodides,8 in which iodide serves as a halogen bond acceptor and iodine is the halogen bond donor.9 At the same time, the study of polyiodide containing PCPs generates wider research interest in electrical conductive materials.Cucurbit[n]urils (abbreviated as CB[n]) are a new class of macrocyclic cavitants with hydrophobic cavity, which can be accessed via two carbonyl-group-fringed portals. Electrostatic potential surface calculation has revealed that the portal regions are electron-rich and become negative, while the outer surface is more positive.10 Investigations have shown that the carbonyl-fringed portals of CB[n] molecules can coordinate to metal cations, preferably to alkali and alkaline earth metal ions.11–13 Its outer surface is proved to be able to interact with anionic and electron rich species, which would provide binding sites to anchor the iodide ions.14 These features make cucurbit[n]uril an ideal candidate to prepare iodide containing CPCs for iodine adsorption, and for superior electrical conductivity properties. We thus design a hybrid system of CB[n] and alkali iodide, in which the alkali cations are expected to bind to the carbonyl-fringed portals of CB[n] through the ion–dipole interaction, while iodide will interact with the positive region of CB[n] (outer surface). In such a way, PCP materials containing discrete iodide moieties can be successful.
In this work, cucurbit[6]uril (CB[6]) as one of the most studied members in the CB[n] family has been employed in the synthesis of iodide containing PCPs. The hydrothermal reaction of CB[6] and sodium iodide affords a crystalline product in high yield. Single crystal structural analysis determines a crystalline PCP material with the formula [(Na2I2CB[6])·8H2O]n (1). As expected, both portals of a CB[6] are coordinated by sodium ions through the ion–dipole interaction (Fig. 1a). These {Na2CB[6]} units are stacked on top of each other in a portal-to-portal fashion, leading to the formation of a supramolecular column. Every four of such paralleled columns construct a 1D channel along the b-axis (Fig. 1c). Within the channels, discrete iodides were encapsulated. The distances between neighboring iodides are 5.49 Å and 6.25 Å, respectively (Fig. 1b). Although these discrete iodide ions in the crystal lattice may not position ideally to anchor iodine molecules, the iodide matrix is able to undergo a dynamic change within the channels to fit iodine molecules in between iodides. Size matching and structural complementarity are crucial for both supramolecular interaction and molecular assembly. This study offers an unusual example to study the interaction of iodides and iodine within a crystal lattice.
 | ||
| Fig. 1 A view of (a) the 1D sodium-CB[6] coordination chain subunit; (b) the discrete iodide ions; and (c) the 3D packing pattern of complex 1, showing alternative 1D sodium-CB[6] chains and the iodide ion matrix. Colour code: C, dark grey; H, green; N, blue; O, red; I, purple; Na, orange. Discrete water molecules were omitted for clarity. | ||
Experimental
Chemicals and materials
All chemicals and reagents were purchased from commercial suppliers (Sigma Aldrich) and used without further purification except for cucurbit[6]uril (CB[6]) which was prepared and purified according to a procedure reported in the literature.15Characterization
Elemental analyses of C, H, and N were carried out on an Elementar Vario EL III analyzer. Raman spectra were collected in the region of 500–100 cm−1 with a Thermo Nicolet Nexus 870 spectrometer equipped with a FT-Raman module (Laser exciting line: Nd-YAg laser emitting at 785 nm). Thermogravimetric analyses (TGAs) were performed under a flow of N2 (100 mL min−1) with a heating rate of 10 °C min−1 using a TA SDT-600 thermogravimetric analyzer. Aluminum oxide crucibles were used for all samples, and the instrument was calibrated using indium as the standard. An empty crucible was used as the reference. A Miniflex600 powder diffractometer was employed for all PXRD measurements with experimental parameters as follows: room temperature, Cu-Kα radiation (λ = 1.54056 Å), reflection mode, 2θ range 5–30°, and step size 0.02° (scanning rate with 2θ at 1° per minute). Electrical conductivity measurements were performed in a quasi-four-electrode direct current (DC) with an online multi-meter (Model 2000, Digital Multimeter, Keithley Instruments Inc., USA).Synthesis of [(Na2I2CB[6])·8H2O]n (1)
Cucurbit[6]uril (20 mg, about 0.02 mmol) and sodium iodide (30 mg, 0.2 mmol) were mixed in 6.0 mL water in a Teflon vessel. The resulting solution was sealed in an autoclave, heated to 120 °C for 3 days and then cooled to room temperature within one day. Colourless rod shape crystals were obtained. The product was filtered and washed with water and dried in air. Yield ca. 90%. Elemental analysis: Calcd for C36H52N24O20Na2I2 (Mr = 1440.7): C, 30.01; H, 3.64; N, 23.33. Found: C, 30.08; H, 4.13; N, 23.38.Crystallography
A crystal of complex 1 suitable for single crystal X-ray diffraction analysis was selected using a polarized light optical microscope. X-ray diffraction data were collected on an Agilent SuperNova diffractometer using Cu-Kα radiation (λ = 1.54184 Å). The structure was solved by direct methods and developed by difference Fourier techniques, both using the SHELXL software package.16 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms on cucurbit[6]urils were placed in geometrically calculated positions and included in the refinement process using a riding model. CIF files contain crystallographic data (CCDC 1472736).Results and discussion
Structural description
X-ray crystallography reveals that complex 1 crystallizes in the orthorhombic space group Pnnm. In the structure of 1, each Na+ cation bind to three carbonyl groups (Na–Ocarbonyl of 2.492(14) Å) and four aqua ligands (Na–Ow of 2.56(3) and 2.13(4) Å). Two of such sodium ions form dimer units by sharing the aqua ligands and connect with CB[6] moieties into a 1D coordination chain structure (Fig. 1a). Discrete iodide anions located in the channel regions are surrounded by {Na2CB[6]} chains, playing both space-filling and charge-compensating roles (Fig. 1c). Separations between two neighboring I− in the iodide matrix are 5.434 Å and 6.185 Å, respectively. In order to insert an I2 molecule between two iodide ions in the form of I−⋯I2⋯I−, the ideal distance of I−⋯I− units is expected to be longer than ca. 10 Å.9 However, the iodides in the lattice of complex 1 are discrete and somehow able to undergo dynamic movement in the ionic channel to fit iodine molecules, resulting in successful insertion of iodine into the iodide matrix. Thus complex 1 can be an unusual example for I2 accommodation via an adjustable I−-containing crystal lattice.X-ray powder diffraction and thermal analysis
The phase purity of complex 1 was identified by powder X-ray diffraction (PXRD) study. The major diffraction peaks (below 20°) of the experimental PXRD pattern match well with the ones simulated from the single crystal data of 1, indicating the phase purity of bulk products (Fig. S3; ESI†). The slight shifts of peaks at high angles may originate from the different collection of diffraction data (room temperature versus 100 K). The thermal stability of complex 1 was studied by thermal analysis in the temperature range of 30–800 °C. As shown in Fig. S2 (ESI†), the first and second weight loss is assigned to the release of lattice water and the coordinated water molecules on sodium ions (experimental 12.53%, calculated 10.0%). The higher weight loss in experimental may be attributable to the loss of water molecules (mainly before 100 °C) absorbed on the surface of the sample. Upon further heating, complex 1 decomposes at a temperature of ca. 450 °C by decomposing and releasing the organic component CB[6], which is in agreement with the thermal stability of the cucurbit[6]uril reported in the literature.10 The thermal stability of complex 1 has been changed significantly upon iodine adsorption. The TG curve of I2@1 (Fig. S2; ESI†) shows continuous weight loss in the temperature range of 30 °C–800 °C. The weight loss can be roughly divided into three stages. The first weight loss from 30 to 100 °C is preliminarily assigned to the release of lattice water, adhesive of water and iodine molecules on the surface of the material. The second weight loss, which is about 25% from 150 °C to 350 °C, can be ascribed to the decomposition of polyiodide and release of iodine. The weight loss matches roughly with the iodine uptake experiment (see the following section), which implies that the iodine adsorption is reversible. Further weight loss after 400 °C can be attributed to the decomposition and release of CB[6].Iodine molecule uptake
In order to evaluate the adsorption efficiency for iodine molecules, complex 1 was used as an absorbent and exposed to the spontaneous vapor of iodine in a sealed vial. The colorless bulk sample of 1 underwent a rapid color change into a brown solid (within 5 seconds) upon I2 vapor exposure, and further into a black colored solid I2@1 after 90 minutes. A rapid color change, from white to brown, can be observed after as short as 5 seconds of exposure. Upon further exposure, the material demonstrates a continuous color change, with brown at two minutes which changes to black after one and a half hour. The color change of the complex is an indication of iodine adsorption and the formation of polyiodide units within the solid material I2@1. After iodine uptake, the weight of the iodide-containing material has increased by ca. 25 wt%, which corresponds to ca. 1.4I2 molecules per formula unit of [(Na2I2CB[6])·8H2O]. The experimental value is in agreement with the ideal value of 1.5 when every two adjacent iodides bind to one iodine molecule. The small discrepancy could be attributable to the shorter distance between iodides observed in the crystal lattice of complex 1, as stated earlier, or possibly due to limited channel space, i.e. the channel is insufficient to allow ideally full iodine binding with the polyiodide units. These results demonstrate that the discrete iodide matrices within the molecular complex are sensitive to iodine vapor and perform fast iodine adsorption, which could promise potential application in nuclear waste treatment where the radioactive gaseous iodine byproduct was released during nuclear fuel reprocessing.5 While, in the iodine uptake experiment, the color of the crystalline sample underwent a drastic change, the shape of crystals was retained, as shown in Fig. 2. The crystallinity of the material has been lost upon iodine adsorption, as demonstrated by PXRD (Fig. S3 in the ESI†). This is, however, evidence confirming that dynamic movement may occur in the crystal lattice, i.e. expansion of I−⋯I− units to allow iodine accommodation. | ||
| Fig. 2 Colour change in the samples of complex 1 upon iodine exposure into I2@1 solid material: (a) at 5 seconds; (b) at 2 minutes; (c) at one and a half hour; and (d) amplification of the picture (c). | ||
Raman spectroscopy
Raman spectroscopy has been used as a useful diagnostic technique in investigating the nature of polyiodides. It could give information about the valence state of the iodine molecule present in polyiodide containing materials. It offers evidence on how iodine molecules are bonded with iodides. In the Raman shift region of 500–100 cm−1, the spectrum of I2@1 (Fig. 3) shows a prominent band peaked at 164 cm−1, which can be assigned to the νI–I symmetric stretch of the iodine molecules that are perturbed by iodide anions on each end via halogen bonding interactions. This proves that the majority of iodine molecules adsorbed into complex 1 are present as iodine molecules and are halogen-bonded by iodide anions, forming a polyiodide structure of [I−⋯I2⋯I−]n. As it is reported that solid iodine has a Raman-active νI–I mode at 180 cm−1, when I2 becomes coordinated to an electron donor, the electron density is donated into the σ* anti-bonding orbital of iodine and the force constant is reduced, resulting in the movement of the νI–I mode to lower wavenumbers.17 Beside this major peak, the minor bands at 112 cm−1 could be assigned to the ν1 symmetrical stretching modes of discrete, linear, and asymmetric triiodide ions.18 This observation means that a small amount of iodine molecules is bonded with iodide to form triiodides I3−. The ν3 antisymmetrical stretching of these triiodides should appear around 142 cm−1, which overlaps with the enormous band at 164 cm−1.18 The asymmetric shape of the major bands in the spectrum corresponds well to the possible forms of interactions that determine both the successful adsorption of I2 in complex 1 and the nature of chemisorption in this process.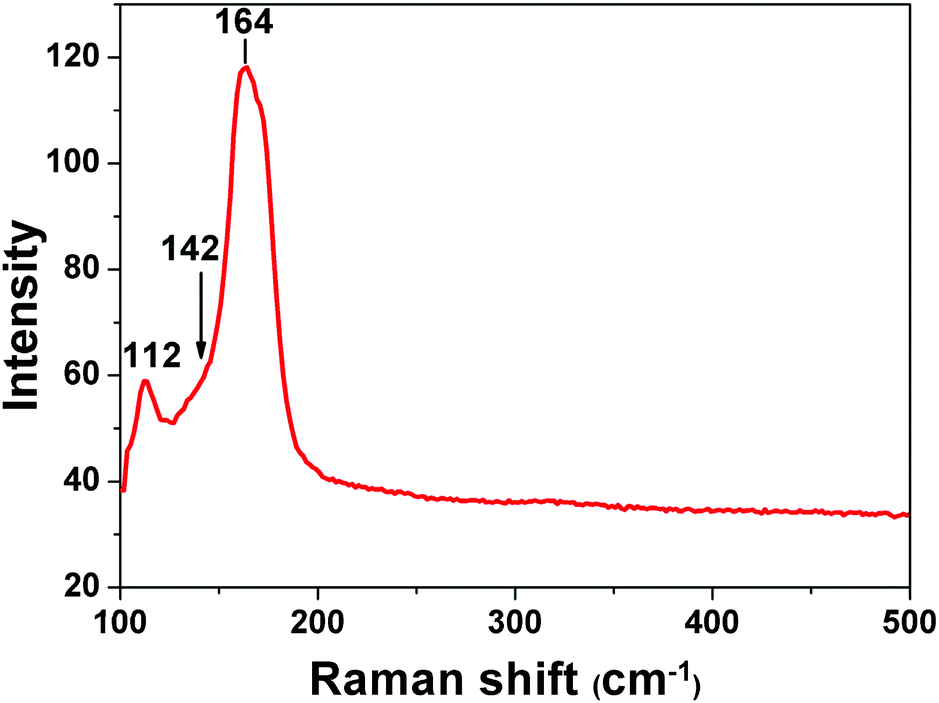 | ||
| Fig. 3 Raman spectrum of I2@1. | ||
Electrical conductivity
The powdered sample of I2@1 was compressed to a pellet with 0.6 mm thickness and 2.5 mm diameter under a presser of 10 MPa. Two sides of the pellet were connected to gold wires (0.04 mm in diameter) by means of gold paste. Electrical resistances of the sample pellet were recorded at room temperature. The electrical conductivity of the sample was deduced from the electrical resistances. The electrical conductivity value (σ) of 7.46 × 10−7 S cm−1 for I2@1 (roughly 10 times the generally accepted value of 7.69 × 10−8 S cm−1 for solid iodine) was obtained.The solid material indeed I2@1 shows enhanced electrical conductivity which demonstrates the initial hypothesis of favorable electron transportation by I2 insertion. The enhanced conductivity of I2@1 is amazingly higher than most of the I2 doped materials except the one documented for a polyiodide containing MOF material, though it is not available for all cases observed by the Zeng group,19 which is somehow beyond comparison for measurement with a single crystal (in their case) compared to that with a bulk phase material (in our study). On the other hand, rational explanations for the conductivity of I2@1 could be the unfavorable charge transportation within the material, which does not, in certain circumstances, follow an electron hopping mechanism over an ion transport mechanism as expected,20,21i.e. a Grotthuss-like charge transfer mechanism of I− + I2 → [I⋯I–I]− ↔ [I–I⋯I]− → I2 + I− or I3− + I2 → [I–I–I⋯I–I]− ↔ [I–I⋯I–I–I]− → I2 + I3− for electron conductivity. In this mechanism, charge transfer requires a high (tri)iodide/iodine packing density in a linear arrangement to guarantee an electronic interaction between iodide, triiodide and iodine without long diffusion lengths;9,22,23 moreover, the unsatisfied crystallinity of I2@1 or the presence of multiple polyiodide units formed in the material i.e. [I−⋯I2⋯I−]n and [I−⋯I2]n is another possible reason for unfavorable charge transportation and for holding back the further enhancement of electrical conductivity. Therefore, the charge transport in the material disfavors an ideal electron hopping mechanism as expected to achieve considerably high conductivity. With these in mind, we sought to seek for more stable porous materials containing a discrete iodide matrix for enhanced iodine adsorption and electrical conductivity.
Conclusions
A crystalline porous coordination polymer (complex 1) containing discrete iodide ions has been prepared by using cucurbit[6]uril and sodium iodide. The iodine uptake using complex 1 as an absorbent demonstrates that discrete iodide ions in the solid complex are ideal binding sites for iodine molecules, leading to the formation of the polyiodide containing material I2@1. Possible binding modes have been rationalized using Raman spectroscopy. Furthermore, the I2@1 material exhibits enhanced electrical conductivity upon I2 adsorption. This strategy could generate general research interest in the preparation of polyiodide containing solid materials for applications in nuclear waste management and electrical conductivity.Acknowledgements
We are grateful for financial support from the 973 Program (Grants 2012CB821705 and 2014CB845605), the NSFC (Grants 21401109, 21450110413, and 21571177), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDA09030), and the State Key Laboratory of Structural Chemistry (Grant 20150023).References
- M.-H. Zeng, Q.-X. Wang, Y.-X. Tan, S. Hu, H.-X. Zhao, L.-S. Long and M. Kurmoo, J. Am. Chem. Soc., 2010, 132, 2561–2563 CrossRef CAS PubMed.
- L.-L. Lv, J. Yang, H.-M. Zhang, Y.-Y. Liu and J.-F. Ma, Inorg. Chem., 2015, 54, 1744–1755 CrossRef CAS PubMed.
- Y. Rachuri, K. K. Bisht and E. Suresh, Cryst. Growth Des., 2014, 14, 3300–3308 CAS.
- S. Parshamoni, S. Sanda, H. S. Jena and S. Konar, Chem. – Asian J., 2015, 10, 653–660 CrossRef CAS PubMed.
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897–8899 CrossRef CAS PubMed.
- C. Falaise, C. Volkringer, J. Facqueur, T. Bousquet, L. Gasnot and T. Loiseau, Chem. Commun., 2013, 49, 10320–10322 RSC.
- Z. M. Wang, Y. J. Zhang, T. Liu, M. Kurmoo and S. Gao, Adv. Funct. Mater., 2007, 17, 1523 CrossRef CAS.
- P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649–1684 CrossRef CAS PubMed.
- A. Abate, M. Brischetto, G. Cavallo, M. Lahtinen, P. Metrangolo, T. Pilati, S. Radice, G. Resnati, K. Rissanene and G. Terraneo, Chem. Commun., 2010, 46, 2724–2726 RSC.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J. K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- W.-J. Chen, D.-H. Yu, X. Xiao, Y.-Q. Zhang, Q.-J. Zhu, S.-F. Xue, Z. Tao and G. Wei, Inorg. Chem., 2011, 50, 6956–6964 CrossRef CAS PubMed.
- J. Lü, J.-X. Lin, M.-N. Cao and R. Cao, Coord. Chem. Rev., 2013, 257, 1334–1356 CrossRef.
- X.-L. Ni, X. Xiao, H. Cong, Q.-J. Zhu, S.-F. Xue and Z. Tao, Acc. Chem. Res., 2014, 47, 1386–1395 CrossRef CAS PubMed.
- D. Bardelang, K. A. Udachin, D. M. Leek, J. C. Margeson, G. Chan, C. I. Ratcliffe and J. A. Ripmeester, Cryst. Growth Des., 2011, 11, 5598–5614 CAS.
- G. M. Sheldrick, SHELXS97, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Congeduti, M. Nardone and P. Postorino, Chem. Phys., 2000, 256, 117–123 CrossRef CAS.
- J. Lin, J. Martí-Rujas, P. Metrangolo, T. Pilati, S. Radice, G. Resnati and G. Terraneo, Cryst. Growth Des., 2012, 12, 5757–5762 CAS.
- Z. Yin, Q.-X. Wang and M.-H. Zeng, J. Am. Chem. Soc., 2012, 134, 4857–4863 CrossRef CAS PubMed.
- S. Cukierman, Biochim. Biophys. Acta, 2006, 1757, 876–885 CrossRef CAS PubMed.
- R. Kawano and M. Watanabe, Chem. Commun., 2005, 16, 2107–2109 RSC.
- L. A. Bengtsson, H. Stegemann, B. Holmberg and H. Füllbier, Mol. Phys., 1991, 73, 283–296 CrossRef CAS.
- H. Stegemann, A. Rohde, A. Rekhe, A. Schnittke and H. Füllbier, Electrochim. Acta, 1992, 37, 379–383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Packing pattern, TGA, and PXRD analyses of complex 1. CCDC 1472736. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00305b |
| This journal is © the Partner Organisations 2016 |