
Stereodivergent and enantioselective total syntheses of isochaetominines A–C and four pairs of isochaetominine C enantiomers: a six-step approach†
Zhong-Yi
Mao
a,
Hui
Geng
a,
Tian-Tian
Zhang
a,
Yuan-Ping
Ruan
a,
Jian-Liang
Ye
a and
Pei-Qiang
Huang
*ab
aDepartment of Chemistry, Fujian Provincial Key Laboratory of Chemical Biology, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, P. R. China. E-mail: pqhuang@xmu.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 30th October 2015
Abstract
The first enantioselective and stereodivergent total syntheses of (−)-isochaetominines A–C and all eight 2,3-cis-stereoisomers of (−)-isochaetominine C, including the natural (+)-14-epi-isochaetominine C, and the proposed structures of (−)-pseudofischerine (2) and (−)-aniquinazoline D (3), have been achieved. The stereodivergent approach relies on the DMDO-initiated divergent tandem reaction to give a separable mixture of two products, a monocyclization product and a diastereomer of isochaetominine C (or a homologue) as a result of double cyclization. An epimerization-free two-step protocol has been developed for the highly diastereoselective transformation of the former product into an isochaetominine-type compound with characteristic 3,14-cis-stereochemistry. As a result of our synthetic efforts, the structures of the natural (−)-pseudofischerine and (−)-aniquinazoline D have been revised both as (−)-isochaetominine C (6).
Introduction
The traditional total synthesis of natural products is target-oriented synthesis (TOS).1 In this context, a synthetic route is designed for a specific target molecule with a defined skeleton, functionality and stereochemistry.2 Many natural products co-exist with their diastereomers.3 Under these circumstances, the total synthesis becomes the construction of multiple diastereomeric targets. Efficient access to different diastereomers of a natural product and/or a bioactive molecule possessing multi-stereocenters is imperative for both structure confirmation4 and the study of structure–activity-relationships.5 Traditional approaches to synthesize different enantioenriched diastereomers of a natural product frequently require the use of different starting materials and/or synthetic routes.6 In addition, not all diastereomers are accessible by a certain method.6 The stereodivergent synthesis7 has been evolved as an efficient approach to access different naturally occurring diastereomers of natural products.8 Among many approaches4–9 that have been developed for the stereodivergent synthesis, the strategy based on the non-selective reactions has attracted less attention.9Non-selective reactions that produce two or more isomeric products in about equal amounts are generally considered to be useless in organic synthesis. However, by carefully planning, the employment of a non-selective reaction in a synthetic route may lead to an efficient stereodivergent approach for the multiple diastereomeric targets synthesis (MDTS). For example, if the isomers produced in a non-selective reaction can all be used for the synthesis of different diastereomeric targets, a non-selective reaction becomes advantageous for stereodivergent synthesis. The success of such a strategy depends on the efficient separation of the isomers, which is also true for other stereodivergent strategies. It is desirable that a non-selective reaction produces a quasi-equal amount of isomers, which is important for both the determination of stereochemistry of natural products4 and the synthesis of stereodiversified libraries.5
Endophytic fungi have emerged as a rich source of bioactive natural products.10 As a typical example, chaetominine-type alkaloids show interesting stereochemical and substituent diversity. The first member, (−)-chaetominine11 (1, Fig. 1) was first isolated from the solid-substrate culture of Chaetomium sp. IFB-E015, an endophytic fungus on apparently healthy Adenophora axilliflora leaves,11a and then from different endophytic fungi.11b,c After the first isolation by Tan et al. in 2006,11a several homologues and diastereomers of (−)-chaetominine have been isolated from different fungi, and characterized as (−)-pseudofischerine (2),12 (−)-aniquinazoline D (3),13 (−)-isochaetominines A–C (4–6),14 and (+)-14-epi-isochaetominine C (7),14 respectively. Thus, except 11-epi-isochaetominine C (8), all other 2,3-cis-diastereomers of isochaetominine C (6) have been reported to be natural products, each as an enantiomer. Moreover, the key structural feature of 14-epi-isochaetominine C (7) is also found in kapakahines [e.g. kapakahines B, F (9, 10)], a group of cyclic peptides isolated from the marine sponge Cribrochalina olemda.15
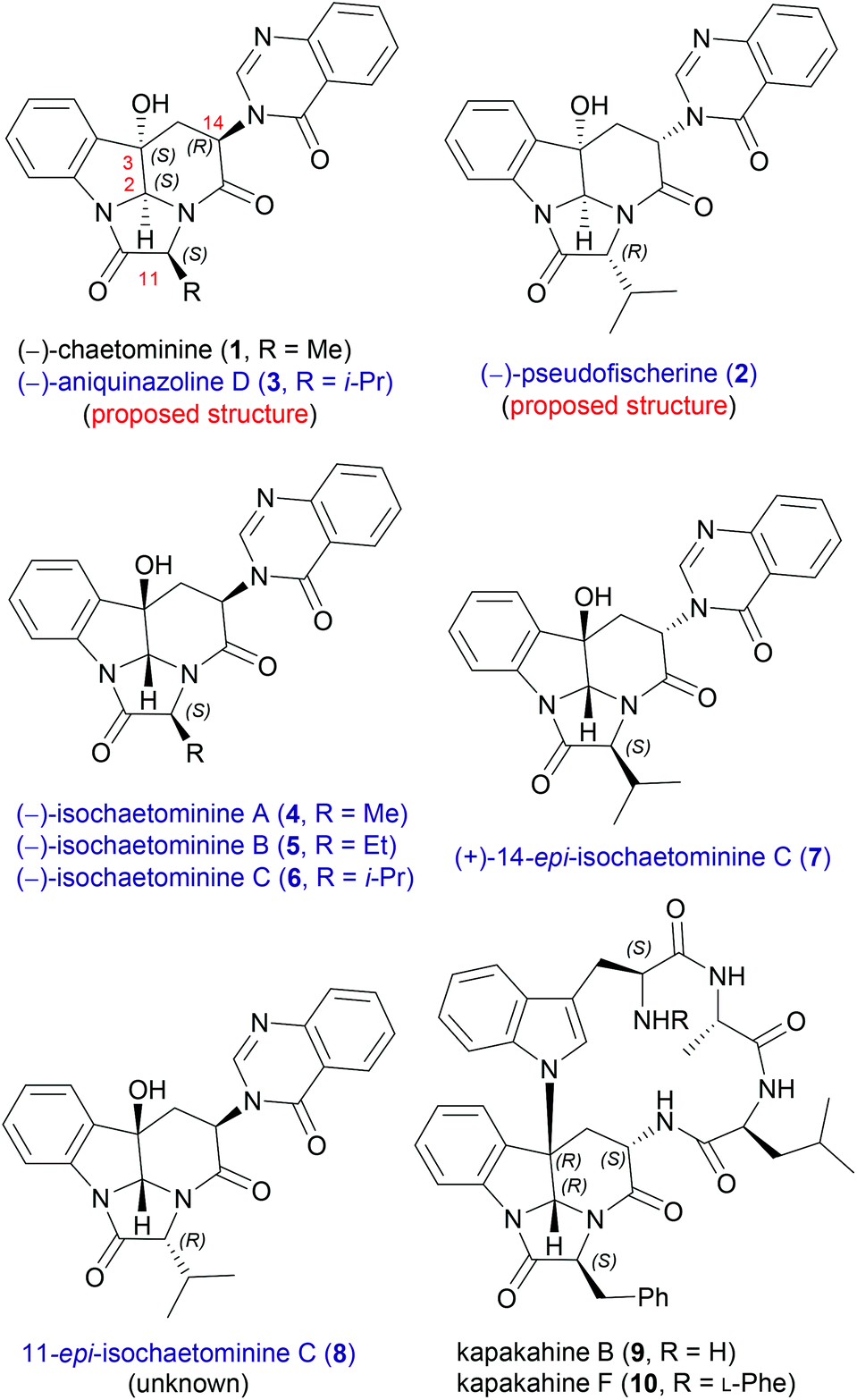 | ||
| Fig. 1 Structures of isochaetominine-type alkaloids and related compounds. | ||
This group of quinazolinone alkaloids16 feature the same singular hexacyclic structure and differ from each other on the stereochemistry and/or the substituent at C-11. The unprecedented framework makes chaetominine an ideal target for exploring novel synthetic strategies,17 which has resulted in several elegant approaches.18 However, to date, only the total syntheses of (−)-chaetominine (1) have been reported18 and synthetic studies on other members of isochaetominine-type alkaloids (2–7) have not appeared.
In connection with our longstanding interests in the development of procedure-economical methodologies for the synthesis of bioactive alkaloids,19 we have recently disclosed a four-step enantioselective total synthesis of (−)-chaetominine (1).18c,f–h We report herein the stereodivergent total synthesis of all of the eight 2,3-cis-diastereoisomers/enantiomers of isochaetominine C (6), and its congeners isochaetominines A and B (4 and 5).
In view of the diastereo- and substituent-diversity of the isochaetominine-type alkaloids, it is highly desirable to develop a unified strategy that is accessible to all the eight 2,3-cis-diastereomers/enantiomers. In our previous approach to (−)-chaetominine (1),18c,f–h the key epoxidation-triggered tandem cyclization reaction yielded two products as a result of non-stereoselective epoxidation. Although the non-selective reaction is unfavorable for the purpose of the TOS of 1, it offers an opportunity to develop an efficient diastereodivergent approach to all the reported 2,3-cis-diastereomers of isochaetominine-type alkaloids (2–7).
Results and discussion
On the basis of our previous strategy for the synthesis of (−)-chaetominine (1),18c,f–h the retrosynthetic analysis of isochaetominine-type alkaloids (2–7) is proposed and shown in Scheme 1. The key element of our strategy resided in the non-diastereoselective epoxidation of easily available quinazolinonyl dipeptide 12 (from D-Trp and L-Ala) to give two diastereomeric epoxides (no shown). The α-epoxide is expected to convert spontaneously to the proposed structure of aniquinazoline D (3) when R is the isopropyl group, while the β-epoxide will form the monocyclization product 11. Compound 11 is an ideal precursor for isochaetominines A–C (4–6). A similar route may be employed for the synthesis of (−)-pseudofischerine (2) from L-Trp and D-Ala. Finally, the syntheses of (+)-14-epi-isochaetominine C (7) would be realizable by the combination of L-Trp with L-Val or L-Ala. It is worth noting that the syntheses of (−)-pseudofischerine and (+)-14-epi-isochaetominine C will also afford other diastereomers 2,3,14-tri-epi-isochaetominine C (ent-8) and 11,14-di-epi-isochaetominine C (ent-3). In addition, the D-Trp and D-Ala combination would provide access to the hitherto unknown enantiomers (−)-14-epi-isochaetominine C (ent-7) and 11-epi-isochaetominine C (8).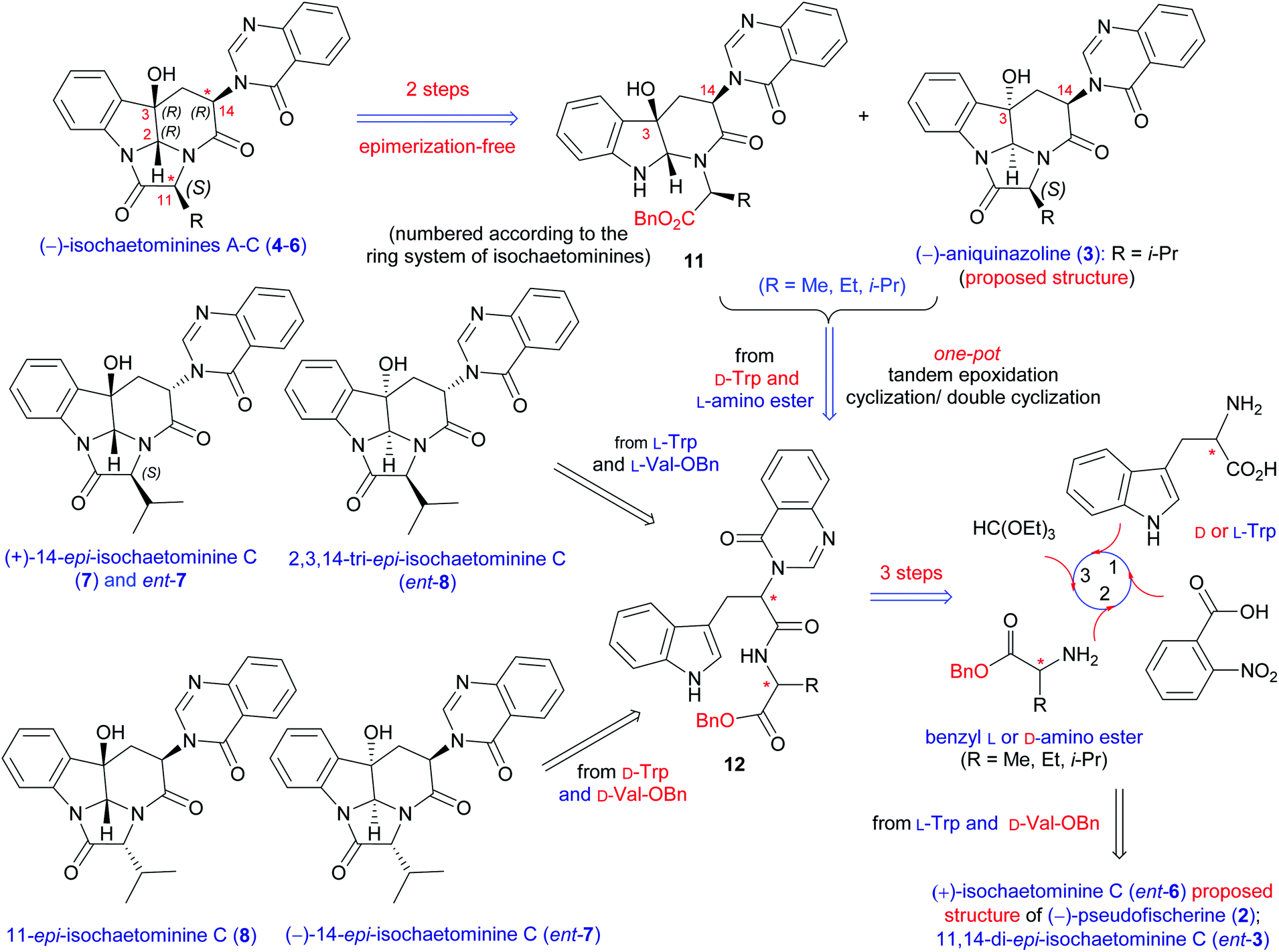 | ||
| Scheme 1 Retrosynthetic analysis of all eight 2,3-cis-stereoisomers of (−)-isochaetominine C and (−)-isochaetominines A, B. | ||
The previous results gained from our synthesis of (−)-chaetominine (1) revealed that only when the hydroxyl group at C-3 and the quinazolinonyl group at C-14 are anti-disposed can the final lactamization occur spontaneously (cf.11versus3 in Scheme 1). In addition, the stereocenter at C-14 is prone to epimerization (cf.11 in Scheme 1).18f–h In a related study, Roche and Tréguier observed during the synthesis of N-Phth-Trp-Phe-OC6F5 that racemization/epimerization was unavoidable.18i Because of this problem, they had to use a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 epimeric mixture of N-Phth-Trp-Phe-OC6F5 in their TOS of 2-fluoro-chaetominine. The subsequent Selectfluor-triggered double annulative cascade reaction resulted in the formation of the tetracyclic core as a mixture of four diastereomers in a ratio of 2:1:1:4 (combined yield: 42%), from which the overall C3–C14 cis/trans ratio was 5:3.18i Hence, the epimerization-free stereoselective formation of the 3,14-cis-stereochemistry found in (−)-pseudofischerine (2) and (−)-isochaetominines A–C (4–6) is challenging. To tackle the epimerization problem, the imidazolinone ring was envisioned to be formed from the corresponding carboxylic acid under mild conditions using a racemization-free peptide coupling reagent.20,21 Thus benzyl valinate was selected as an amino acid component hoping to cleave the benzyl group under epimerization-free conditions at a later stage.
:1 epimeric mixture of N-Phth-Trp-Phe-OC6F5 in their TOS of 2-fluoro-chaetominine. The subsequent Selectfluor-triggered double annulative cascade reaction resulted in the formation of the tetracyclic core as a mixture of four diastereomers in a ratio of 2:1:1:4 (combined yield: 42%), from which the overall C3–C14 cis/trans ratio was 5:3.18i Hence, the epimerization-free stereoselective formation of the 3,14-cis-stereochemistry found in (−)-pseudofischerine (2) and (−)-isochaetominines A–C (4–6) is challenging. To tackle the epimerization problem, the imidazolinone ring was envisioned to be formed from the corresponding carboxylic acid under mild conditions using a racemization-free peptide coupling reagent.20,21 Thus benzyl valinate was selected as an amino acid component hoping to cleave the benzyl group under epimerization-free conditions at a later stage.
We started our investigation by developing a diastereodivergent synthesis of the proposed structure of (−)-aniquinazoline D (3) and (−)-isochaetominine C (6). (−)-Aniquinazoline D (3) was isolated by Wang et al. from the culture of Aspergillus nidulans MA-143, an endophytic fungus obtained from the leaves of the marine mangrove plant Rhizophora stylosa.13 The relative stereochemistry was determined by the NOESY technique, while the absolute configuration of C-11 was elucidated by Marfey's method, which led them to assume the absolute configuration of (−)-aniquinazoline D (3) as 2S,3S,11S,14R. The authors also showed that aniquinazoline D exhibited potent lethality against brine shrimp (LD50 = 3.42 μM), much more effective than the positive control colchicine (LD50 = 88.4 μM). (−)-Isochaetominines A–C (4–6) and (+)-14-epi-isochaetominine C (7) were isolated very recently by Oh, Shin and co-workers from the solid-substrate culture of an Aspergillus sp. fungus collected from marine-submerged decaying wood from Korea.14 These alkaloids showed weak inhibition against Na+/K+-ATPase (IC50 = 78, 20, 38, and 57 μM, respectively).
The synthesis started from N-aroylation of D-tryptophan (Trp) (o-nitrobenzoyl chloride, THF, 1 M NaOH, 0 °C, 2 h)18f (Scheme 2). Coupling of the resulting (R)-14 with benzyl L-valinate p-toluenesulfonic acid salt [i-BuOCOCl/N-methylmorpholine (NMM), THF, 20 °C, 12 h] produced the dipeptide derivative 15c in 93% yield. Treatment of 15c with Zn/TiCl4 and trimethyl orthoformate in THF22 at 0 °C (ref. 18f) afforded the quinazolinone derivative 12c in 95% yield. We next carried out the key oxidative cyclization of 12c. Thus, epoxidation of 12c with dimethyldioxirane (DMDO)23 in acetone at −78 °C, followed by treatment of the resulting sensitive epoxide intermediates anti-16c and syn-16c with K2CO3 in MeOH at −15 °C, produced the proposed structure of (−)-aniquinazoline D (3) in 33% yield and the monocyclization product 11c in 41% yield. With 11c in hand, we were in a position to close the last ring to form 6. Since 11c is prone to epimerization because of the cis-stereochemistry of the OH group at C-3 and the quinazolinonyl group at C-14, a careful selection of reaction conditions for the subsequent transformations is required.18f–h Pleasingly, removal of the benzyl group of 11c under catalytic hydrogenation conditions (H2, 10% Pd/C, MeOH, r.t., 2 h) and subsequent lactam formation using Ye's coupling reagent (DEPBT)21 led to the formation of isochaetominine C (6) in 77% yield without noticeable epimerization. Interestingly, cyclization could also be promoted using oxalyl chloride–Hünig base (DIPEA) and a catalytic amount of DMF24 (CH2Cl2, −10 °C). The spectral data (1H and 13C NMR) and the sign of the optical rotation of our synthetic product are in full agreement with those reported for the natural product. However, our product, obtained in high purity as colorless crystals (m.p. 166–168 °C), displayed a lower value of specific rotation {[α]25D −71.0 (c 1.0, MeOH)} compared with that reported for the natural product {[α]25D −90 (c 0.6, MeOH)14}. These differences could be attributed to the minute quantities isolated from the natural source (a pale yellow amorphous solid, no m.p. data reported) and the relatively low purity of the isolated sample as indicated by the reported 1H and 13C NMR spectra.14
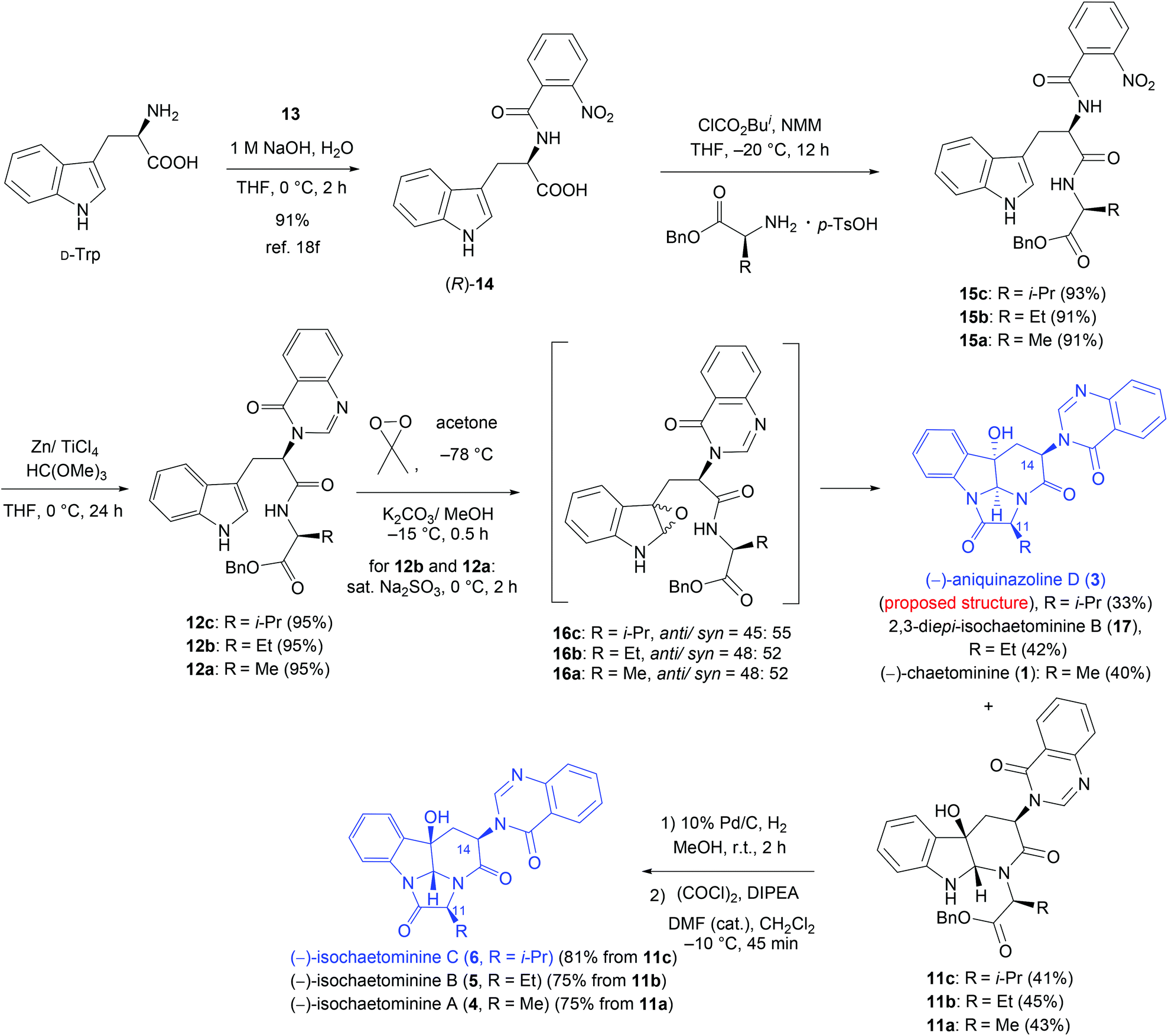 | ||
| Scheme 2 Diastereodivergent syntheses of (−)-isochaetominines A−C (4–6) and the proposed structure of (−)-aniquniazoline D (2). | ||
A careful comparison of the 1H and 13C NMR data of our synthetic product 3 with those reported for the natural (−)-aniquinazoline D showed that they are different. The structure of our synthetic product 3 {colorless crystals, m.p. 302–303 °C; [α]25D −50.7 (c 0.5, MeOH)} was confirmed by single-crystal X-ray diffraction analysis (Fig. 2). Thus, the structure assigned by Wang et al.13 for natural (−)-aniquinazoline D is incorrect. Interestingly, the 1H and 13C NMR data of natural (−)-aniquinazoline D fully matched those of both natural14 and our synthetic (−)-isochaetominine C (6) although the values of specific rotations are different {(−)-aniquinazoline D: yellowish solid, [α]20D −33 (c 0.37, MeOH);13 (−)-isochaetominine C (6): [α]25D −90 (c 0.6, MeOH)14}. Based on these results, we concluded that the structure of the natural (−)-aniquinazoline D should be revised as that shown for (−)-isochaetominine C (6).
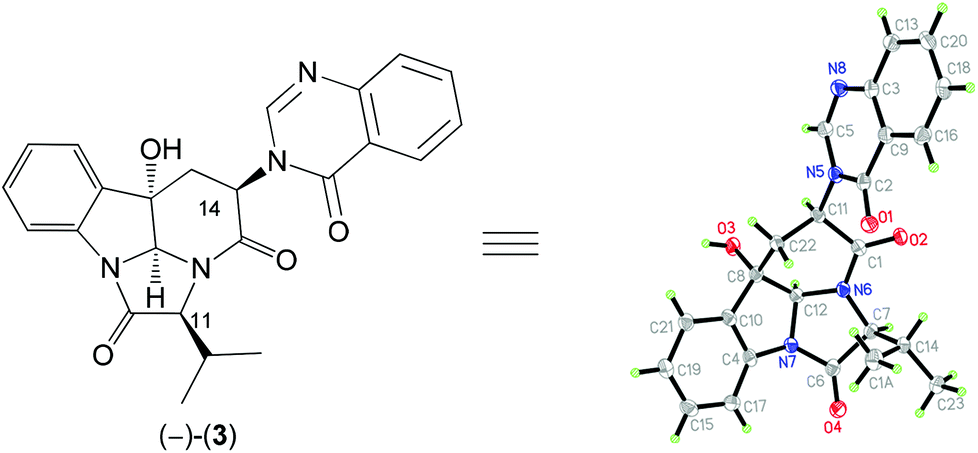 | ||
| Fig. 2 X-ray crystal structure of synthetic (−)-3. | ||
With the mild epimerization-free route established for the synthesis of isochaetominine C (6), we proceeded to the syntheses of isochaetominines B and A (4 and 5) by following the similar five-step synthetic sequence (Scheme 2). Thus, by substituting the benzyl L-valinate p-toluenesulfonic acid salt with its ethyl or methyl homologue, 2,3-di-epi-isochaetominine B (17), isochaetominine B (5), chaetominine (1), and isochaetominine A (4) were obtained without incidents as colorless crystals (m.p., 17: 302–304 °C; 5: 173–175 °C; 1: 301–302 °C; 4: 184–186 °C). The spectral data (1H and 13C NMR) and the sign of the specific rotation of our synthetic products fully matched those reported for the natural products. As in the cases of isochaetominine C, our synthetic products 5 {[α]25D −49.0 (c 1.0, MeOH)} and 4 {[α]25D −23.0 (c 1.0, MeOH)} displayed a lower value of specific rotation compared with the corresponding value reported for the natural isochaetominine B (5) {[α]25D −73 (c 0.6, MeOH)14} and isochaetominine A (4) {[α]25D −63 (c 0.5, MeOH)14}, respectively. Once again, such differences are probably caused by the relatively low purity of the natural isolates.
To further demonstrate the versatility of our stereodivergent strategy, we turned to investigate the enantiodivergent synthesis of 14-epi-isochaetominine C (7) and ent-7, as well as 2,3,14-tri-epi-isochaetominine C (ent-8). To this end, natural L-Trp was o-nitro-benzoylated18g and the resulting L-Trp-derivative (S)-14 was coupled with benzyl L-valinate p-toluenesulfonic acid salt to give 18 in 91% yield (Scheme 3). The LVT-promoted reductive condensation of 18 with trimethyl orthoformate in THF at 0 °C provided quinazolinone 19 in 96% yield. Epoxidation of 19 with DMDO followed by work-up with K2CO3/MeOH at −15 °C afforded the double cyclization product (+)-14-epi-isochaetominine C (7) in 35% yield along with the monocyclization product 20 in 44% yield. The structure of our synthetic product 7 {colorless crystals, m.p. 323–325 °C; [α]25D +48.0 (c 1.0, MeOH); Lit.14 [α]25D +33 (c 0.7, MeOH)} was confirmed by single-crystal X-ray diffraction analysis (Fig. 3). The spectral (1H and 13C NMR) data and the sign of specific rotation of our synthetic product matched those reported for the natural product.
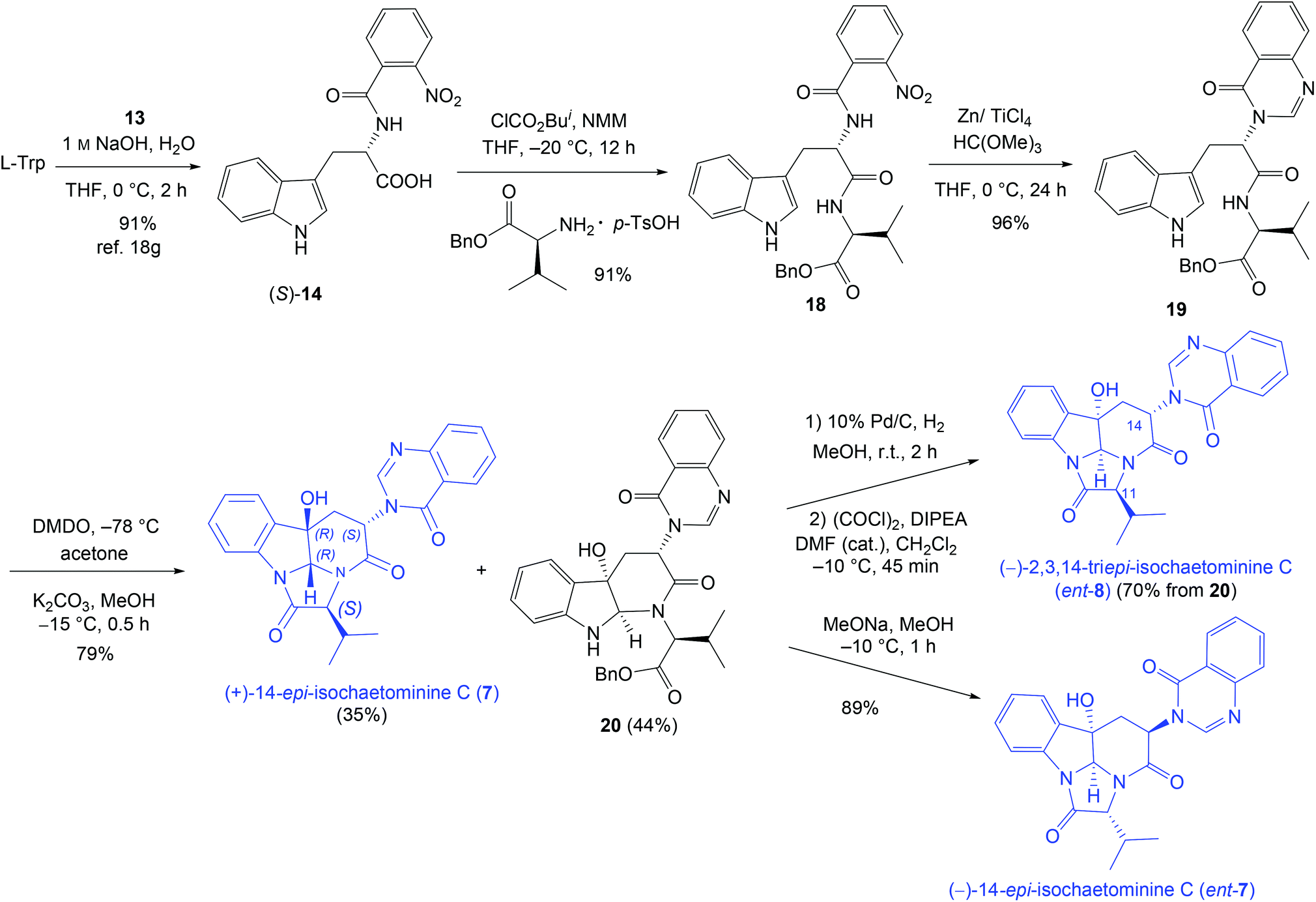 | ||
| Scheme 3 Enantio- and diastereo-divergent synthesis of (+)-14-epi-isochaetominine (7), ent-7, and 2,3,14-tri-epi-isochaetominine C (ent-8). | ||
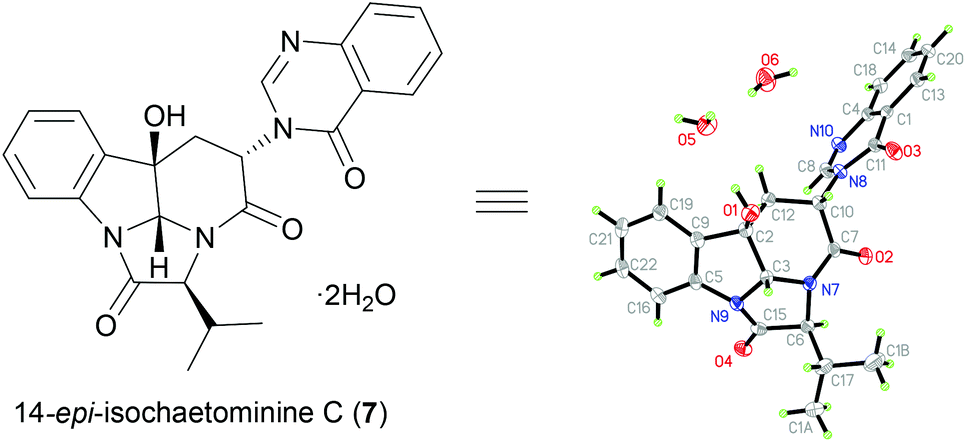 | ||
| Fig. 3 X-ray crystal structure of synthetic (+)-14-epi-isochaetominine C·2H2O. | ||
Debenzylation of 20 followed by cyclization led to 2,3,14-tri-epi-isochaetominine C (ent-8) in 70% yield over two steps. On the other hand, treatment of 20 with MeONa/MeOH at −10 °C for 1 h led to bis-epimerization at C-11 and C-14 and cyclization affording ent-7 in 89% yield. This approach thus constitutes an enantio- and diastereo-divergent synthesis of three diastereomers/enantiomers of isochaetominine C.
We next turned our attention to (−)-pseudofischerine (2), which was isolated from a culture of the fungus Neosartorya pseudofischeri S. W. Peterson obtained from agricultural soil collected in Thailand.12 Although the absolute configuration has not been determined experimentally, the authors suggested that it was the one shown in Fig. 1 and proposed that 2 was biosynthesized from L-tryptophan, anthranilic acid, and D-valine. Our synthesis commenced with the preparation of the dipeptide derivative 22 from L-Trp and D-Val-OBn (Scheme 4). The DMDO-initiated tandem reaction of 22 produced 11,14-di-epi-isochaetominine C (ent-3) in 33% yield, along with 23 in 41% yield. Successive debenzylation and cyclization of 23 yielded the structure proposed for (−)-pseudofischerine (2), namely, the antipode of the alkaloid isochaetominine C (ent-6), in 81% yield. The 1H and 13C NMR data of our synthetic compound 2 are identical with those reported for (−)-pseudofischerine as well as isochaetominine C (6). However, the sign and magnitude of the specific rotation of our synthetic product {2: [α]20D +76.8 (c 1.0, CHCl3)} are different from those reported for the natural (−)-pseudofischerine {[α]20D −16.9 (c 0.18, CHCl3)}.12 Because the reported optical rotation data of natural (−)-pseudofischerine and natural (−)-isochaetominine C (6) have been recorded in different solvents, which prevents a direct comparison of those data to determine their enantiomeric relationship. To clarify this issue, we measured the optical rotations of our two synthetic enantiomers 2 (ent-6) and 6 in both MeOH and CHCl3 {2: [α]25D +71.0 (c 1.0, MeOH); [α]20D +76.8 (c 1.0, CHCl3); 6: [α]25D −71.0 (c 1.0, MeOH); [α]20D −76.8 (c 1.0, CHCl3); natural isochaetominine C (6): [α]25D −90 (c 0.6, MeOH)14}. These results allowed us to conclude that natural (−)-pseudofischerine and (−)-isochaetominine C (6) are the same, namely, the structure of (−)-pseudofischerine should be revised to that shown for (−)-isochaetominine C (6).
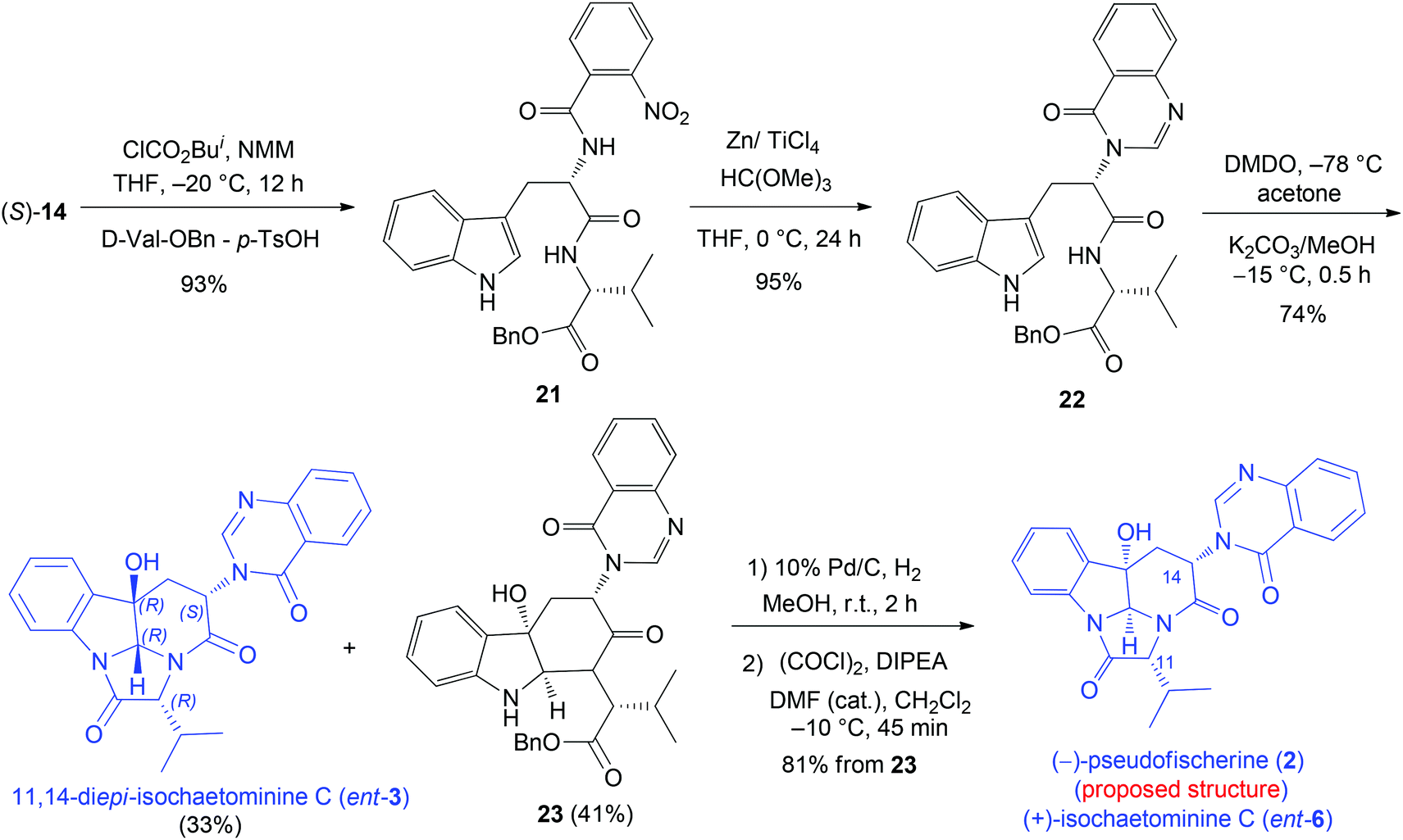 | ||
| Scheme 4 Diastereodivergent synthesis of 11,14-di-epi-isochaetominine C (ent-3) and the proposed structure of (−)-pseudofischerine (2). | ||
Finally, the D-Trp – D-Val-OBn combination was investigated leading to the synthesis of (−)-14-epi-isochaetominine C (ent-7) and (+)-11-epi-isochaetominine C (8) in three and five steps respectively (Scheme 5) from the same tryptophan derivative (R)-14.
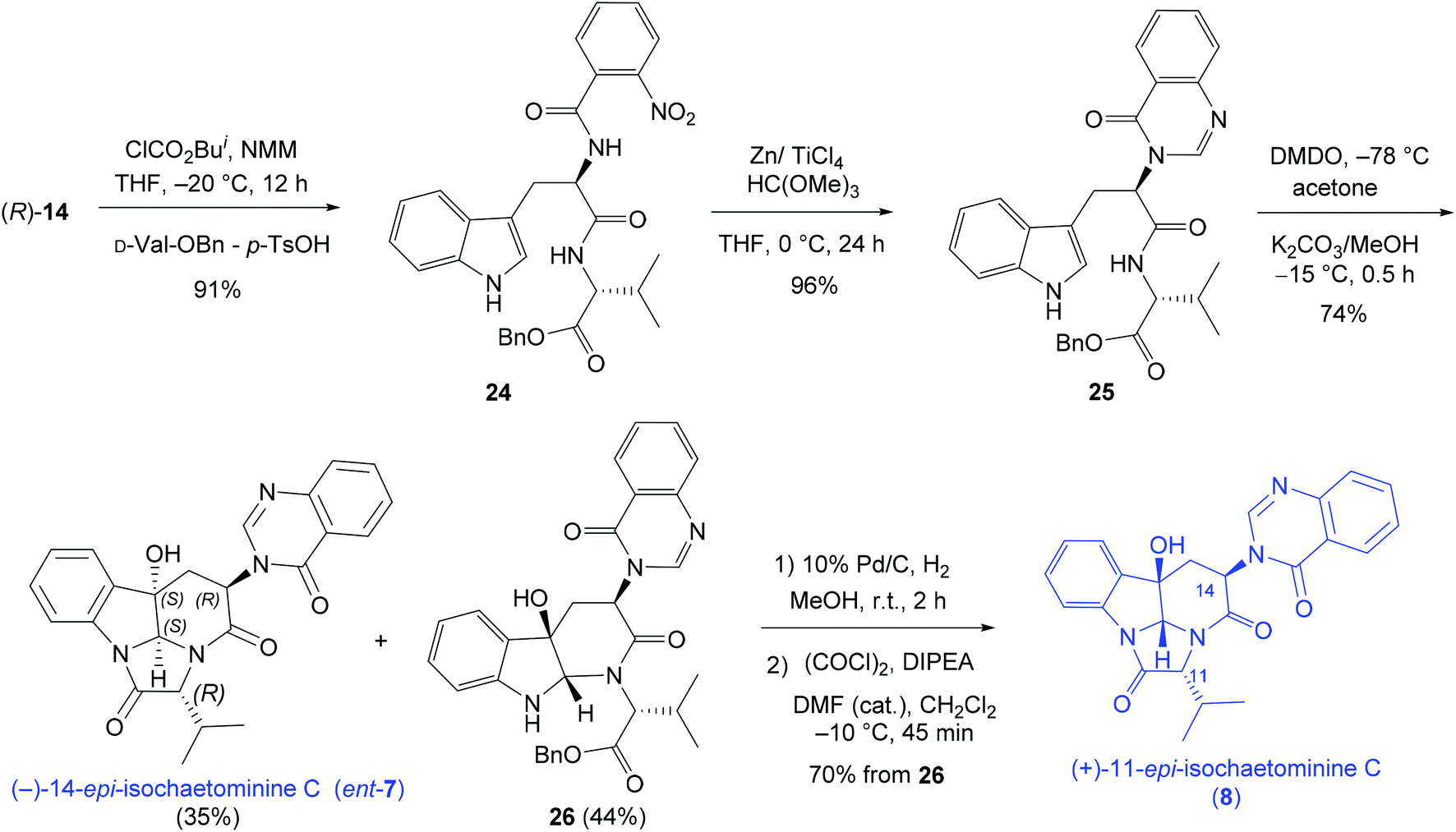 | ||
| Scheme 5 Diastereodivergent synthesis of (−)-14-epi-isochaetominine C (ent-7) and (+)-11-epi-isochaetominine C (8). | ||
Since the magnitudes of specific rotations of all our synthetic products are significantly different from those reported for the natural products, it was necessary to determine the enantiopurities of the synthetic samples. HPLC analyses on a chiral stationary phase (see the ESI†) of (−)-isochaetominine A (6), (+)-14-epi-isochaetominine C (7), the proposed structures of (−)-pseudofischerine (2 = ent-6) and (−)-aniquinazoline D (3), hitherto unknown 11-epi-isochaetominine C (8), ent-3, ent-7, and ent-8, showed that the enantiomeric excesses of all compounds are >99%.
Conclusion
In summary, a unified strategy has been developed for the highly enantioselective total syntheses of the proposed structures of (−)-pseudofischerine (2) and (−)-aniquinazoline D (3), (−)-isochaetominines A–C (4–6), (+)-14-epi-isochaetominine C (7), as well as other four hitherto unknown stereoisomers of isochaetominine C. The non-diastereoselective epoxidation reaction led to the formation of two diastereomers for each of the six-step synthetic sequences. In addition, in combination with a bis-epimerization at C-11 and C-14 of 20 and tandem cyclization, three diastereomers were obtained in a seven-step route. All eight 2,3-cis-stereoisomers of (−)-isochaetominine C have been synthesized from the combination of four amino acids/esters, D-Trp, L-Trp, L-Val-OBn, and D-Val-OBn. The sensitive 3,14-cis-stereochemistry of isochaetominines A–C has been established by the use of a mild and epimerization-free protocol involving the use of benzyl α-amino esters (e.g. Val-OBn) and a lactamization reaction. It is worth noting that Ye's peptide coupling reagent afforded advantages of operational simplicity in the lactamization step. Importantly, the structures of natural (−)-pseudofischerine (2) and (−)-aniquinazoline D (3) have been revised both to (−)-isochaetominine C (6) based on our synthetic efforts. Although neither (+)-11-epi-isochaetominine C (8) nor its antipode ent-8 has been isolated from natural sources, the ready availability of both 8 and ent-8 by chemical synthesis allowed the assumption that an enantiomer of 11-epi-isochaetominine C is a natural product. Finally, our strategy for the synthesis of 14-epi-isochaetominine C (7) could serve as an inspiration for the development of an efficient enantioselective synthesis of the structurally related natural products such as kapakahines B and F25 (9 and 10).
Experimental section
General methods
1H NMR and 13C NMR spectra were recorded on a Bruker 400 or Bruker 500 (1H/400 or 500 MHz, 13C/100 or 125 MHz) spectrometer. Chemical shifts are expressed in parts per million (δ) relative to an internal standard of residual chloroform (7.26 ppm for 1H NMR and 77.0 ppm for 13C NMR). Data for 1H NMR are reported as chemical shifts (multiplicity, coupling constant, number of proton). ESI-Mass spectra were recorded on a Bruker Dalton ESquire 3000 plus LC-MS apparatus. Optical rotations were measured with a Perkin-Elmer 341 automatic polarimeter or an Anton Paar MCP 500 polarimeter. Melting points were determined on a Büchi M560 Automatic Melting Point apparatus. Infrared spectra were recorded with a Nicolet Avatar 330 FT-IR spectrometer using film or KBr pellet techniques.Silica gel (300–400 mesh) was used for flash column chromatography, eluting (unless otherwise stated) with ethyl-acetate/petroleum ether (PE) (60–90 °C) mixture. THF was distilled over sodium benzophenone ketyl under N2. Dichloromethane was distilled over calcium hydride under N2.
General procedure for step 1
The preparations of (R)-14 from D-tryptophan18f and (S)-14 from L-tryptophan18g by N-aroylation have been described in our previous reports.18f–hGeneral procedure for step 2 (general procedure 2)
To a stirring solution of 14 (5.09 mmol) in THF (20 mL) at −20 °C were added successively N-methylmorpholine (0.84 mL, 7.63 mmol) and iBuOCOCl (0.74 mL, 5.60 mmol). After being stirred at −20 °C under N2 for 15 min, the resulting mixture was added slowly to a solution of an amino acid benzyl ester p-toluenesulfonic acid salt (10.18 mmol) and N-methylmorpholine (1.68 mL, 15.28 mmol) in THF (36 mL) at −78 °C. The resulting mixture was stirred for 12 h at −20 °C. The reaction was quenched with a saturated aqueous solution of NH4Cl (20 mL). The resulting mixture was diluted with water (100 mL) and the organic phase was separated. The aqueous phase was extracted with EtOAc (3 × 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc:PE = 3:2) to give the corresponding dipeptide derivative.
General procedure for step 3 (general procedure 3)
To a suspension of zinc powder (776 mg, 11.94 mmol) in THF (50 mL) was added TiCl4 (0.66 mL, 5.98 mmol). The resulting mixture was stirred for 1 h at 50 °C, and cooled to 0 °C. A THF (10 mL) solution of a tryptophan-derived dipeptide derivative (1.50 mmol) and trimethyl orthoformate (0.66 mL, 5.98 mmol) was added. The resulting mixture was stirred for 24 h at 0 °C. The reaction mixture was quenched with brine (10 mL) and the resulting mixture was stirred for 2 h. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3 × 30 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc:PE = 1:3 to DCM:MeOH = 40:1) to give the corresponding quinazolinonyl dipeptide derivative.
General procedure A for step 4 (general procedure 4-A)
To a solution of a quinazolinonyl dipeptide derivative (0.38 mmol) in anhydrous acetone (2 mL) was added a 0.04 M solution of DMDO in acetone (19 mL, 0.76 mmol) at −78 °C. After being stirred for 1 h, a saturated aqueous solution of Na2SO3 (10 mL) was added and the resulting mixture was stirred for 2 h at 0 °C. The reaction was quenched with a saturated aqueous solution of NH4Cl (10 mL) and concentrated under reduced pressure. To the resulting residue was added H2O (50 mL), and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine (2 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc:PE = 1:1) to give a chaetominine/isochaetominine-type compound and a monocyclization product.
:PE = 1:1).
(−)-Chaetominine (1): colorless crystals, m.p. 301–302 °C (EtOAc) [lit. 161–163 °C;11a 288–290 °C (MeOH)18f]; [α]20D −49.7 (c 0.5, MeOH) {lit. [α]20D −70 (c 0.48, MeOH);11a [α]20D −49.4 (c 0.26, MeOH);18a [α]20D −49.7 (c 0.48, MeOH)18f}. The IR, 1H NMR, 13C NMR, and MS data are identical to those reported previously.18f
Compound 11a: white solid, m.p. 154–156 °C (EtOAc); [α]20D −145.1 (c 1.0, CHCl3); IR (film) vmax: 3365, 2930, 1738, 1677, 1610, 1475, 1321, 1244, 1185, 1140 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.18 (dd, J = 8.2, 0.8 Hz, 1H), 7.69 (td, J = 7.8, 1.2 Hz, 1H), 7.64–7.56 (m, 2H), 7.43 (ddd, J = 8.2, 7.8, 0.9 Hz, 1H), 7.38–7.26 (m, 6H), 7.13 (td, J = 7.8, 0.9 Hz, 1H), 6.78 (t, J = 7.4 Hz, 1H), 6.57 (d, J = 7.8 Hz, 1H), 5.36–5.05 (m, 6H), 4.10 (s, 1H), 2.88 (dd, J = 12.5, 12.5 Hz, 1H), 2.48 (dd, J = 12.5, 4.0 Hz, 1H), 1.53 (d, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.7, 169.5, 160.6, 148.2, 147.2, 145.4, 135.1, 134.4, 130.8, 129.1, 128.7 (2C), 128.6, 128.2 (2C), 127.3, 127.1, 126.9, 123.6, 121.8, 120.2, 110.2, 80.5, 79.3, 67.3, 52.2, 40.7, 16.3 (one carbon was not observed due to slow rotation at the C9–N bond);18a MS (ESI) m/z 533 (M + Na+, 100%), HRMS (ESI, m/z) calcd for C29H26N4O5Na [M + Na]+: 533.1795, found: 533.1795.
:PE = 1:1).
2,3-Di-epi-isochaetominine B (17): colorless crystals, m.p. 302–304 °C (EtOAc); [α]20D −47.7 (c 1.0, MeOH); IR (film) vmax: 3443, 1732, 1656, 1476, 1277, 1191, 1137 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.30 (br s, 1H), 8.17 (d, J = 1.3 Hz, 1H), 7.87 (ddd, J = 7.7, 7.6, 1.5 Hz, 1H), 7.71 (d, J = 6.9 Hz, 1H), 7.58 (t, J = 7.4 Hz, 1H), 7.54–7.47 (m, 2H), 7.44 (td, J = 7.6, 1.2 Hz, 1H), 7.25 (td, J = 7.5, 1.0 Hz, 1H), 6.70 (br s, 1H), 6.18–5.70 (br s, 1H), 5.58 (s, 1H), 4.65 (t, J = 3.5 Hz, 1H), 2.90 (dd, J = 12.8, 12.8 Hz, 1H), 2.63 (dd, J = 12.8, 3.0 Hz, 1H), 2.49–2.38 (m, 1H), 1.97–1.80 (m, 1H), 0.69 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 172.2, 165.5 (br), 160.3, 147.6, 146.6 (br), 139.1, 136.5, 134.9, 130.2, 127.5, 127.4, 126.5, 125.6, 125.2, 121.5, 114.4, 82.6, 76.7, 64.1, 50.8 (br), 38.1, 20.0, 8.9 (br); MS (ESI) m/z 439 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C23H20N4O4Na [M + Na]+: 439.1377, found: 439.1380.
Compound 11b: white solid, m.p. 113–115 °C (EtOAc); [α]20D −146.7 (c 1.0, CHCl3); IR (film) vmax: 3442, 2932, 1733, 1671, 1612, 1470, 1321, 1238, 1176 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.18 (dd, J = 7.8, 1.3 Hz, 1H), 7.68 (ddd, J = 8.2, 7.2, 1.5 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.50–7.39 (m, 2H), 7.38–7.30 (m, 5H), 7.25 (d, J = 7.4 Hz, 1H), 7.10 (ddd, J = 8.2, 7.8, 1.1 Hz, 1H), 6.74 (dd, J = 7.4, 7.2 Hz, 1H), 6.54 (d, J = 8.0 Hz, 1H), 5.40–5.10 (m, 6H), 4.30 (s, 1H), 2.88 (dd, J = 12.5, 12.5 Hz, 1H), 2.43 (dd, J = 12.5, 3.9 Hz, 1H), 2.10–1.98 (m, 1H), 1.85–1.71 (m, 1H), 1.08 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.5, 170.4, 160.5, 148.2, 147.2, 145.3, 135.0, 134.3, 130.7, 129.3, 128.7 (2C), 128.6, 128.3 (2C), 127.2, 127.0, 127.0, 123.6, 121.8, 120.0, 110.0, 80.1, 79.5, 67.3, 57.1, 41.2, 23.6, 10.4 (one carbon was not observed due to slow rotation at the C9–N bond);18a MS (ESI) m/z 547 (M + Na+, 100%), HRMS (ESI, m/z) calcd for C30H28N4O5Na [M + Na]+: 547.1952, found: 547.1954.
General procedure B for step 4 (general procedure 4-B)
To a solution of a quinazolinonyl dipeptide derivative in anhydrous acetone (2 mL) was added a 0.04 M solution of DMDO in acetone (19 mL, 0.76 mmol) at −78 °C. After being stirred for 1 h, K2CO3/MeOH (94 mg/10 mL) was added and the resulting mixture was warmed up to −15 °C over 30 minutes. The reaction mixture was quenched with a saturated aqueous solution of NH4Cl (10 mL) and the resulting mixture was concentrated under reduced pressure. To the resulting residue was added H2O (50 mL), and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine (2 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc:PE = 1:1) to give an isochaetominine-type compound and a monocyclization product.
:PE = 1:1).
Compound 3 [the proposed structure of (−)-aniquinazoline D13]: colorless crystals, m.p. 302–303 °C (EtOAc); [α]20D −50.7 (c 0.5, MeOH); IR (film) vmax: 3446, 2965, 1738, 1642, 1610, 1475, 1275, 1192, 1137 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.30 (br s, 1H), 8.16 (d, J = 1.3 Hz, 1H), 7.86 (ddd, J = 7.7, 7.6, 1.5 Hz, 1H), 7.70 (d, J = 6.9 Hz, 1H), 7.57 (t, J = 7.4 Hz, 1H), 7.53–7.46 (m, 2H), 7.43 (td, J = 7.6, 1.2 Hz, 1H), 7.24 (td, J = 7.5, 1.0 Hz, 1H), 6.68 (br s, 1H), 6.16–5.68 (br s, 1H), 5.52 (s, 1H), 4.56 (d, J = 2.8 Hz, 1H), 3.17 (m, 1H), 2.91 (dd, J = 12.8, 12.8 Hz, 1H), 2.62 (dd, J = 12.8, 3.0 Hz, 1H), 1.13 (d, J = 7.3 Hz, 3H), 0.79 (d, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 171.9, 166.3 (br), 160.6, 147.9, 146.6 (br), 139.5, 136.5, 135.1, 130.5, 127.8, 127.7, 126.8, 125.8, 125.4, 121.8, 114.6, 82.6, 77.0, 68.0, 51.2 (br), 38.6, 25.8, 18.3, 16.6 (br); MS (ESI) m/z 453 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C24H22N4O4Na [M + Na]+: 453.1533, found: 453.1536; conditions for the chiral HPLC analysis: Chiralpak AD-H (n-hexane/EtOH, 30:70), flow rate = 0.8 mL min−1, Rt = 9.9 min, respectively. The enantiomeric excess was determined to be >99%.
Crystallographic data for compound 3: C24N4O4H22, M = 430.46 g mol−1, crystal size 0.3 × 0.2 × 0.1 mm3, orthorhombic, space group P212121, a = 7.6889(2) Å, b = 15.5382(5) Å, c = 16.4272(6) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 1962.58(11) Å3, Z = 4, ρcalc = 1.457 g cm−3, μ = 0.832 mm−1, λ = 1.54184 Å, T = 99.8(5) K, θ range = 3.92–68.84°, reflections collected 4748, independent reflections 3186 (Rint = 0.0469, Rsigma = 0.0597), 292 parameters. The structure was solved by direct methods and refined by goodness-of-fit on F2 (1.037); final R indices [I > 2σ(I)] R1 = 0.0413 and wR2 = 0.0982; largest diff. peak/hole 0.18/−0.23 e Å−3. CCDC 1423418 contains the supplementary crystallographic data for this structure.
Compound 11c: white solid, m.p. 296–298 °C (EtOAc); [α]20D −180.1 (c 0.5, CHCl3); IR (film) vmax: 3388, 2924, 1731, 1680, 1610, 1472, 1321, 1229, 1175 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.19 (dd, J = 8.0, 0.8 Hz, 1H), 7.70 (ddd, J = 7.8, 7.6, 1.0 Hz, 1H), 7.62 (dd, J = 8.0, 0.8 Hz, 1H), 7.57 (br s, 1H), 7.44 (ddd, J = 7.7, 7.5, 0.9 Hz, 1H), 7.39–7.31 (m, 5H), 7.27 (dd, J = 7.2, 0.9 Hz, 1H), 7.15 (ddd, J = 7.6, 7.5, 0.8 Hz, 1H), 6.79 (dd, J = 7.4, 7.3 Hz, 1H), 6.56 (d, J = 7.8 Hz, 1H), 5.47 (d, J = 4.4 Hz, 1H), 5.35–5.07 (m, 3H), 5.01 (d, J = 10.4 Hz, 1H), 4.93 (d, J = 4.2 Hz, 1H), 3.66 (s, 1H), 2.89 (dd, J = 13.0, 13.0 Hz, 1H), 2.47 (dd, J = 13.0, 3.7 Hz, 1H), 2.30–2.18 (m, 1H), 1.13 (d, J = 6.7 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.6, 170.5, 160.5, 148.3, 147.2, 145.2, 134.9, 134.3, 130.8, 129.1, 128.7 (2C), 128.6, 128.5 (2C), 127.2, 127.1, 127.0, 123.7, 121.8, 120.2, 110.2, 80.1, 79.8, 67.2, 60.8, 41.2, 28.9, 19.4, 18.7 (one carbon was not observed due to slow rotation at the C9–N bond);18a MS (ESI) m/z 561 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C31H30N4O5Na [M + Na]+: 561.2108, found: 561.2115.
:PE = 1:1).
(+)-14-epi-Isochaetominine C (7): white solid, m.p. 323–325 °C (EtOAc); [α]25D +48.0 (c 1.0, MeOH) {lit. [α]25D +33 (c 0.7, MeOH)14}; IR (film) vmax: 3406, 1716, 1690, 1603, 1443, 1325, 1267, 1224 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.22 (br s, 1H), 8.19 (dd, J = 7.9, 1.3 Hz, 1H), 7.87–7.83 (ddd, J = 7.8, 7.6, 1.2 Hz, 1H), 7.70 (dd, J = 7.8, 0.9 Hz, 1H), 7.59 (ddd, J = 7.9, 7.6, 0.9 Hz, 1H), 7.50 (dd, J = 7.8, 1.2 Hz, 1H), 7.47 (dd, J = 7.9, 0.9 Hz, 1H), 7.42 (ddd, J = 7.8, 7.4, 1.1 Hz, 1H), 7.24 (ddd, J = 7.5, 7.4, 1.0 Hz, 1H), 6.74 (br s, 1H), 5.99 (dd, J = 13.0, 2.9 Hz, 1H), 5.78 (s, 1H), 4.38 (d, J = 6.6 Hz, 1H), 2.94 (dd, J = 13.0, 13.0 Hz, 1H), 2.49 (dd, J = 13.0, 3.5 Hz, 1H), 2.30–2.21 (m, 1H), 1.09 (d, J = 6.5 Hz, 3H), 1.05 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 169.7, 167.2, 160.0, 147.3, 146.7, 137.6, 137.5, 134.7, 129.7, 127.2, 127.2, 126.4, 125.5, 124.5, 121.0, 114.7, 84.5, 76.7, 69.6, 49.1, 38.2, 30.3, 19.1, 19.0; MS (ESI) m/z 453 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C24H22N4O4Na [M + Na]+: 453.1533, found: 453.1539; conditions for the chiral HPLC analysis: Chiralpak AD-H (n-hexane/EtOH, 30:70), flow rate = 0.8 mL min−1, Rt = 17.0 min, respectively. The enantiomeric excess was determined to be >99%.
Crystallographic data for (+)-14-epi-isochaetominine C (7)·2H2O: C24N4O6H26, M = 466.50 g mol−1, crystal size 0.2 × 0.1 × 0.05 mm3, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.3200(6) Å, b = 10.5489(6) Å, c = 12.1772(7) Å, α = 95.818(5)°, β = 90.490(5)°, γ = 111.805(6)°, V = 1104.43(12) Å3, Z = 2, ρcalc = 1.4027 g cm−3, μ = 0.849 mm−1, λ = 1.54184 Å, T = 173.00(14) K, θ range = 3.65–60.18°, reflections collected 5671, independent reflections 3246 (Rint = 0.0442, Rsigma = 0.0662), 315 parameters. The structure was solved by direct methods and refined by goodness-of-fit on F2 (0.939); final R indices [I > 2σ(I)] R1 = 0.0380 and wR2 = 0.0881; largest diff. peak/hole 0.27/−0.27 e Å−3. CCDC 1423426 contains the supplementary crystallographic data for this structure.
, a = 9.3200(6) Å, b = 10.5489(6) Å, c = 12.1772(7) Å, α = 95.818(5)°, β = 90.490(5)°, γ = 111.805(6)°, V = 1104.43(12) Å3, Z = 2, ρcalc = 1.4027 g cm−3, μ = 0.849 mm−1, λ = 1.54184 Å, T = 173.00(14) K, θ range = 3.65–60.18°, reflections collected 5671, independent reflections 3246 (Rint = 0.0442, Rsigma = 0.0662), 315 parameters. The structure was solved by direct methods and refined by goodness-of-fit on F2 (0.939); final R indices [I > 2σ(I)] R1 = 0.0380 and wR2 = 0.0881; largest diff. peak/hole 0.27/−0.27 e Å−3. CCDC 1423426 contains the supplementary crystallographic data for this structure.
Compound 20: white solid, m.p. 121–122 °C (EtOAc); [α]20D +98.7 (c 1.0, CHCl3); IR (film) vmax: 3356, 2914, 2850, 1732, 1681, 1607, 1476, 1239, 1198, 1130 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.19 (dd, J = 8.0, 0.8 Hz, 1H), 7.70 (ddd, J = 7.8, 7.6, 1.0 Hz, 1H), 7.62 (dd, J = 8.0, 0.8 Hz, 1H), 7.57 (br s, 1H), 7.44 (ddd, J = 7.7, 7.5, 0.9 Hz, 1H), 7.39–7.31 (m, 5H), 7.27 (dd, J = 7.2, 0.9 Hz, 1H), 7.15 (ddd, J = 7.6, 7.5, 0.8 Hz, 1H), 6.79 (dd, J = 7.4, 7.3 Hz, 1H), 6.56 (d, J = 7.8 Hz, 1H), 5.47 (d, J = 4.4 Hz, 1H), 5.35–5.07 (m, 3H), 5.01 (d, J = 10.4 Hz, 1H), 4.93 (d, J = 4.2 Hz, 1H), 3.66 (s, 1H), 2.89 (dd, J = 13.0, 13.0 Hz, 1H), 2.47 (dd, J = 13.0, 3.7 Hz, 1H), 2.30–2.18 (m, 1H), 1.13 (d, J = 6.7 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.1, 170.0, 160.1, 147.9, 147.3, 144.9, 135.0, 134.4, 130.8, 128.9, 128.7 (2C), 128.7, 128.6 (2C), 127.3, 127.2, 126.9, 123.8, 121.7, 120.9, 110.5, 82.4, 80.0, 67.6, 63.6, 41.9, 29.3, 20.1, 19.5 (one carbon was not observed due to slow rotation at the C9–N bond);18a MS (ESI) m/z 561 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C31H30N4O5Na [M + Na]+: 561.2108, found: 561.2108.
:PE = 1:1).
11,14-Di-epi-isochaetominine C (ent-3): colorless crystals, m.p. 302–303 °C (EtOAc); [α]20D +50.7 (c 0.5, MeOH); IR (film) vmax: 3407, 2962, 1732, 1655, 1613, 1476, 1198, 1133, 1076 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.30 (br s, 1H), 8.16 (d, J = 1.3 Hz, 1H), 7.86 (ddd, J = 7.7, 7.6, 1.5 Hz, 1H), 7.70 (d, J = 6.9 Hz, 1H), 7.57 (t, J = 7.4 Hz, 1H), 7.53–7.46 (m, 2H), 7.43 (td, J = 7.6, 1.2 Hz, 1H), 7.24 (td, J = 7.5, 1.0 Hz, 1H), 6.68 (br s, 1H), 6.16–5.68 (br s, 1H), 5.52 (s, 1H), 4.56 (d, J = 2.8 Hz, 1H), 3.17 (m, 1H), 2.91 (dd, J = 12.8, 12.8 Hz, 1H), 2.62 (dd, J = 12.8, 3.0 Hz, 1H), 1.13 (d, J = 7.3 Hz, 3H), 0.79 (d, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 171.9, 166.3 (br), 160.6, 147.9, 146.6 (br), 139.5, 136.5, 135.1, 130.5, 127.8, 127.7, 126.8, 125.8, 125.4, 121.8, 114.6, 82.6, 77.0, 68.0, 51.2 (br), 38.6, 25.8, 18.3, 16.6 (br); MS (ESI) m/z 453 (M + Na+, 100%), HRMS (ESI, m/z) calcd for C24H22N4O4Na [M + Na]+: 453.1533, found: 453.1533; conditions for the chiral HPLC analysis: Chiralpak AD-H (n-hexane/EtOH, 30:70), flow rate = 0.8 mL min−1, Rt = 11.8 min, respectively. The enantiomeric excess was determined to be >99%.
Compound 23: white solid, m.p. 296–298 °C (EtOAc); [α]20D +180.1 (c 0.5, CHCl3); IR (film) vmax: 3388, 2924, 1731, 1680, 1610, 1472, 1321, 1229, 1175 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.19 (dd, J = 8.0, 0.8 Hz, 1H), 7.70 (ddd, J = 7.8, 7.6, 1.0 Hz, 1H), 7.62 (dd, J = 8.0, 0.8 Hz, 1H), 7.57 (br s, 1H), 7.44 (ddd, J = 7.7, 7.5, 0.9 Hz, 1H), 7.39–7.31 (m, 5H), 7.27 (dd, J = 7.2, 0.9 Hz, 1H), 7.15 (ddd, J = 7.6, 7.5, 0.8 Hz, 1H), 6.79 (dd, J = 7.4, 7.3 Hz, 1H), 6.56 (d, J = 7.8 Hz, 1H), 5.47 (d, J = 4.4 Hz, 1H), 5.35–5.07 (m, 3H), 5.01 (d, J = 10.4 Hz, 1H), 4.93 (d, J = 4.2 Hz, 1H), 3.66 (s, 1H), 2.89 (dd, J = 13.0, 13.0 Hz, 1H), 2.47 (dd, J = 13.0, 3.7 Hz, 1H), 2.30–2.18 (m, 1H), 1.13 (d, J = 6.7 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.7, 170.5, 160.5, 148.3, 147.2, 145.2, 134.8, 134.4, 130.9, 129.0, 128.7 (2C), 128.6, 128.5 (2C), 127.3, 127.1, 127.0, 123.7, 121.8, 120.2, 110.2, 80.1, 79.8, 67.3, 60.7, 41.2, 28.9, 19.4, 18.7 (one carbon was not observed due to slow rotation at the C9–N bond);18a MS (ESI) m/z 561 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C31H30N4O5Na [M + Na]+: 561.2108, found: 561.2110.
:PE = 1:1).
14-epi-Isochaetominine C (ent-7): white solid, m.p. 323–325 °C (EtOAc); [α]25D −48.0 (c 1.0, MeOH); IR (film) vmax: 3406, 1716, 1690, 1603, 1443, 1325, 1267, 1224 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.22 (br s, 1H), 8.19 (dd, J = 7.9, 1.3 Hz, 1H), 7.87–7.83 (ddd, J = 7.8, 7.6, 1.2 Hz, 1H), 7.70 (dd, J = 7.8, 0.9 Hz, 1H), 7.59 (ddd, J = 7.9, 7.6, 0.9 Hz, 1H), 7.50 (dd, J = 7.8, 1.2 Hz, 1H), 7.47 (dd, J = 7.9, 0.9 Hz, 1H), 7.42 (ddd, J = 7.8, 7.4, 1.1 Hz, 1H), 7.24 (ddd, J = 7.5, 7.4, 1.0 Hz, 1H), 6.74 (br s, 1H), 5.99 (dd, J = 13.0, 2.9 Hz, 1H), 5.78 (s, 1H), 4.38 (d, J = 6.6 Hz, 1H), 2.94 (dd, J = 13.0, 13.0 Hz, 1H), 2.49 (dd, J = 13.0, 3.5 Hz, 1H), 2.30–2.21 (m, 1H), 1.09 (d, J = 6.5 Hz, 3H), 1.05 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 169.7, 167.2, 160.0, 147.3, 146.7, 137.6, 137.5, 134.7, 129.7, 127.2, 127.2, 126.4, 125.5, 124.5, 121.0, 114.7, 84.5, 76.7, 69.6, 49.1, 38.2, 30.3, 19.1, 19.0; MS (ESI) m/z 453 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C24H22N4O4 [M + Na]+: 453.1533, found: 453.1535; conditions for the chiral HPLC analysis: Chiralpak AD-H (n-hexane/EtOH, 30:70), flow rate = 0.8 mL min−1, Rt = 28.7 min, respectively. The enantiomeric excess was determined to be >99%.
Compound 26: white solid, m.p. 121–122 °C (EtOAc); [α]20D −98.7 (c 1.0, CHCl3); IR (film) vmax: 3356, 2914, 2850, 1732, 1681, 1607, 1476, 1239, 1198, 1130 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.19 (dd, J = 8.0, 0.8 Hz, 1H), 7.70 (ddd, J = 7.8, 7.6, 1.0 Hz, 1H), 7.62 (dd, J = 8.0, 0.8 Hz, 1H), 7.57 (br s, 1H), 7.44 (ddd, J = 7.7, 7.5, 0.9 Hz, 1H), 7.39–7.31 (m, 5H), 7.27 (dd, J = 7.2, 0.9 Hz, 1H), 7.15 (ddd, J = 7.6, 7.5, 0.8 Hz, 1H), 6.79 (dd, J = 7.4, 7.3 Hz, 1H), 6.56 (d, J = 7.8 Hz, 1H), 5.47 (d, J = 4.4 Hz, 1H), 5.35–5.07 (m, 3H), 5.01 (d, J = 10.4 Hz, 1H), 4.93 (d, J = 4.2 Hz, 1H), 3.66 (s, 1H), 2.89 (dd, J = 13.0, 13.0 Hz, 1H), 2.47 (dd, J = 13.0, 3.7 Hz, 1H), 2.30–2.18 (m, 1H), 1.13 (d, J = 6.7 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.1, 170.0, 160.1, 147.9, 147.3, 144.9, 135.0, 134.4, 130.8, 128.9, 128.7 (2C), 128.7, 128.6 (2C), 127.3, 127.2, 126.9, 123.8, 121.7, 120.9, 110.5, 82.4, 80.0, 67.6, 63.6, 41.9, 29.3, 20.1, 19.5 (one carbon was not observed due to slow rotation at the C9–N bond);18a MS (ESI) m/z 561 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C31H30N4O5Na [M + Na]+: 561.2108, found: 561.2112.
General procedure A for step 5 (general procedure 5-A)
A suspension of a quinazolinonyl dipeptide derivative (0.10 mmol) and 10% Pd/C (8 mg) in methanol (2 mL) was stirred under an atmosphere of H2 for 2 h at room temperature. The reaction mixture was filtered through a Celite pad and the residue was washed with methanol. The filtrate was concentrated under reduced pressure. Without further purification, the residue was directly treated with Ye's reagent (DEPBT)21 (63 mg, 0.21 mmol) and DIPEA (0.035 mL, 0.21 mmol) in anhydrous CH2Cl2 (3 mL) for 8 h at room temperature. The reaction was quenched with water (5 mL) at 0 °C, and the resulting mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (2 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc:PE = 1:1) to give the corresponding isochaetominine-type compound.
General procedure B for step 5 (general procedure 5-B)
A suspension of a quinazolinonyl dipeptide derivative (0.10 mmol) and 10% Pd/C (8.00 mg) in methanol (2 mL) was stirred under an atmosphere of H2 for 2 h at room temperature. The mixture was filtered through a Celite pad and the solid was washed with methanol. The filtrate was concentrated under reduced pressure. Without further purification, the residue was directly treated with (COCl)2 (0.013 mL, 0.15 mmol) and a catalytic amount of DMF in anhydrous CH2Cl2 (3 mL) for 5 minutes at −10 °C. To the resulting mixture was added DIPEA (0.035 mL, 0.21 mmol) and the mixture was stirred for 45 min. The reaction was quenched with water (5 mL) at −10 °C, and the mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (2 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: EtOAc:PE = 1:1) to give the corresponding isochaetominine-type compound.
:PE = 1:1) as colorless crystals. M.p. 184–186 °C (EtOAc); [α]25D −23.0 (c 1.0, MeOH) {lit.14 [α]25D −63 (c 0.5, MeOH)}; IR (film) vmax: 3410, 2925, 1728, 1674, 1608, 1477, 1383, 1327, 1290, 1136 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.28 (s, 1H), 8.18 (dd, J = 8.0, 1.1 Hz, 1H), 7.87 (ddd, J = 7.8, 7.8, 1.3 Hz, 1H), 7.71 (dd, J = 7.9, 0.8 Hz, 1H), 7.59 (ddd, J = 7.9, 7.9, 0.9 Hz, 1H), 7.51 (dd, J = 7.6, 0.9 Hz, 1H), 7.44 (dd, J = 8.1, 1.1 Hz, 1H), 7.42 (ddd, J = 7.6, 7.4, 1.0 Hz, 1H), 7.24 (ddd, J = 7.5, 7.4, 1.2 Hz, 1H), 6.29 (br s, 1H), 5.92 (s, 1H), 4.91 (dd, J = 7.0, 5.0 Hz, 1H), 4.62 (q, J = 7.3 Hz, 1H), 2.98 (dd, J = 14.2, 7.0 Hz, 1H), 2.73 (dd, J = 14.2, 5.2 Hz, 1H), 1.60 (d, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 174.4, 164.2, 159.9, 147.6, 146.7, 139.8, 135.8, 134.8, 130.1, 127.4, 127.2, 126.2, 125.3, 124.7, 121.5, 114.1, 82.9, 74.1, 59.7, 55.6, 34.7, 13.9; MS (ESI) m/z 425 (M + Na+, 100%), HRMS (ESI, m/z) calcd for C22H18N4O4Na [M + Na]+: 425.1220, found: 425.1224.
:PE = 1:1) as colorless crystals. M.p. 173–175 °C (EtOAc); [α]25D −49.0 (c 1.0, MeOH) {lit.14 [α]25D −73 (c 0.6, MeOH)}; IR (film) vmax: 3435, 2079, 1643, 1477, 1381, 1326, 1290, 1136 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.30 (s, 1H), 8.18 (dd, J = 7.9, 1.0 Hz, 1H), 7.87 (ddd, J = 7.8, 7.8, 1.3 Hz, 1H), 7.71 (dd, J = 7.9, 0.8 Hz, 1H), 7.59 (ddd, J = 7.9, 7.9, 0.9 Hz, 1H), 7.52 (dd, J = 7.6, 0.9 Hz, 1H), 7.43 (dd, J = 7.6, 1.0 Hz, 1H), 7.42 (ddd, J = 7.4, 7.2, 1.0 Hz, 1H), 7.25 (ddd, J = 7.5, 7.4, 1.2 Hz, 1H), 6.28 (br s, 1H), 5.86 (s, 1H), 4.90 (dd, J = 7.3, 5.2 Hz, 1H), 4.47 (dd, J = 9.2, 5.9 Hz, 1H), 2.98 (dd, J = 14.2, 7.5 Hz, 1H), 2.73 (dd, J = 14.2, 5.3 Hz, 1H), 2.11–1.93 (m, 2H), 1.11 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 174.0, 164.6, 159.8, 147.5, 146.7, 139.9, 135.4, 134.7, 130.0, 127.3, 127.2, 126.2, 125.2, 124.6, 121.4, 114.1, 83.3, 74.0, 65.3, 55.6, 34.4, 21.7, 10.9; MS (ESI) m/z 439 (M + Na+, 100%), HRMS (ESI, m/z) calcd for C23H20N4O4Na [M + Na]+: 439.1377, found: 439.1381.
:PE = 1:1).
Following the general procedure 5-B, the reaction of compound 11c (54 mg, 0.10 mmol) gave compound 6 (34 mg, yield: 81%) (eluent: EtOAc:PE = 1:1) as colorless crystals. M.p. 166–168 °C (EtOAc); [α]25D −71.0 (c 1.0, MeOH); [α]20D −76.8 (c 1.0, CHCl3) {lit. [α]20D −33 (c 0.37, MeOH);13 [α]25D −90 (c 0.6, MeOH);14 [α]20D −16.9 (c 0.18, CHCl3);12}; IR (film) vmax: 3404, 2962, 2917, 1732, 1684, 1607, 1476, 1325, 1197, 1181 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.30 (s, 1H), 8.18 (dd, J = 7.9, 1.2 Hz, 1H), 7.87 (ddd, J = 7.7, 7.5, 1.5 Hz, 1H), 7.71 (dd, J = 7.9, 0.9 Hz, 1H), 7.59 (ddd, J = 7.9, 7.6, 1.0 Hz, 1H), 7.54 (dd, J = 7.1, 1.0 Hz, 1H), 7.47 (dd, J = 7.6, 1.0 Hz, 1H), 7.43 (ddd, J = 7.4, 7.2, 1.0 Hz, 1H), 7.26 (ddd, J = 7.4, 7.2, 1.0 Hz, 1H), 6.29 (br s, 1H), 5.81 (s, 1H), 4.85 (dd, J = 8.0, 5.3 Hz, 1H), 4.21 (d, J = 9.3 Hz, 1H), 3.00 (dd, J = 14.0, 8.0 Hz, 1H), 2.77 (dd, J = 14.0, 5.3 Hz, 1H), 2.43–2.46 (m, 1H), 1.15 (d, J = 6.7 Hz, 3H), 1.12 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 173.9, 164.9, 159.8, 147.5, 146.9, 140.3, 134.9, 134.7, 130.1, 127.4, 127.2, 126.2, 125.2, 124.6, 121.5, 114.2, 83.7, 73.8, 69.4, 55.7, 34.1, 28.2, 20.2, 18.7; MS (ESI) m/z 453 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C24H22N4O4Na [M + Na]+: 453.1533, found: 453.1535; conditions for the chiral HPLC analysis: Chiralpak AD-H (n-hexane/isopropanol, 70:30), flow rate = 0.8 mL min−1, Rt = 21.4 min, respectively. The enantiomeric excess was determined to be >99%.
:PE = 1:1) as a white solid. M.p. 179–181 °C (EtOAc); [α]20D −105.0 (c 1.0, CHCl3); IR (film) vmax: 3395, 2964, 1732, 1681, 1607, 1476, 1328, 1262, 1185 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.25 (dd, J = 8.2, 1.1 Hz, 1H), 8.01 (s, 1H), 7.77 (ddd, J = 7.9, 7.7, 1.4 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.57 (d, J = 8.1 Hz, 1H), 7.51 (ddd, J = 7.5, 7.4, 1.0 Hz, 1H), 7.40–7.30 (m, 2H), 7.16 (ddd, J = 7.6, 7.5, 0.7 Hz, 1H), 5.80 (s, 1H), 4.85 (d, J = 5.2 Hz, 1H), 4.49 (d, J = 3.0 Hz, 1H), 4.21 (s, 1H), 3.33–3.20 (m, 1H), 2.87 (dd, J = 15.4, 1.3 Hz, 1H), 2.46 (dd, J = 15.4, 6.3 Hz, 1H), 1.24 (d, J = 7.2 Hz, 3H), 0.83 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2, 163.4, 162.1, 147.7, 145.5, 138.6, 135.9, 135.0, 130.7, 128.0, 127.5, 126.7, 125.7, 124.0, 121.7, 115.3, 84.2, 75.6, 68.6, 58.9, 39.2, 25.2, 17.8, 16.2; MS (ESI) m/z 453 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C24H22N4O4Na [M + Na]+: 453.1533, found: 453.1534; conditions for the chiral HPLC analysis: Chiralpak AD-H (n-hexane/EtOH, 30:70), flow rate = 0.8 mL min−1, Rt = 28.1 min, respectively. The enantiomeric excess was determined to be >99%.
:PE = 1:1) as colorless crystals. M.p. 166–168 °C (EtOAc); [α]25D +71.0 (c 1.0, MeOH); [α]20D +76.8 (c 1.0, CHCl3); IR (film) vmax: 3404, 2962, 1732, 1684, 1607, 1476, 1198, 1181, 1076 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.30 (s, 1H), 8.18 (dd, J = 7.9, 1.2 Hz, 1H), 7.87 (ddd, J = 7.7, 7.5, 1.5 Hz, 1H), 7.71 (dd, J = 7.9, 0.9 Hz, 1H), 7.59 (ddd, J = 7.9, 7.6, 1.0 Hz, 1H), 7.54 (dd, J = 7.1, 1.0 Hz, 1H), 7.47 (dd, J = 7.6, 1.0 Hz, 1H), 7.43 (ddd, J = 7.4, 7.2, 1.0 Hz, 1H), 7.26 (ddd, J = 7.4, 7.2, 1.0 Hz, 1H), 6.29 (br s, 1H), 5.81 (s, 1H), 4.85 (dd, J = 8.0, 5.3 Hz, 1H), 4.21 (d, J = 9.3 Hz, 1H), 3.00 (dd, J = 14.0, 8.0 Hz, 1H), 2.77 (dd, J = 14.0, 5.3 Hz, 1H), 2.43–2.46 (m, 1H), 1.15 (d, J = 6.7 Hz, 3H), 1.12 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 173.9, 164.9, 159.8, 147.5, 146.9, 140.3, 134.9, 134.7, 130.1, 127.4, 127.2, 126.2, 125.2, 124.6, 121.5, 114.2, 83.7, 73.8, 69.4, 55.7, 34.1, 28.2, 20.2, 18.7; MS (ESI) m/z 453 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C24H22N4O4Na [M + Na]+: 453.1533, found: 453.1532; conditions for the chiral HPLC analysis: Chiralpak AD-H (n-hexane/isopropanol, 70:30), flow rate = 0.8 mL min−1, Rt = 14.2 min, respectively. The enantiomeric excess was determined to be >99%.
:PE = 1:1) as colorless crystals. M.p. 179–181 °C (EtOAc); [α]20D +105.0 (c 1.0, CHCl3); IR (film) vmax: 3395, 2964, 1732, 1681, 1607, 1476, 1328, 1262, 1185 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.25 (dd, J = 8.2, 1.1 Hz, 1H), 8.01 (s, 1H), 7.77 (ddd, J = 7.9, 7.7, 1.4 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.57 (d, J = 8.1 Hz, 1H), 7.51 (ddd, J = 7.5, 7.4, 1.0 Hz, 1H), 7.40–7.30 (m, 2H), 7.16 (ddd, J = 7.6, 7.5, 0.7 Hz, 1H), 5.80 (s, 1H), 4.85 (d, J = 5.2 Hz, 1H), 4.49 (d, J = 3.0 Hz, 1H), 4.21 (s, 1H), 3.33–3.20 (m, 1H), 2.87 (dd, J = 15.4, 1.3 Hz, 1H), 2.46 (dd, J = 15.4, 6.3 Hz, 1H), 1.24 (d, J = 7.2 Hz, 3H), 0.83 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2, 163.4, 162.1, 147.7, 145.5, 138.6, 135.9, 135.0, 130.7, 128.0, 127.5, 126.7, 125.7, 124.0, 121.7, 115.3, 84.2, 75.6, 68.6, 58.9, 39.2, 25.2, 17.8, 16.2; MS (ESI) m/z 453 (M + Na+, 100%); HRMS (ESI, m/z) calcd for C24H22N4O4Na [M + Na]+: 453.1533, found: 453.1535; conditions for the chiral HPLC analysis: Chiralpak AD-H (n-hexane/EtOH, 30:70), flow rate = 0.8 mL min−1, Rt = 47.0 min, respectively. The enantiomeric excess was determined to be >99%.
:PE = 1:1) to give (−)-14-epi-isochaetominine C (ent-7) (38 mg, yield: 89%) as a white solid. The physical and spectral data of ent-7 thus obtained are in full agreement with those of ent-7 prepared from 25.
Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (21332007 and 21472153) and the Program for Changjiang Scholars and Innovative Research Team in the University of Ministry of Education, China.Notes and references
-
K. C. Nicolaou and S. A. Snyder, Classics in total synthesis, Wiley-VCH, Weinheim, 1996 Search PubMed
.
-
E. J. Corey and X.-M. Cheng, The logic of chemical synthesis, John Wiley & Sons Inc., 1989 Search PubMed
-
(a) P. Bayón and M. Figueredo, Chem. Rev., 2013, 113, 4680 CrossRef PubMed
- For examples of the synthesis of chlorofusin and its all seven chromophore diastereomers, see:
(a) H.-B. Qiu, W.-J. Qian, S.-M. Yu and Z.-J. Yao, Tetrahedron, 2015, 71, 370 CrossRef CAS
-
(a) B. J. Ayers, A. F. G. Glawar, R. F. Martínez, N. Ngo, Z. Liu, G. W. J. Fleet, T. D. Butters, R. J. Nash, C.-Y. Yu, M. R. Wormald, S. Nakagawa, I. Adachi, A. Kato and S. F. Jenkinson, J. Org. Chem., 2014, 79, 3398 CrossRef CAS PubMed
- M. A. Schafroth, G. Zuccarello, S. Krautwald, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2014, 53, 13898 CrossRef CAS PubMed
- For recent reviews/highlights, see:
(a) L. C. Miller and R. Sarpong, Chem. Soc. Rev., 2011, 40, 4550 RSC
-
(a) T. O. Schrader and M. L. Snapper, J. Am. Chem. Soc., 2002, 124, 10998 CrossRef CAS PubMed
-
(a) L. Pérez, R. Alibés, P. de March, F. Busqué, M. Figueredo and J. Font, J. Org. Chem., 2013, 78, 4483 CrossRef PubMed
- For recent reviews on endophytic fungi derived bioactive natural products, see:
(a) X. Zhang, W. Wei and R.-X. Tan, Sci. China: Chem., 2015, 58, 1097 CrossRef CAS
-
(a) R. H. Jiao, S. Xu, J. Y. Liu, H. M. Ge, H. Ding, C. Xu, H. L. Zhu and R. X. Tan, Org. Lett., 2006, 8, 5709 CrossRef CAS PubMed
- E. Amnat, K. Anake, B. Céline, M. Véronique, M. Leka, L. Florence, S. Artur, K. Robert and H. Werner, Planta Med., 2012, 78, 1767 CrossRef PubMed
- C.-Y. An, X.-M. Li, C.-S. Li, M.-H. Wang, G.-M. Xu and B.-G. Wang, Mar. Drugs, 2013, 11, 2682 CrossRef PubMed
- L. Liao, M. You, B. K. Chung, D.-C. Oh, K.-B. Oh and J. Shin, J. Nat. Prod., 2015, 78, 349 CrossRef CAS PubMed
-
(a) Y. Nakao, B. K. S. Yeung, Y. W. Yoshida and P. J. Scheuer, J. Am. Chem. Soc., 1995, 117, 8271 CrossRef CAS
- For excellent reviews on quinazolinone alkaloids, see:
(a) U. A. Kshirsagar, Org. Biomol. Chem., 2015, 13, 9336 RSC
- For an excellent review on the total syntheses based on the strategies of dearomatization of indoles, see: S. P. Roche, J. J. Y. Tendoung and B. Treguier, Tetrahedron, 2015, 71, 3549 CrossRef CAS
- For enantioselective syntheses of chaetominine, see:
(a) B. B. Snider and X. X. Wu, Org. Lett., 2007, 9, 4913 CrossRef CAS PubMed
- For selected recent examples, see:
(a) P.-Q. Huang, Y. Wang, S.-P. Luo, H. Geng, Y.-P. Ruan and A.-E Wang, Tetrahedron Lett., 2015, 56, 1255 CrossRef CAS
- For reviews on peptide coupling reagents, see:
(a) Y. Hamada and T. Shioiri, Chem. Rev., 2005, 105, 4441 CrossRef CAS PubMed
-
(a) C.-X. Fan, X.-L. Hao and Y.-H. Ye, Synth. Commun., 1996, 26, 1455 CrossRef CAS
-
(a) D. Q. Shi, L. C. Rong, J. X. Wang, Q. Y. Zhuang, X. S. Wang and H. W. Hu, Tetrahedron Lett., 2003, 44, 3199 CrossRef CAS
-
(a) R. W. Murray, Chem. Rev., 1989, 89, 1187 CrossRef CAS
-
(a) H. H. Bosshard, R. Mory, M. Schmid and H. Zollinger, Helv. Chim. Acta, 1959, 42, 1653 CrossRef CAS
- For enantioselective syntheses of kapakahines B, E and F, see:
(a) T. Newhouse, C. A. Lewis and P. S. Baran, J. Am. Chem. Soc., 2009, 131, 6360 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all new compounds and compounds 2–7. Chiral HPLC diagrams of compounds 3, 6, 7, 8, ent-3, ent-6, ent-7 and ent-8, crystallographic structure files for 3 and 7 (CIF). CCDC 1423418 and 1423426. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00298b |
| This journal is © the Partner Organisations 2016 |