
Iridium(III)- and rhodium(III)-catalyzed coupling of anilines with α-diazoesters via chelation-assisted C–H activation†
Guo-Dong
Tang
ab,
Cheng-Ling
Pan
*b and
Xingwei
Li
*b
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
bLaboratory of Multi Scale Materials and Molecular Catalysis, School of Materials Science and Engineering, Anhui University of Science and Technology, Huainan, 232001, China. E-mail: clpan@aust.edu.cn; xwli@dicp.ac.cn
First published on 17th November 2015
Abstract
Iridium and rhodium complexes exhibited complementary activity in the coupling of N-functionalized anilines with α-diazoesters via C–H activation. The coupling with α-diazo β-ketoesters was realized under Ir(III) catalysis to afford ester-functionalized indoles. In contrast, coupling with α-diazomalonates under Rh(III) catalysis afforded alkylation products without any annulation. Mechanistic studies have been performed and an iridacycle has been isolated as an intermediate.
Indoles are important building blocks in synthetic organic chemistry. They are also a key structure motif widely present in natural products and pharmaceuticals that have exhibited important biological activity.1 Consequently, increasing efforts have been devoted to the synthesis of indoles. Arguably, the Fischer indole synthesis2 and the Larock indole synthesis3 are among the most commonly employed methods. However, the Fischer indole synthesis required the usage of a strong acid under harsh conditions, which may pose issues of functional group compatibility. On the other hand, the Larock indole synthesis required the employment of 2-iodonated anilines and generated a stoichiometric amount of inorganic wastes.
In order to achieve high efficiency and high atom-economy, the C–H activation strategy has been increasingly employed in recent years, especially in the synthesis of heterocycles.4 This strategy takes advantage of the ubiquitous C–H bond in unactivated arenes as the direct source. In 2008, Fagnou and coworkers reported a highly efficient rhodium(III)5-catalyzed oxidative synthesis of indoles starting from readily available acetanilides and alkynes via C–H activation.6 Inspired by this work, a number of Rh(III)-catalyzed syntheses of nitrogen heterocycles such as indoles and isoquinolines, among others, have been reported.7 In addition, the synthesis of indoles has also been extended to catalysis by other transition metals.8 To address the limitation of using a stoichiometric amount of an oxidant, Glorius reported in 2013 the employment of arenes bearing an oxidizing hydrazine directing group9 for indole synthesis.10
Very recently, Yu, Li, Rovis, and others demonstrated that Rh(III) and Ir(III) catalysts can effect the coupling of arenes with (activated) diazoesters via C–H activation, leading to alkylation, in which the diazo compounds act as a C1 source (carbene precursor).11 Subsequently, the groups of Glorius, Bolm, Wang, Zeng, and others reported that these diazoesters can function as a C2 source in that the electrophilic carbonyl group in the diazoesters can further participate in cyclization reactions under redox-neutral conditions.12 In the case of α-diazoacetylacetates, a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond was incorporated into the newly formed heterocycles. This amounts to the same type of product as was obtained under the oxidative cyclization conditions using alkynes (Scheme 1). Although Rh(III)-catalyzed C–H activation-coupling with α-diazoacetylacetates has been reported, Ir(III) catalysts have been rarely used. In addition, the synthesis of indoles via this redox-neutral strategy remains limited.11e,13 Thus, it is necessary to explore this type of indole synthesis using readily available N-phenyl-2-aminopyridines, although we and others have previously investigated the C–H activation of this substrate in the coupling with unsaturated molecules.14 We now report the Ir(III)- and Rh(III)-catalyzed coupling between N-phenyl-2-aminopyridines and different activated diazoesters.
C double bond was incorporated into the newly formed heterocycles. This amounts to the same type of product as was obtained under the oxidative cyclization conditions using alkynes (Scheme 1). Although Rh(III)-catalyzed C–H activation-coupling with α-diazoacetylacetates has been reported, Ir(III) catalysts have been rarely used. In addition, the synthesis of indoles via this redox-neutral strategy remains limited.11e,13 Thus, it is necessary to explore this type of indole synthesis using readily available N-phenyl-2-aminopyridines, although we and others have previously investigated the C–H activation of this substrate in the coupling with unsaturated molecules.14 We now report the Ir(III)- and Rh(III)-catalyzed coupling between N-phenyl-2-aminopyridines and different activated diazoesters.
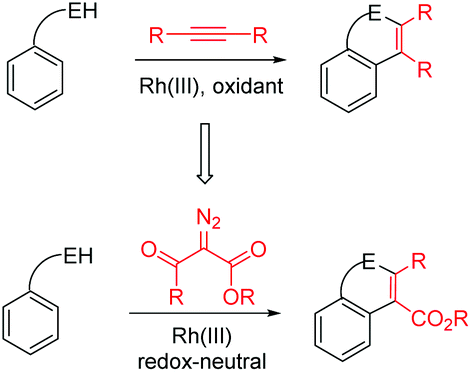 | ||
| Scheme 1 Annulative coupling using alkynes versus α-diazoesters. | ||
We embarked on our studies with the optimization of the coupling of N-phenyl-2-aminopyridine (1a) with ethyl α-diazoacetylacetate (Table 1). The reaction proceeded well when catalyzed by [IrCp*Cl2]2 in the presence of CsOAc and HOAc using water as a green solvent even after 1 h (entry 1).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Time | Yieldb |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.45 mmol), [IrCp*Cl2]2 (2.5 mol%), solvent (3 mL), 100 °C, 5 h, under air. b Isolated yields based on 1. c No CsOAc was used. d No AcOH was used. e No catalyst was used. f [RhCp*Cl2]2 was used as a catalyst. | |||
| 1 | H2O | 1 h | 75% |
| 2 | H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (2:1) :EtOH (2:1) |
1 h | 82% |
| 3 | H2O:EtOH (2:1) |
5 h | 89% |
| 4 | EtOH | 5 h | 53% |
| 5c | H2O:EtOH (2:1) |
5 h | N.R. |
| 6d | H2O:EtOH (2:1) |
5 h | 62% |
| 7e | H2O | 5 h | N.R. |
| 8f | H2O | 5 h | 80% |
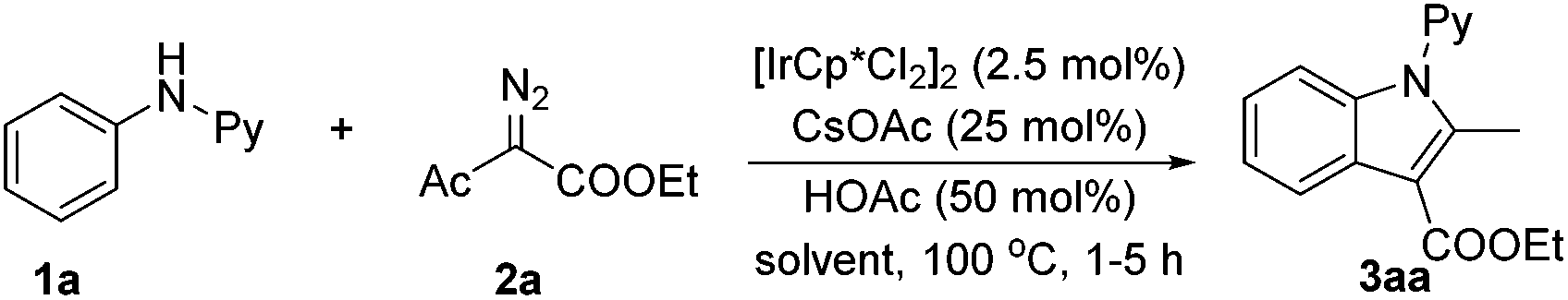
The coupled product 3aa was identified as an ester-functionalized indole. The yield was further improved to 82% when a mixed solvent of water/EtOH was used (v/v = 2:1). Prolonging the reaction time to 5 h led to the isolation of 3aa in 89% yield (entry 3), and the reaction could be performed under air without loss of the isolated yield of the product. Screening also indicated that using EtOH as the sole solvent only afforded inferior results (entry 4). The catalytic amount of CsOAc proved necessary, and omission of HOAc also gave rise to reduced yield (entry 6). Control experiments also confirmed that the metal catalyst is necessary (entry 7). While a slightly lower yield was obtained using the rhodium congener as a catalyst (entry 8), we stuck to iridium catalysis due to the relatively low cost of iridium and the rarity of iridium(III)-catalyzed C–H activation of arenes.15
Having established the optimal conditions, we next explored the scope of generality of this coupling system (Scheme 2). N-Phenyl-2-aminopyridines bearing electron-donating, -withdrawing, and halogen groups at the para position in the benzene ring all coupled smoothly with ethyl α-diazoacetylacetate in consistently high efficiency, and the indole products were isolated in 67–87% yield (3ba–3ga). Introduction of different meta substituents is also well-tolerated, and in all cases C–H functionalization occurred at the less hindered ortho site in high regioselectivity (3ha–3la and 3qa). The reaction also tolerated substrates bearing ortho substituents (3ma–3pa), indicative of the tolerance of the steric effect caused by these ortho substituents. The diazo ester is not limited to ethyl α-diazoacetylacetate, and several other esters bearing different substituents also reacted in comparable or even higher efficiency (3mb, 3bc, and 3bd). In addition to the 2-pyridyl directing group, the directing group has also been successfully extended to a 2-pyrimidyl and the reaction proceeded with equally high efficiency regardless of the substitution pattern of the phenyl ring (3ra–3va). All these results indicated that the catalytic system has the advantage of high efficiency, high selectivity, and good functional group tolerance.
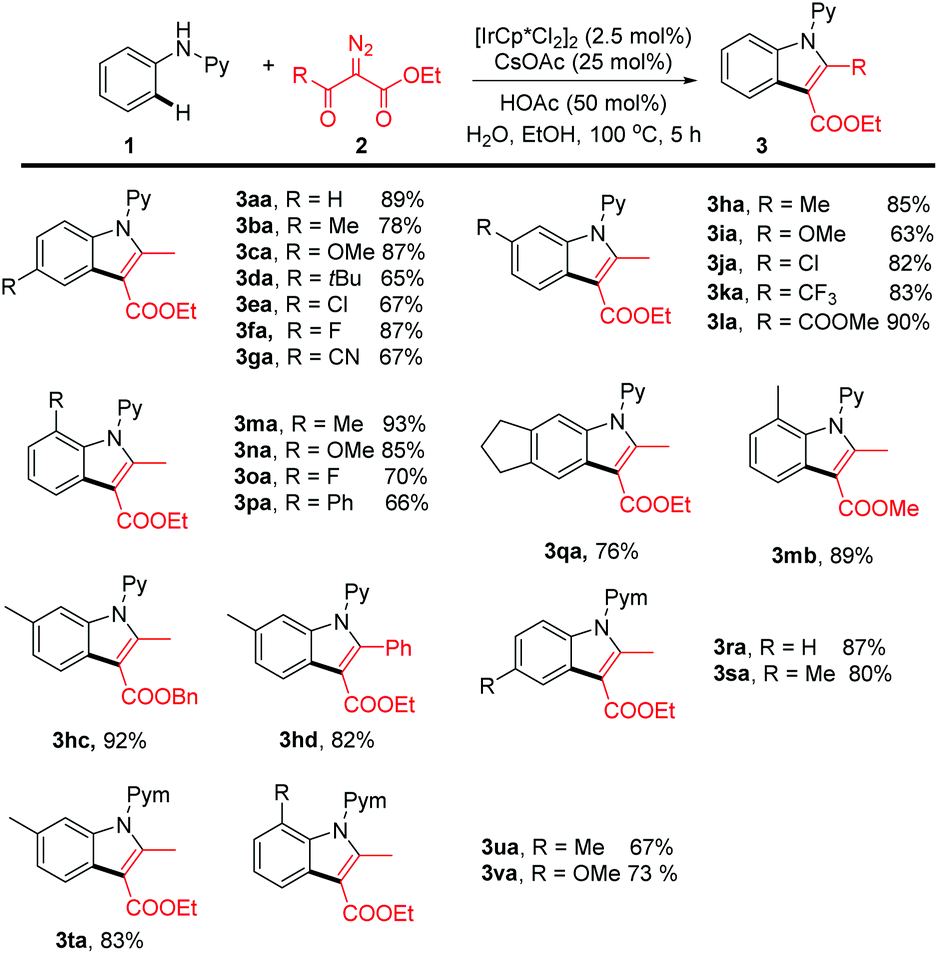 | ||
| Scheme 2 Substrate scope of Ir-catalyzed annulative synthesis of indoles. Reaction conditions: 1 (0.3 mmol), diazoester (0.45 mmol), [IrCp*Cl2]2 (2.5 mol%), CsOAc (25 mol%), HOAc (50 mol%), H2O (2 mL), EtOH (1 mL), 100 °C, 5 h, under air. Isolated yield after column chromatography. | ||
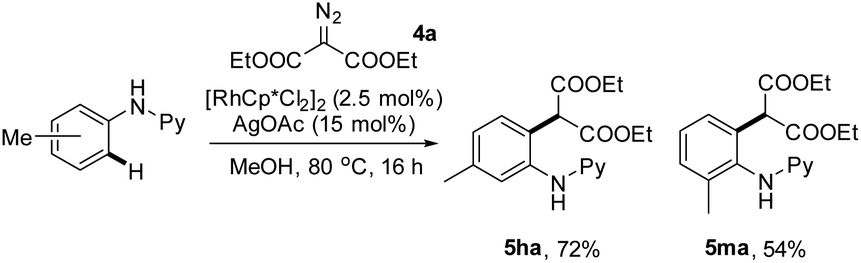
To further define the scope of α-diazo esters, diethyl α-diazomalonate (4a) has been applied as a coupling partner. However, essentially no desired coupling occurred under the iridium-catalyzed conditions. We next resorted to rhodium(III)-catalysis. By following the conditions that we previously reported,11c the coupling of N-phenyl-2-aminopyridines with diethyl α-diazomalonate afforded the ortho alkylation product in good yield without any cyclization (eqn (1)).
 | (1) |
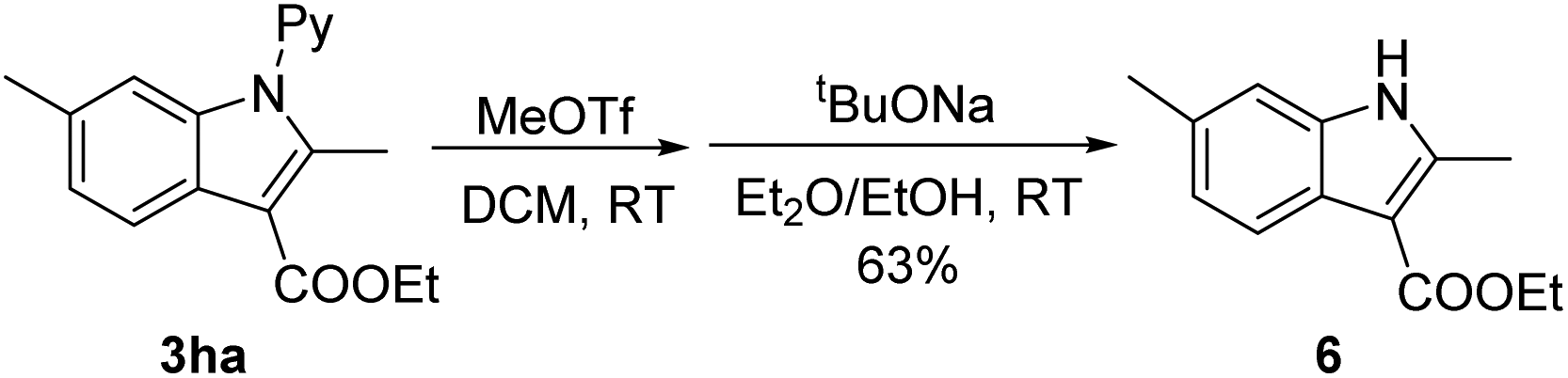
To demonstrate the removability of the directing group, derivatization of a coupled product (3ha) has been performed. Treatment of 3ha with MeOTf led to N-methylation. Subsequent base-treatment in the presence of EtOH afforded the corresponding unprotected NH indole 6 in 63% overall yield (eqn (2)). This result indicates the synthetic utility of our coupling method, and synthesis of 6 using other methods is not trivial.16
 | (2) |
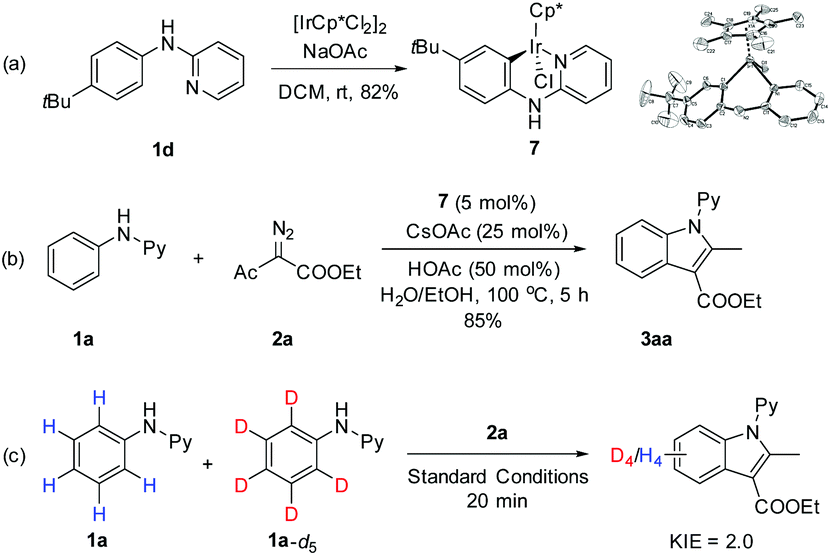
Mechanistic studies have been performed on the coupling of N-phenyl-2-aminopyridines with α-diazoacetylacetate (Scheme 3). To explore the relevancy of C–H activation and the interactions between the arene substrate and the iridium catalyst, a stoichiometric reaction has been performed between 1d and [IrCp*Cl2]2 in the presence of a base.17 An iridacycle 7 was isolated in 82% yield and was fully characterized by X-ray crystallography. When 7 was used as a catalyst for the coupling of 1a and 2a under otherwise the same conditions, the product 3aa was isolated in 85% yield, a yield closely comparable to that obtained under the original conditions using [IrCp*Cl2]2. These results pointed to a conclusion that the reaction occurred via a C–H activation pathway and 7 is a likely intermediate or a direct precursor. To further understand the C–H activation process, the kinetic isotope effect (KIE) has been measured. The KIE result obtained via intermolecular competition using an equimolar mixture of 1a and 1a-d5 under low conversion gave kH/kD = 2.0. This suggests that C–H activation is probably involved in the turnover-limiting step.
 | ||
| Scheme 3 Mechanistic studies. | ||
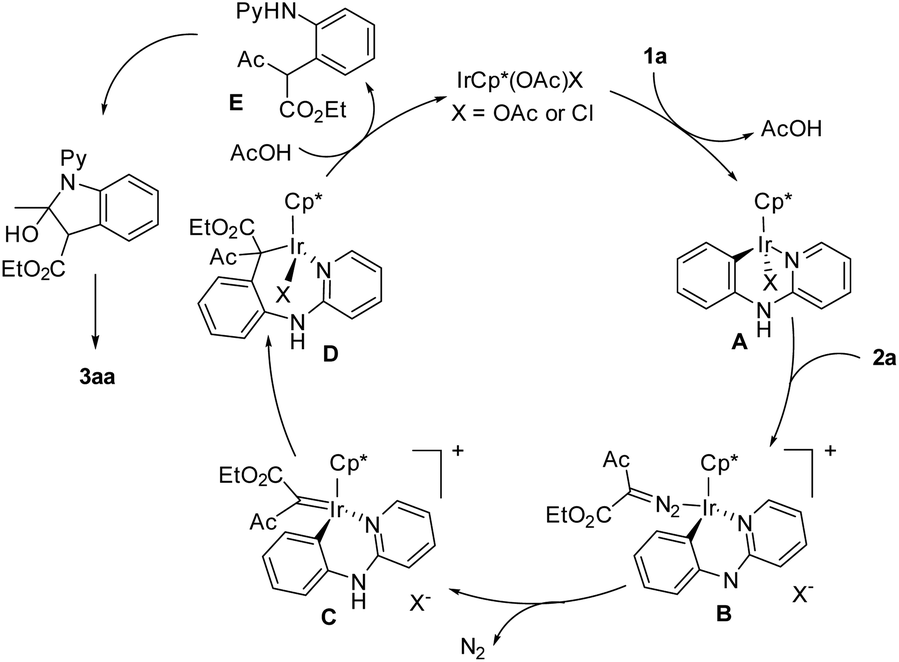
On the basis of these preliminary results and literature precedents, a plausible mechanism is proposed (Scheme 4). Starting from a IrCp*(OAc)X (X = OAc or Cl) catalyst, coordination of the pyridyl nitrogen and subsequent cyclometalation afforded iridacycle A. The incoming diazoester 2a undergoes coordination to afford B. Notably, the coordination of 2a requires dissociation of the X ligand because of the coordination saturation of A. This accounts for the role of a highly polar solvent (water and EtOH), which assists the dissociation of the X ligand and stabilizes the separated charges. In addition, the coordination and C–H activation of 1a may also require the dissociation of the X ligand if a concerned metalation–deprotonation mechanism is followed. Intermediate B is proposed to undergo denitrogenation to afford a carbene intermediate C. Migratory insertion of the Rh–C bond into the carbene gives an iridium alkyl species D, and protonolysis of the Ir–C bond generates an alkylated intermediate E and returns the active catalyst. Intermediate E is proposed to undergo nucleophilic cyclization followed by dehydration to eventually furnish the indole product 3aa. In contrast to the high reactivity of diazoacetylacetates, the reaction stopped at the alkylation stage when a diazomalonate was used.
 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, we have achieved highly efficient couplings between N-phenyl-2-aminopyridines and activated diazoesters. The reactions proceeded via a C–H activation pathway, and iridium and rhodium catalysts offered complementary activity. The coupling with α-diazo β-ketoesters proceeded under iridium catalysis to give indole products as a result of C–H activation, subsequent alkylation, and eventual nucleophilic cyclization. In contrast, the coupling with α-diazomalonates only afforded the alkylation product and high efficiency was realized only with a rhodium(III) catalyst. The pyridyl directing group can be readily removed. This operationally simple method offers direct access to functionalized indoles starting from simple and readily available arenes and may find applications in the synthesis of complex structures.Acknowledgements
This work was supported by the NSFC (no. 21272231, 21402190, and 21472186) and the Dalian Institute of Chemical Physics, Chinese Academy of Sciences.Notes and references
-
(a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed
; (b) S. B. Herzon and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 5342 CrossRef CAS PubMed
-
(a) B. Robinson, Chem. Rev., 1963, 63, 373 CrossRef
-
(a) Y. Monguchi, S. Mori, S. Aoyagi, A. Tsutsui, T. Maegawa and H. Sajiki, Org. Biomol. Chem., 2010, 8, 3338 RSC
- Selected reviews:
(a) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed
-
(a) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC
- D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474 CrossRef CAS PubMed
-
(a) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050 CrossRef CAS PubMed
-
(a) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed
-
(a) F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977 CrossRef CAS PubMed
- D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed
-
(a) T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed
-
(a) J. Shi, J. Zhou, Y. Yan, J. Jia, X. Liu, H. Song, H. E. Xu and W. Yi, Chem. Commun., 2015, 51, 668 RSC
- W. Ai, X. Yang, Y. Wu, X. Wang, Y. Li, Y. Yang and B. Zhou, Chem. – Eur. J., 2014, 20, 17653 CrossRef CAS PubMed
-
(a) W. Song and L. Ackermann, Chem. Commun., 2013, 49, 6638 RSC
-
(a) J. Ryu, J. Kwak, K. Shin, D. Lee and S. Chang, J. Am. Chem. Soc., 2013, 135, 12861 CrossRef CAS PubMed
- R. Odani, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2014, 53, 10784 CrossRef CAS PubMed
- L. Li, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 3492 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. CCDC 1430299. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00316d |
| This journal is © the Partner Organisations 2016 |