
Dearomative C–C and C–N bond cleavage of 2-arylindoles: transition-metal-free access to 2-aminoarylphenones†
Shuang
Luo
*,
Ziwei
Hu
and
Qiang
Zhu
*
State Key Laboratory of Respiratory Disease, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, 190 Kaiyuan Avenue, Guangzhou 510530, China. E-mail: luo_shuang@gibh.ac.cn; zhu_qiang@gibh.ac.cn
First published on 15th January 2016
Abstract
A transition-metal-free conversion of 2-arylindoles to 2-aminoarylphenones, using environmentally benign O2 as the sole oxidant, has been developed. This novel oxidative dearomatization process involves cleavage of both C–C and C–N bonds followed by new C–C and C–O bond formation. The C2 carbon of an indole scaffold was released in the form of CO2.
In recent years, much attention has been paid to chemoselective C–C bond cleavage followed by new C–C and/or C–heteroatom bond formation; since it enables a great number of readily available substances in chemical transformations.1 In general, certain classes of molecules such as carbonyl derivatives, restrained small rings, alkenes or alkynes, etc., in combination with transition-metal catalysts are applied in this process due to the relatively high energy barrier for C–C bond dissociation.2 Meanwhile, dearomatization of aromatic compounds is also a thermodynamically unfavourable transformation in which transition-metal catalysts or hypervalent iodine reagents are normally required.3 Notably, in most of the reported dearomatization reactions, the frameworks of the starting material are maintained in the dearomatized products. Merging the two important classes of reactions together, that is cleavage of the C–C bond(s) inbuilt in an aromatic system in order to lead to dearomatized products, is scarce in the literature.
One major class of C–C bond cleavage reaction proceeds through one C–C bond activation followed by the introduction of a new group from another reactant (a, Scheme 1).2h,j Thus, the part excised is unavoidably wasted. Another form of the reaction involves two bond cleavage including at least one C–C bond, and then two of the three resulting parts are reconnected. That is, a small portion of the substrate is extruded from the parent molecule (b, Scheme 1). For example, transition-metal-catalyzed decarboxylation and decarbonylation reactions are the representatives of this class, in which carbon dioxide and carbon monoxide are extruded from the substrates, respectively (c, Scheme 1).4 However, an extrusion reaction involving a non-carbonyl substrate is rare, especially under transition-metal-free conditions. In a related study, we reported an uncommon hypervalent iodine(III)-promoted tandem demethylenation/C–H cycloamination process of N-benzyl-2-aminopyridines via C–C and C–N bond cleavage.5 Recently, Laha et al. developed a novel conversion of 10,11-dihydro-5H-dibenzo[b,e][1,4]-diazepines to phenazines through transition-metal-free tandem oxidative extrusion of the benzylic methylene group.6 Cui et al. reported a metal-free synthesis of diaryl-1,2-diketones by the extrusion of a carbon atom of alkynones.7 Herein, we present a transformation of 2-arylindoles to unexpected products, which were identified as 2-aminoarylphenones, through dearomative C–C and C–N bond cleavage using environmentally benign oxygen as the sole oxidant (d, Scheme 1). In this process, three chemical bonds (2 C–C and 1 C–N) are cleaved and one new C–C bond and one C–O bond are generated in one step. It is well known that 2-aminoarylphenones are unique precursors for heterocyclic synthesis,8 and traditional methods for their syntheses include Friedel–Crafts acylation,9 palladium-catalyzed carbonylation reactions,10 and addition reactions of Grignard reagents to nitriles.11 However, all of these presented procedures have some disadvantages, including lack of regio-selectivity, use of transition metals or poor functional group tolerance. The development of an efficient and general synthetic route is highly desirable.
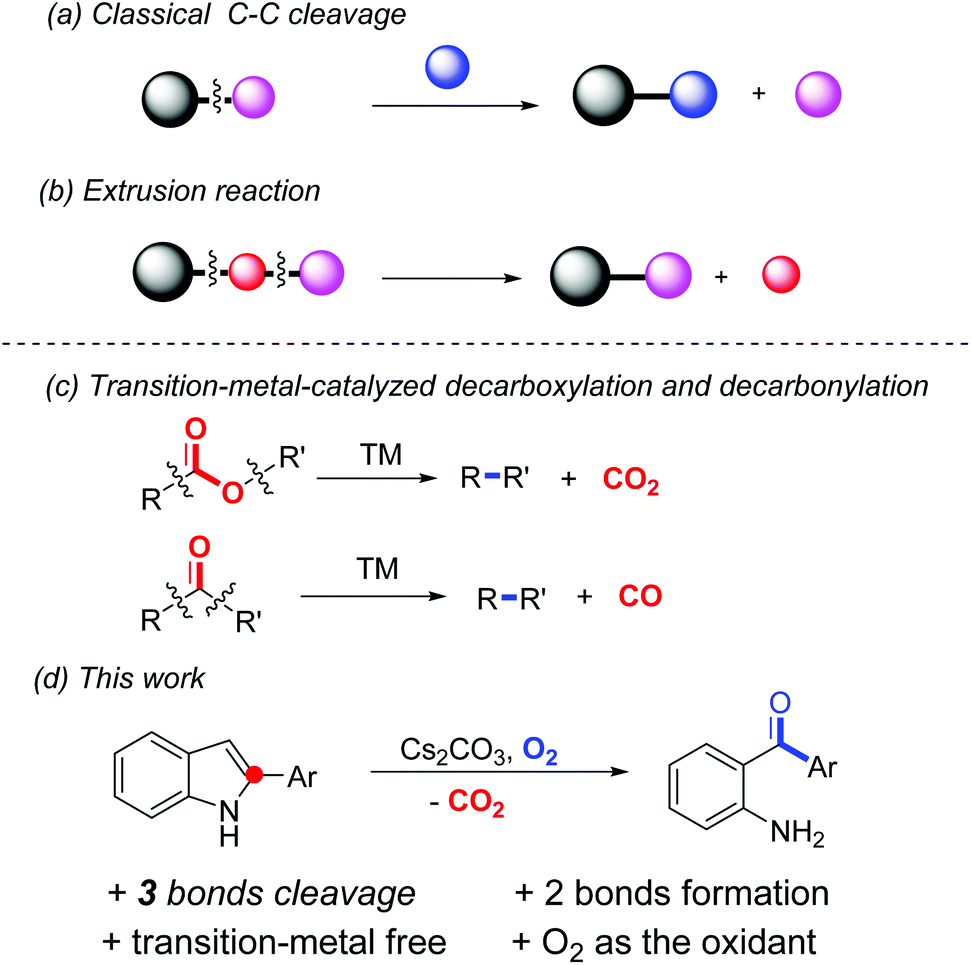 | ||
| Scheme 1 C–C bond cleavage reactions. | ||
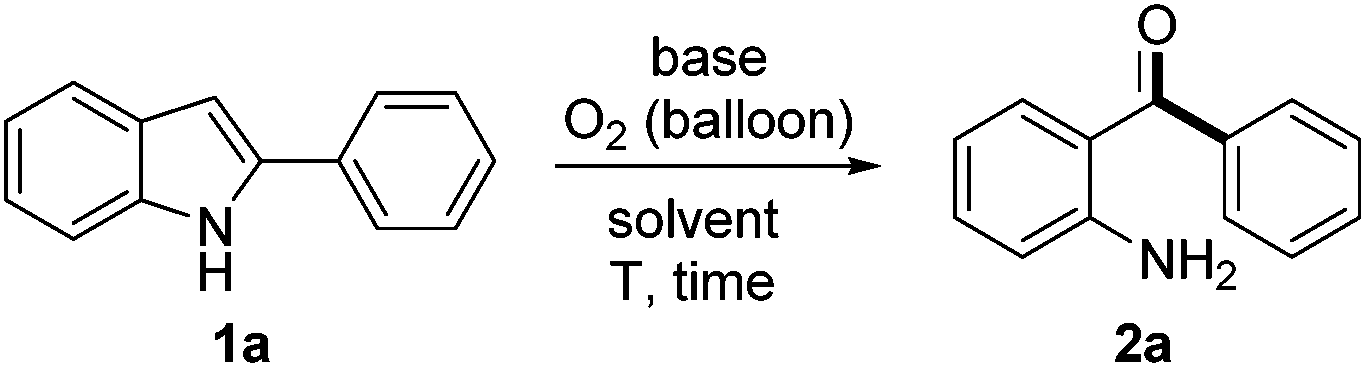
Initially, treatment of 2-phenylindole 1a in the presence of 2.0 equiv. of Cs2CO3 in DMSO (used as received) under an O2 (1 atm) atmosphere at 120 °C afforded an unexpected product which was identified as 2-aminobenzophenone 2a (51% yield, entry 1, Table 1). When the reaction was performed in toluene, dioxane, DCE or acetonitrile, 1a was completely recovered (entries 2–5). However, 1a was decomposed in NMP and 2a could be obtained in only 10–20% yields in DMF or DMA, which indicated the crucial role played by DMSO (entries 6–8). Screening of bases demonstrated that Cs2CO3 was the most effective in promoting the reaction (entries 9–12). It is worth noting that the yield of 2a decreased significantly in anhydrous DMSO. The importance of H2O was further proven by adding 10 equiv. of H2O to anhydrous DMSO, which gave a comparable yield of 2a (see ESI† for details). Shortening the reaction time from 18 h to 16 h was beneficial (56%, entry 14). It was intriguing that when the reaction tube was sealed in air and then equipped with an oxygen balloon rather than pure O2, 2a was isolated in an improved 69% yield (entry 15). Changing the reaction tube to a smaller one (10 mL) under the same procedure described in entry 15, the isolated yield of 2a finally improved to 73% (entry 16).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Base (2.0 eq.) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), solvent (1 mL), base (2.0 equiv.), degassed with O2, 120 °C, 18 h, O2 balloon, in a 50 mL Schlenk tube. b NMR yields with CH2Br2 as an internal standard. Numbers in the parentheses are isolated yields. c Anhydrous DMSO with 100 mg of 4 Å MS. d 16 h. e Without degassing with O2. f Reaction was run in a 10 mL Schlenk tube. | |||
| 1 | DMSO | Cs2CO3 | 51 |
| 2 | Toluene | Cs2CO3 | nr |
| 3 | Dioxane | Cs2CO3 | nr |
| 4 | DCE | Cs2CO3 | nr |
| 5 | MeCN | Cs2CO3 | nr |
| 6 | NMP | Cs2CO3 | 0 |
| 7 | DMF | Cs2CO3 | 20 |
| 8 | DMA | Cs2CO3 | 10 |
| 9 | DMSO | K2CO3 | 0 |
| 10 | DMSO | Na2CO3 | nr |
| 11 | DMSO | CsOAc | nr |
| 12 | DMSO | LiOH | 9 |
| 13c | DMSO | Cs2CO3 | 38 |
| 14d | DMSO | Cs2CO3 | 56 |
| 15d,e | DMSO | Cs2CO3 | 73 (69) |
| 16d,e,f | DMSO | Cs2CO3 | 78 (73) |
With the optimal reaction conditions in hand, the substrate scope was then investigated as summarized in Scheme 2. Substrates with various substituents on the 2-phenyl group, regardless of their electron-donating (Me, tBu and OMe) or electron-withdrawing (F, Cl, Br and phenyl) properties, could undergo the transformation in moderate to good yields. In general, 2-arylindoles with meta substituents gave high yields than the same group in the para position (e.g., 71% for 2g and 40% for 2f, 70% for 2l and 60% for 2m). However, 2-arylindole bearing a strong electron-withdrawing CF3 group (2k) was less compatible under reaction conditions. The substrate with an ortho Me group on the phenyl ring afforded 2b in 58% yield. Moreover, naphthyl and thienyl substituents were also tolerated under these oxidative conditions and provided the desired products in 57% and 55% yields, respectively (2o and 2p). Unfortunately, no product was obtained when 2-alkylindoles or 2-alkenylindoles were subjected to the reaction.
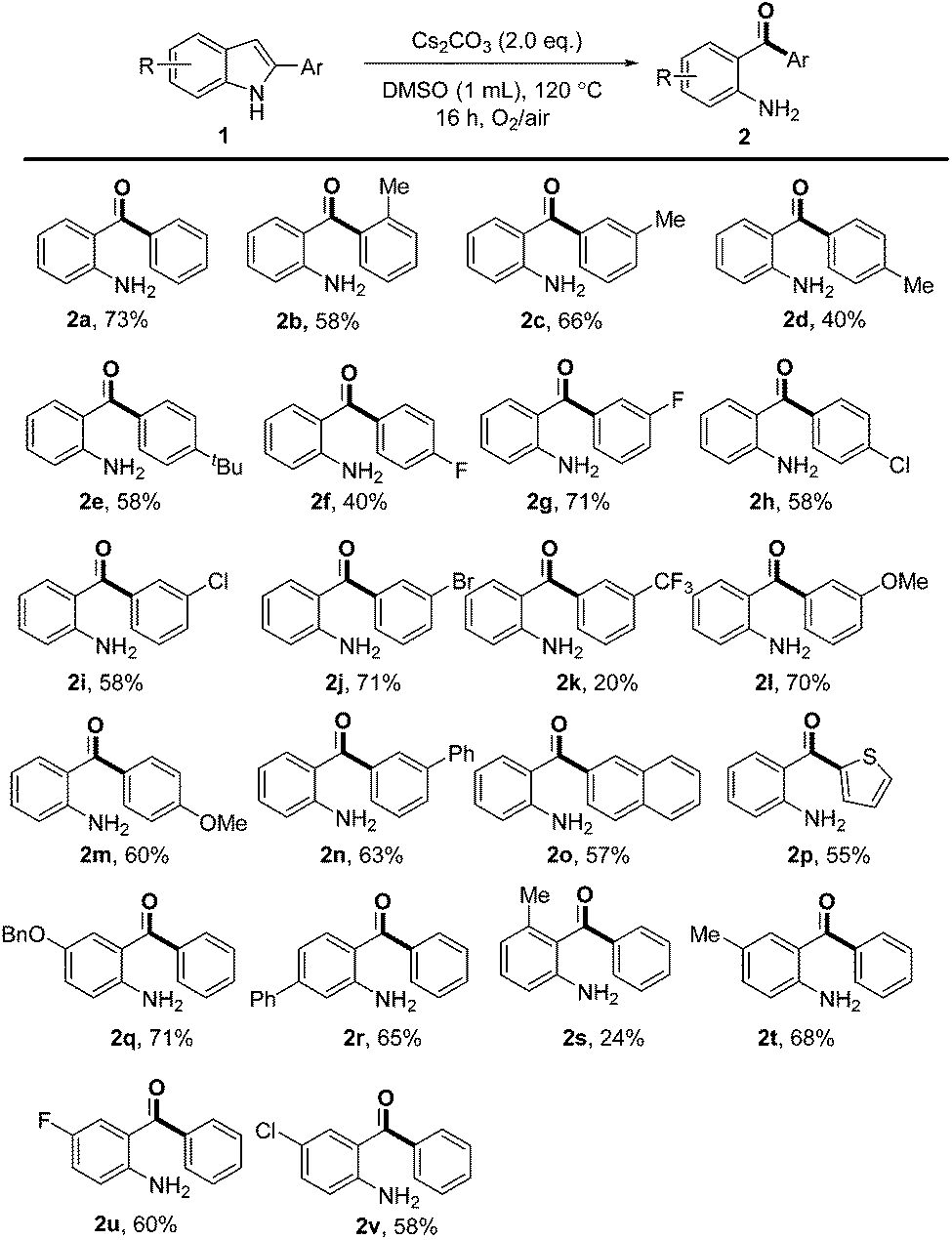 | ||
| Scheme 2 Substrate scope. Reaction conditions: 1 (0.2 mmol), Cs2CO3 (0.4 mmol), DMSO (used as received, 1.0 mL), 10 mL Schlenk tube, sealed in air, O2 balloon, 120 °C, 16 h. | ||
Then, substrates with different functional groups on the indole ring were also investigated. The corresponding products were obtained in moderate to good yields irrespective of the electronic nature of the substituents on the 5- or 6-position of the indole ring (2q, 2r, 2t, 2u and 2v). However, a low yield of 24% was obtained when a methyl group was located at the 4-position of the indole ring, probably due to the steric hindrance (2s). The structures of products 2r and 2s clearly showed that the anilinic carbon linked to the carbonyl moiety was the one which was originally connected to C3 of indole.
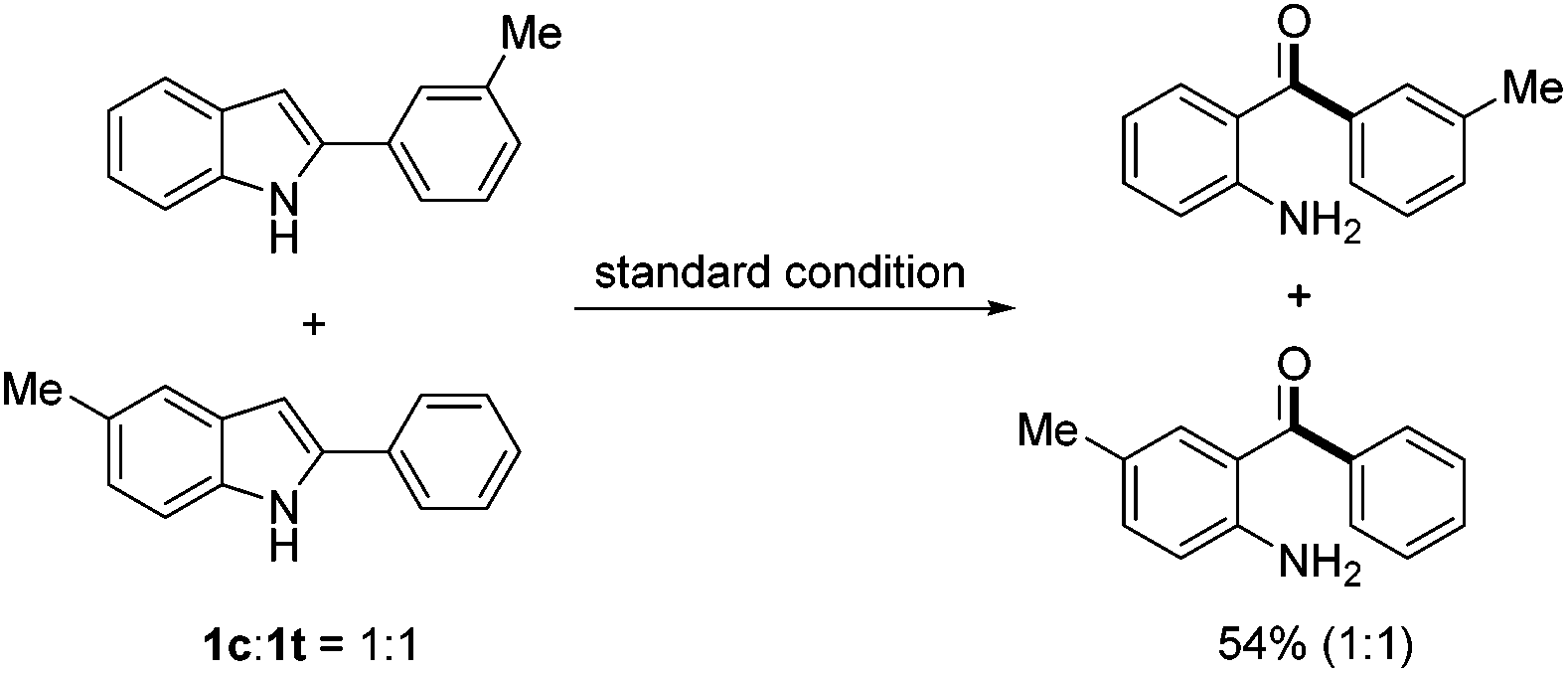
To gain more insight into the reaction mechanism, a crossover experiment using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2-(m-tolyl)-1H-indole (1c) and 5-methyl-2-phenyl-1H-indole (1t) was conducted under the standard conditions. No crossover product was detected by NMR analysis of the reaction products, which indicated that an intramolecular pathway was involved (eqn (1)). To confirm which carbon atom (C2 or C3) was extruded during the reaction, 2-phenyl-2-[13C]indole (13C-1a) was prepared and subjected to the standard conditions. No 13C enriched 2a was isolated, demonstrating undoubtedly that the indole C2 carbon was extruded during the process (eqn (2)).
:1 mixture of 2-(m-tolyl)-1H-indole (1c) and 5-methyl-2-phenyl-1H-indole (1t) was conducted under the standard conditions. No crossover product was detected by NMR analysis of the reaction products, which indicated that an intramolecular pathway was involved (eqn (1)). To confirm which carbon atom (C2 or C3) was extruded during the reaction, 2-phenyl-2-[13C]indole (13C-1a) was prepared and subjected to the standard conditions. No 13C enriched 2a was isolated, demonstrating undoubtedly that the indole C2 carbon was extruded during the process (eqn (2)).
 | (1) |
 | (2) |
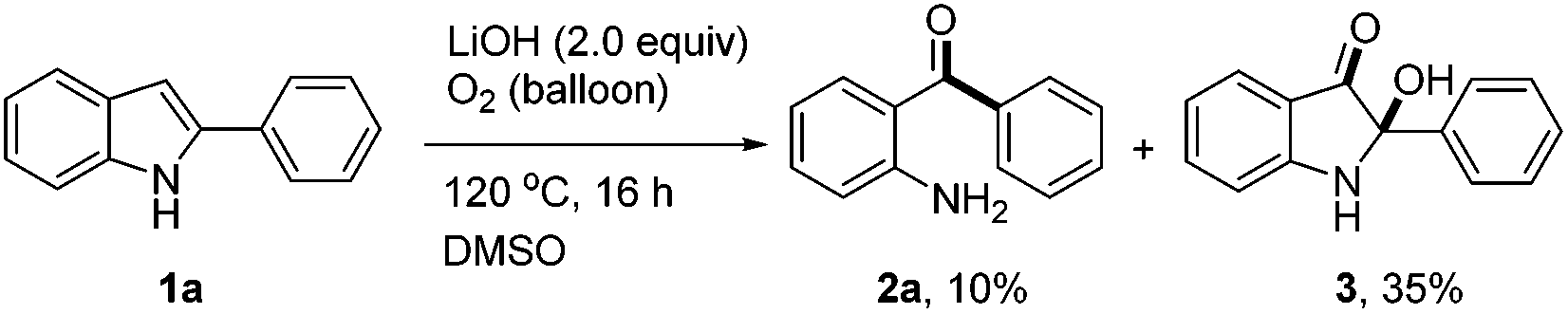
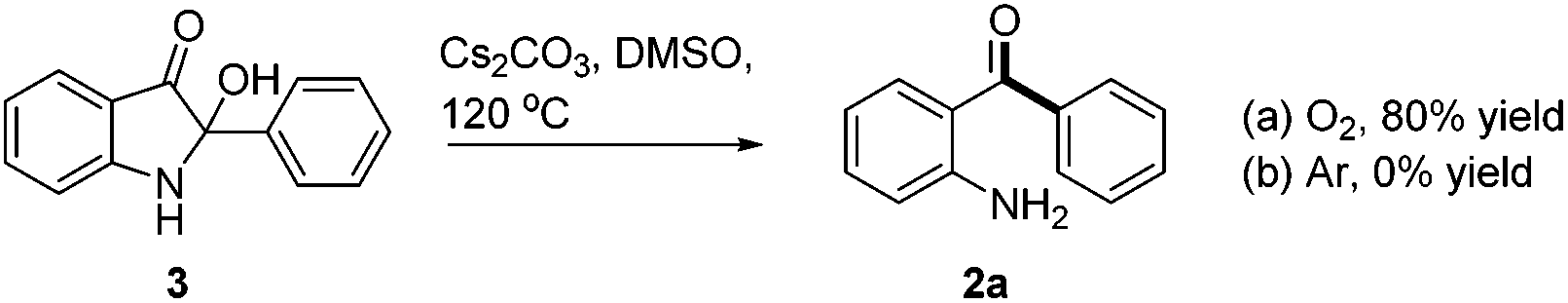
During the optimization of the reaction conditions, we found that when LiOH was used as a base, the desired product 2a was isolated in low yield (10%) along with a byproduct 3 (35% yield, eqn (3)). Subjecting compound 3 to the standard reaction led to the desired product 2a in 80% yield, which suggested that compound 3 might be a reaction intermediate (eqn (4)(a)). It was interesting to find out that compound 3 was stable in the absence of O2 (eqn (4)(b)). In addition, CO2 was generated by passing the gas through the reaction tube into clear limewater which turned cloudy during the test (see ESI† for details).7
 | (3) |
 | (4) |
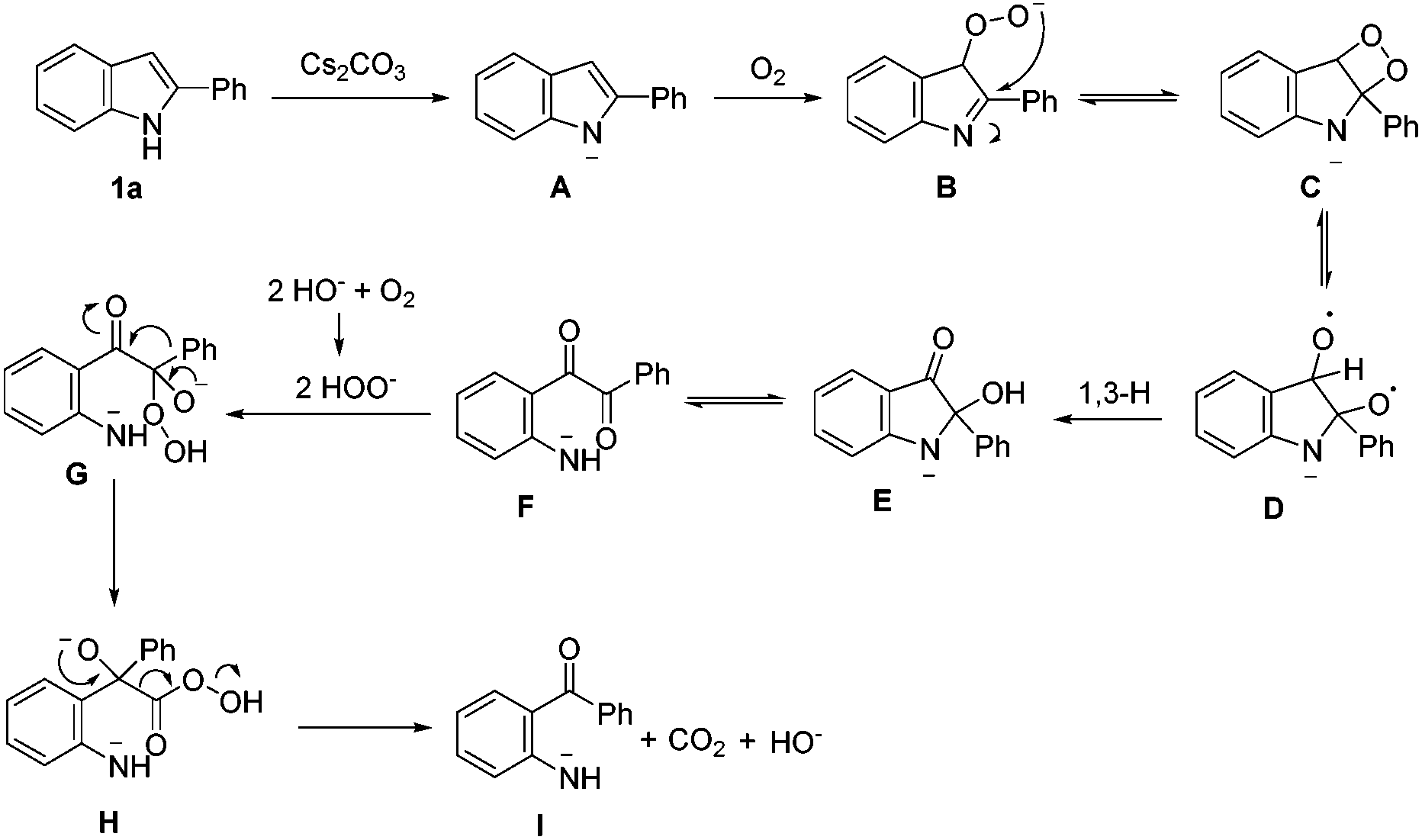
Based on the results obtained, a plausible mechanistic pathway was proposed for this uncommon oxidative cleavage reaction (Scheme 3). A peroxy anion B is formed by the deprotonated substrate in the presence of a base and oxygen. Then intramolecular nucleophilic attack on the C–N double bond results in a four-membered ring C. Subsequently, intermediate D is formed by a homolytic cleavage of the peroxy bond, followed by the 1,3-H shift to give intermediate E, which can be protonated to afford 3. Then, C–N bond cleavage in the aminoacetal moiety of E generates a diaryl-1,2-diketone F, which undergoes nucleophilic attack by the in situ-formed peroxy anion to afford intermediate G. Finally, aryl group migration and subsequent elimination of carbon dioxide give intermediate I, which is protonated to afford the final product.
 | ||
| Scheme 3 Proposed reaction mechanism. | ||
In summary, we have developed an unexpected, novel and environmentally benign method for the conversion of 2-arylindoles to 2-aminoarylphenones under transition-metal-free conditions using oxygen as the sole oxidant. The reaction proceeds through the cleavage of two C–C bonds and one C–N bond followed by new C–C and C–O bond formation. A reaction using 13C labeled 2-arylindole revealed that the C2 carbon of an indole scaffold was released in the form of CO2.
Acknowledgements
We are grateful for the financial support of this work by National Science Foundation of China (21202167, 21402203, 21472190, and 21532009).Notes and references
- (a) K. C. Bishop, Chem. Rev., 1976, 76, 461 CrossRef CAS; (b) P. L. Watson and G. W. Parshall, Acc. Chem. Res., 1985, 18, 51 CrossRef CAS; (c) R. H. Crabtree, Chem. Rev., 1985, 85, 245 CrossRef CAS; (d) P. W. Jennings and L. L. Johnson, Chem. Rev., 1994, 94, 2241 CrossRef CAS; (e) R. H. Crabtree, in Chemistry of Alkanes and Cycloalkanes, ed. S. Patai and Z. Rappoport, Wiley, New York, 1994, p. 653 Search PubMed; (f) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (g) T. Takahashi, M. Kotora, R. Hara and Z. Xi, Bull. Chem. Soc. Jpn., 1999, 72, 2591 CrossRef CAS; (h) M. Murakami and Y. Ito, In Topics in Organometallic Chemistry, ed. S. Murai, Springer, Berlin, Germany, 1999, pp. 97–129 and references therein Search PubMed; (i) T. Takahashi and K.-I. Kanno, Top. Organomet. Chem., 2004, 8, 217 CrossRef; (j) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (k) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed; (l) Topics in Current Chemistry: C–C Bond Activation, ed. G. B. Dong, Springer, Berlin, Germany, 2014, vol. 346 Search PubMed.
- (a) C. H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (b) Y. J. Park, J. W. Park and C. H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed; (c) T. Niwa, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2007, 46, 2643 CrossRef CAS PubMed; (d) D. V. Gribkov, S. J. Pastine, M. Schnurch and D. Sames, J. Am. Chem. Soc., 2007, 129, 11750 CrossRef CAS PubMed; (e) A. M. Dreis and C. J. Douglas, J. Am. Chem. Soc., 2008, 131, 412 CrossRef PubMed; (f) M. T. Wentzel, V. J. Reddy, T. K. Hyster and C. J. Douglas, Angew. Chem., Int. Ed., 2009, 48, 6121 CrossRef CAS PubMed; (g) J. Wang, W. Chen, S. Zuo, L. Liu, X. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 12334 CrossRef CAS PubMed; (h) J. Wang, B. Liu, H. Zhao and J. Wang, Organometallics, 2012, 31, 8598 CrossRef CAS; (i) H. M. Ko and G. B. Dong, Nat. Chem., 2014, 6, 739 CrossRef CAS PubMed; (j) R. Zeng and G. B. Dong, J. Am. Chem. Soc., 2015, 137, 1408 CrossRef CAS PubMed; (k) H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2011, 133, 15244 CrossRef CAS PubMed; (l) K. hen, H. Li, Y. Li, X.-S. Zhang, Z.-Q. Lei and Z.-J. Shi, Chem. Sci., 2012, 3, 1645 RSC; (m) Z.-Q. Lei, F. Pan, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, Y.-X. Li, J. Sun and Z.-J. Shi, J. Am. Chem. Soc., 2015, 137, 5012 CrossRef CAS PubMed.
- (a) A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917 CrossRef CAS PubMed; (b) S. P. Roche and J. A. Porco Jr., Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS PubMed; (c) D. S. Wang, Q. A. Chen, S. M. Lu and Y. G. Zhou, Chem. Rev., 2012, 112, 2557 CrossRef CAS PubMed; (d) C. X. Zhao, C. Zheng and S. L. You, Acc. Chem. Res., 2014, 47, 2558 CrossRef PubMed.
- (a) C. Zhang and D. Seidel, J. Am. Chem. Soc., 2010, 132, 1798 CrossRef CAS PubMed; (b) J. D. Weaver, B. J. Ka, D. K. Morris, W. Thompson and J. A. Tunge, J. Am. Chem. Soc., 2010, 132, 12179 CrossRef CAS PubMed; (c) F. Zhang and M. F. Greaney, Angew. Chem., Int. Ed., 2010, 49, 2768 CrossRef CAS PubMed; (d) B. M. Trost, B. Schäffner, M. Osipov and D. A. A. Wilton, Angew. Chem., Int. Ed., 2011, 50, 3548 CrossRef CAS PubMed; (e) P. Hu, M. Zhang, X. Jie and W. Su, Angew. Chem., Int. Ed., 2012, 51, 227 CrossRef CAS PubMed; (f) R. Shang, D.-S. Ji, L. Chu, Y. Fu and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 4470 CrossRef CAS PubMed; (g) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429 CrossRef CAS PubMed; (h) P. Fristrup, M. Kreis, A. Palmelund, P. Norrby and R. Madsen, J. Am. Chem. Soc., 2008, 130, 5206 CrossRef CAS PubMed; (i) M. Murakami, H. Amii, K. Shigeto and Y. Ito, J. Am. Chem. Soc., 1996, 118, 8285 CrossRef CAS; (j) A. E. Roa, V. Salazar, J. López-Serrano, E. Ońate, M. Paneque and M. L. Poveda, Organometallics, 2012, 31, 716 CrossRef CAS; (k) A. E. Roa, V. Salazar, J. López-Serrano, E. Ońate, J. G. Alvarado-Rodríguez, M. Paneque and M. L. Poveda, Organometallics, 2012, 31, 3185 CrossRef CAS.
- D. Liang, Y. He, L. Liu and Q. Zhu, Org. Lett., 2013, 15, 3476 CrossRef CAS PubMed.
- J. K. Laha, K. S. S. Tummalapalli and A. Gupta, Org. Lett., 2014, 16, 4392 CrossRef CAS PubMed.
- X. Wang, G. Cheng, J. Shen, X. Yang, M. Wei, Y. Feng and X. Cui, Org. Chem. Front., 2014, 1, 1001 RSC.
- (a) T. Chanda, R. K. Verma and M. S. Singh, Chem. – Asian J., 2012, 7, 778 CrossRef CAS PubMed; (b) S. K. Panja and S. Saha, RSC Adv., 2013, 3, 14495 RSC; (c) D. Zhao, Q. Shen and J.-X. Li, Adv. Synth. Catal., 2015, 357, 339 CrossRef CAS.
- I. Ivanov, S. Nikolova and S. S. Abeghe, Synth. Commun., 2006, 36, 1405 CrossRef CAS.
- J. Chen, L. Ye and W. Su, Org. Biomol. Chem., 2014, 12, 8204 CAS.
- J. Zhang, D. Zhu, C. Yu, C. Wan and Z. Wang, Org. Lett., 2010, 12, 2841 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c5qo00394f |
| This journal is © the Partner Organisations 2016 |