
Primary alkyl bis-catecholato silicates in dual photoredox/nickel catalysis: aryl- and heteroaryl-alkyl cross coupling reactions†‡
Christophe
Lévêque
a,
Ludwig
Chenneberg
a,
Vincent
Corcé
a,
Jean-Philippe
Goddard
*b,
Cyril
Ollivier
*a and
Louis
Fensterbank
*a
aInstitut Parisien de Chimie Moléculaire, UMR CNRS 8232, Sorbonne Universités UPMC Univ Paris 06. 4 Place Jussieu, CC 229, F-75252 Paris Cedex 05, France. E-mail: cyril.ollivier@upmc.fr; louis.fensterbank@upmc.fr; Fax: +33(0)144277360
bLaboratoire de Chimie Organique et Bioorganique EA 4566, Université de Haute-Alsace, Ecole Nationale Supérieure de Chimie de Mulhouse. 3 Bis rue Alfred Werner, 68093 Mulhouse Cedex, France. E-mail: jean-philippe.goddard@uha.fr
First published on 15th February 2016
Abstract
Primary alkyl bis-catecholato silicates have been successfully engaged with aryl and heteroaryl bromide substrates in photoredox/nickel dual catalysis to provide aryl- and heteroaryl-alkyl cross coupling products. The scope of the transformation is wide and the process appears to be tolerant of various functional groups present. Of note, most examples rely on the challenging use of highly reactive primary radicals which constitutes a significant advance in these cross coupling reactions.
Photoredox/nickel dual catalysis1,2 has recently emerged as a novel opportunity to form C–C bonds and rapidly couple functionalized fragments for the elaboration of more complex molecules.3 A key advantage of this strategy is to merge the mildness of visible light photoredox catalysis for the generation of hot radical entities4 with the robustness of nickel catalyzed cross coupling reactions.5 While the electrophilic partner involved in the oxidative addition to nickel has so far consisted of aryl halides and related substrates,6 more functional variation has been brought on the radical source. To the best of our knowledge, all studies have involved α-amino,2b,6,7 α-O,2b,6 α-phenyl,2b,8 α-oxo,9 and secondary6 carboxylic acids, mixed anhydrides,10 dimethylaniline,2b benzyl,2a,11 secondary2a,9,12 and activated (α-O) primary trifluoroborates.13 More recently, potassium and ammonium bis-catecholato silicates have been reported by us14 as a valuable source of C-centered radicals upon visible light photooxidation using Ir[(dF(CF3)ppy)2(bpy)](PF6) as the catalyst. The silicate substrates offer neat advantages over the related trifluoroborates in terms of solubility, byproducts (no gas release, no fluoride, no boron) and the scope of the possibly generated radicals (from stabilized alkyl ones to highly reactive primary ones). As a perspective, it was also found that they could be engaged in photoredox/nickel dual catalysis of Csp2–Csp3 cross-coupling reactions, notably involving unstabilized primary radicals.14a,15
The limited sampling of these preliminary findings drove us to gain more insight into this transformation and define its scope by varying both partners (silicates and arylbromides, see Scheme 1). Subsequent to the first report of these cross coupling reactions by us,14a the group of Molander described similar transformations of related ammonium bis-catecholato secondary and primary alkyl silicates with aryl- and heteroaryl-bromides.16 Herein, we focus on primary radical intermediates and bring a complementary picture to these highly powerful reactions.
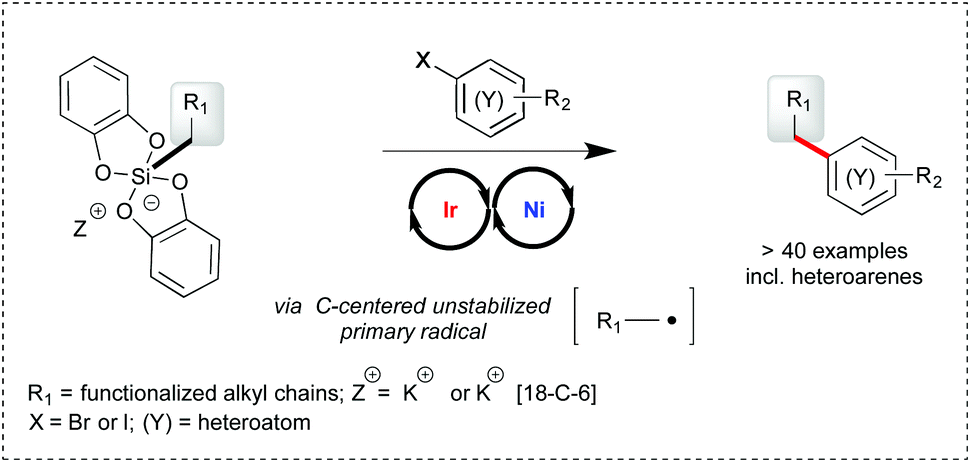 | ||
| Scheme 1 Photoredox/nickel dual catalysis involving primary radicals from silicates. | ||
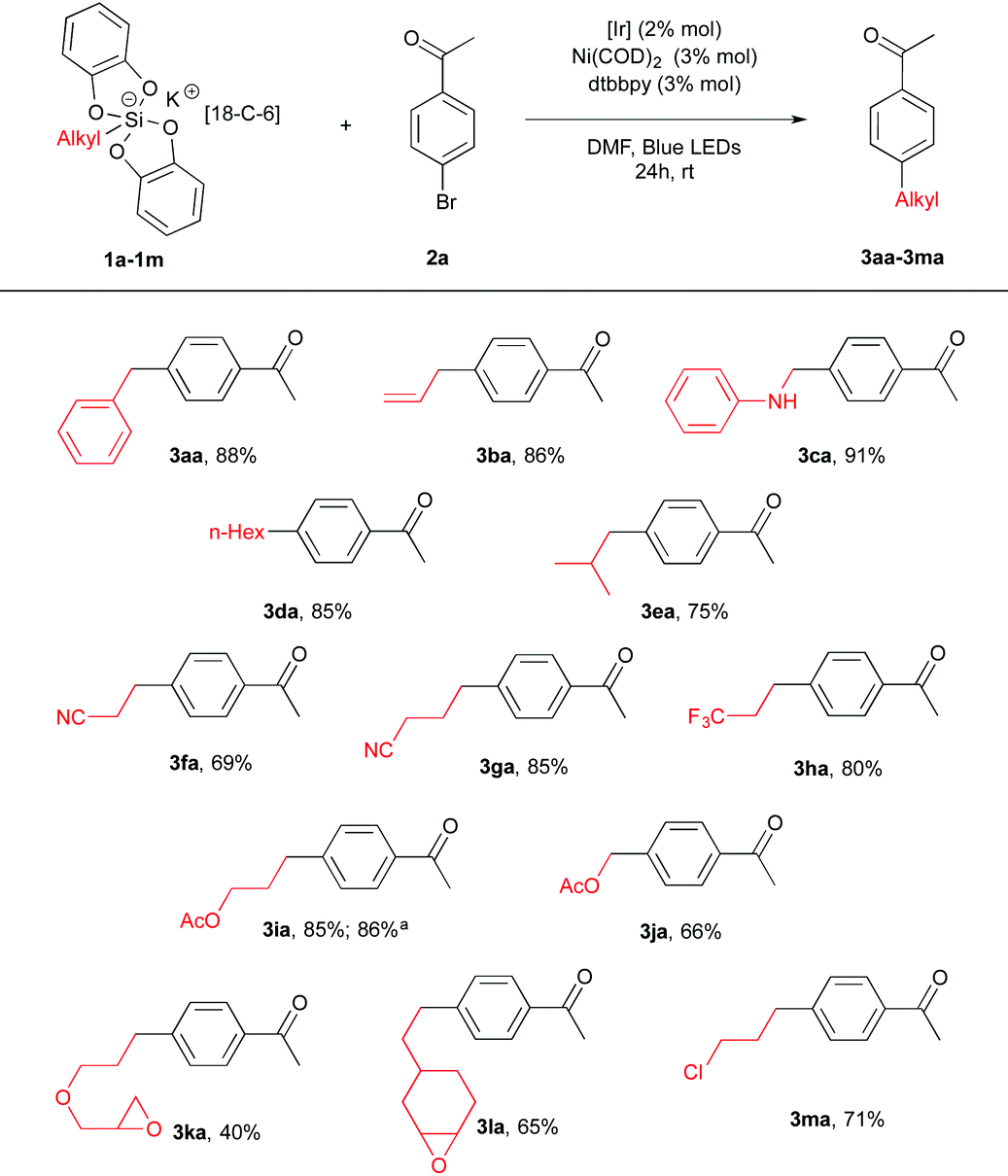
We first prepared a series of potassium silicates 1. As found before,14 the latter are rendered rock stable by admixing the 18-C-6 crown ether additive. Nevertheless, we could show that this additive is not necessary to promote homolytic reactivity (see below). A preliminary screening of conditions with a given arylbromide, namely 4-bromoacetophenone 2a led us to the following protocol. To a DMF solution of silicate 1 (1.5 equiv., 0.1 M) in the presence of Ir[(dF(CF3)ppy)2(bpy)](PF6) (2 mol%), Ni(COD)2 (3 mol%) and dtbbpy (3 mol%) 4-bromoacetophenone 2a (1 equiv.) was added as a ligand. After stirring for 24 h at rt, excellent yields of coupling products 3aa–3ca were obtained from benzyl, allyl and α-amino silicates (1a–c). Gratifyingly, less stabilized radicals could also be involved furnishing the corresponding coupling products 3 in 40–85% yields bearing various functional groups such as an ester (3ia, 3ja), a nitrile (3fa, 3ga), an oxirane (3ka, 3la) or a chloride (3ma). Interestingly, a comparison was made with the corresponding benzyltrifluoroborate precursor and showed under our conditions a more productive reaction (adduct 3aa) from the silicate 1a (88% vs. 58% yield). Of note, potassium silicate with no 18-C-6 1i′ also provided a good yield (86%) of the coupling product 3ia (Table 1).
| a Starting from the corresponding potassium silicate with no 18-C-6 1i′ and 2a. |
|---|
|
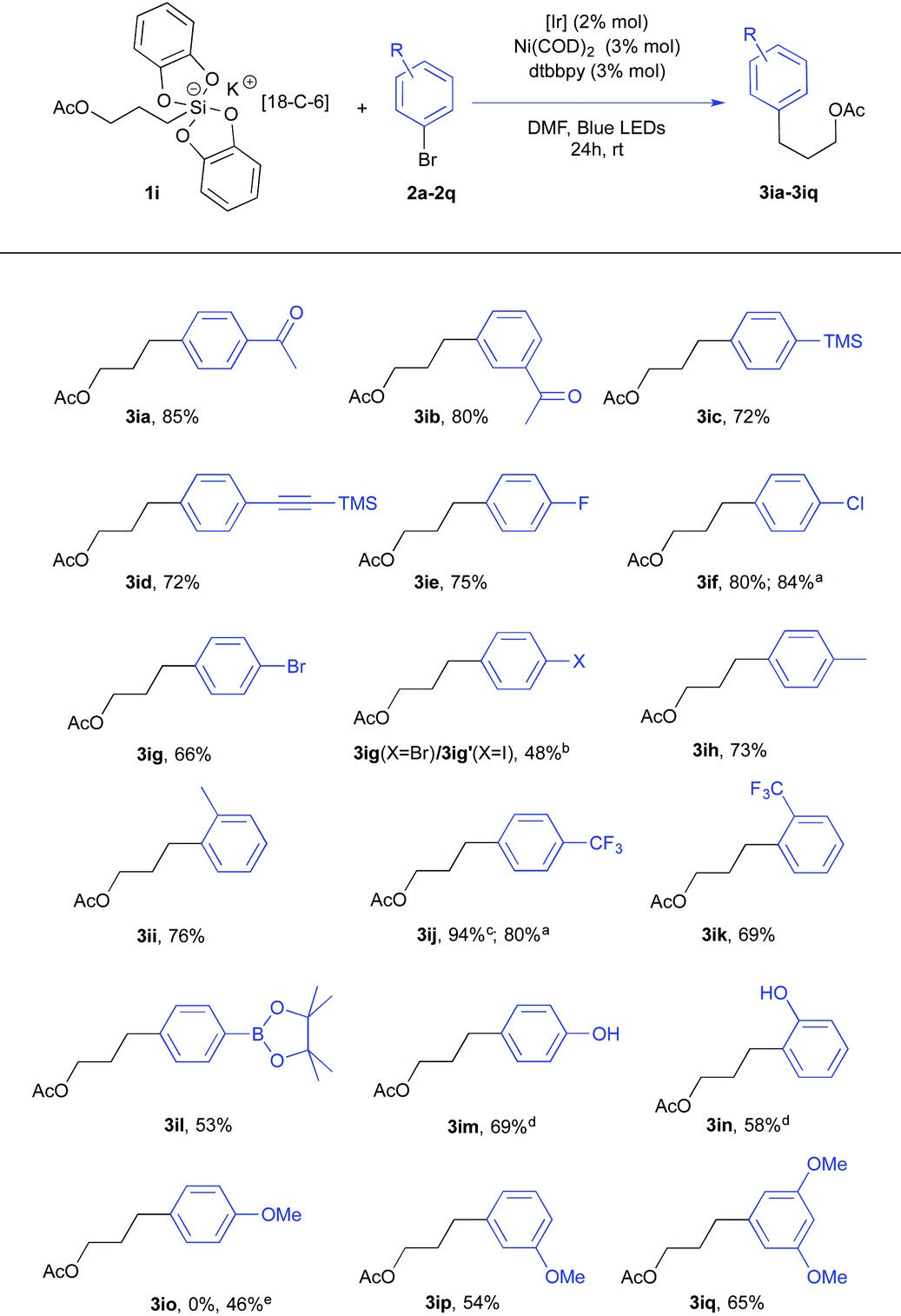
Using the same conditions, a series of arylbromides 2a–2q with varied substitution patterns could be used in conjunction with acetate silicate 1i or 1i′ (Table 2). First, the reaction scope proved to be quite wide. Second, a closer examination of these results revealed interesting features. For instance, the difference of reactivity between a Csp2–Br and a Csp2–I bond was investigated with 1-bromo-4-iodobenzene 2g. Not surprisingly, a preferred oxidative addition took place at the carbon-iodide bond17 giving a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of products 3ig (X = Br) and 3ig′ (X = I). This appeared quite opportune for electron enriched aryl substrates.18 While meta-bromo anisoles (2p and 2q) could be effectively used (adducts 3ip and 3iq), only the starting material was recovered with ortho-bromoanisole and para-bromoanisole. The reluctant behavior of para-bromoanisole could be partly fixed by using the corresponding iodo derivative since the cross coupling product 3io was obtained albeit in modest yield (46%). These findings contrast with Molander's report16a in which electron rich substrates proved to be competent. Their catalytic mixture (5 mol% NiCl2(dme), 5 mol% dtbbpy, and 2 mol% Ru(bpy)3(PF6)2) differs from ours (3 mol% Ni(COD)2, 3 mol% dtbbpy and 2 mol% Ir[(dF(CF3)ppy)2(bpy)](PF6)) implying interesting metal and ligand effects that we are now investigating.18
:1 ratio of products 3ig (X = Br) and 3ig′ (X = I). This appeared quite opportune for electron enriched aryl substrates.18 While meta-bromo anisoles (2p and 2q) could be effectively used (adducts 3ip and 3iq), only the starting material was recovered with ortho-bromoanisole and para-bromoanisole. The reluctant behavior of para-bromoanisole could be partly fixed by using the corresponding iodo derivative since the cross coupling product 3io was obtained albeit in modest yield (46%). These findings contrast with Molander's report16a in which electron rich substrates proved to be competent. Their catalytic mixture (5 mol% NiCl2(dme), 5 mol% dtbbpy, and 2 mol% Ru(bpy)3(PF6)2) differs from ours (3 mol% Ni(COD)2, 3 mol% dtbbpy and 2 mol% Ir[(dF(CF3)ppy)2(bpy)](PF6)) implying interesting metal and ligand effects that we are now investigating.18
|
a Starting from the corresponding potassium silicate with no 18-C-6 1i′.
b Starting from 1-bromo-4-iodobenzene. A 10:1 mixture of 3ig and 3ig′ was isolated.
c 2 mol% Ru(bpy)3(PF6)2 was used instead of 2 mol% [lr].
d Pinacolborane directly oxidized before purification (H2O2, NaOH for 30 min at 0 °C).
e Starting from 4-iodoanisole.
|
|---|
|
Remarkably, a pinacol boronate function could also be tolerated under these conditions11 as illustrated by the formation of 3il that could be utilized for further coupling reactions. Nevertheless, direct isolation of this product after column chromatography was detrimental to the yield. Instead, when the crude reaction mixture was oxidized (NaOH, H2O2), a higher yield (69% vs. 53% for 3il) of the phenol product 3im was recorded. A similar protocol was applied to give the phenol product 3in.
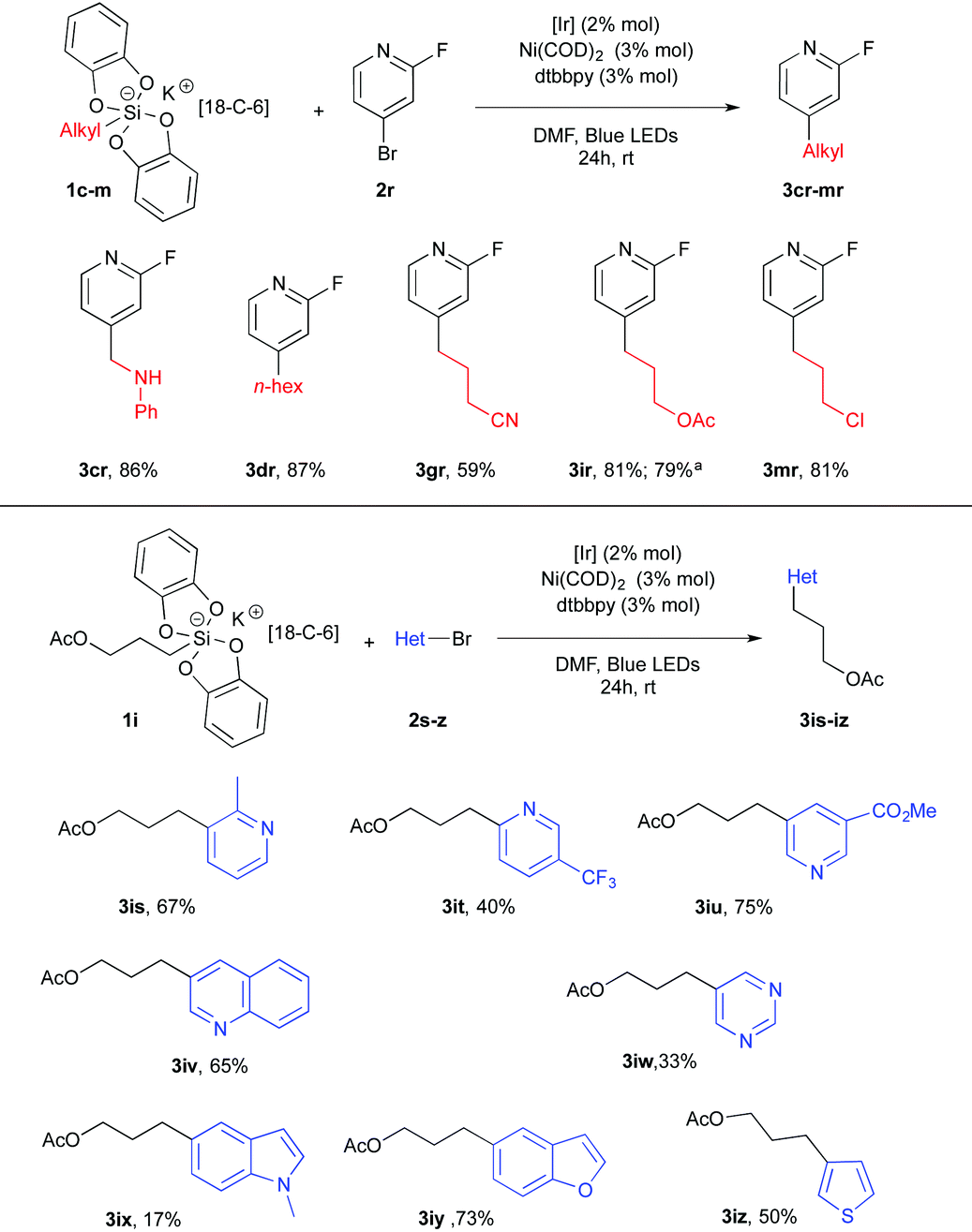
Heterocyclic bromides completed the scope of this transformation and established its high synthetic potential. 2-Fluoro-4-bromopyridine 2r was used as a partner of the dual catalysis in the presence of silicates 1c–m and delivered a small library of new alkylpyridines 3cr–3mr in satisfactory yields (Table 3). Other pyridyl systems 2s–u proved to be competent for this cross-coupling transformation. The corresponding products 3is–3iu were obtained in a 40–75% yield range. Finally, various heterocyclic systems such as quinoline 2v, pyrimidine 2w, indole 2x, benzofuran 2y and thiophene 2z were used and yielded the corresponding cross coupling adducts 3 from mitigated to fair yields.19
| a Starting from the corresponding potassium silicate with no 18-C-6 1i′. |
|---|
|
The proposed catalytic cycle is outlined in Scheme 2. It is well established that excitation of the photoredox catalyst Ir[(dF(CF3)ppy)2(bpy)](PF6) produces a long-lived photoexcited state *Ir[(dF(CF3)ppy)2(bpy)](PF6).4 The latter acts as a strong oxidant (E[*IrIII/IrII] = +1.32 V vs. SCE in MeCN)20 and undergoes a single electron transfer (SET) with bis-catecholato silicates 1 (Eox between +0.34 and +0.87 V vs. SCE in DMF)14a generating a primary alkyl radical which interacts with a Ni species. However, the mechanism of this dual catalysis is still in debate.9,21 All proposals converge toward a Ni(III) intermediate which upon reductive elimination delivers the cross coupling product and a Ni(I) intermediate complex, the latter being further reduced (E[NiII/Ni0] = −1.2 V vs. SCE in DMF)22 by SET from the iridium(II) (E[IrIII/IrII] = −1.37 V vs. SCE in MeCN)20 to generate a Ni(0) complex. From this zero-valent nickel entity, two possibilities have been advanced. Initial oxidative addition of the ArX gives a Ni(II) species that would trap the nucleophilic radical intermediate. Alternatively, an alkyl Ni(I) species would result from the trapping of the radical intermediate by Ni(0) followed by oxidative addition of the ArX to the Ni(III) intermediate (Scheme 2). Some of our results in Table 2 with the formation of 3ig and 3io highlight the importance of the oxidative addition step. However, this will necessitate more investigation to clarify that matter.
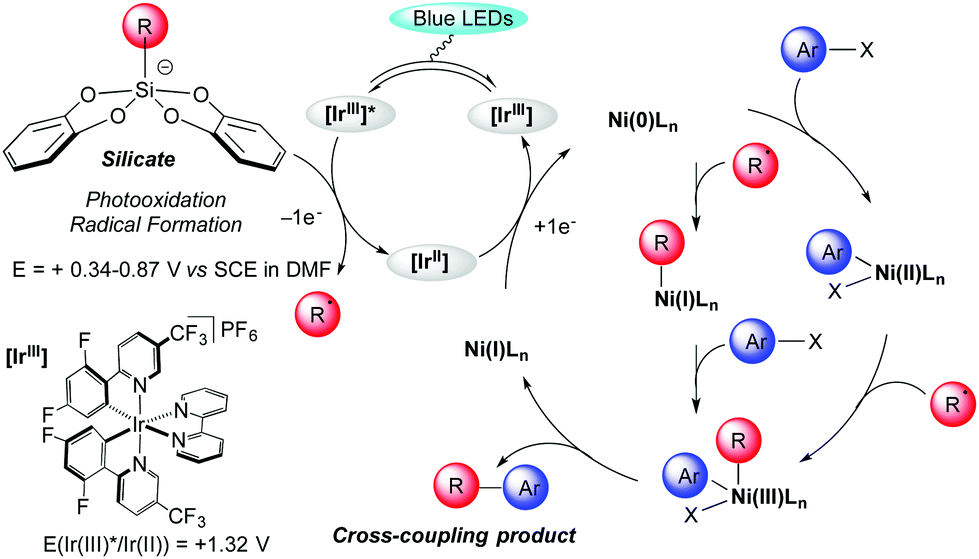 | ||
| Scheme 2 Proposed mechanism of the photoredox/nickel dual catalysis using silicates. | ||
In conclusion, this study illustrates that bis-catecholato silicates are very reliable partners for photoredox/nickel dual catalysis. We have focused our efforts on the use of highly reactive primary radicals which so far has remained an uncharted territory. Our generally successful results in terms of yields and structure scope, since notably heteroatomic functions and heterocyclic platforms do not appear as limiting factors in these transformations, augur well for the application of this methodology in different settings.
Acknowledgements
We warmly thank CNRS, UPMC, UHA, IUF, MSER (ASF PHD grant to CL), LABEX Michem (ANR-11-IDEX-0004-02), La Région Martinique (PhD grant to LC), ANR NHCX (11-BS07-008, postdoc grant to VC). COST Action CM1201 is gratefully acknowledged.Notes and references
- For a recent review on dual catalysis, see: (a) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874 CrossRef CAS PubMed. For recent reviews on dual photoredox/nickel catalysis, see: (b) C. Vila, ChemCatChem, 2015, 7, 1790 CrossRef CAS; (c) J. J. Murphy and P. Melchiorre, Nature, 2015, 524, 297 CrossRef CAS PubMed; (d) Y.-Y. Gui, L. Sun, Z.-P. Lu and D.-G. Yu, Org. Chem. Front., 2016 10.1039/c5qo00437c.
- For seminal reports, see: (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terret, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed.
- For C–O bond formation, see: (a) J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed. For C–P bond formation, see: (b) J. Xuan, T.-T. Zeng, J. R. Chen, L. Q. Lu and W. J. Xiao, Chem. – Eur. J., 2015, 21, 4962 CrossRef CAS PubMed.
- For reviews and books, see: (a) J. M. R. Narayanaman and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (b) F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859 CrossRef; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (d) M. A. Ischay and T. P. Yoon, Eur. J. Org. Chem., 2012, 3359 CrossRef CAS; (e) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (g) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed; (h) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727 CrossRef; (i) T. Koike and M. Akita, Synlett, 2013, 2492 CrossRef CAS; (j) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed; (k) Chemical Photocatalysis, ed. B. König, DeGruyter, Berlin, 2013 Search PubMed; (l) Photochemically generated intermediates in Synthesis, ed. A. Albini and M. Fagnoni, John Wiley & Sons, Hoboken, 2013 Search PubMed.
- For reviews on nickel-catalyzed coupling reactions, see: (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed; (b) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed; (c) V. P. Ananikov, ACS Catal., 2015, 5, 1964 CrossRef CAS.
- For vinylation, see: A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624 CrossRef CAS PubMed.
- M. El Khatib, R. A. M. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254 CrossRef CAS PubMed.
- D. Ryu, D. N. Primer, J. C. Tellis and G. A. Molander, Chem. – Eur. J., 2016, 22, 120 CrossRef CAS PubMed.
- L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929 CrossRef CAS PubMed.
- C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 11938 CrossRef CAS PubMed.
- Y. Yamashita, J. C. Tellis and G. A. Molander, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12026 CrossRef CAS PubMed.
- D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195 CrossRef CAS PubMed.
- I. Karakaya, D. N. Primer and G. A. Molander, Org. Lett., 2015, 17, 3294 CrossRef CAS PubMed.
- (a) V. Corcé, L. M. Chamoreau, E. Derat, J.-P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed., 2015, 54, 11414 CrossRef PubMed; (b) For the oxidation of silicates with organic reagents, see also: L. Chenneberg, C. Lévêque, V. Corcé, A. Baralle, J.-P. Goddard, C. Ollivier and L. Fensterbank, Synlett, 2016 DOI:10.1055/s-0035-1561337.
- A primary carboxylic (5-phenylvaleric acid) has also been used as unstabilized radical precursor, providing the cross-coupling product but in 11% yield, see ref. 6.
- (a) M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475 CrossRef CAS PubMed; (b) See also for vinylation: N. R. Patel, C. B. Kelly, M. Jouffroy and G. A. Molander, Org. Lett., 2016 DOI:10.1021/acs.orglett.6b00024.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, 2009 Search PubMed.
- Cross coupling of 1i with 2o using, 3 mol% of NiCl2·dme and 3 mol% of dtbbpy, and 2 mol% of Ir[(dF(CF3)ppy)2 (bpy)](PF6) provided 81% of 3io. For a recent discussion about the importance of the oxidation state of the nickel precatalyst, see: M. S. Oderinde, M. Frenette, D. W. Robbins, B. Aquila and J. W. Johannes, J. Am. Chem. Soc., 2016, 138, 1760 CrossRef CAS PubMed.
- The poor yields observed on 3it, 3iw and 3ix remain unexplained, since the crude products looked clean for each reaction.
- D. Hanss, J. C. Freys, G. Bernardinelli and O. S. Wenger, Eur. J. Inorg. Chem., 2009, 4850 CrossRef CAS.
- (a) M. S. Oderinde, A. Varela-Alvarez, B. Aquila, D. W. Robbins and J. W. Johannes, J. Org. Chem., 2015, 80, 7642 CrossRef CAS PubMed; (b) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896 CrossRef CAS PubMed; (c) See also for a recent contribution: J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C.-M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016 DOI:10.1021/jacs.6b00250.
- M. Durandetti, M. Devaud and J. Perichon, New J. Chem., 1996, 20, 659 CAS.
Footnotes |
| † Dedicated to John Murphy |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00014b |
| This journal is © the Partner Organisations 2016 |