
Manganese(II)-catalyzed modular synthesis of isoquinolines from vinyl isocyanides and hydrazines†
Hong
Mao
a,
Mingchun
Gao
a,
Bingxin
Liu
a and
Bin
Xu
*abc
aDepartment of Chemistry, Innovative Drug Research Center, Shanghai University, Shanghai 200444, China. E-mail: xubin@shu.edu.cn; Fax: +86-21-66132830; Tel: +86-21-66132830
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
cShanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, Shanghai 200062, China
First published on 22nd February 2016
Abstract
An efficient manganese(II)-catalyzed oxidative radical cascade reaction was developed for the modular synthesis of multi-substituted isoquinolines from easily accessible vinyl isocyanides and hydrazines. Pyrrolo[1,2-a]quinoxalines and phenanthridines could also be afforded efficiently by this method and a wide range of alkyl, (hetero)aryl and alkoxycarbonyl substitutions could be easily introduced.
Isoquinolines represent one of the most important molecules in pharmaceutical and agrochemical industries due to their biological and pharmacological activities as therapeutic agents in medicinal chemistry1,2 and natural products,3 and for this reason the practical synthesis of the core skeleton is of considerable interest in organic synthesis.4,5 Following the earlier established traditional reactions such as Bischler–Napieralski, Pomeranz–Fritsch and Pictet–Spengler,4 many synthetic methods have been developed recently to construct isoquinoline frameworks.5 Among them, one of the most common synthetic strategies was attributed to C–H bond activation through transition metal catalyzed cyclization of alkynes with amines,5c aryl hydrazones,5b aryl imines,5a,d,h–j azides,5f benzamidines,5g or oximes.5e However, the reaction products are usually less substituted or lack diversity and the use of noble metals (Pd, Rh, Ru) would limit their applications in synthetic chemistry although these reactions have overcome many drawbacks of the previously reported reactions such as the use of strong acidic conditions and elevated temperatures.
Manganese salts, which exist widely in nature, are ideal catalysts or promotors in organic synthesis due to their inexpensiveness, chemical stabilities and special reactivity properties.6 However, as a first-row transition metal, manganese has been less studied except for some well-known oxidants like high-valent species KMnO4 and MnO2. Recently, promising and wide applications of manganese reagents have been explored based on their existence in various valences of manganese. For example, Wang and co-workers reported a MnBr(CO)5 catalyzed dehydrogenative annulation of imines and alkynes,7 which suggested that Mn(I) could be an efficient catalyst to replace noble metals such as palladium or rhodium in the field of C–H bond activation. More typically, Mn(III) salts are recognized as efficient promotors in radical reactions due to their oxidation tendency.8 In this context, the use of stoichiometric amounts of manganese reagents in some reactions will limit their application, and result in the unwanted production of quantities of waste metals in the meantime. Although Mn(II)-catalyzed reactions have been developed in cross coupling reactions9a–c and asymmetric oxidations,9d,e it is still rare for manganese compounds to act as radical initiators catalytically in comparison with the use of a stoichiometric amount of manganese in radical reactions.9f,g Therefore, from the viewpoint of sustainable development, green chemistry and the fact that obtaining an efficient catalytic turnover remains challenging, the development of manganese-catalyzed reactions, especially using the cheaper Mn(II) salts, will be highly desirable and of great value.
Isocyanides are uniquely versatile C1 building blocks and have been widely applied in the synthesis of various complicated compounds.10 They are not only capable of reacting with nucleophiles and electrophiles, but also can serve as effective radical acceptors. Although 2-isocyanobiphenyls have been fully reported for the synthesis of phenanthridines with various radical precursors through the somophilic isocyanide insertion reaction,8a,11 the chemistry of vinyl isocyanides remains largely unexplored and few examples have been demonstrated to construct useful isoquinolines from them.12 An elegant example was observed in the visible light-promoted annulation in the catalysis of noble iridium.12a Recently, we have demonstrated the feasible synthesis of isoquinolines through Mn(II)/O2-promoted vinyl isocyanide insertion with organoboronic acids (Scheme 1),13 however, two equivalents of manganese were necessary to accelerate the reaction and limited substrates could be applied as no desired 2-alkyl substituted products could be obtained from the corresponding alkyl boronic acids. In continuation of our recent research interest in isocyanide chemistry,13–15 herein, we disclose an oxidative radical cascade reaction which, to the best of our knowledge, represents the first example of an Mn(II)-catalyzed protocol for the synthesis of multi-substituted isoquinolines from easily accessible vinyl isocyanides and various hydrazines,16 whereby the formation of two C–C bonds was involved in one step (Scheme 1). For the given approach, various functional groups including aryl, alkyl, alkoxycarbonyl and acyl could be introduced efficiently from hydrazines onto the C1-position of isoquinolines in good to excellent yields.
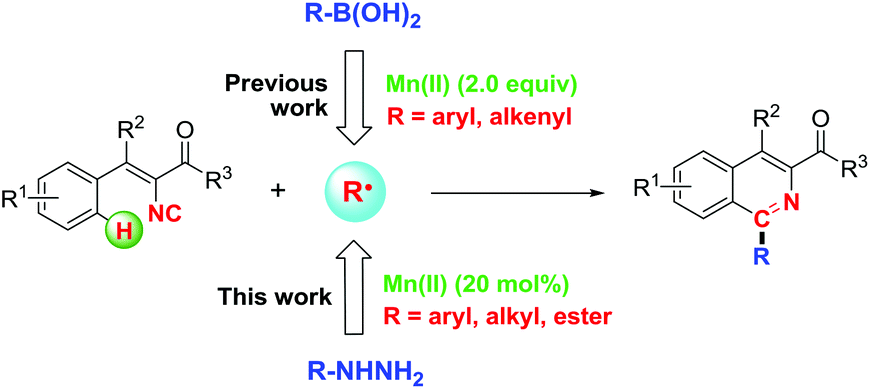 | ||
| Scheme 1 Mn(II)-catalyzed radical cascade reaction of vinyl isocyanides. | ||
At the outset of this investigation, we commenced our study by exploring the catalytic reaction of vinyl isocyanide 1a with phenylhydrazine 2a in the presence of manganese(II) acetate tetrahydrate in DMF using TBPB as an oxidant under a nitrogen atmosphere at 70 °C. Intriguingly, the desired product 3a was isolated in 31% yield after reaction for 23 h (Table 1, entry 1). Various solvents were examined next, which demonstrated that CH3CN was the most suitable solvent in this catalytic system (entries 2–6). Other oxidants such as TBHP and DTBP also worked in the reaction and afforded 3a in a relatively low yield compared with TBPB (entries 7 and 8). Not much better results were observed when the reaction temperature was increased or decreased (entries 9 and 10), and a nitrogen atmosphere was preferred after the screening of atmospheres (entries 11 and 12). Extensive optimization studies of the amount of Mn(OAc)2·4H2O (entries 13 and 14), phenyl hydrazine (entries 15 and 16) and TBPB (entry 17) as well as other manganese catalysts (entries 18–20) suggested that the use of 20 mol% of Mn(OAc)2·4H2O and TBPB in CH3CN solvent at 70 °C under a nitrogen atmosphere turned out to be the best choice and afforded the target product 3a in 82% yield (entry 6). A low yield of 3a (30%) was afforded in the absence of Mn(II) catalyst, which indicated that the manganese catalyst was crucial for this cyclization reaction (entry 21).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Oxidant | Atmos. | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), 2a (1.6 mmol), catalyst (20 mol%), oxidant (2.4 mmol) in solvent (3.0 mL), performed in a nitrogen-purged tube at 70 °C, 23 h. TBHP = tert-butyl hydroperoxide. TBPB = tert-butyl peroxybenzoate. DTBP = di-tert-butyl peroxide. b Isolated yield. c 2a (0.8 mmol) was used. d 2a (1.2 mmol) was used. e TBPB (1.6 mmol) was used. | ||||||
| 1 | Mn(OAc)2·4H2O (20) | DMF | TBPB | N2 | 70 | 31 |
| 2 | Mn(OAc)2·4H2O (20) | THF | TBPB | N2 | 70 | 38 |
| 3 | Mn(OAc)2·4H2O (20) | EtOH | TBPB | N2 | 70 | 24 |
| 4 | Mn(OAc)2·4H2O (20) | Toluene | TBPB | N2 | 70 | 42 |
| 5 | Mn(OAc)2·4H2O (20) | PhCN | TBPB | N2 | 70 | 40 |
| 6 | Mn(OAc) 2 ·4H2O (20) | CH 3 CN | TBPB | N 2 | 70 | 82 |
| 7 | Mn(OAc)2·4H2O (20) | CH3CN | TBHP | N2 | 70 | 52 |
| 8 | Mn(OAc)2·4H2O (20) | CH3CN | DTBP | N2 | 70 | 18 |
| 9 | Mn(OAc)2·4H2O (20) | CH3CN | TBPB | N2 | 80 | 74 |
| 10 | Mn(OAc)2·4H2O (20) | CH3CN | TBPB | N2 | 60 | 77 |
| 11 | Mn(OAc)2·4H2O (20) | CH3CN | TBPB | Air | 70 | 56 |
| 12 | Mn(OAc)2·4H2O (20) | CH3CN | TBPB | O2 | 70 | 63 |
| 13 | Mn(OAc)2·4H2O (15) | CH3CN | TBPB | N2 | 70 | 65 |
| 14 | Mn(OAc)2·4H2O (10) | CH3CN | TBPB | N2 | 70 | 63 |
| 15 | Mn(OAc)2·4H2O (20) | CH3CN | TBPB | N2 | 70 | 53c |
| 16 | Mn(OAc)2·4H2O (20) | CH3CN | TBPB | N2 | 70 | 57d |
| 17 | Mn(OAc)2·4H2O (20) | CH3CN | TBPB | N2 | 70 | 77e |
| 18 | Mn(OAc)3·2H2O (20) | CH3CN | TBPB | N2 | 70 | 66 |
| 19 | Mn(acac)3 (20) | CH3CN | TBPB | N2 | 70 | 72 |
| 20 | Mn(acac)2 (20) | CH3CN | TBPB | N2 | 70 | 65 |
| 21 | — | CH3CN | TBPB | N2 | 70 | 30 |
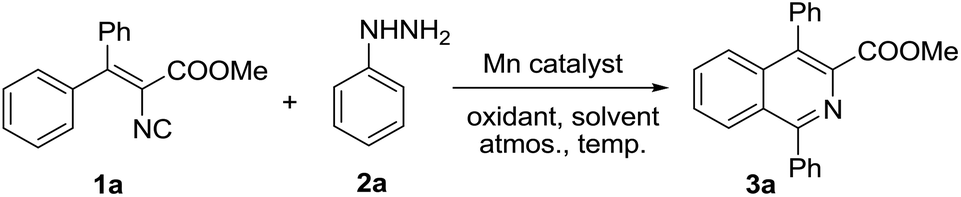
With the optimized reaction conditions established, the scope of hydrazine substrates was next examined (Table 2). Gratifyingly, the catalytic reaction proceeded smoothly with products afforded in moderate to good yields no matter whether the electron-donating (3b, 3d, 3g, 3k) or electron-withdrawing groups (3c, 3e–3f, 3h–3j) were on the aryl ring of the C1-position of products. Substrates bearing ortho- (3b–3c), meta- (3d–3e), para- (3f–3k) groups or with multi-substitutions (3l) all proceeded well. Furthermore, halo-substituted phenylhydrazines (3c, 3e–3f, 3h–3i, 3l) also accomplished the reactions smoothly with the halo-atoms still remaining in the isoquinolines, which appeared to be the key intermediates for further transformation of these products in the classic cross coupling reactions. To our delight, hydrazines with a heterocyclic substituent (3m) or an ester group (3n) were also proved to be suitable components to react with isocyanide 1a under the optimized conditions. It should be noted that 1-alkyl substituted isoquinolines (3o–3s) could be obtained in moderate to good yields, which would significantly expand the application scope of the reaction and provide a good complement and improvement to our previous work, where only aryl and limited alkenyl boronic acids could be used.13 Additionally, alkyl hydrazines worked smoothly, not only for those compounds with a linear (3o, 3s) or tertiary carbon center (3p), but also with cycloalkyl groups (3q–3r). 1-Phenethyl quinoline (3s) was achieved in 58% yield, however, no desired benzyl substituted isoquinoline product could be isolated from benzylhydrazine, while the corresponding benzoyl substituted product (3t) was isolated in 20% yield instead. The reason may be the rapid oxidation of the generated 1-benzyl isoquinoline to the unusual 1-benzoyl product under the reaction conditions,17a while currently we cannot rule out the possibility of the oxidative transformation of the instable benzyl radical to the corresponding benzoyl radical during the reaction.17b–e
| a Reaction conditions: the reaction was carried out with 1a (0.4 mmol), 2a–t (1.6 mmol), Mn(OAc)2·4H2O (0.08 mmol) and TBPB (2.4 mmol) in CH3CN (3.0 mL) at 70 °C under a nitrogen atmosphere. b Isolated yield. c tert-Butylhydrazine hydrochloride or cyclopentylhydrazine hydrochloride was used in the presence of NaHCO3 (5.0 equiv.). d Benzylhydrazine was used as the substrate. |
|---|
|
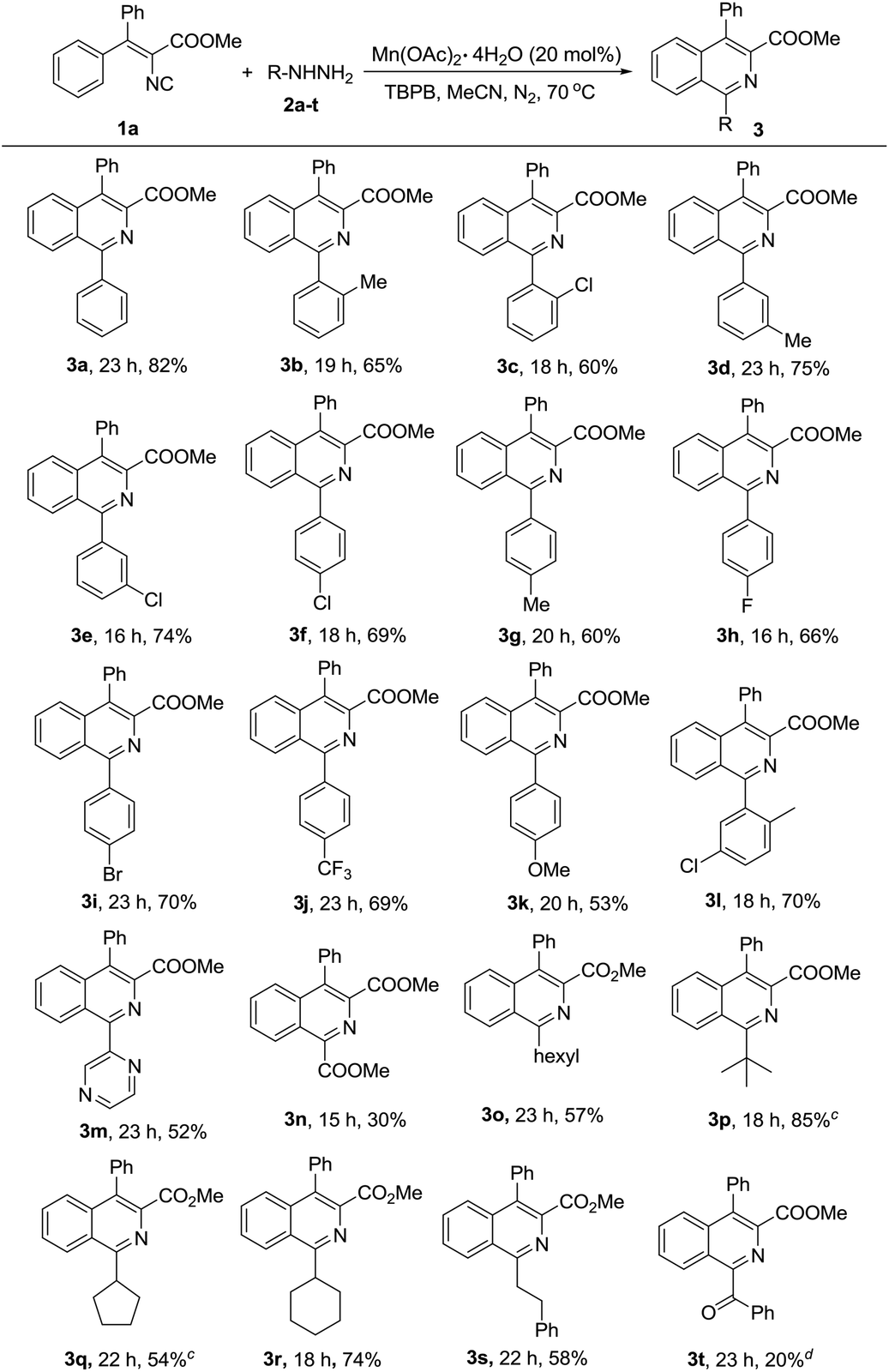
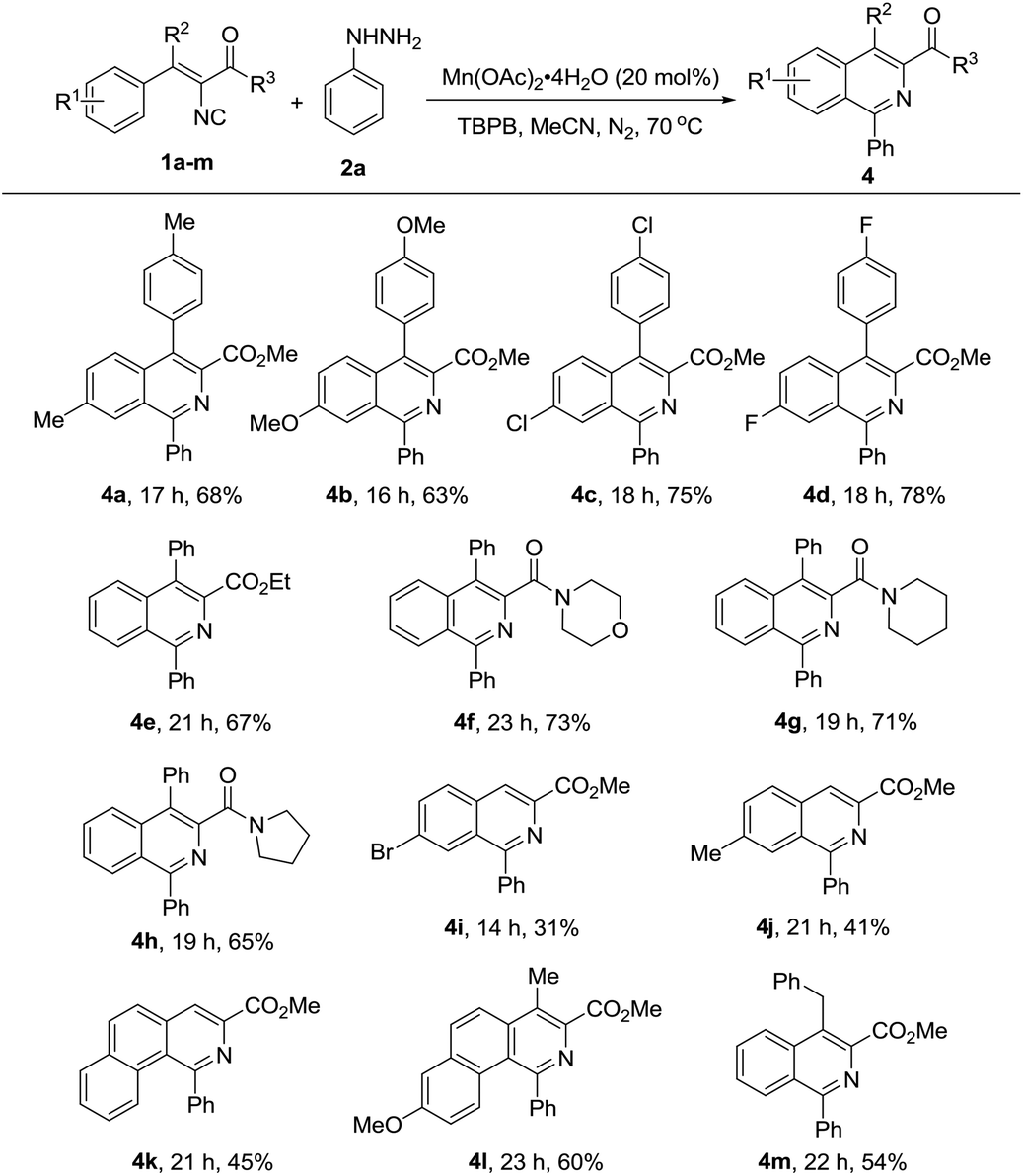
To further study the generality and scope of this reaction, a variety of substituted vinyl isocyanides were investigated with phenyl hydrazine. As illustrated in Table 3, substrates with ester (1a–1e, 1i–1m) and amide (1f–1h) substituents would proceed efficiently to afford the corresponding isoquinolines in moderate to good yields. Notably, vinyl isocyanides derived from diaryl ketones (4a–4h), alkyl aryl ketones (4l–4m) and aryl aldehydes (4i–4k) all proceeded smoothly under the optimized conditions to obtain the corresponding products, regardless of their different electronic properties and substitution positions. Furthermore, the product type was not limited to isoquinolines, fused rings such as benzo[h]isoquinoline derivatives could also be produced successfully (4k and 4l).
This newly established protocol was not limited to vinyl isocyanides, the substrate scope could also be extended further to include aryl isocyanides providing direct access to the corresponding pyrrolo[1,2-a]quinoxalines (5a–5b) and phenanthridine products (5c–5d) in moderate to good yields (Scheme 2), which will further illustrate the broad substrate scope and product diversity of this reaction.
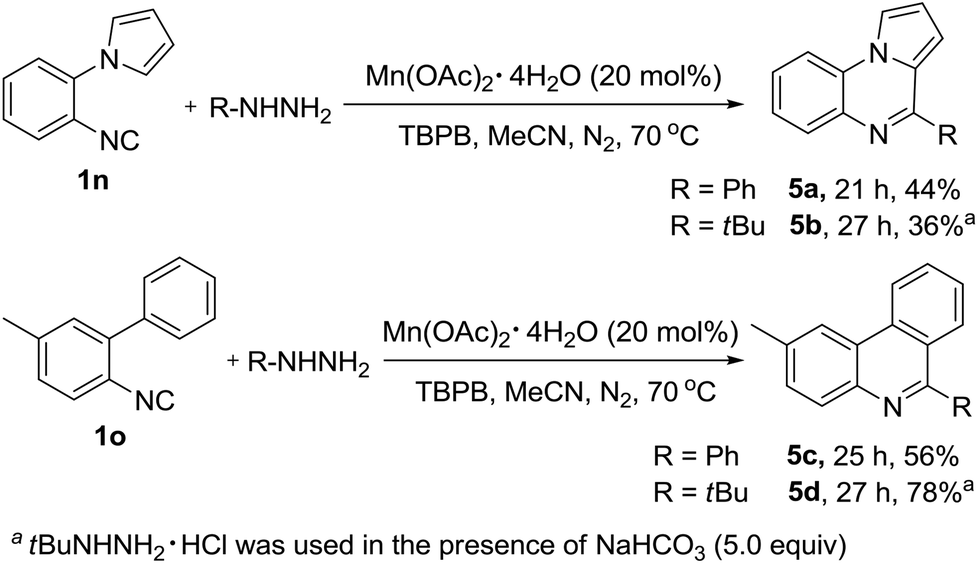 | ||
| Scheme 2 Construction of other heterocyclic skeletons. | ||
To understand the mechanism of this Mn(II)-catalyzed cyclization reaction, preliminary mechanistic investigation was carried out, as shown in Scheme 3. The reaction afforded 3a in only 8% yield with the addition of 2,2,6,6-tetramethyl-piperidine-1-oxy (TEMPO) under the standard conditions, and the addition of p-nitrophenol caused the reaction to become sluggish and decreased the product yield to 22%, which implied that the reaction may experience a single electron transfer pathway.
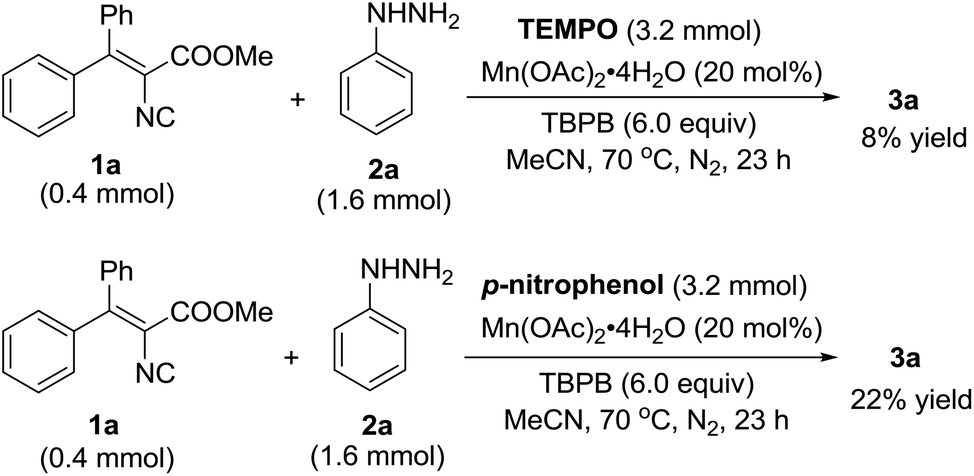 | ||
| Scheme 3 Prevention reaction by radical inhibitors. | ||
On the basis of the above-mentioned experimental observations, a plausible mechanism was proposed for this radical cascade reaction (Scheme 4). Initially, the tert-butoxy radical was generated from Mn(II) and TBPB by a single electron transfer process, together with the formation of Mn(III) and benzoate anions.18 The given Mn(III) was then reacted with phenylhydrazine 2a to afford the phenylhydrazine radical A through sequential single electron transfer and proton transfer steps, and the Mn(II) catalyst was regenerated to complete the catalytic cycle. With the assistance of the tert-butoxy radical, the phenyl radical was formed through stepwise hydrogen abstractions with the concomitant release of nitrogen gas.19 The produced phenyl radical then underwent intermolecular addition to isocyanide 1a to form the imidoyl radical B, which subsequently gave the cyclohexadienyl type radical C by an intramolecular attack of the imidoyl radical on the aromatic ring. Finally, the isoquinoline 3a could be afforded by another hydrogen abstraction from intermediate C in the presence of the tert-butoxy radical with the release of tBuOH.
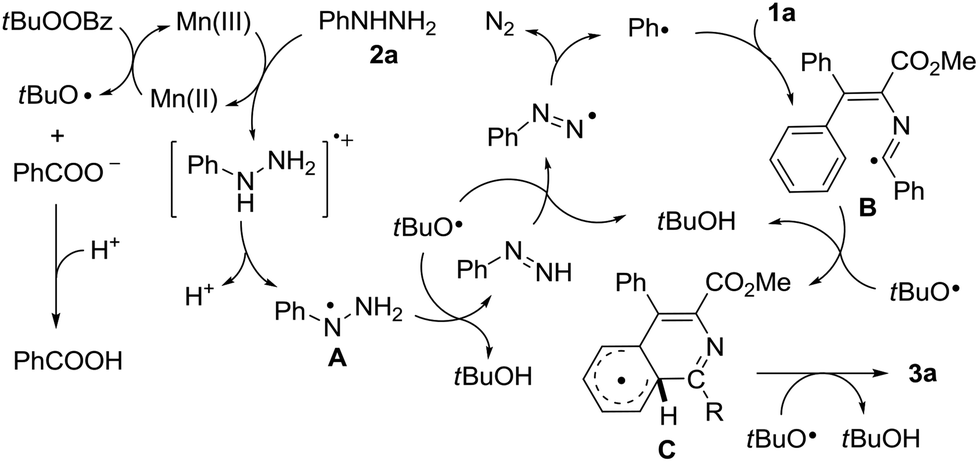 | ||
| Scheme 4 Plausible mechanism for the synthesis of 3a from 1a. | ||
In conclusion, we have developed a manganese(II)-catalyzed cascade radical reaction from easily accessible vinyl isocyanides and hydrazines. This approach offers a unique strategy and alternative route for the convenient preparation of pharmacologically interesting isoquinolines in moderate to good yields with a wide substrate scope, good functionality tolerance and efficient synthesis modularity. The present manganese catalyst system was also successfully applied to the synthesis of valuable pyrrolo[1,2-a]quinoxalines and phenanthridines and a wide range of alkyl, (hetero)aryl and alkoxycarbonyl substitutions could be easily introduced by this method. Further research into applications of this catalytic system is now underway in our laboratory.
We thank the National Natural Science Foundation of China (no. 21272149), and Innovation Program of Shanghai Municipal Education Commission (no. 14ZZ094) for financial support. Prof. Hongmei Deng's (Laboratory for Microstructures, SHU) help with NMR spectroscopy measurements is highly appreciated. This work was partly supported by Shanghai Key Laboratory of High Temperature Superconductors (no. 14DZ2260700).
Notes and references
- For selected books, see: (a) R. Alajarín and C. Burgos, in Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. J. Vaquero and J. Barluenga, Wiley-VCH, Weinheim, 2011, vol. 1, ch. 17, p. 1527 Search PubMed; (b) T. Eicher and S. Hauptmann, The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications, Wiley-VCH, Weinheim, 2nd edn, 2003 Search PubMed; (c) J. A. Joule and K. Mills, Heterocyclic Chemistry, Blackwell Science, Oxford, 4th edn, 2000 Search PubMed.
- (a) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444 RSC; (b) A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS PubMed; (c) C. W. Lim, O. Tissot, A. Mattison, M. W. Hooper, J. M. Brown, A. R. Cowley, D. I. Hulmes and A. J. Blacker, Org. Process Res. Dev., 2003, 7, 379 CrossRef CAS; (d) F. Dzierszinski, A. Coppin, M. Mortuaire, E. Dewailly, C. Slomianny, J.-C. Ameisen, F. DeBels and S. Tomavo, Antimicrob. Agents Chemother., 2002, 46, 3197 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Chen, M. Sajjad, Y. Wang, C. Batt, H. A. Nabi and R. K. Pandey, ACS Med. Chem. Lett., 2010, 2, 136 CrossRef PubMed; (b) B. Reux, T. Nevalainen, K. H. Raitio and A. M. P. Koskinen, Bioorg. Med. Chem., 2009, 17, 4441 CrossRef CAS PubMed; (c) J. E. van Muijlwijk-Koezen, H. Timmerman, H. van der Goot, W. M. P. B. Menge, J. F. von Drabbe Künzel, M. de Groote and A. P. IJzerman, J. Med. Chem., 2000, 43, 2227 CrossRef CAS PubMed; (d) S. C. Beasley, N. Cooper, L. Gowers, J. P. Gregory, A. F. Haughan, P. G. Hellewell, D. Macari, J. Miotla, J. G. Montana, T. Morgan, R. Naylor, K. A. Runcie, B. Tuladhar and J. B. H. Warneck, Bioorg. Med. Chem. Lett., 1998, 8, 2629 CrossRef CAS PubMed.
- (a) A. Pictet and T. Spengler, Chem. Ber., 1911, 44, 2030 CrossRef CAS; (b) A. Bischler and B. Napieralski, Ber. Dtsch. Chem. Ges., 1893, 26, 1903 CrossRef; (c) P. Fritsch, Ber. Dtsch. Chem. Ges., 1893, 26, 419 CrossRef; (d) C. Pomeranz, Monatsh. Chem., 1893, 14, 116 CrossRef.
- For selected examples on isoquinoline synthesis via C–H activation, see: (a) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 4950 CrossRef CAS PubMed; (b) W. Liu, X. Hong and B. Xu, Synthesis, 2013, 2137 CAS; (c) P. Villuendas and E. P. Urriolabeitia, J. Org. Chem., 2013, 78, 5254 CrossRef CAS PubMed; (d) J. Jayakumar, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2012, 51, 197 CrossRef CAS PubMed; (e) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Org. Lett., 2012, 14, 3032 CrossRef CAS PubMed; (f) Y.-F. Wang, K. K. Toh, J.-Y. Lee and S. Chiba, Angew. Chem., Int. Ed., 2011, 50, 5927 CrossRef CAS PubMed; (g) X. Wei, M. Zhao, Z. Du and X. Li, Org. Lett., 2011, 13, 4636 CrossRef CAS PubMed; (h) P. C. Too, S. H. Chua, S. H. Wong and S. Chiba, J. Org. Chem., 2011, 76, 6159 CrossRef CAS PubMed; (i) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed; (j) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050 CrossRef CAS PubMed.
- For reviews, see: (a) P. Saisaha, J. W. de Boerb and W. R. Browne, Chem. Soc. Rev., 2013, 42, 2059 RSC; (b) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1926 RSC; (c) R. A. Layfield, Chem. Soc. Rev., 2008, 37, 1098 RSC.
- R. He, Z. Huang, Q. Zheng and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 4950 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed; (b) A. W. J. Logan, J. S. Parker, M. S. Hallside and J. W. Burton, Org. Lett., 2012, 14, 2940 CrossRef CAS PubMed; (c) J. Zhou, G. Zhang, J. Zou and W. Zhang, Eur. J. Org. Chem., 2011, 3412 CrossRef CAS; (d) H. Nishino, K. Ishida, H. Hashimoto and K. Kurosawa, Synthesis, 1996, 888 CrossRef CAS.
- For selected examples of Mn(II)-catalyzed cross coupling reactions with Grignard reagents, see: (a) G. Cahiez, O. Gager and F. Lecomte, Org. Lett., 2008, 10, 5255 CrossRef CAS PubMed; (b) G. Cahiez, A. Moyeux, J. Buendia and C. Duplais, J. Am. Chem. Soc., 2007, 129, 13788 CrossRef CAS PubMed; (c) G. Cahiez, D. Luart and F. Lecomte, Org. Lett., 2004, 6, 4395 CrossRef CAS PubMed. For selected examples see Mn(II)-catalyzed asymmetric oxidations, see: (d) W. Dai, J. Li, B. Chen, G. Li, Y. Lv, L. Wang and S. Gao, Org. Lett., 2013, 15, 5658 CrossRef CAS PubMed; (e) W. Dai, J. Li, G. Li, H. Yang, L. Wang and S. Gao, Org. Lett., 2013, 15, 4138 CrossRef CAS PubMed. For selected examples of Mn(II)-catalyzed radical reactions, see: (f) H. C. Fisher, O. Berger, F. Gelat and J. Montchamp, Adv. Synth. Catal., 2014, 356, 1199 CrossRef CAS; (g) O. Tayama, A. Nakano, T. Iwahama, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2004, 69, 5494 CrossRef CAS PubMed.
- For recent reviews on isocyanide, see: (a) V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698 CrossRef CAS PubMed; (b) G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257 RSC; (c) T. Vlaar, E. Ruijter, B. U. Maes and R. V. Orru, Angew. Chem., Int. Ed., 2013, 52, 7084 CrossRef CAS PubMed; (d) S. Lang, Chem. Soc. Rev., 2013, 42, 4867 RSC; (e) V. Nenajdenko, Isocyanide Chemistry: Applications in Synthesis and Material Science, Wiley-VCH, Weinheim, 2012 Search PubMed; (f) R. M. Wilson, J. L. Stockdill, X. Wu, X. Li, P. A. Vadola, P. K. Park, P. Wang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2012, 51, 2834 CrossRef CAS PubMed; (g) M. Tobisu and N. Chatani, Chem. Lett., 2011, 40, 330 CrossRef CAS.
- (a) Z. Xia, J. Huang, Y. He, J. Zhao, J. Lei and Q. Zhu, Org. Lett., 2014, 16, 2546 CrossRef CAS PubMed; (b) J. Liu, C. Fan, H. Yin, C. Qin, G. Zhang, X. Zhang, H. Yi and A. Lei, Chem. Commun., 2014, 50, 2145 RSC; (c) L. Gu, C. Jin, J. Liu, H. Ding and B. Fan, Chem. Commun., 2014, 50, 4643 RSC; (d) B. Zhang, C. Daniliuc and A. Studer, Org. Lett., 2014, 16, 250 CrossRef CAS PubMed; (e) J. Cao, T. Zhu, S. Wang, Z. Gu, X. Wang and S. Ji, Chem. Commun., 2014, 50, 6439 RSC; (f) L. Wang, W. Sha, Q. Dai, X. Feng, W. Wu, H. Peng, B. Chen and J. Cheng, Org. Lett., 2014, 16, 2088 CrossRef CAS PubMed; (g) T. Xiao, L. Li, G. Lin, Q. Wang, P. Zhang, Z. Mao and L. Zhou, Green Chem., 2014, 16, 2418 RSC; (h) Q. Wang, X. Dong, T. Xiao and L. Zhou, Org. Lett., 2013, 15, 4846 CrossRef CAS PubMed; (i) B. Zhang, C. Mück-Lichtenfeld, C. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 10792 CrossRef CAS PubMed; (j) Y. Cheng, H. Jiang, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 5520 CrossRef CAS PubMed; (k) D. Leifert, C. Daniliuc and A. Studer, Org. Lett., 2013, 15, 6286 CrossRef CAS PubMed; (l) H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2013, 52, 13289 CrossRef CAS PubMed.
- (a) H. Jiang, Y. Cheng, R. Wang, Y. Zhang and S. Yu, Chem. Commun., 2014, 50, 6164 RSC; (b) Y. Cheng, X. Yuan, H. Jiang, R. Wang, J. Ma, Y. Zhang and S. Yu, Adv. Synth. Catal., 2014, 356, 2859 CrossRef CAS; (c) P. Qian, B. Du, J. Zhou, H. Mei, J. Han and Y. Pan, RSC Adv., 2015, 5, 64961 RSC.
- H. Wang, Y. Yu, X. Hong and B. Xu, Chem. Commun., 2014, 50, 13485 RSC.
- For review on inert bond activation with isocyanides, see: H. Wang and B. Xu, Chin. J. Org. Chem., 2015, 35, 588 CrossRef CAS.
- (a) M. Gao, Y. Li, Y. Gan and B. Xu, Angew. Chem., Int. Ed., 2015, 54, 8795 CrossRef CAS PubMed; (b) G. Qian, X. Hong, B. Liu, H. Mao and B. Xu, Org. Lett., 2014, 16, 5294 CrossRef CAS PubMed; (c) T. Fang, Q. Tan, Z. Ding, B. Liu and B. Xu, Org. Lett., 2014, 16, 2342 CrossRef CAS PubMed; (d) X. Hong, H. Wang, G. Qian, Q. Tan and B. Xu, J. Org. Chem., 2014, 79, 3228 CrossRef CAS PubMed; (e) X. Huang, S. Xu, Q. Tan, M. Gao, M. Li and B. Xu, Chem. Commun., 2014, 50, 1465 RSC; (f) S. Xu, X. Huang, X. Hong and B. Xu, Org. Lett., 2012, 14, 4614 CrossRef CAS PubMed.
- (a) S. Tšupova and U. Mäeorg, Heterocycles, 2014, 88, 129 CrossRef; (b) S. Choudhary, M. K. Muthyala, K. Parang and A. Kumar, Org. Chem. Front., 2014, 1, 683 RSC; (c) Y. Wang, J. Xu, Y. Gu and S. Tian, Org. Chem. Front., 2014, 1, 812 RSC; (d) T. Zhu, T. Wei, S. Wang and S. Ji, Org. Chem. Front., 2015, 2, 259 RSC; (e) U. Ragnarsson, Chem. Soc. Rev., 2001, 30, 205 RSC; (f) E. F. Rothgery, Hydrazine and its Derivatives, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, New York, 4th edn, 2004 Search PubMed; (g) Encyclopedia of reagents for organic synthesis, ed. L. A. Paquette, Wiley, Chichester, 1995 Search PubMed.
- For selected examples for the formation of benzoyl substituted products, see: (a) M. Wan, H. Lou and L. Liu, Chem. Commun., 2015, 51, 13953 RSC; (b) F. Xiong, C. Qian, D. Lin, W. Zeng and X. Lu, Org. Lett., 2013, 15, 5444 CrossRef CAS PubMed; (c) Z. Xu, B. Xiang and P. Sun, RSC Adv., 2013, 3, 1679 RSC; (d) Y. Wu, P. Y. Choy, F. Mao and F. Y. Kwong, Chem. Commun., 2013, 49, 689 RSC; (e) Z. Yin and P. Sun, J. Org. Chem., 2012, 77, 11339 CrossRef CAS PubMed.
- C. J. Easton, K. Kociuba and S. C. Peters, J. Chem. Soc., Chem. Commun., 1991, 1475 RSC.
- (a) X. Xu, Y. Tang, X. Li, G. Hong, M. Fang and X. Du, J. Org. Chem., 2014, 79, 446 CrossRef CAS PubMed; (b) C. Pan, J. Han, H. Zhang and C. Zhu, J. Org. Chem., 2014, 79, 5374 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, characterization data and copies of the 1H, 13C and 19F NMR spectra for all compounds. See DOI: 10.1039/c6qo00048g |
| This journal is © the Partner Organisations 2016 |