DOI:
10.1039/C6QO00117C
(Research Article)
Org. Chem. Front., 2016,
3, 852-855
CuBr2-promoted cyclization and bromination of arene–alkynes: C–Br bond formation via reductive elimination of Cu(III) species†
Received
24th March 2016
, Accepted 12th May 2016
First published on 13th May 2016
Abstract
A CuBr2-promoted cyclization and bromination of arene–alkyne substrates has been developed, affording 9-bromophenanthrene derivatives highly efficiently. This reaction provides a novel and efficient protocol for C–Br bond formation from an inorganic bromine source via the reductive elimination of the Cu(III) species.
Introduction
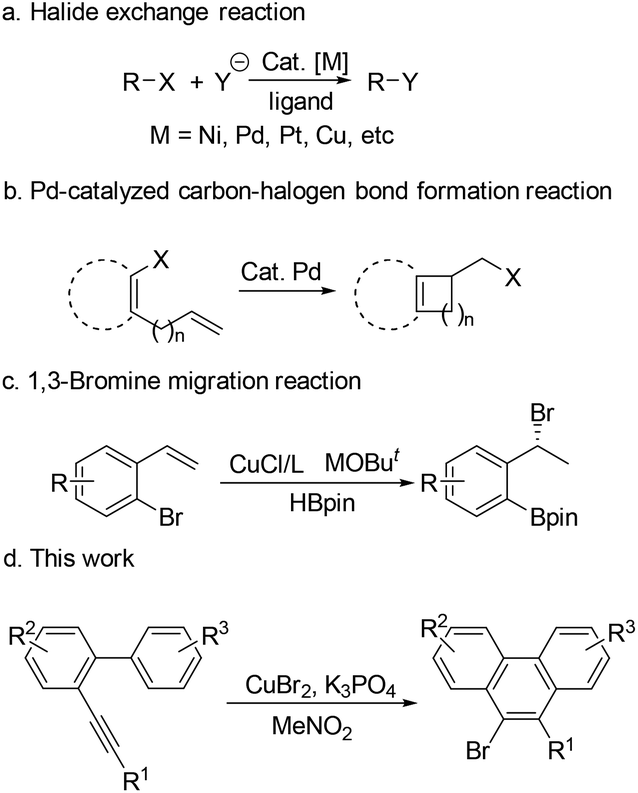
Halides are ubiquitous building blocks in organic synthesis, which can be used for lots of transformations.1 They are also present in a large range of natural products,2 pharmaceuticals,3 and agrochemicals.4 Therefore, highly efficient carbon–halogen bond construction methods have received continuous attention from chemists.5 Research studies on transition-metal-catalyzed carbon–halogen bond formation have a rich history. The halide exchange reaction is one of the most studied examples in this field, which has been applied in both academic and industrial processes.5c,6 In such transformations, an initial organic halide is required (Scheme 1a). The carbon–halogen bond can also be formed by reductive elimination of the Pd(II) species, in which sterically hindered ligands are necessary7 (Scheme 1b). Recently, the Cu-catalyzed 1,3-bromine migration reaction in the presence of MOBut has been reported, which provides another route for the construction of the C–Br bond8 (Scheme 1c). Given the great importance of organic halides in modern chemistry, new protocols for preparing organic halides from inorganic halides are highly desired. Herein, we wish to report our recent observation on the CuBr2-promoted transformation of arene–alkynes into 9-bromophenanthrenes, which presents a new way to build C–Br bonds via the reductive elimination of Cu(III) species using the inorganic bromide salt as the bromine source (Scheme 1d).
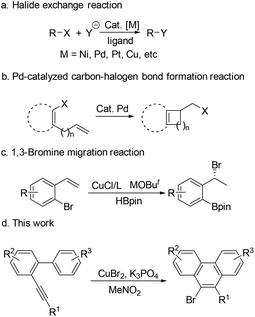 |
| | Scheme 1 Some carbon–halogen bond formation reactions. | |
Results and discussion
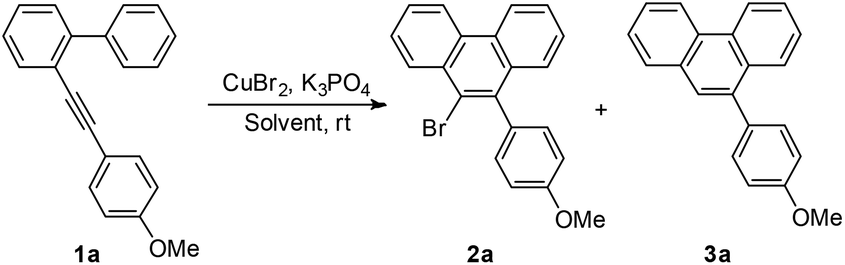
Initially, to optimize the reaction conditions, 2-((4-methoxyphenyl)ethynyl)-1,1′-biphenyl 1a was chosen as the model substrate and CuBr2 was applied as the bromine source. In the first step, the solvent effect was examined carefully. No reaction occurred in triethylamine or ethyl acetate (entries 1 and 2, Table 1). Reactions in acetone, DCM, DCE, EtOH, THF, MeOH, DMF, or MeCN proceeded slowly to yield the desired product 2a in low yields (entries 3–10, Table 1). Encouragingly, when MeNO2 was used as the solvent, a relatively higher reaction speed and yield were observed. However, the chemoselectivity was not satisfying, since the non-brominated byproduct 3a was also formed (entry 11, Table 1). Considering that the byproduct 3a might result from the in situ generated proton (vide infra), K3PO4 was added to neutralize the proton and therefore to improve the selectivity. In the presence of 0.2 equiv. of K3PO4, the yield increased, but the selectivity decreased slightly (entry 12, Table 1). When 0.5 equiv. of K3PO4 was added, the selectivity dramatically rose up to >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, however, the reaction speed was much slower (entry 13, Table 1). Next, more CuBr2 was used (entries 14 and 15, Table 1). To our great delight, when 3 equiv. of CuBr2 was applied, 2a was yielded quantitatively as the sole product (entry 15, Table 1). Thus, Condition A (CuBr2 (3 equiv.), K3PO4 (0.5 equiv.), and CH3NO2) was applied for the following studies. At >99:1, however, the reaction speed was much slower (entry 13, Table 1). Then we tried to add more CuBr2 (entries 14 and 15, Table 1). To our great delight, when 3 equiv. of CuBr2 was applied, 2a was yielded quantitatively as the sole product (entry 15, Table 1). Attempts for a catalytical reaction were then carried out using CuBr2 (10 mol%) and a cheap halogen source NaBr (6 equiv.) under argon (entry 16, Table 1) or oxygen (entry 17, Table 1) atmosphere. Disappointingly, no reaction took place at all. During the whole optimization process, we had been seeking the Cu(I) product/species for any possible information on the mechanism. However, all attempts failed. Thus, we finally conducted the reaction using CuBr instead of CuBr2 under an argon (entry 18, Table 1) or oxygen (entry 19, Table 1) atmosphere. No reaction occurred under an argon atmosphere. A trace amount of the desired product was formed under an oxygen atmosphere. These results might indicate that CuBr cannot catalyze this reaction, however, when Cu(I) was oxidized by oxygen into Cu(II), the reaction could be initiated, although the efficiency was much lower than the standard conditions. Thus, Condition A (CuBr2 (3 equiv.), K3PO4 (0.5 equiv.), and MeNO2) was applied for the following studies.
:1, however, the reaction speed was much slower (entry 13, Table 1). Next, more CuBr2 was used (entries 14 and 15, Table 1). To our great delight, when 3 equiv. of CuBr2 was applied, 2a was yielded quantitatively as the sole product (entry 15, Table 1). Thus, Condition A (CuBr2 (3 equiv.), K3PO4 (0.5 equiv.), and CH3NO2) was applied for the following studies. At >99:1, however, the reaction speed was much slower (entry 13, Table 1). Then we tried to add more CuBr2 (entries 14 and 15, Table 1). To our great delight, when 3 equiv. of CuBr2 was applied, 2a was yielded quantitatively as the sole product (entry 15, Table 1). Attempts for a catalytical reaction were then carried out using CuBr2 (10 mol%) and a cheap halogen source NaBr (6 equiv.) under argon (entry 16, Table 1) or oxygen (entry 17, Table 1) atmosphere. Disappointingly, no reaction took place at all. During the whole optimization process, we had been seeking the Cu(I) product/species for any possible information on the mechanism. However, all attempts failed. Thus, we finally conducted the reaction using CuBr instead of CuBr2 under an argon (entry 18, Table 1) or oxygen (entry 19, Table 1) atmosphere. No reaction occurred under an argon atmosphere. A trace amount of the desired product was formed under an oxygen atmosphere. These results might indicate that CuBr cannot catalyze this reaction, however, when Cu(I) was oxidized by oxygen into Cu(II), the reaction could be initiated, although the efficiency was much lower than the standard conditions. Thus, Condition A (CuBr2 (3 equiv.), K3PO4 (0.5 equiv.), and MeNO2) was applied for the following studies.
Table 1 Optimization of the reaction conditionsa
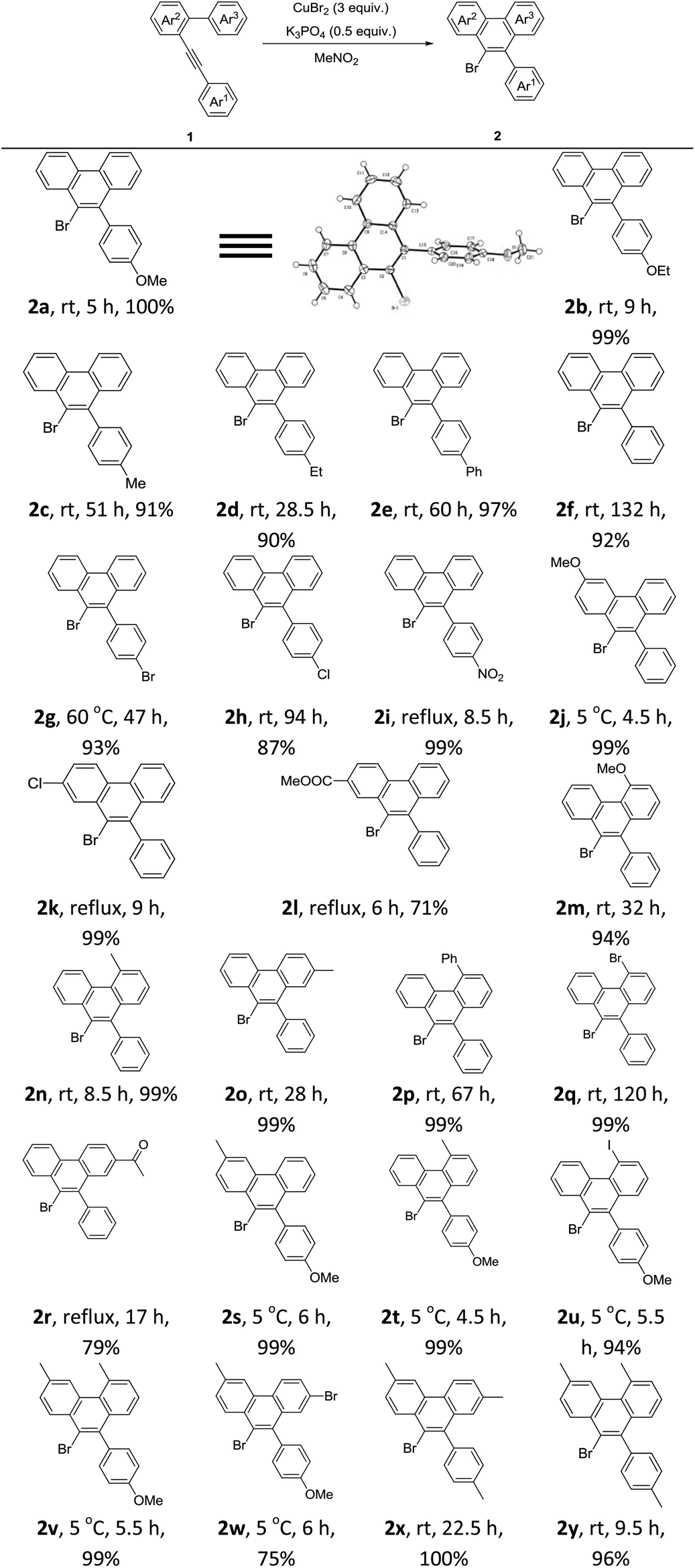
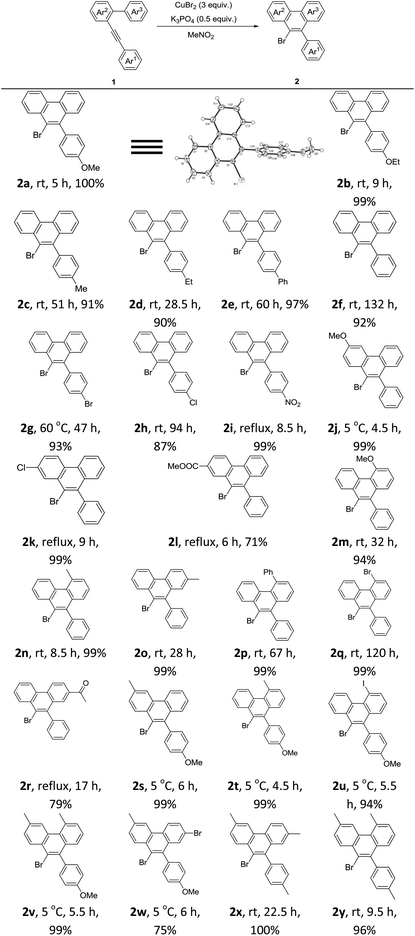
After the above optimization, we paid attention to investigate the scope of this reaction. A series of mono-, di-, or tri-substituted arene–alkynes were applied under Condition A. Some typical results are summarized in Table 2. First, the electronic effect of Ar1 was studied carefully. With strong electron-donating groups, such as methoxy and ethyoxy, the corresponding products were generated in excellent yields. The exact structure of product 2 was determined by the X-ray diffraction study of 2a.9 The reaction speed decreased, when this aryl ring turned to be electron-deficient. With strong electron-withdrawing groups, like the nitro group, a much higher temperature was required. Then the electronic effects of Ar2 and Ar3 were examined. Similar results were observed. The reactions of highly electron-deficient substrates should be carried out under refluxing conditions. Next, some di- or tri-substituted arene–alkynes were applied under Condition A, which showed a very nice reactivity affording the corresponding 9-bromophenanthrenes in good to excellent yields. Notably, for highly electron-deficient substrates, a high reaction temperature was necessary. However for highly electron-rich reactants, a low reaction temperature was required. Most importantly, in all the tested cases, 2 was formed as the only product.
Table 2 Scope of this reactiona
|
A solution of 1 (0.2 mmol), K3PO4 (0.5 equiv.), and CuBr2 (3 equiv.) in anhydrous MeNO2 (3 mL) was stirred under an argon atmosphere. Isolated yield was reported.
|
|
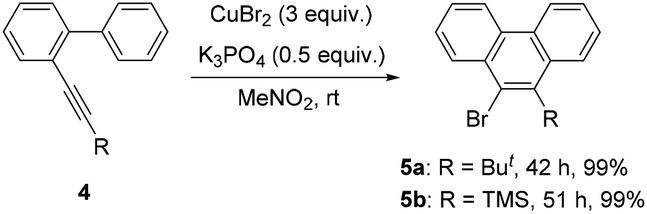
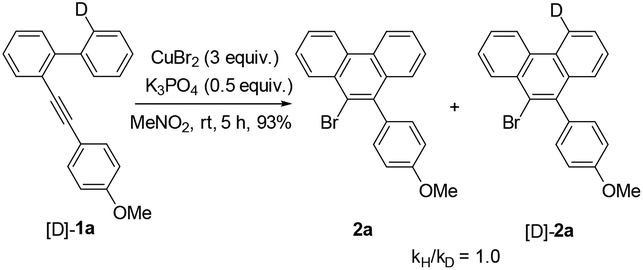
Further studies indicated that aliphatic substrates also showed very nice reactivities (Scheme 2). Notably, 9-bromo-10-trimethylsilylphenanthrene 5b was afforded in nearly quantitative yield. Given that TMS is another nice group for many transformations,10 some other more complicated phenanthrene derivatives might be easily prepared from this product. To gain more mechanistic insights, a competition reaction using [D]-1a was conducted (Scheme 3). The observed primary kinetic isotopic effect (kH/kD = 1.0) was low, which implied that the C–H bond cleavage was not the rate-limiting step. Thus, this reaction should not start from C–H bond activation.
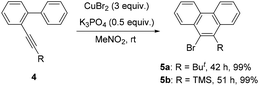 |
| | Scheme 2 The reaction of aliphatic substrates. | |
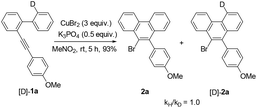 |
| | Scheme 3 Kinetic isotopic effect experiment. | |
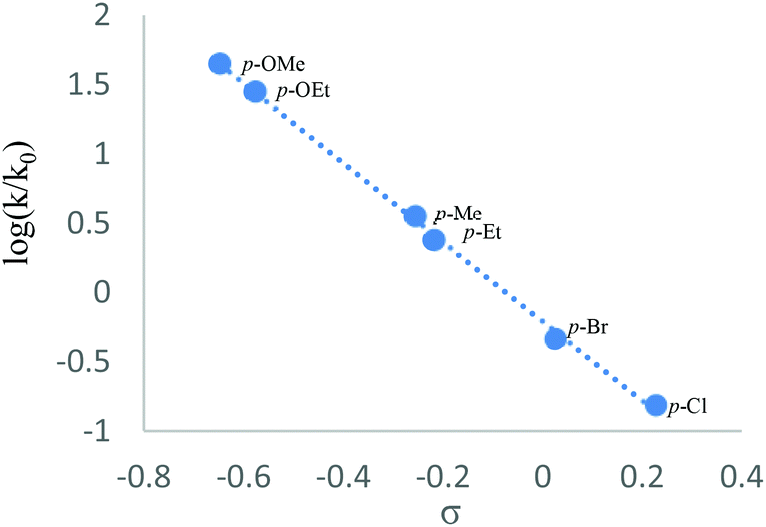
Next, the CuBr2-promoted competitive reactions of some typical para-substituted substrates were carried out. The observed reactivity order for these reactions is as follows: p-OMe (kX/kH = 44.43) > p-OEt (28.00) > p-Me (3.54) > p-Et (2.38) > p-Br (0.46) > p-Cl (0.15).11 As shown in Fig. 1, a linear correlation with a slope of −2.9 was obtained. This negative value indicated that the reaction might take place via a positively charged transition state.12
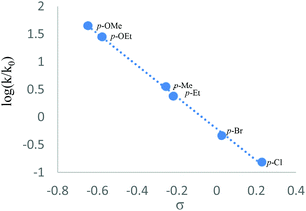 |
| | Fig. 1 Hammett plot. | |
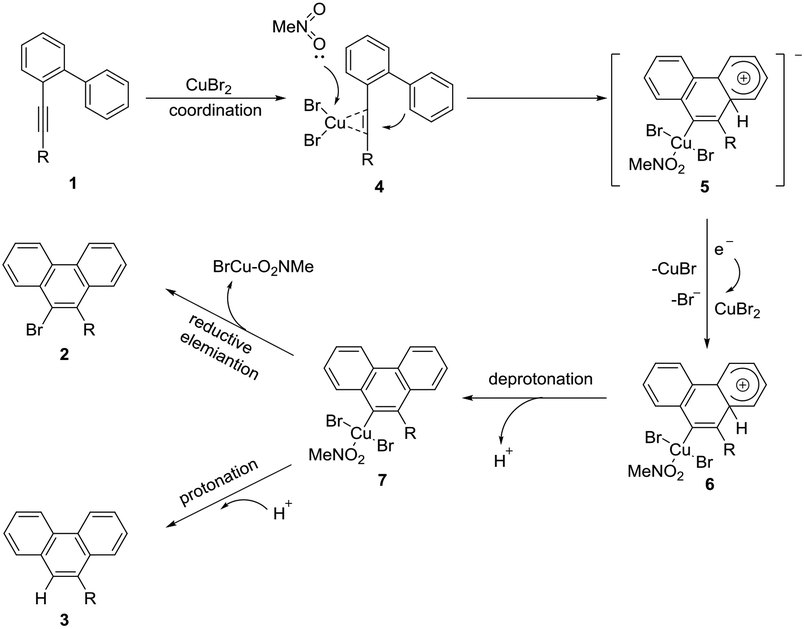
Based on the above results and literature precedents,13,14 a possible mechanism was proposed as shown in Scheme 4. First, CuBr2 coordinated with the alkyne moiety of 1. Then the activated C–C triple bond was nucleophilically attacked by the intramolecular aryl ring as a molecule of MeNO2 coordinated at the copper center to yield the cyclized alkenyl square planar Cu(II) anion 4. Subsequently, single electron transfer from 4 to a molecule of CuBr2 afforded the Cu(III) species 6, CuBr, and the bromide ion. In this step, at least two equiv. of CuBr2 were required to enable the whole transformation. This might be an explanation for why the excess amount of CuBr2 is necessary. The following deprotonation resulted in the neutral aryl Cu(III) species 7 which could be protonated to give byproduct 3. In the presence of K3PO4, the in situ generated proton was neutralized. Therefore, only reductive elimination of the Cu(III)13,14 species 7 took place to form the brominated product 2.
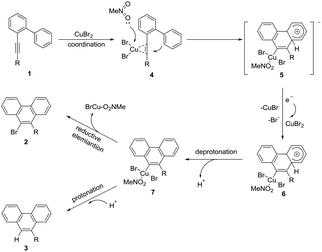 |
| | Scheme 4 Proposed mechanism. | |
Conclusions
In summary, a novel protocol for the construction of the C–Br bond from an inorganic bromine source via reductive elimination of the Cu(III) species has been developed. No organic ligand is required in this reaction. A series of 9-bromophenanthrenes have been synthesized from arene–alkynes, showing broad scope and high potential in organic synthesis.
Acknowledgements
We greatly acknowledge the financial support from the National Basic Research Program of China (973 Program, 2012CB720300), the International Science & Technology Cooperation Program of China (2014DFE40130), the Shanghai Rising-Star Program (14QA1400500), the medical guiding program of Shanghai Science Technology Commission (14411965200), and the Natural Science Foundation of Shanghai Science Technology Commission (12ZR1425600).
Notes and references
-
A. Vigalok, C-X Bond Formation, in Topics in Organometallic Chemistry, ed. P. Bichler, M. S. Eisen, D. S. Glueck, L. Hintermann, A. W. Kaspi, J. A. Love, K. Muñiz, A. N. Vedernikov and A. Vigalok, John Wiley & Sons, New York, 1973, pp. 39–184 Search PubMed.
-
(a) D. R. M. Smith, S. Grüschow and R. J. M. Goss, Curr. Opin. Chem. Biol., 2013, 17, 276 CrossRef CAS PubMed;
(b) K. Benkendorff, Mar. Drugs, 2013, 11, 1370 CrossRef CAS PubMed;
(c) P. M. Pauletti, L. S. Cintra, C. G. Braguine, A. A. D. S. Filho, M. L. A. E. Silva, W. R. Cunha and A. H. Januário, Mar. Drugs, 2010, 8, 1526–1549 CrossRef CAS PubMed;
(d) C. Wagner, M. El Omari and G. M. König, J. Nat. Prod., 2009, 72, 540 CrossRef CAS PubMed;
(e) C. S. Neumann, D. G. Fujimori and C. T. Walsh, Chem. Biol., 2008, 15, 99 CrossRef CAS PubMed.
-
(a) M. Gobinath, N. Subramanian and V. Alagarsamy, J. Saudi Chem. Soc., 2015, 19, 282 CrossRef;
(b) M. K. Reddy, Y. J. Rao and G. L. D. Krupadanam, J. Saudi Chem. Soc., 2015, 19, 372 CrossRef;
(c) A. Shah and D. Raj, J. Saudi Chem. Soc., 2015, 19, 73 CrossRef;
(d) A. Jain, L. S. Duvvuri, S. Farah, N. Beyth, A. J. Domb and W. Khan, Adv. Healthcare Mater., 2014, 3, 1969 CrossRef CAS PubMed;
(e) C. Kim and Y. Cha, Amino Acids, 2014, 46, 89 CrossRef CAS PubMed;
(f) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed;
(g) J. Bégué and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992 CrossRef;
(h) A. S. Christophersen, Toxicol. Lett., 2000, 112–113, 127 CrossRef CAS PubMed.
-
(a) T. Fujiwara and D. O. Hagan, J. Fluorine Chem., 2014, 167, 16 CrossRef CAS;
(b) P. Jeschke, Pest Manage. Sci., 2010, 66, 10 CrossRef CAS PubMed.
-
(a) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS PubMed;
(b) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef CAS PubMed;
(c) T. D. Sheppard, Org. Biomol. Chem., 2009, 7, 1043 RSC.
-
(a) M. Chen, S. Ichikawa and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 263 CrossRef CAS PubMed;
(b) H. Kuniyasu, A. Sanagawa, T. Nakajima, T. Iwasaki, N. Kambe, K. Bobuatong and M. Ehara, J. Organomet. Chem., 2014, 769, 34 CrossRef CAS;
(c) Z. Zhang, F. Wang, X. Mu, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2013, 52, 7549 CrossRef CAS PubMed;
(d) H. Kuniyasu, A. Sanagawa, D. Nakane, T. Iwasaki, N. Kambe, K. Bobuatong, Y. Lu and M. Ehara, Organometallics, 2013, 32, 2026 CrossRef CAS;
(e) A. A. Cant, R. Bhalla, S. L. Pimlott and A. Sutherland, Chem. Commun., 2012, 48, 3993 RSC;
(f) X. Feng, Y. Qu, Y. Han, X. Yu, M. Bao and Y. Yamamoto, Chem. Commun., 2012, 48, 9468 RSC;
(g) M. B. Thathagar and G. Rothenberg, Org. Biomol. Chem., 2006, 4, 111 RSC;
(h) S. H. Yang, C. S. L. A., 1987, 52, 691 CAS;
(i) R. G. R. Bacon and H. A. O. Hill, J. Org. Chem., 1964, 1097 CAS.
-
(a) C. M. Le, P. J. C. Menzies, D. A. Petrone and M. Lautens, Angew. Chem., Int. Ed., 2015, 54, 254 CrossRef CAS PubMed;
(b) D. A. Petrone, H. Yoon, H. Weinstabl and M. Lautens, Angew. Chem., Int. Ed., 2014, 53, 7908 CrossRef CAS PubMed;
(c) D. A. Petrone, M. Lischka and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 10635 CrossRef CAS PubMed;
(d) H. Liu, C. Li, D. Qiu and X. Tong, J. Am. Chem. Soc., 2011, 133, 6187 CrossRef CAS PubMed.
-
(a) R. Van Hoveln, B. M. Hudson, H. B. Wedler, D. M. Bates, G. Le Gros, D. J. Tantillo and J. M. Schomaker, J. Am. Chem. Soc., 2015, 137, 5346 CrossRef CAS PubMed;
(b) S. C. Schmid, R. Van Hoveln, J. W. Rigoli and J. M. Schomaker, Organometallics, 2015, 34, 4164 CrossRef CAS;
(c) R. D. Grigg, R. Van Hoveln and J. M. Schomaker, J. Am. Chem. Soc., 2012, 134, 16131 CrossRef CAS PubMed.
- For crystal data of 2a, see ESI.† CCDC 1438272.
-
(a) K. Gondo, J. Oyamada and T. Kitamura, Org. Lett., 2015, 17, 4778 CrossRef CAS PubMed;
(b) M. Yonemoto-Kobayashi, K. Inamoto and Y. Kondo, Chem. Lett., 2014, 43, 477 CrossRef CAS;
(c) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Chem. – Eur. J., 2012, 18, 2931 CrossRef CAS PubMed;
(d) L. T. Ball, G. C. Lloyd- Iones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed.
-
(a) C. G. Swain and E. C. Lupton, J. Am. Chem. Soc., 1968, 90, 4328 CrossRef CAS;
(b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- J. Xiang, R. Yuan, R. Wang, N. Yi, L. Lu, H. Zou and W. He, J. Org. Chem., 2014, 79, 11378 CrossRef CAS PubMed.
-
(a) S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756 CrossRef CAS PubMed;
(b) D. Yu, J. Shao, R. He and M. Li, Chin. Chem. Lett., 2015, 26, 564 CrossRef CAS;
(c) Á. Sinai, D. Vangel, T. Gáti, P. Bombicz and Z. Novák, Org. Lett., 2015, 17, 4136 CrossRef PubMed;
(d) N. Xin and S. Ma, Eur. J. Org. Chem., 2012, 3806 CrossRef CAS.
-
(a) B. Yao, Z.-L. Wang, H. Zhang, D.-X. Wang, L. Zhao and M.-X. Wang, J. Org. Chem., 2012, 77, 3336 CrossRef CAS PubMed;
(b) A. Casitas, M. Canta, M. Solà, M. Costas and X. Ribas, J. Am. Chem. Soc., 2011, 133, 19386 CrossRef CAS PubMed;
(c) B. Yao, D.-X. Wang, Z.-T. Huang and M.-X. Wang, Chem. Commun., 2009, 2899 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1438272. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00117c |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.