
A versatile strategy for difunctionalization of carbon–carbon multiple bonds by photoredox catalysis
Takashi
Koike
* and
Munetaka
Akita
*
Laboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, R1-27, 4259 Nagatsuta-cho, Midori-ku, Yokohama, 226-8503, Japan. E-mail: koike.t.ad@m.titech.ac.jp; makita@res.titech.ac.jp
First published on 31st May 2016
Abstract
Photoredox catalysis has emerged as a strong synthetic tool for radical reactions. In particular, a variety of regioselective difunctionalizations of carbon–carbon multiple bonds through SET photoredox processes (oxidative quenching cycle) have been accomplished by an appropriate choice of an electrophilic radical precursor and a nucleophile. The basic concept, representative results and future prospect are the topics of this article.
 Takashi Koike | Takashi Koike was born in 1977 in Toyama, Japan. He received his doctoral degree from Tokyo Institute of Technology in 2005 under the guidance of Prof. Takao Ikariya. After his graduate career, he joined Professor Robert H. Grubbs’ group in California Institute of Technology, USA as a postdoctoral research scholar. In 2007 he returned to Tokyo Institute of Technology as an assistant professor of Chemical Resources Laboratory. In 2016, he was posted to Institute of Innovative Research. His research interests are in synthetic organic chemistry and organometallic chemistry. Recently, his efforts have been directed to the development of visible-light-driven photocatalysis. |
 Munetaka Akita | Munetaka Akita is Professor and Director of Laboratory for Chemistry and Life Science at Tokyo Institute of Technology. He received his master's and PhD degrees from Kyoto University (with Prof. Makoto Kumada) and Osaka University (with Prof. Akira Nakamura), respectively. In 1984 he moved to Tokyo Institute of Technology as a research associate and was appointed as a professor in 2002. Recently, his research interests involve in application of carbon-rich organometallics to molecular devices, photoredox catalysis, and supramolecular systems based on anthracene. |
Difunctionalization of compounds with carbon–carbon multiple bonds such as alkenes and alkynes, which are abundant carbon feedstocks, is a fundamental transformation in organic synthetic chemistry for construction of structurally diverse molecules by simple protocols.1 In particular, regiospecific introduction of two different functional groups across unsaturated bonds is still a challenging topic. The Kharasch reaction, i.e., atom transfer radical addition (ATRA), is a classical radical functionalization of alkenes (Scheme 1).2 In general, initial radical addition is directed to the terminal position to form a more stable radical intermediate. Thus, radical reactions can be a promising strategy for regioselective functionalization of carbon–carbon double bonds, if designed appropriately.
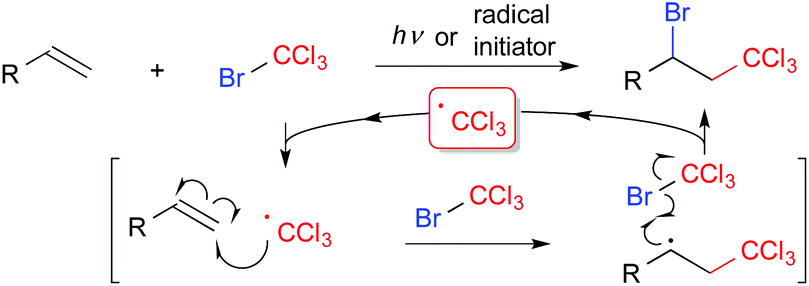 | ||
| Scheme 1 Kharasch reaction. | ||
For the past several years, photoredox catalysis with organic dyes and metal complexes has emerged as a versatile tool for radical reactions because they catalyze single-electron-transfer (SET) processes under mild reaction conditions, i.e., visible light (including sunlight) at room temperature.3 Sequential SET processes mediated by photoredox catalysis are shown in Scheme 2, where [Ru(bpy)3]2+ is used as a typical example for the catalyst. The reaction is initiated by photoexcitation to generate the triplet excited species *[Ru(bpy)3]2+ with a hole (1e-oxidant) and a high-energy electron (1e-reductant). In the presence of an electron donor (D), a SET process from D to *[Ru(bpy)3]2+ forms the cationic radical of D (D+˙) and the reduced metal species [Ru(bpy)3]+, which then undergoes 1e-reduction of an electron acceptor (A) to form the anionic radical of A (A−˙) and the ground-state metal catalyst [Ru(bpy)3]2+. Thus, during one catalytic cycle, the catalyst undergoes sequential reductive and oxidative SET processes with external substrates (D → D+˙ & A → A−˙) to make the system “redox-neutral”. This catalytic cycle initiated by the reduction process of the catalyst is called “reductive quenching cycle”.
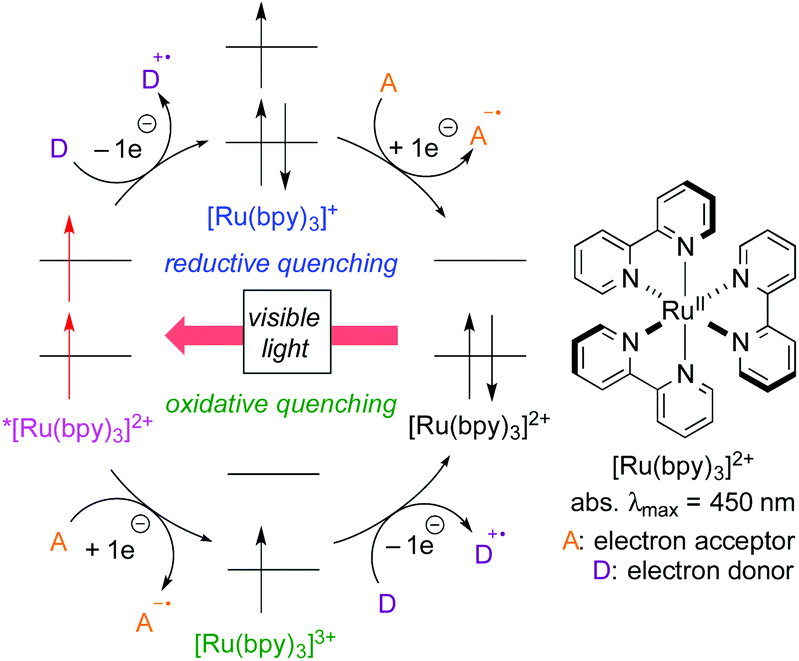 | ||
| Scheme 2 SET photoredox processes mediated by [Ru(bpy)3]2+. | ||
Another catalytic cycle initiated by the oxidation process of the catalyst called “oxidative quenching cycle” is also viable. It turns out that the photoredox catalysis can be a “green” catalytic system, because it requires neither addition of a sacrificial redox reagent (redox-neutral) nor energy from fossil fuel when sunlight is used. A key factor in designing the catalytic system is the appropriate choice of the photocatalyst and the radical precursor, taking their redox potentials into consideration. Both quenching processes can be applied to difunctionalization of carbon–carbon unsaturated bonds.3i We directed our attention to the oxidative quenching cycle mediated by a photoredox catalyst (PC), especially difunctionalization of olefins as shown in Scheme 3. The first SET process from the excited catalyst (*PC) to an electrophilic radical precursor may cause R–X bond cleavage to generate radical species (˙R) and the highly oxidized catalyst (PC+). The resultant radical species (˙R) adds to olefin in a highly regioselective manner. Subsequently, the second SET process from the radical intermediate to the highly oxidized species (PC+) produces the carbocationic intermediate. In the presence of an appropriate nucleophile, the carbocationic intermediate would be captured by the nucleophile, resulting in difunctionalized products. In this “article”, photoredox-catalyzed redox-neutral difunctionalization of carbon–carbon multiple bonds developed by our group will be the major topic. The choice of the radical precursor and nucleophile is a key to success. Then, future prospect of this research field will be discussed.
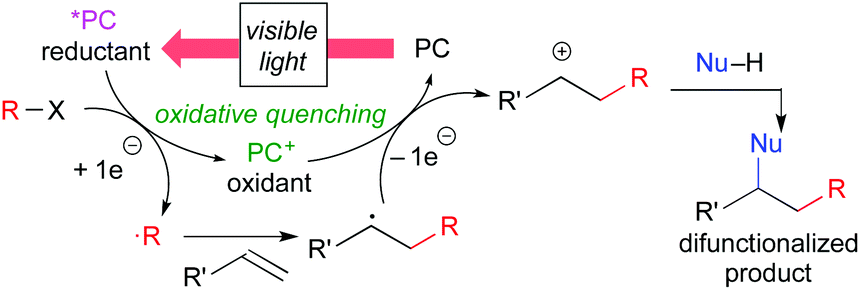 | ||
| Scheme 3 Difunctionalization through the oxidative quenching cycle. | ||
Fluoroalkyl groups have attracted great interest, especially in the fields of pharmaceuticals and agrochemicals because of the improvement of ADME.4 Surprisingly, versatile fluoroalkylative difunctionalization of carbon–carbon multiple bonds has not been well-developed until quite recently.5 In 2011, Stephenson et al. reported that photoredox catalysis could induce ATRA of CF3I to alkenes, leading to iodotrifluoromethylation (Scheme 4).6a,b In 2014, Cho et al. developed photoredox-catalyzed iodotrifluoromethylation of alkynes.6d Later, a number of studies on ATRA-type fluoroalkylation by photoredox catalysis have been reported so far.6
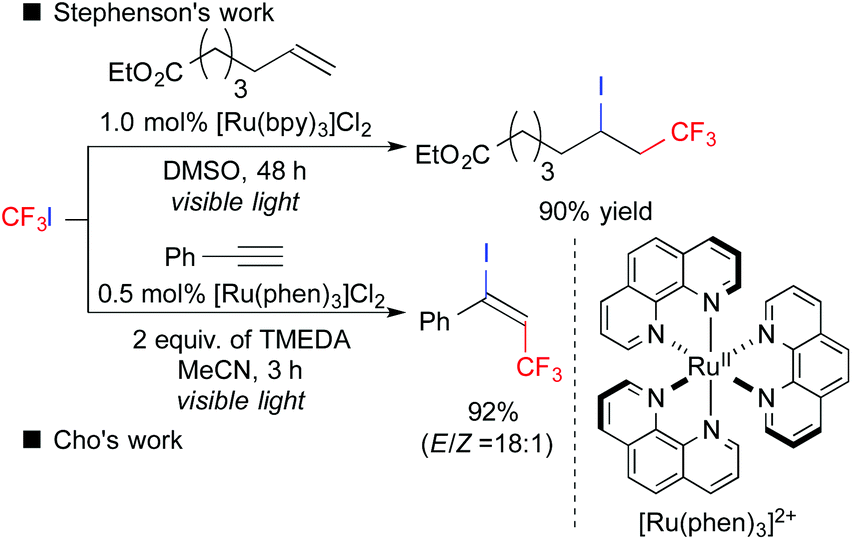 | ||
| Scheme 4 Photocatalytic ATRA-type trifluoromethylation. | ||
In the ATRA-type reactions, transfer of a halogen atom is very efficient. To expand variations of fluoroalkylative difunctionalization, we used a halogen-free fluoroalkyl radical precursor to develop novel and versatile transformations. We chose electrophilic CF3 reagents such as the Umemoto reagent7a and Togni reagent,7b which are easy-to-handle and solid reagents. In addition, they have redox potentials enough to undergo SET from the photoexcited [Ru(bpy)3]2+ and fac-[Ir(ppy)3] catalysts, i.e., the reagents serve as electron acceptors. In fact, we found that the photocatalytic reaction of aromatic alkenes with the electrophilic CF3 reagents in the presence of H2O, alcohols, carboxylic acids, organic nitrile, or DMSO readily induced three-component oxy-, amino- and keto-trifluoromethylation, respectively (Scheme 5).8a,b,d When alkenes bear nucleophilic functional groups such as carboxylic acid and amide, trifluoromethylative cyclization proceeds smoothly.8c,e In these reactions, generation of the α-CF3-carbocationic intermediate through the oxidative quenching cycle is the point.
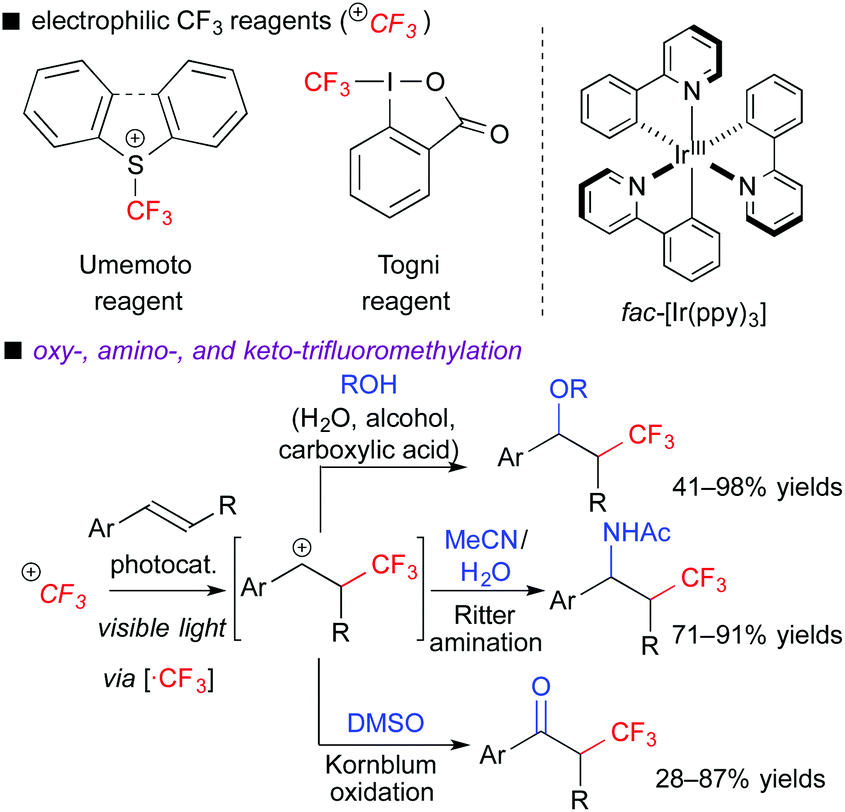 | ||
| Scheme 5 Trifluoromethylative difunctionalization of alkenes. | ||
In addition, this reaction system can also be applied to stereoselective trifluoromethanesulfonyloxy-trifluoromethylation of aromatic alkynes. The obtained CF3-alkenyl triflate can be used as an electrophile for palladium-catalyzed cross coupling reactions. As a result, one-pot stereoselective synthesis of tetrasubstituted CF3-alkenes without the need for isolation of the triflate intermediate was achieved (Scheme 6).9 Our concept can be expanded to other trifluoromethylative difunctionalization. The groups of Gouverneur, Masson, Glorius, and others developed hydro- and carbo-trifluoromethylation (Scheme 7).10 In the hydrotrifluoromethylation, hydrogen abstraction from MeOH is proposed.
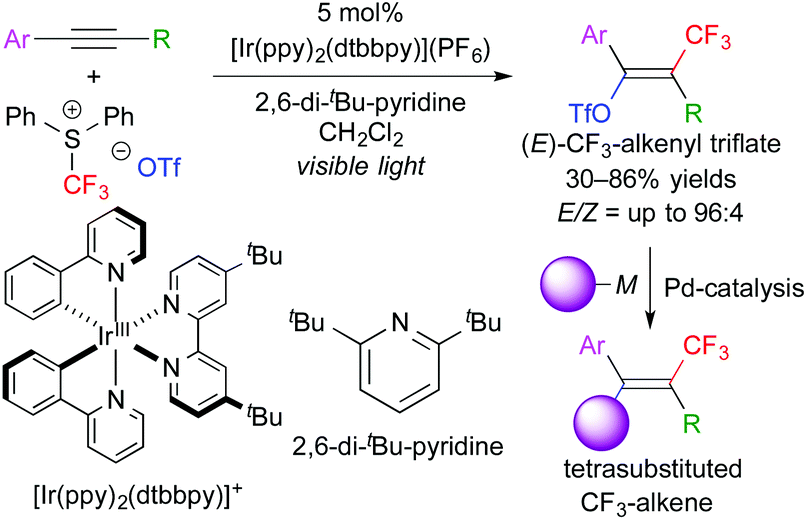 | ||
| Scheme 6 One-pot stereoselective synthesis of CF3-alkenes through trifluoromethanesulfonyloxytrifluoromethylation of alkynes. | ||
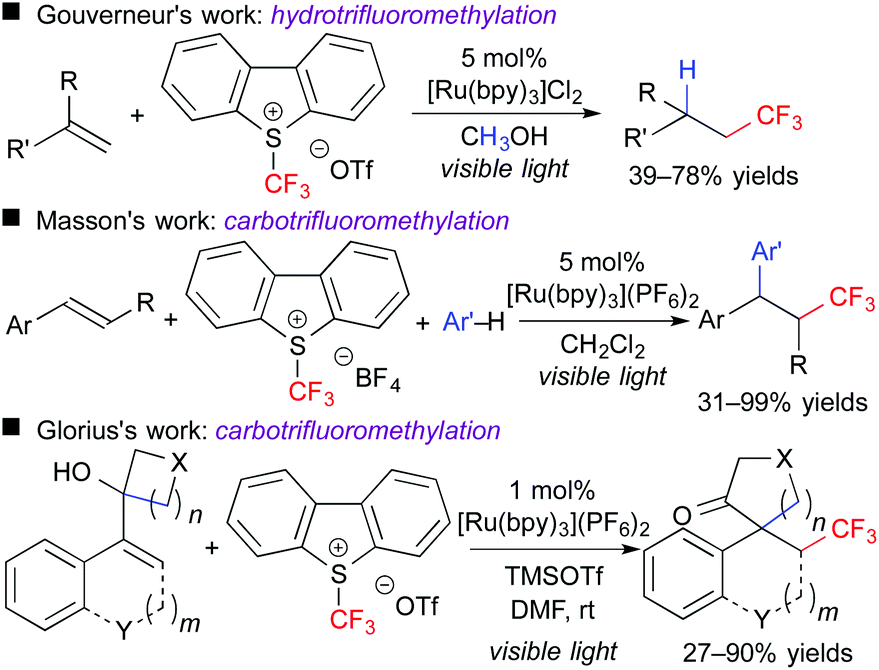 | ||
| Scheme 7 Selected examples of photocatalytic hydro- and carbo-trifluoromethylation. | ||
Furthermore, we recently found that N-tosyl-S-difluoromethyl-S-phenylsulfoximine (Hu reagent) serves as a difluoromethyl radical precursor in the presence of a photoredox catalyst. Under the reaction conditions similar to those of the above-mentioned oxy-trifluoromethylation, oxy-difluoromethylation of aromatic alkenes was performed (Scheme 8).11 It is noteworthy that these reactions exhibit highly regiocontrolled outcomes.
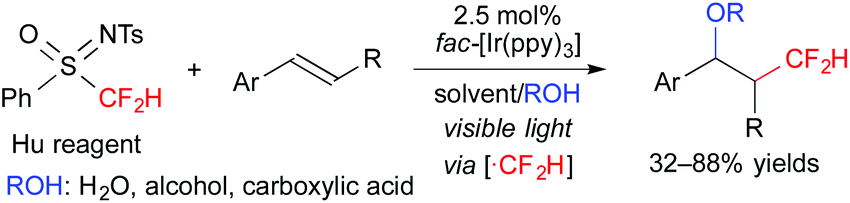 | ||
| Scheme 8 Photocatalytic oxydifluoromethylation of aromatic alkenes. | ||
Reports on fluoromethylative difunctionalization through an oxidative quenching cycle strongly suggest that related difunctionalization of olefins involving carbon-centered radicals is feasible. Aryldiazonium salt is a well-known aryl radical precursor and an electron acceptor. Indeed, the use of aryldiazonium and related aryliodonium salt in place of fluoromethylating reagents leads to the corresponding arylative difunctionalization such as oxy-, carbo-, amino-, and keto-arylation.12 More recently, the groups of Glorius and Toste reported that integration of photoredox catalysis and gold catalysis provided a highly beneficial effect on arylative difunctionalization of unactivated alkenes (Scheme 9).12b,g It should be noted that merger with another catalytic system is promising to expand the scope of photoredox catalysis.3f
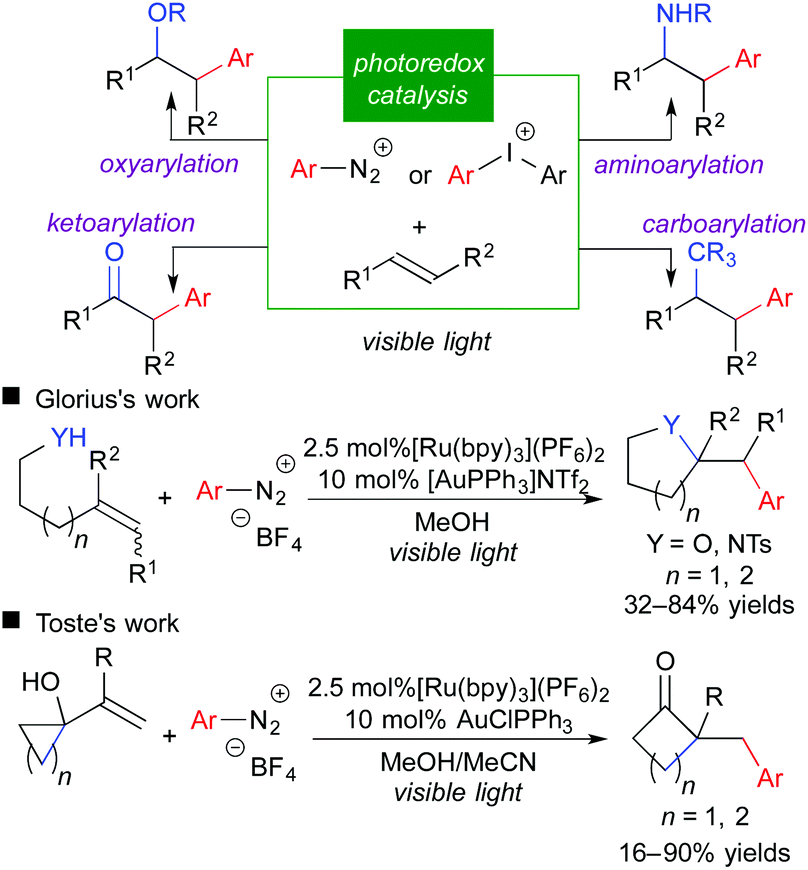 | ||
| Scheme 9 Photocatalytic arylative difunctionalization. | ||
Photoredox-catalyzed reactions involving hetero-atom-centered radical species have been rapidly spreading for the past few years.13 Only if an appropriate radical precursor is used, the present concept, i.e., difunctionalization of unsaturated bonds through the oxidative quenching cycle, could be expanded to, for example, N-centered radical species (Scheme 10).14 In 2015, we found that 1-aminopyridinium reagent serve as an electron acceptor and an N-centered radical precursor by the action of a photoredox catalyst. Under the reaction conditions similar to those of the above-mentioned oxy-trifluoromethylation, aminohydroxylation of olefins efficiently proceeds to give valuable 1,2-aminoalcohol scaffolds.14a Almost at the same time, the group of Yu reported the ATRA-type haloamination of olefins with haloamine.14b Greaney et al. developed three-component azidation of aromatic alkenes in the presence of copper photocatalyst using the Zhadankin reagent.14c In addition, the group of Leonori showed that an oxime bearing electron-deficient aryl group can undergo SET from the photoexcited eosin Y, leading to intramolecular radical hydroimination.14d These results suggest that radical aminative difunctionalization through SET photoredox processes is also feasible by appropriate design of the reaction system.
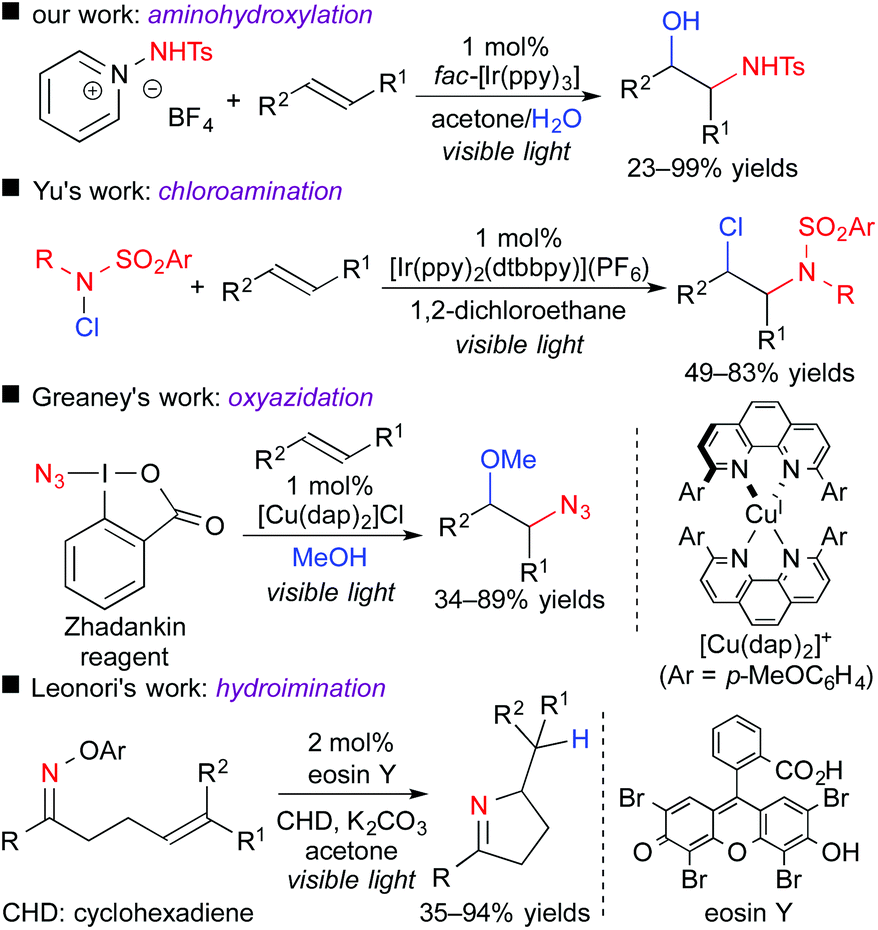 | ||
| Scheme 10 Photocatalytic aminative difunctionalization. | ||
Photoredox catalysis, especially the redox-neutral oxidative quenching cycle, has become a useful strategy for difunctionalization of carbon–carbon multiple bonds. In particular, appropriate design of the electrophilic radical precursor and choice of the nucleophile enable a regiocontrolled three-component reaction. This strategy is really versatile. In the near future, the present photoredox-catalyzed protocol will be expanded not only to functionalized complex alkenes and alkynes but also to dienes and cumulenes. In addition, the development of asymmetric difunctionalization has remained a challenging topic. To design an asymmetric system, a highly programmed merger of photoredox catalysis with an appropriate asymmetric catalysis would be needed.
Notes and references
- For selected reviews, see: (a) K. Muñiz, Chem. Soc. Rev., 2004, 33, 166 RSC; (b) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083 RSC; (c) J. P. Wolfe, Synlett, 2008, 2913 CrossRef CAS; (d) S. R. Chemler, Org. Biomol. Chem., 2009, 7, 3009 RSC; (e) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (f) Y. Shimizu and M. Kanai, Tetrahedron Lett., 2014, 55, 3727 CrossRef CAS; (g) J. R. Coombs and J. P. Morken, Angew. Chem., Int. Ed., 2016, 55, 2636 CrossRef CAS PubMed.
- (a) M. S. Kharasch, E. V. Jensen and W. H. Urry, Science, 1945, 102, 128 CAS; (b) M. S. Kharasch, E. V. Jensen and W. H. Urry, J. Am. Chem. Soc., 1947, 69, 1100 CrossRef CAS; (c) M. S. Kharasch, B. M. Kuderna and W. Urry, J. Org. Chem., 1948, 13, 895 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (e) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed; (f) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (g) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562 RSC; (h) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS; (i) M.-Y. Cao, X. Ren and Z. Lu, Tetrahedron Lett., 2015, 56, 3732 CrossRef CAS.
- (a) T. Hiyama, Organofluorine Compounds: Chemistry and Applications, Springer, Berlin, 2000 Search PubMed; (b) Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Blackwell, Chichester, 2009 Search PubMed; (c) P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2013 Search PubMed.
- (a) J. Xu, X. Liu and Y. Fu, Tetrahedron Lett., 2014, 55, 585 CrossRef CAS; (b) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (c) E. Merino and C. Nevado, Chem. Rev. Soc., 2014, 43, 6598 RSC; (d) T. Koike and M. Akita, J. Fluorine Chem., 2014, 167, 30 CrossRef CAS; (e) T. Besset, T. Poisson and X. Pannecoucke, Eur. J. Org. Chem., 2015, 2765 CrossRef CAS; (f) P. Gao, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Chem. – Eur. J., 2015, 21, 7648 CrossRef CAS PubMed; (g) C. Alonso, E. M. de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed.
- (a) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef CAS PubMed; (b) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef CAS PubMed; (c) S. H. Oh, Y. R. Malpani, N. Ha, Y.-S. Jung and S. B. Han, Org. Lett., 2014, 16, 1310 CrossRef CAS PubMed; (d) N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539 CrossRef CAS PubMed; (e) E. Yoshioka, S. Kohtani, E. Tanaka, Y. Hata and H. Miyabe, Tetrahedron, 2015, 7, 773 CrossRef; (f) D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999 CrossRef CAS PubMed; (g) X.-J. Tang and W. R. Dolbier Jr., Angew. Chem., Int. Ed., 2015, 54, 4246 CrossRef CAS PubMed.
- (a) T. Umemoto, Chem. Rev., 1996, 96, 1757 CrossRef CAS PubMed; (b) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed.
- (a) Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567 CrossRef CAS PubMed; (b) Y. Yasu, T. Koike and M. Akita, Org. Lett., 2013, 15, 2136 CrossRef CAS PubMed; (c) Y. Yasu, Y. Arai, R. Tomita, T. Koike and M. Akita, Org. Lett., 2014, 16, 780 CrossRef CAS PubMed; (d) R. Tomita, Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2014, 53, 7144 CrossRef CAS PubMed; (e) N. Noto, K. Miyazawa, T. Koike and M. Akita, Org. Lett., 2015, 17, 3710 CrossRef CAS PubMed.
- R. Tomita, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2015, 54, 12923 CrossRef CAS PubMed.
- For selected reports, see: (a) S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Médebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505 CrossRef CAS PubMed; (b) P. Xu, J. Xie, Q. Xue, C. Pan, Y. Cheng and C. Zhu, Chem. – Eur. J., 2013, 19, 14039 CrossRef CAS PubMed; (c) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Chem. Commun., 2014, 50, 14197 RSC; (d) B. Sahoo, J.-L. Li and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 11577 CrossRef CAS PubMed.
- Y. Arai, R. Tomita, G. Ando, T. Koike and M. Akita, Chem. – Eur. J., 2016, 22, 1262 CrossRef CAS PubMed.
- For selected reports, see: (a) P. Schroll, D. P. Hari and B. König, ChemistryOpen, 2012, 1, 130 CrossRef CAS PubMed; (b) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed; (c) W. Fu, F. Xu, Y. Fu, M. Zhu, J. Yu, C. Xu and D. Zou, J. Org. Chem., 2013, 78, 12202 CrossRef CAS PubMed; (d) G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2013, 15, 4398 CrossRef CAS PubMed; (e) D. P. Hari, T. Hering and B. König, Angew. Chem., Int. Ed., 2014, 53, 725 CrossRef PubMed; (f) W. Guo, H.-G. Cheng, L.-Y. Chen, J. Xuan, Z.-J. Feng, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Adv. Synth. Catal., 2014, 356, 2787 CrossRef CAS; (g) X.-Z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed; (h) C.-J. Yao, Q. Sun, N. Rastogi and B. König, ACS Catal., 2015, 5, 2935 CrossRef CAS; (i) M. Bu, T. F. Niu and C. Cai, Catal. Sci. Technol., 2015, 5, 830 RSC.
- (a) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977 CrossRef PubMed; (b) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044 RSC , and references therein.
- (a) K. Miyazawa, T. Koike and M. Akita, Chem. – Eur. J., 2015, 21, 11677 CrossRef CAS PubMed; (b) Q. Qin, D. Ren and S. Yu, Org. Biomol. Chem., 2015, 13, 10295 RSC; (c) G. Fumagalli, P. T. G. Rabet, S. Boyd and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11481 CrossRef CAS PubMed; (d) J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2016 |