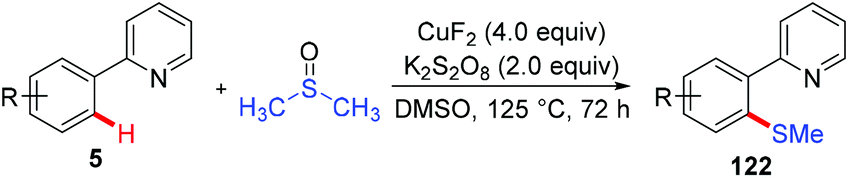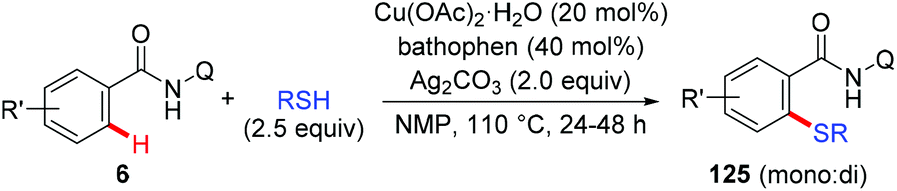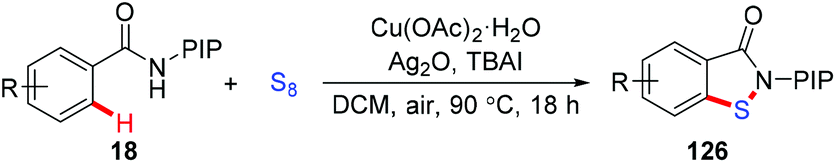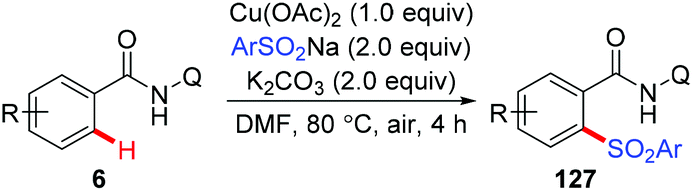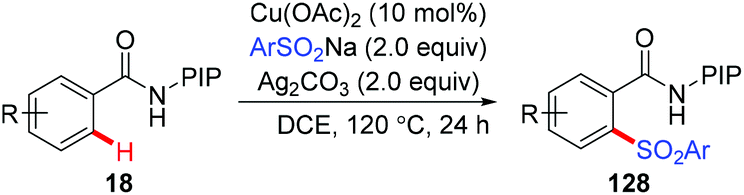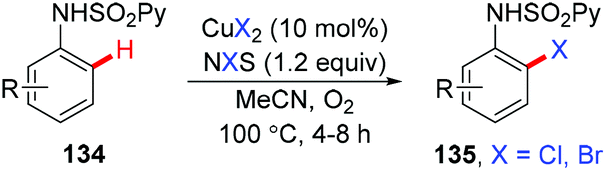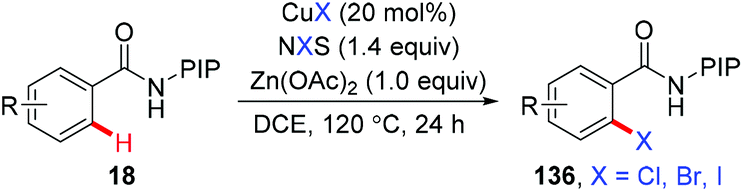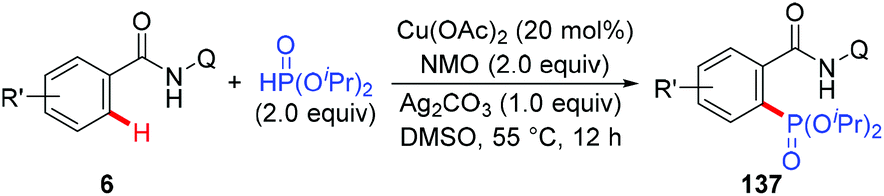DOI:
10.1039/C6QO00156D
(Review Article)
Org. Chem. Front., 2016,
3, 1028-1047
Recent advances in copper-mediated chelation-assisted functionalization of unactivated C–H bonds
Received
13th April 2016
, Accepted 16th June 2016
First published on 20th June 2016
Abstract
Transition metal-catalyzed direct functionalization of C–H bonds has recently emerged as a powerful strategy for the construction of carbon–carbon and carbon–heteroatom bonds. Among various metals employed, base metal copper has attracted significant attention owing to its relatively low cost, abundance, and versatile reactivity. This review aims to comprehensively summarize the recent advances in copper-mediated (both stoichiometric and catalytic) chelation-assisted functionalization of unactivated C–H bonds.
 Wei-Hao Rao | Wei-Hao Rao was born in Henan Province, China in 1985. He received his B.S. degree in 2008 and M.S. degree in 2011 from Henan Normal University. Then he pursued his Ph.D. degree in the laboratory of Prof. Bing-Feng Shi in the Department of Chemistry, Zhejiang University. After receiving the PhD, he then joined the College of Chemistry and Chemical Engineering, Xinyang Normal University. His current research is focused on C–H bond activation. |
 Bing-Feng Shi | Bing-Feng Shi received his B.S. degree from Nankai University in 2001 and a Ph.D. degree from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the guidance of Professor Biao Yu in 2006. After being a postdoctoral fellow at the University of California, San Diego (2006–2007), he moved to The Scripps Research Institute working with Professor Jin-Quan Yu as a research associate. In 2010, he joined the Department of Chemistry at Zhejiang University as a professor. His research focus is on transition-metal-catalyzed C(sp3)–H functionalization and its application in the synthesis of natural products. |
1. Introduction
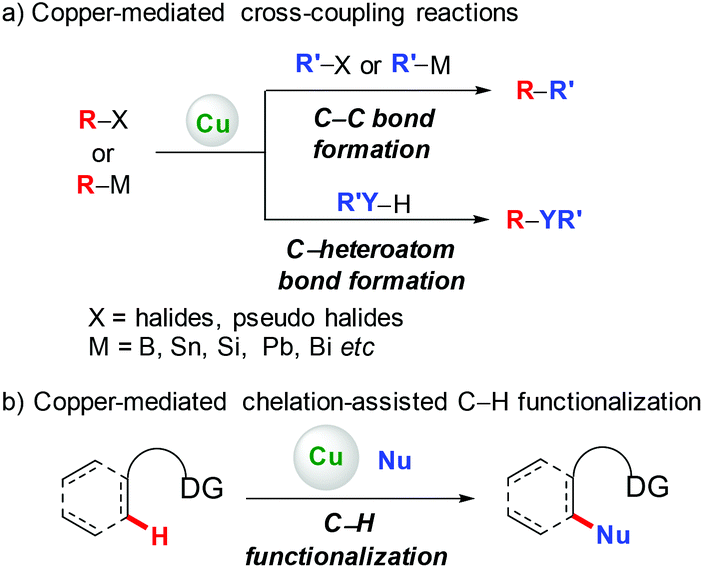
Since the pioneering work of Ullmann and Goldberg,1 copper-mediated cross-coupling reactions have been extensively applied in organic synthesis (Scheme 1a).2 Traditionally, these methods rely on the use of prefunctionalized starting materials (such as organohalides/pseudohalides, or organometallic compounds), which require tedious synthetic operation, and thus reduce the overall efficiency of these transformations. To avoid these disadvantages, transition metal-catalyzed direct functionalization of C–H bonds has recently proved to be one of the most efficient and straightforward complements, since it obviates the prefunctionalization of the starting materials, which makes the reaction more step- and atom-economical.3 Among the various transition metals employed, base metal copper has attracted significant attention owing to its relatively low cost, abundance, and versatile reactivity.2,4 Early developed methods based on copper-mediated direct functionalization of C–H bonds are only scoped to electronically activated (hetero)aryl C–H bonds, C–H bonds adjacent to a heteroatom, and benzylic C(sp3)–H bonds. These elegant examples have appeared in several relevant reviews and will not be discussed in this review.4
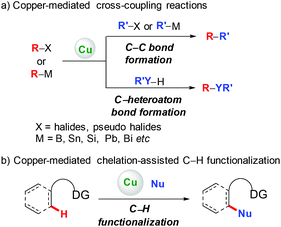 |
| | Scheme 1 Copper-mediated cross-coupling and C–H functionalization. | |
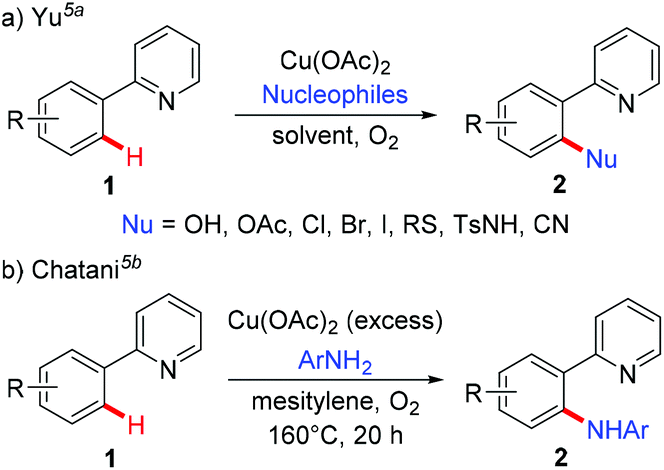
To address the difficulty in the functionalization of unactivated C–H bonds and the issue of regioselectivity, directing groups have been introduced into C–H functionalization.3 In 2006, the Yu group5a and the Chatani group5b independently reported copper-mediated pyridyl-directed C–H functionalization of 2-arylpyridines (Scheme 2). Recently, the 8-aminoquinoline (AQ) bidentate auxiliary, which was first introduced by Daugulis,6 has been used in copper-mediated C–H functionalization. Inspired by these elegant studies, significant advances have recently been made in copper-mediated (both stoichiometric and catalytic) chelation-assisted functionalization of unactivated C–H bonds, which provide practical benefits towards the construction of carbon–carbon and carbon–heteroatom bonds. This review aims to comprehensively summarize recent advances in this area and serve as a handy reference for those interested in using copper-mediated C–H functionalization in organic synthesis (Scheme 1b). For clarity, this review is classified by the type of bond formation, including C–C, C–N, C–O, and other carbon–heteroatom bonds formation.
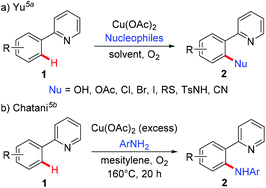 |
| | Scheme 2 Copper-mediated pyridyl-assisted functionalization of C(sp2)–H bonds. | |
2. Carbon–carbon bond formation
2.1 Arylation
Copper-mediated diaryl compound synthesis was first demonstrated by the dehydrogenative homocoupling of two identical arenes.5a,7 The cross-coupling with other arylation partners was first demonstrated by Miura and co-workers, where a Cu(OAc)2-mediated pyridyl- or pyrimidyl-directed dehydrogenative heteroarylation of C(sp2)–H bonds was achieved (Scheme 3).8a They found that the choice of copper(II) acetate was critical to the success of this reaction and other divalent copper salts completely failed to give the heteroarylated products. PivOH could dramatically improve the reaction efficiency. Good tolerance of a wide spectrum of functional groups was also observed. However, the coupling partner was limited to 1,3-azoles and caffeine containing acidic C–H bonds. Another drawback is the use of unremovable directing groups, such as pyridine and pyrimidine. To overcome these drawbacks, the same research group further developed a copper-mediated dehydrogenative heteroarylation of benzamides using 8-aminoquinoline as the directing group, which could be easily introduced and removed as a protecting group (Scheme 4a).8b Moreover, the oxidative dehydrogenative heteroarylation of amides bearing a picolinamide (PA) auxiliary was accomplished by the same group under nearly identical conditions in 2013 (Scheme 4b).8c
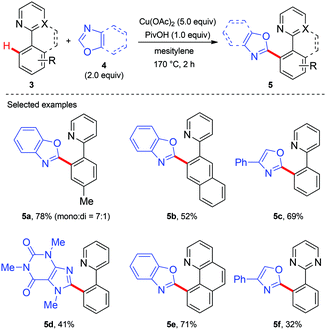 |
| | Scheme 3 Copper-mediated pyridine or pyrimidine-directed oxidative heteroarylation of C(sp2)–H bonds with 1,3-azoles. | |
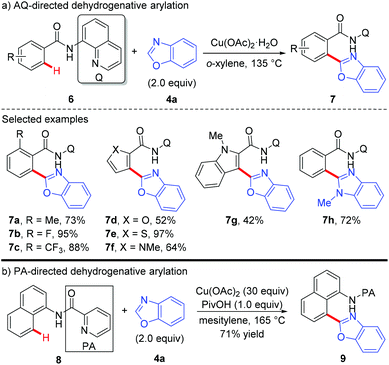 |
| | Scheme 4 Copper-mediated dehydrogenative heteroarylation of C(sp2)–H bonds with 1,3-azoles. | |
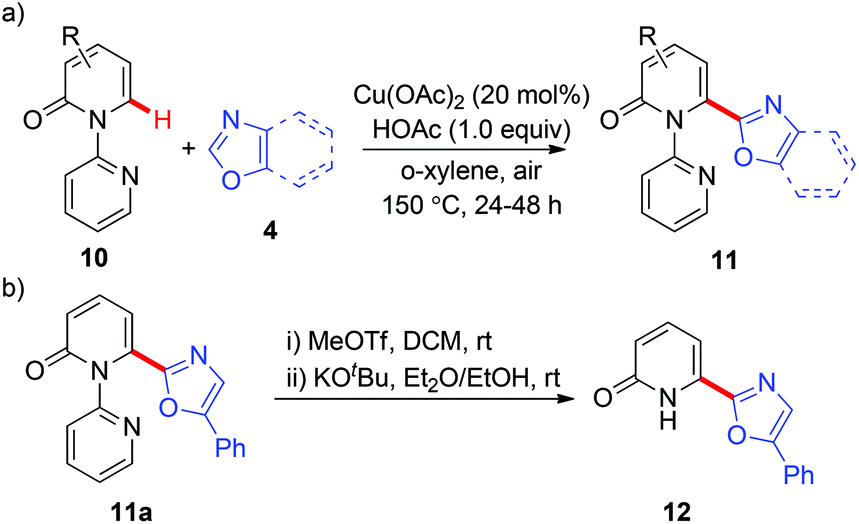
These heteroarylation reactions were enabled by excess copper salts, which hampered its synthetic application. In the related investigations of copper-mediated C6-selective dehydrogenative heteroarylation of 2-pyridones with 1,3-azoles, Miura and co-workers disclosed that in certain cases, the dehydrogenative heteroarylation in the presence of catalytic copper acetate (20 mol%) could be feasible with molecular oxygen in air as an efficient terminal oxidant (Scheme 5a).8d Notably, the 2-pyridyl auxiliary could be readily removed at room temperature, affording the desired heteroarylated 2-pyridone 12 with a free N–H group (Scheme 5b).
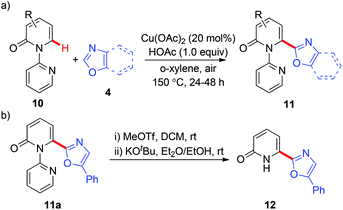 |
| | Scheme 5 Copper-catalyzed pyridine-directed heteroarylation of 2-pyridones with 1,3-azoles. | |
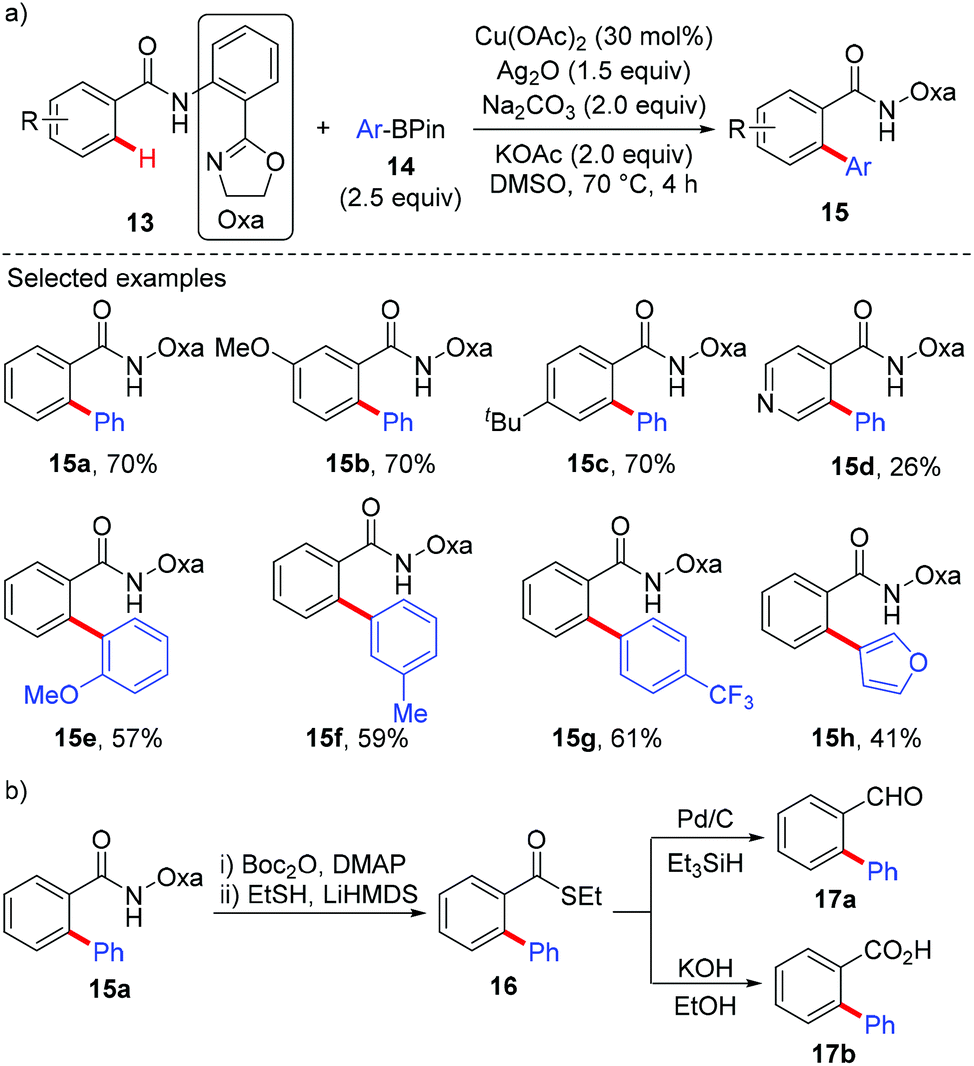
In 2014, Dai and Yu reported a copper-catalyzed arylation of aromatic amides with arylboron reagents using a readily removable amide-oxazoline directing group (Scheme 6).9a Both the benzamides (13) and the arylboron reagents (14) tolerate a broad range of functional groups. To demonstrate the synthetic utility, the arylated product 15a was further transformed into its thioester 16. The thioester can then be converted to the aldehyde 17a and carboxylic acid 17b in good yields (Scheme 6b). Very recently, Tan and co-workers reported a copper-mediated arylation of benzamides with arylboronic acids using the AQ directing group.9b
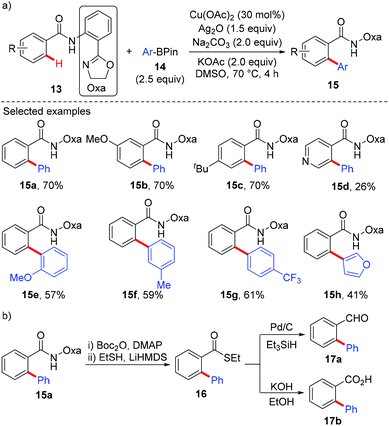 |
| | Scheme 6 Copper-catalyzed oxa-directed arylation of C(sp2)–H bonds with ArBpin. | |
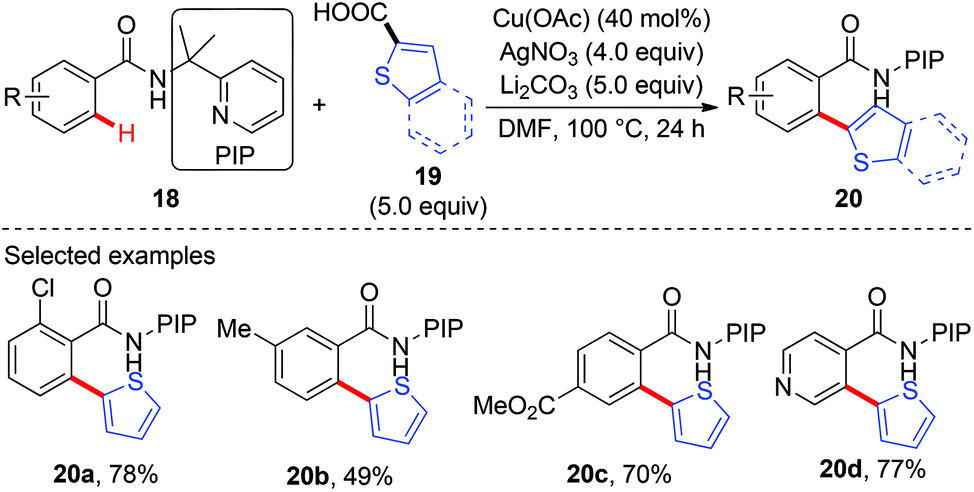
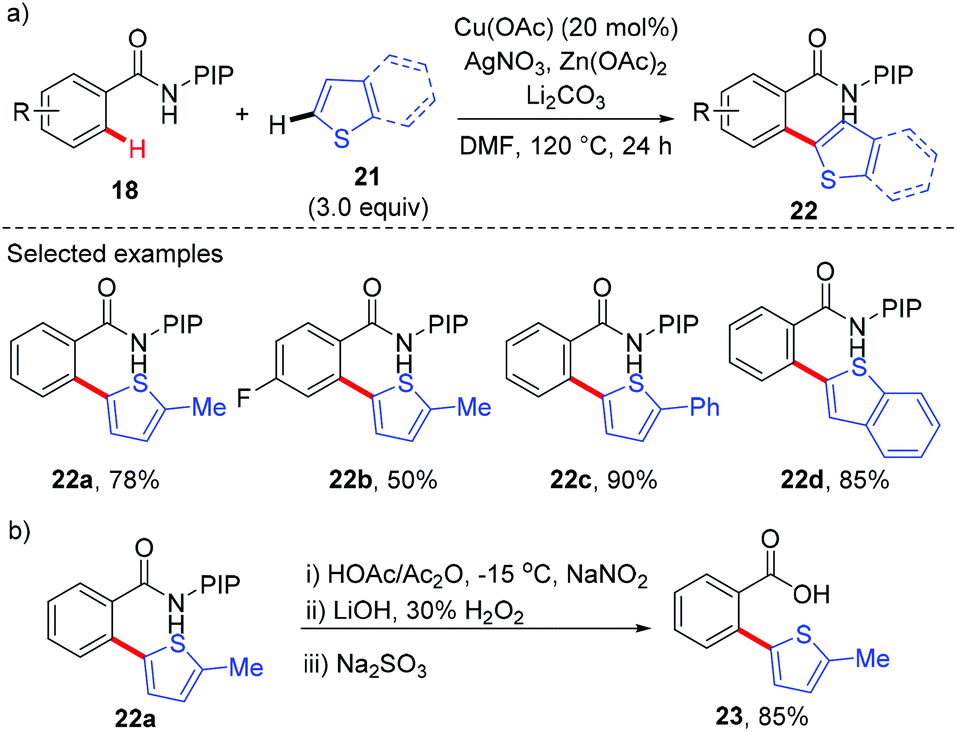
Recently, our group has developed a removable PIP (2-pyridinylisopropyl) bidentate directing group,10 which has shown to be effective in base-metal catalyzed C–H functionalization.11 In 2015, we reported the copper-catalyzed, PIP-directed arylation of benzamides using aromatic carboxylic acids as arylation reagents (Scheme 7).12a Kinetic isotope effects disclosed that either C–H cleavage was the turnover-limiting step or the turnover-limiting step occurred prior to C–H cleavage, and reactions with radical scavengers suggested that radical intermediates might not be involved in the reaction. Further mechanistic studies revealed that the reaction proceeded via a protodecarboxylative/dehydrogenative arylation sequence other than direct decarboxylative arylation. Enlightened by these results, we finally realized a copper-catalyzed dehydrogenative arylation of benzamides with thiophenes using PIP as a bidentate directing group (Scheme 8a).12b Notably, the reaction was scalable and tolerated a wide range of functional groups. The PIP directing group could be easily removed by a nitrosylation/hydrolysis sequence (Scheme 8b).
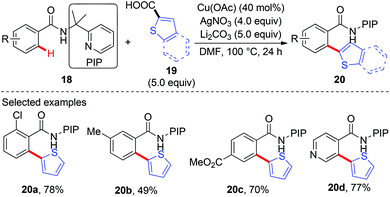 |
| | Scheme 7 Copper-catalyzed protodecarboxylative/dehydrogenative arylation of C(sp2)–H bonds with 2-thiophenecarboxylic acids. | |
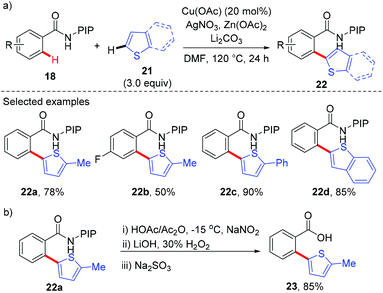 |
| | Scheme 8 Copper-catalyzed oxidative dehydrogenative arylation of C(sp2)–H bonds with thiophenes. | |
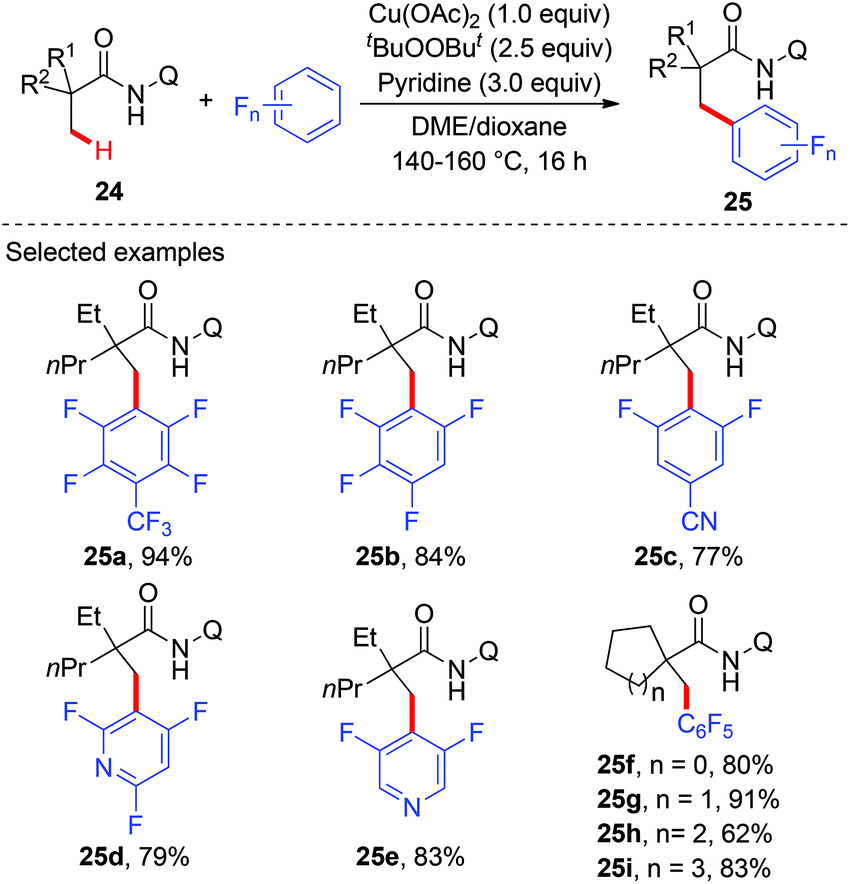
Although several examples of direct arylation of aryl C–H bonds have been developed through the copper-mediated chelation-assisted strategy, the strategy was limited to the arylation of C(sp2)–H bonds. Recently, the Ge group described a copper-mediated dehydrogenative arylation of aminoquinoline amides 24 with polyfluoroarenes as the coupling partners (Scheme 9).13 However, the scope of aliphatic amides 24 was limited to substrates possessing the quaternary carbon center adjacent to a carbonyl group.
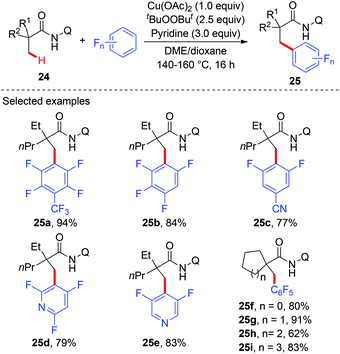 |
| | Scheme 9 Copper-mediated oxidative dehydrogenative arylation of C(sp3)–H bonds with polyfluoroarenes. | |
2.2 Alkynylation
Aryl alkynes are among the most important structural motifs in natural products, pharmaceuticals, and materials.14 Undoubtedly, the oxidative coupling of aryl C–H bonds with terminal alkynes would be an ideal strategy to access these compounds from the viewpoint of atom- and step-economy.15 In a pioneering study of the mechanism and applications of aryl-Cu(III) complexes, Wang and co-workers reported the first C(aryl)–C(alkynyl) bond formation via the cross-coupling of alkynyllithium reagents with several structurally well-defined aryl-Cu(III) intermediates 2 (Scheme 10).16a This transformation provides an unique synthetic route to access alkynylated azacalixaromatics, which can serve as key building blocks to various tailor-made functional macrocyclic host molecules for supramolecular chemistry. It is worth noting that the seminal studies regarding the structurally well-defined aryl-Cu(III) species by Wang, Stahl and Ribas have greatly deepened the mechanistic insights.16
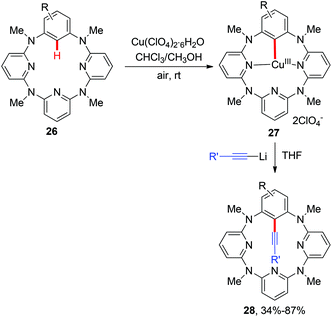 |
| | Scheme 10 Copper-mediated alkynylation of C(sp2)–H bonds with terminal alkynes. | |
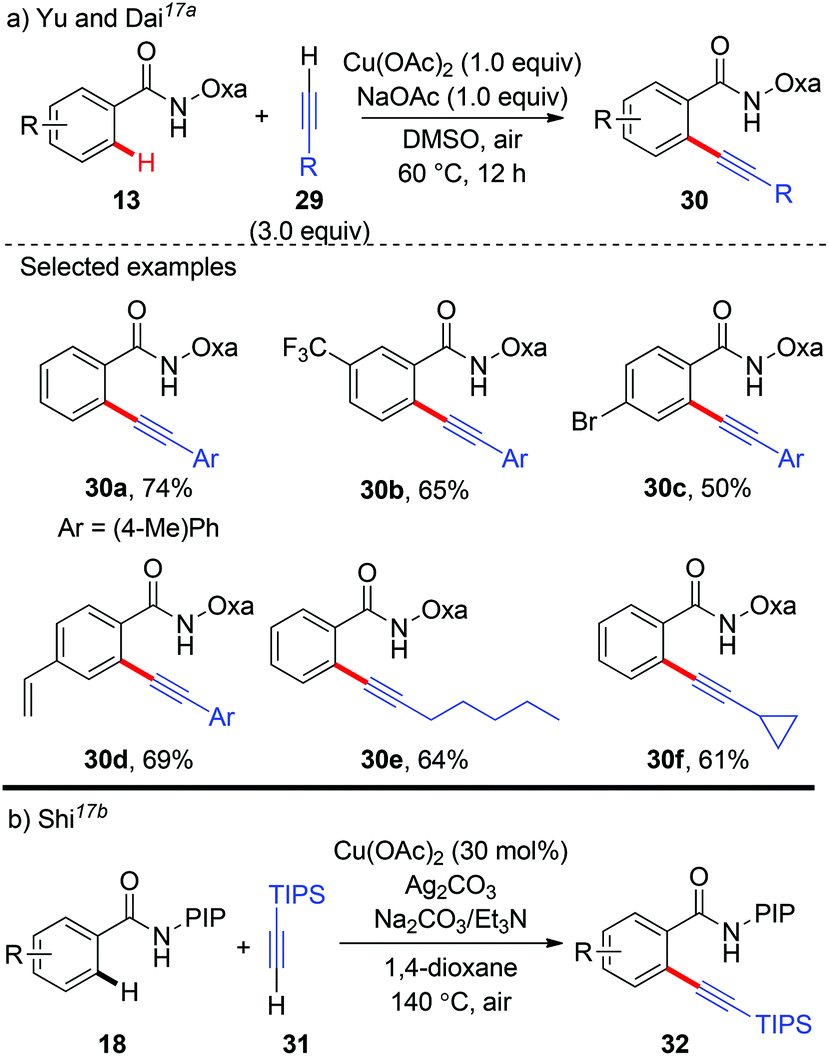
In 2014, Yu and Dai reported the synthesis of aryl alkynes via a copper-mediated dehydrogenative alkynylation of C(sp2)–H bonds with the assistance of an amide-oxazoline group (Scheme 11a).17a The method demonstrated good generality to both benzamides and terminal alkynes, and both aromatic amides and heteroaromatic amides could be alkynylated under mild reaction conditions, thus providing an alternative pathway to traditional Sonogashira coupling. In the same year, our group independently described a Cu(OAc)2-catalyzed oxidative annulation of benzamides bearing a PIP auxiliary with terminal alkynes (Scheme 11b).17b As expected, an external oxidant was necessary for the catalytic cycle, and screening experiments showed that Ag2CO3 as the terminal oxidant was an effective ingredient. Also, the presence of Et3N as a co-base was beneficial for the transformation since it might facilitate the deprotonation of the terminal alkyne. A broad range of (hetero)aromatic amides could even be alkynylated. However, the terminal alkyne was limited to TIPS-alkyne 31. We rationalized that a copper-mediated basic-ligand-enabled concerted metalation/deprotonation (CMD) rather than a copper-mediated electrophilic aromatic substitution (SEAr) pathway was likely involved in the alkynylation.
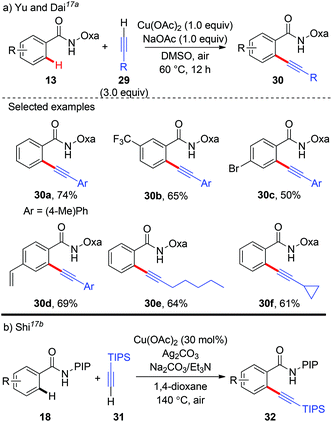 |
| | Scheme 11 Copper-mediated alkynylation of C(sp2)–H bonds with terminal alkynes. | |
2.3 Trifluoromethylation
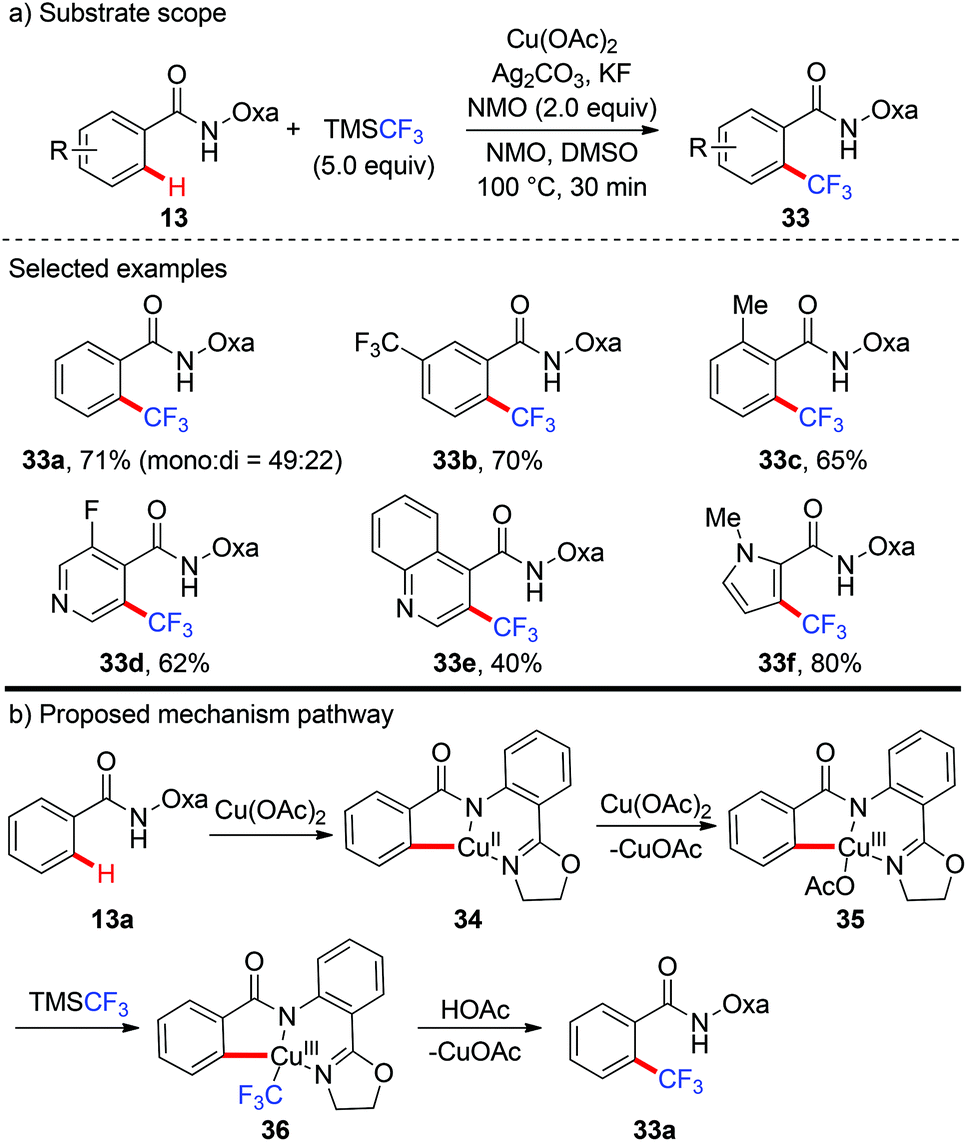
In 2014, the first copper-mediated ortho-trifluoromethylation of benzamides with TMSCF3 employing the amide-oxazoline directing group was reported by Yu and Dai (Scheme 12).18 Stoichiometric Cu(OAc)2 and excess Ag2CO3 were essential to the success of this transformation. Nevertheless, excess N-methyl morpholine oxide (NMO) as the oxidant was necessary. Various electron-rich and electron-deficient benzamides underwent trifluoromethylation efficiently to deliver the trifluoromethylated products. It was worth noting that this protocol was compatible with a variety of heterocycles, including pyridines, pyrroles, benzofurans and quinolone-derived substrates. The significant isotope effects and negligible effect of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) on the reaction rendered a radical pathway unlikely. Preliminary mechanistic studies suggested that a copper-mediated C–H activation pathway rather than an electrophilic aromatic substitution (SEAr) or radical pathway was involved in the reaction (Scheme 12b). Oxazoline-directed cupration of benzamide 13a with Cu(OAc)2via C–H activation followed by disproportionation of Cu(II) species with Cu(OAc)2 afforded a chelated copper(III) species 35. Subsequent transmetallation of this Cu(III) species with TMSCF3 generated the Cu(III)–CF3 intermediate 36 which underwent reductive elimination to afford the desired product 33a.
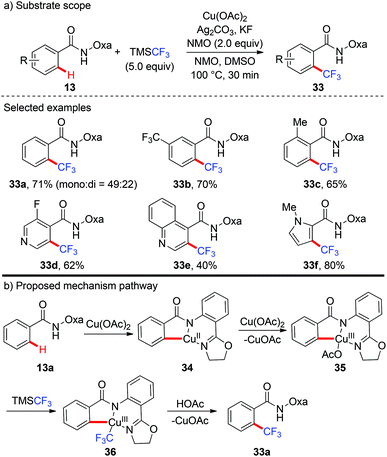 |
| | Scheme 12 Copper-mediated ortho-trifluoromethylation of C(sp2)–H bonds with TMSCF3. | |
2.4 Tandem C–H functionalization/annulation
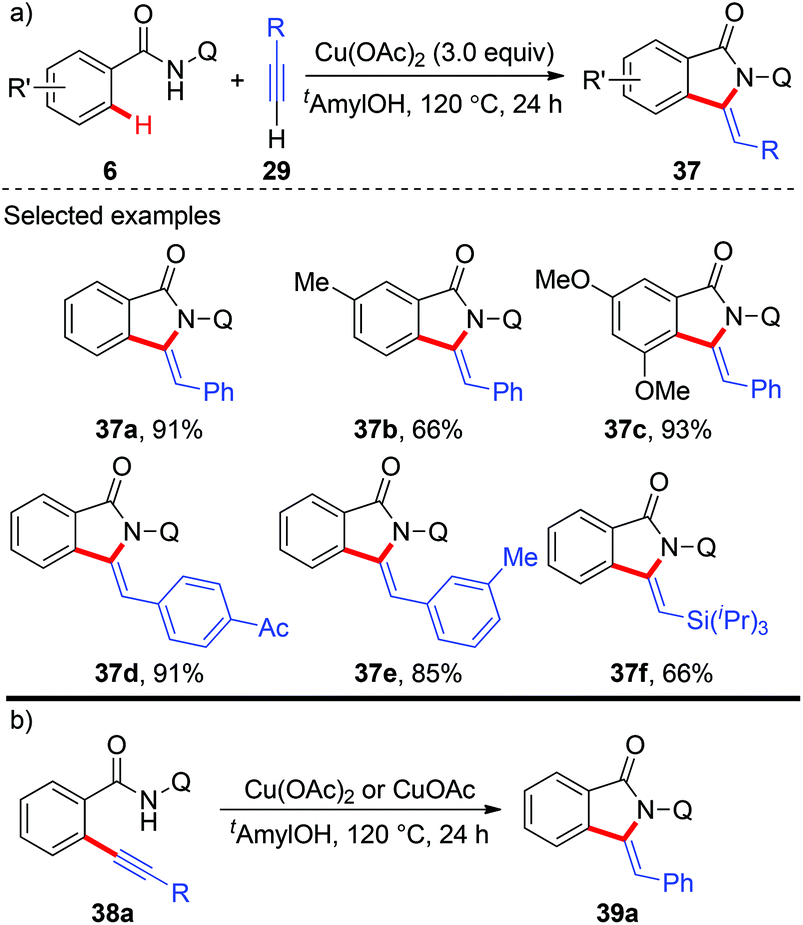
In 2014, copper-mediated tandem alkynylation/annulation of arenes with terminal alkynes was first reported by You and co-workers (Scheme 13a).19a After a short screening of copper salts, copper acetate proved to be the best choice, giving the desired oxidative annulation products in moderate to high yields. The protocol featured a wide substrate scope, high functional group tolerance and exclusive chemo-, regio- as well as stereoselectivity. Moreover, they found that ortho-alkynyl benzamide 38a could be transformed into the corresponding annulation product 39a in the presence of either Cu(OAc)2 or CuOAc, indicating that the domino reaction might proceed via an oxidative alkynylation/intramolecular annulation sequence (Scheme 13b). Shortly after, the Huang and Yu group independently reported the same tandem oxidative alkynylation/annulation reaction.19b
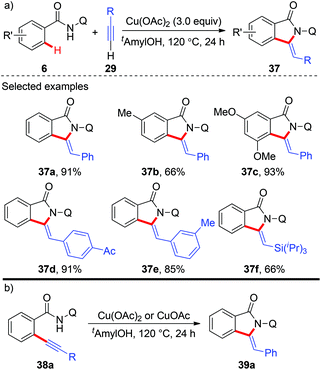 |
| | Scheme 13 Copper-mediated AQ-directed tandem oxidative alkynylation and annulations of arenes with terminal alkynes. | |
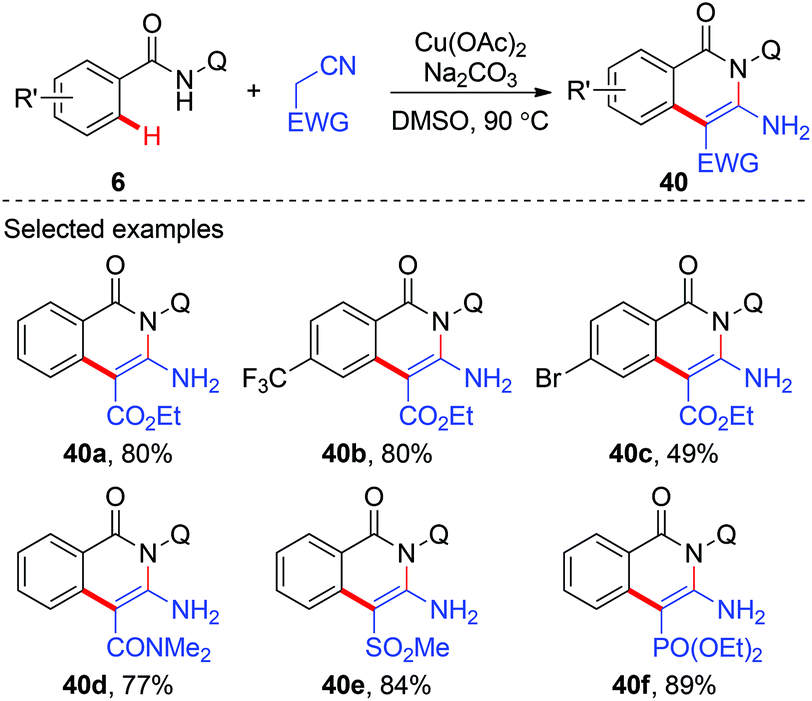
In 2014, Liu and co-workers reported a copper-mediated annulation of benzamides with ethyl cyanoacetate using AQ as the directing group (Scheme 14).20 A number of isoquinolinone compounds 40 were synthesized via this tandem C(sp2)–C(sp3)/C(sp3)–N bond formation sequence. Apart from ethyl cyanoacetate, a variety of cyano substrates, such as cyano substituted amides, phosphonates, and methylsulfonyls, could also be used as coupling partners.
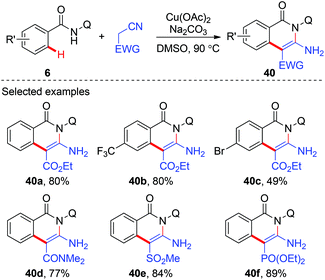 |
| | Scheme 14 Copper-mediated annulation of benzamides with cyano substrates. | |
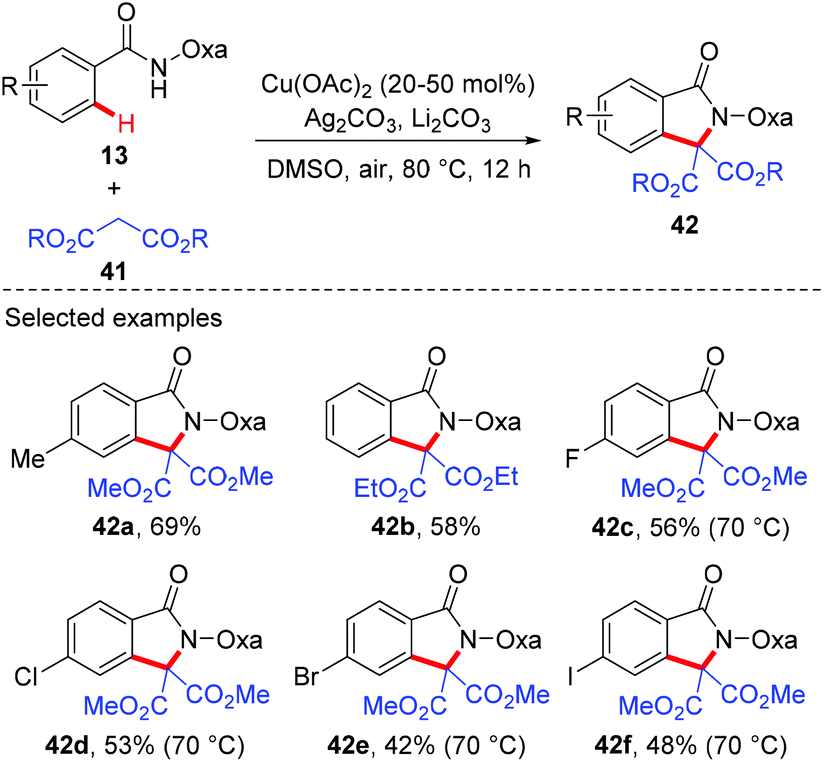
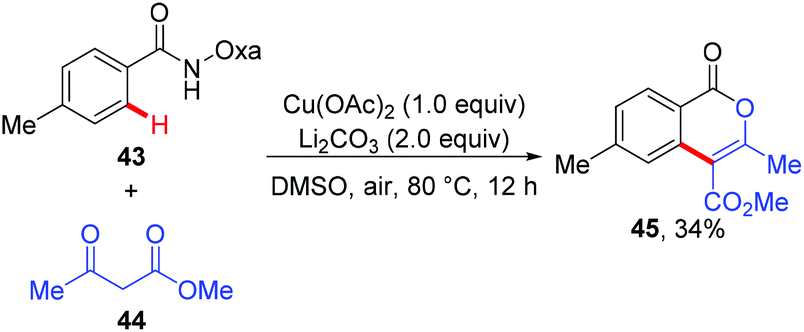
Since the amide-oxazoline directing group has demonstrated versatile reactivity in copper-mediated C–H functionalization, the Yu group rationalized that the same directing group might promote the coupling of aromatic C–H bonds with malonates under copper catalysis (Scheme 15).21 As expected, after extensive screening of reaction parameters, the desired products could be obtained in moderate to high yields with Cu(OAc)2 and Ag2CO3 as the optimal catalyst and oxidant respectively. As such, this method presents an alternative tool for the construction of isoindolinone scaffolds 42. Interestingly, the reaction of benzamide 43 with 3-oxobutanoate 44 produced methyl 3,6-dimethyl-1-oxo-1H-isochromene-4-carboxylate 45via the enolate O-acylation following oxidative C–H coupling (Scheme 16).
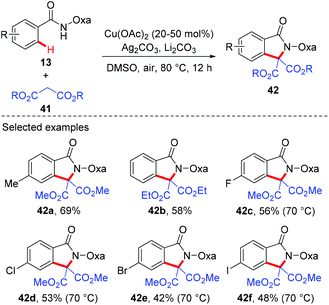 |
| | Scheme 15 Copper-catalyzed coupling of aromatic C–H bonds with malonates. | |
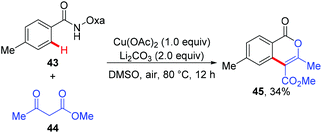 |
| | Scheme 16 Coupling of the benzamide derivative with 3-oxobutanoate. | |
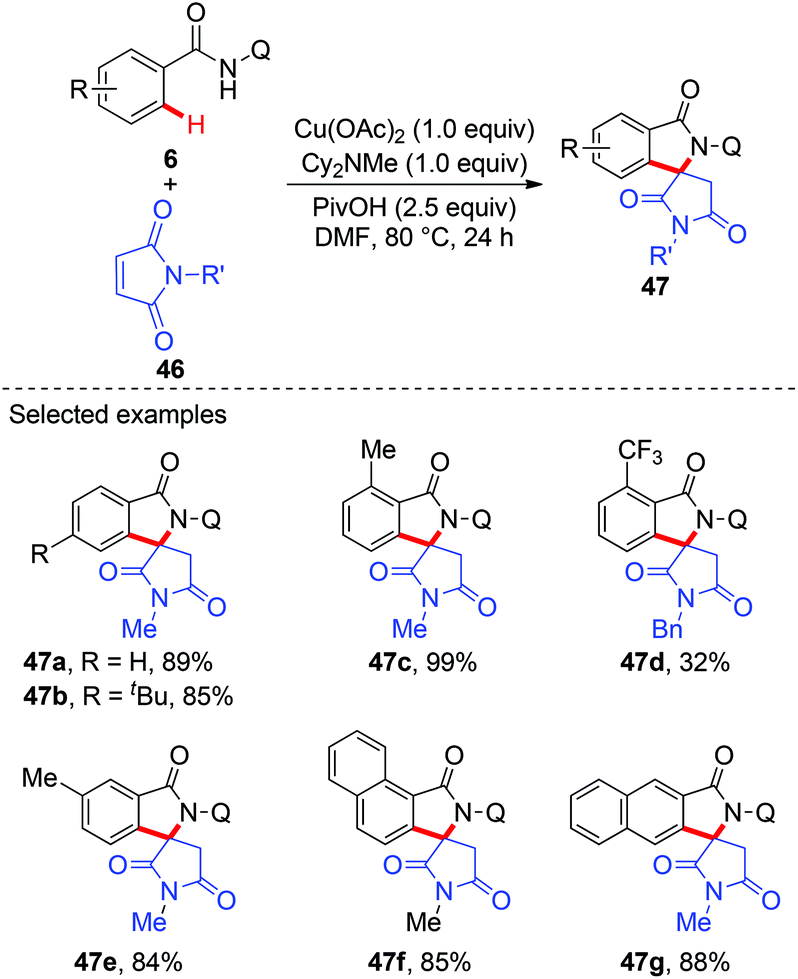
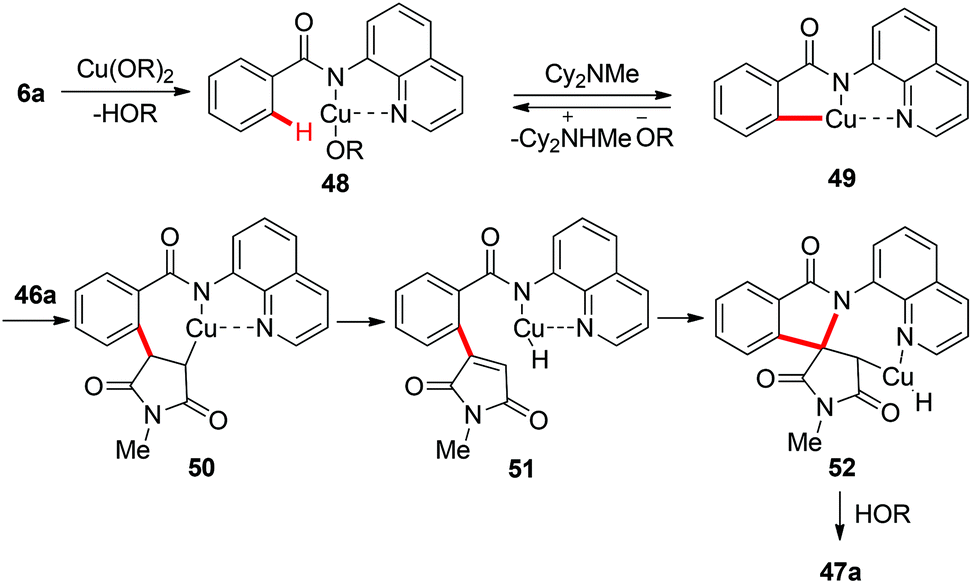
Although a variety of carbon-based nucleophiles mentioned above have been developed as the coupling partners for copper-mediated C–H bond functionalization, the use of accessible alkenes as the coupling partners remained a great challenge. In 2015, Miura et al. reported the copper-mediated oxidative coupling of benzamides bearing 8-aminoquinoline with maleimides (Scheme 17).22a Cy2NMe was crucial for the success of this reaction. Based on the observations of the reaction in the absence of Cy2NMe, they proposed that Cy2NMe not only facilitated the C–H cleavage of benzamides, but also accelerated other critical elementary steps. A plausible reaction pathway was proposed as shown in Scheme 18.
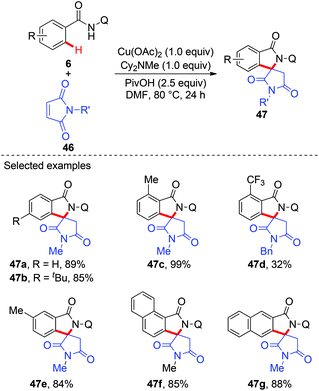 |
| | Scheme 17 Copper-mediated oxidative coupling of benzamides with maleimides. | |
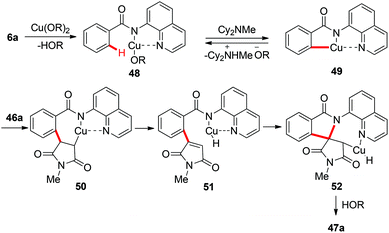 |
| | Scheme 18 Plausible mechanism. | |
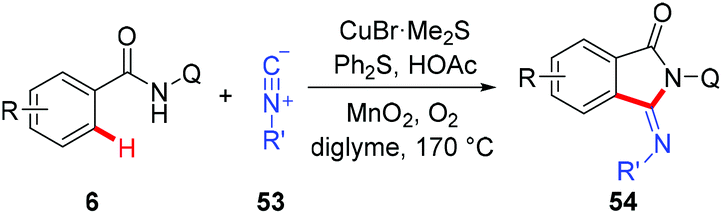
Recently, a copper-catalyzed formal [4 + 1] cycloaddition of benzamides and isonitriles via AQ-directed C–H cleavage has also been developed by Miura and co-workers (Scheme 19).22b The corresponding 3-iminoisoindolinones 54 were exclusively delivered in good yields. The use of diphenyl sulfide as an additive demonstrated a unique acceleration effect on the transformation. Although the exact role of Ph2S remained to be elucidated, the authors considered that the presence of diphenyl sulfide could suppress the formation of inactive, isonitrile-overcoordinated copper species.
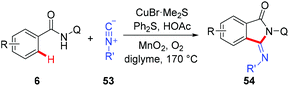 |
| | Scheme 19 Copper-catalyzed 8-aminoquinoline-directed [4 + 1] cycloaddition of benzamides and isonitriles. | |
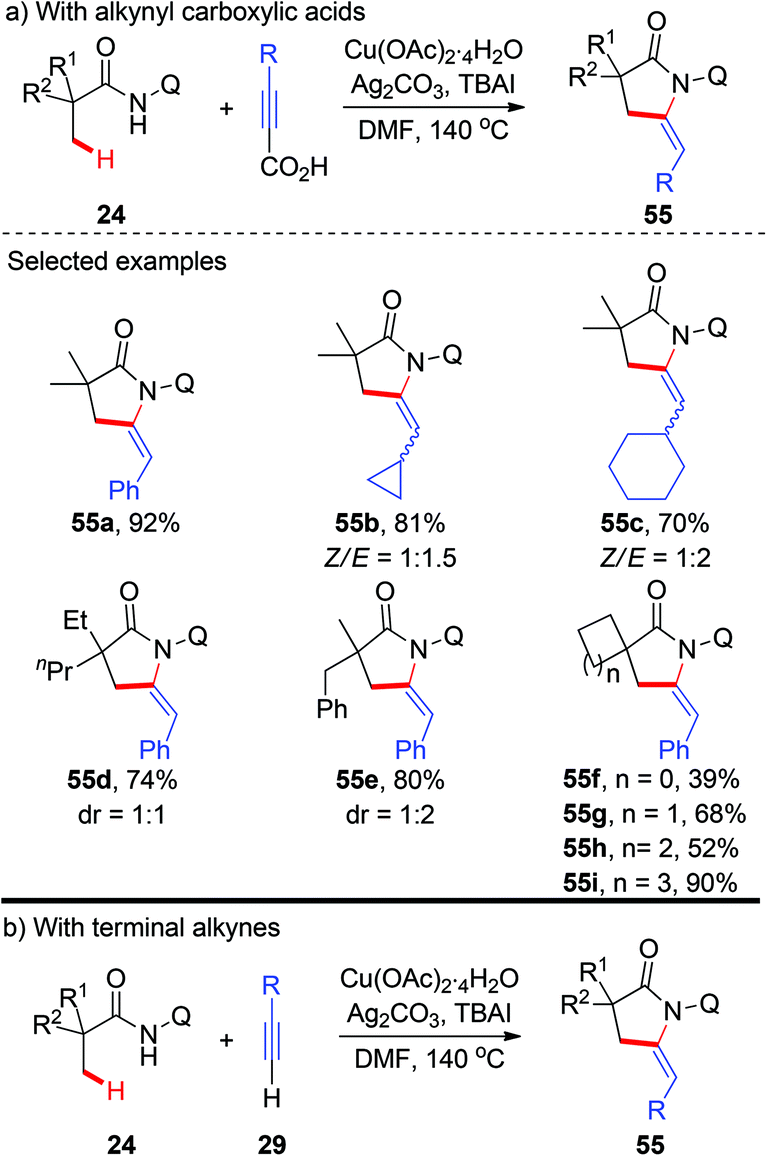
More recently, a copper-catalyzed AQ-directed tandem oxidative alkynylation/annulation of aliphatic amides for the synthesis of pyrrolidones was demonstrated by Zhang and co-workers (Scheme 20a).23 A wide range of alkynyl carboxylic acids were used as coupling partners. Consistent with previous reports, high selectivity of β-methyl groups over methylene groups was observed. The use of TBAI as an additive could significantly improve the efficiency, and preliminary results support that TBAI acts as a phase transfer catalyst. Interestingly, they also found that terminal alkynes could also be used in this transformation, albeit with less efficiency (Scheme 20b).
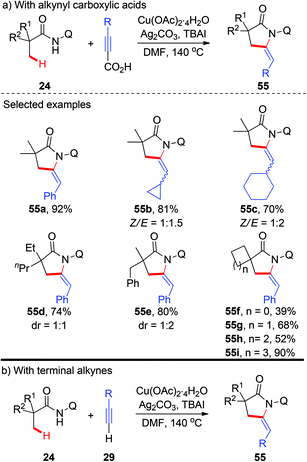 |
| | Scheme 20 Copper-catalyzed AQ-directed oxidative alkynylation/annulation of C(sp3)–H bonds with alkynyl carboxylic acids or terminal alkynes. | |
2.5 Cyanation
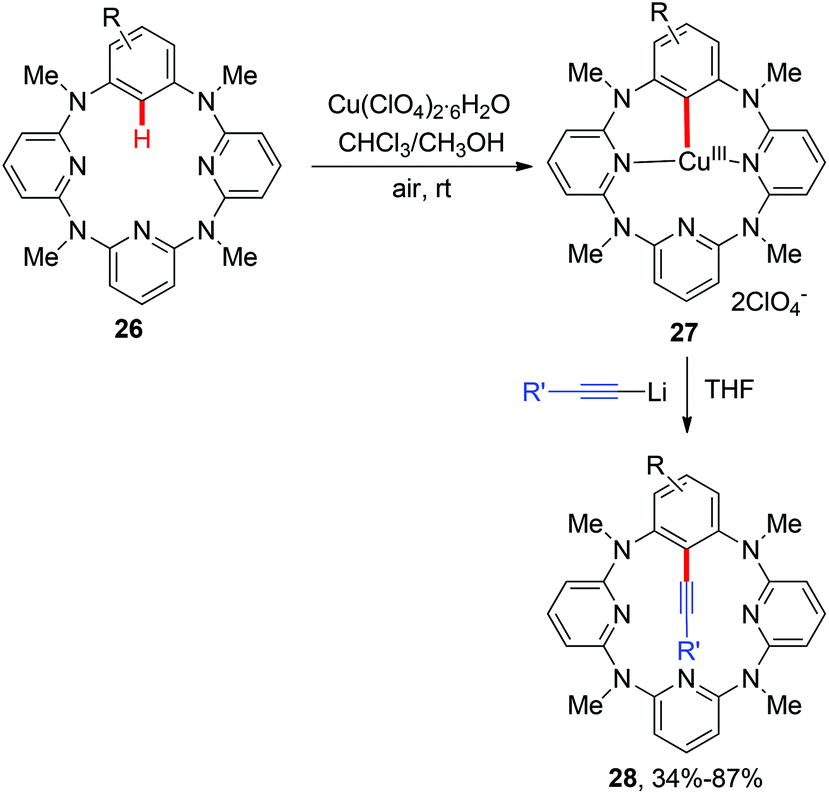
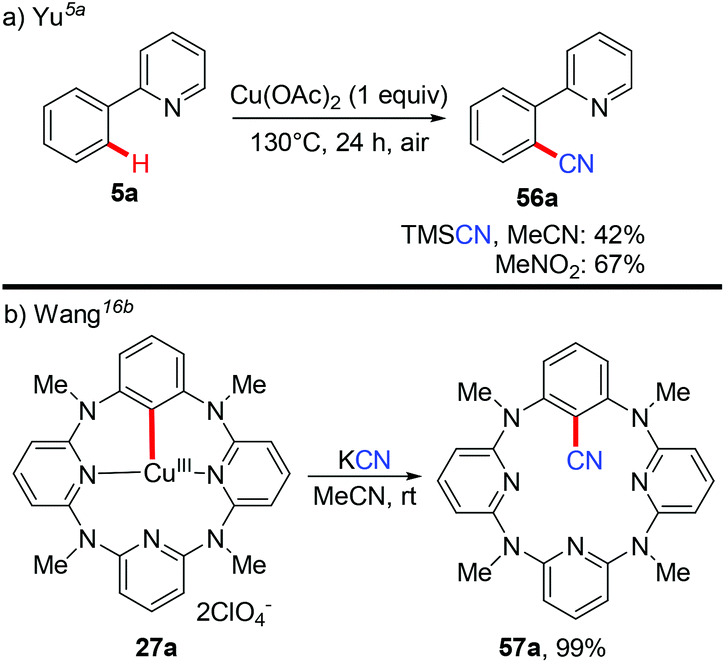
Aryl nitriles are important structural motifs, due to their ubiquitous presence in bioactive organic molecules and chemical transformations. Therefore, the synthesis of aryl nitriles has attracted much attention.24 In 2006, Yu's group reported the first Cu-mediated ortho-cyanation of 2-phenylpyridine (Scheme 21a).5a Besides the use of TMSCN as a normal “CN” source, they also found that nitromethane (CH3NO2) could also work as a “CN” source. However, the mechanism of the cyanation with CH3NO2 as the cyanide anion surrogate is still unclear. In 2009, Wang disclosed that the aryl-Cu(III) complex 27a reacted with KCN at ambient temperature to afford the corresponding cyano-functionalized tetraazacalix[1]arene[3]pyridine 57a in an almost quantitative yield (Scheme 21b).16b
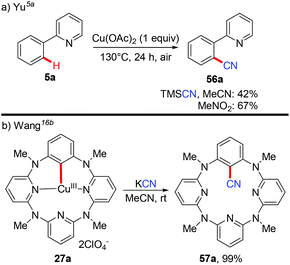 |
| | Scheme 21 Copper-mediated ortho-cyanation of C(sp2)–H bonds with TMSCN, MeNO2 or KCN. | |
Several examples of copper-mediated pyridyl-directed cyanation of C(sp2)–H bonds with various cyanide anion surrogates were reported in the following years.25 In 2012, Wang et al. used benzyl nitrile as a safe and operational benign cyanide anion surrogate for aerobic copper-mediated cyanation of 2-arylpyridines (Scheme 22).25a In the study, several by-products including benzaldehyde, benzoic acid, N,N-dimethylbenzamide, cis-/trans-2,3-diphenylfumaronitrile, and the free cyanide anion were detected in the reaction mixture and these by-products were considered to be generated from the aerobic C–H oxidation step of benzyl nitrile catalyzed by copper salt. Moreover, the authors proposed that the cyanation step proceeded via a single electron transfer (SET) pathway.
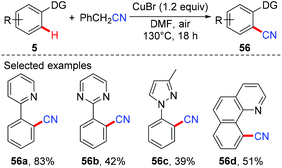 |
| | Scheme 22 Copper-mediated cyanation of C(sp2)–H bonds with benzyl nitrile. | |
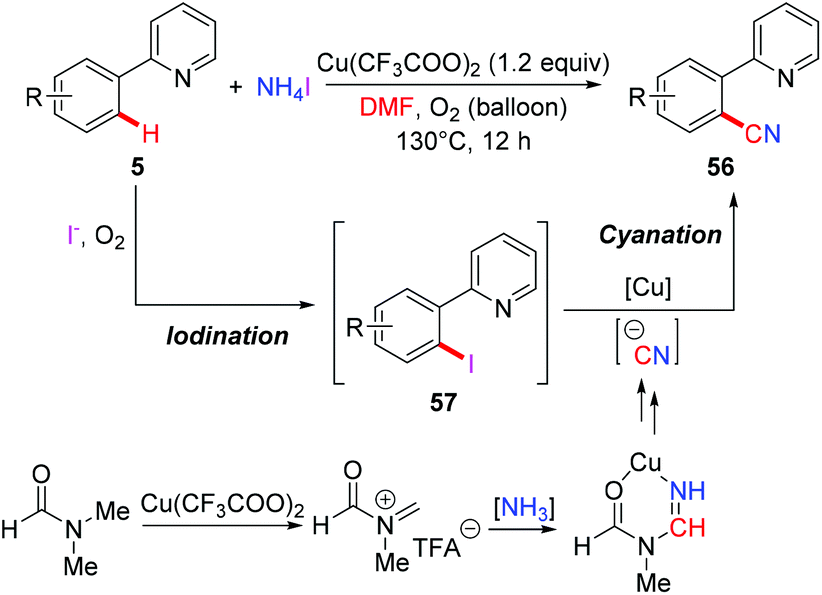
The Chang group employed a NH4I/DMF system as the combined source of a cyano unit for copper-mediated ortho-cyanation of 2-arylpyridines (Scheme 23).25b They proposed that ammonium iodide played a dual role: as a source of iodide for the formation of iodine and as a donor of the “N” unit of the cyano moiety. It was believed that 2-arylpyridines might undergo iodination through a radical-cation pathway leading to the iodinated intermediates 57, which underwent subsequent cyanation to furnish the desired cyanated products 56. The cyanide anion is postulated to be generated from the copper-mediated reaction of ammonia and DMF.26a,b
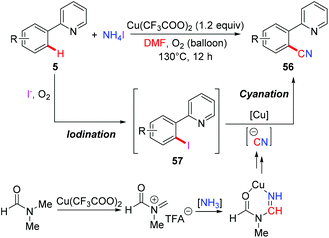 |
| | Scheme 23 Copper-mediated ortho-cyanation of 2-arylpyridines with NH4I and DMF. | |
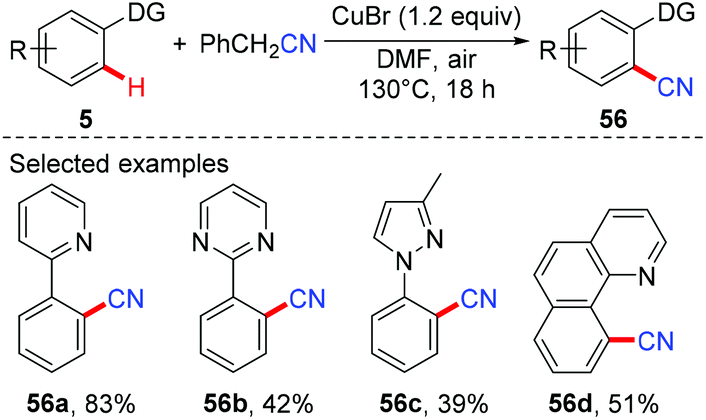
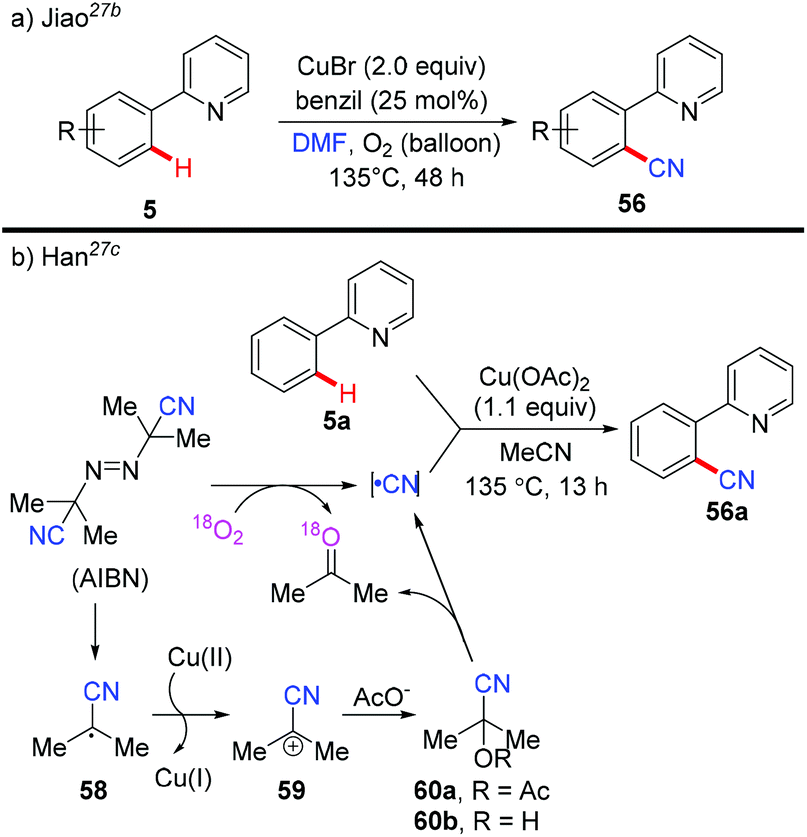
The Jiao group reported a Pd-catalyzed direct cyanation of indoles and benzofurans employing DMF as both the reagent and solvent in 2011.27a Unlike Chang and Cheng's work,26 they found that both the “C” and “N” units of the cyano group are derived from DMF. Inspired by these results, Jiao and co-workers successfully achieved copper-mediated cyanation of 2-arylpyridines employing DMF as both the reagent and solvent, providing a complementary strategy to access aryl nitriles (Scheme 24a).27b Although the mechanism is not clear yet, preliminary studies suggested that the mechanism of the present protocol was totally different from that of Chang's reactions.25b
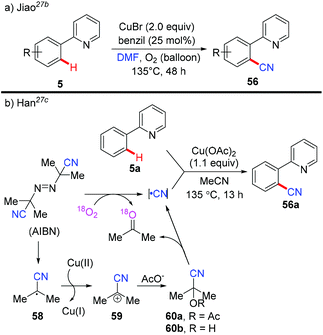 |
| | Scheme 24 Copper-catalyzed ortho-cyanation of 2-arylpyridines: (a) DMF as a cyanide source; (b) AIBN as a free-radical “CN” source. | |
In 2013, Han et al. reported a copper-mediated pyridyl-directed C(sp2)–H cyanation using azobisisobutyronitrile (AIBN) as a free-radical “CN” source (Scheme 24b).27c The 18O-labeled experiments confirmed that acetone was mainly generated by the reaction of O2 and AIBN. They also observed the formation of acetyl acetone cyanohydrin 60a by analysis of the reaction mixture with ESI-MS. X-ray photoelectron spectroscopic (XPS) analyses revealed the reduction of Cu(OAc)2 to the Cu(I) species by AIBN with or without 2-phenylpyridine 5a. Based on these results, the generation of the cyano group was proposed as shown in Scheme 24b.
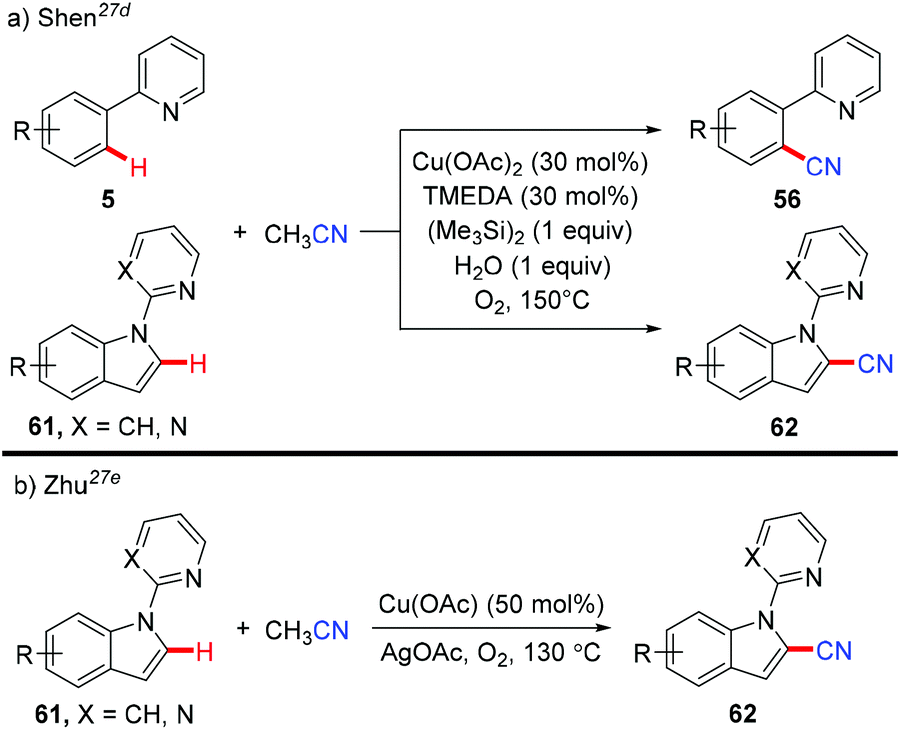
In 2013, Shen et al. reported a copper-catalyzed cyanation of the aromatic C–H bond using acetonitrile as a cyanide source (Scheme 25a).27d The authors hypothesized that hexamethyldisilane may only serve as a Lewis acid to activate the C–CN bond cleavage of acetonitrile to afford the copper cyanide species, which subsequently reacted with the aromatic C–H bond to yield a new aromatic C–CN bond.
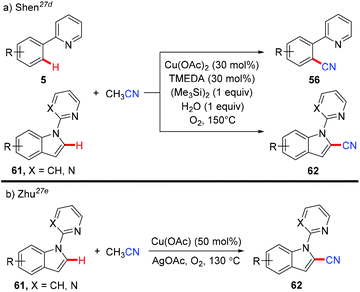 |
| | Scheme 25 Copper-catalyzed cyanation with MeCN. | |
In the same year, Zhu and co-workers also reported a copper-mediated C2-cyanation of indoles employing acetonitrile as the cyanide source (Scheme 25b).27e The presence of either the pyridine or pyrimidine auxiliary in the substrates was crucial for the C2-selectivity.
3. Carbon–nitrogen bond formation
3.1 Intermolecular amination
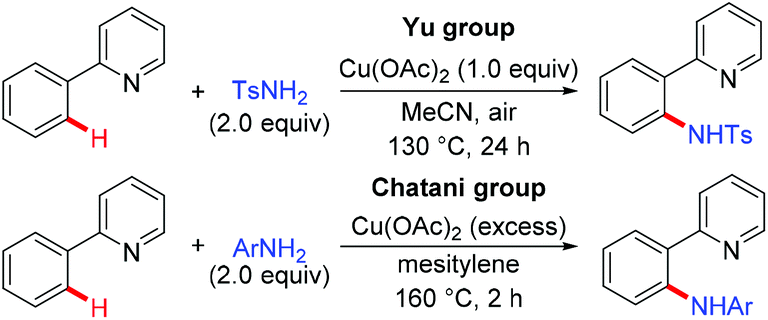
In 2006, Yu's group and Chatani's group independently developed an aerobic copper-mediated C(sp2)–H bond amination using pyridine as the directing group (Scheme 26).5 Subsequently, several different protocols for the copper-mediated ortho-amination of 2-phenylpyridines have been developed.28 These precedents indicated that copper-mediated direct amination of unactivated C–H bonds was feasible with the assistance of a suitable directing group. Furthermore, the reaction of well-defined Cu(III) complexes with N-nucleophiles to form C–N bonds has been well established.16
 |
| | Scheme 26 Pioneering work on copper-mediated pyridyl-directed amination of C(sp2)–H bonds by Yu and Chatani. | |
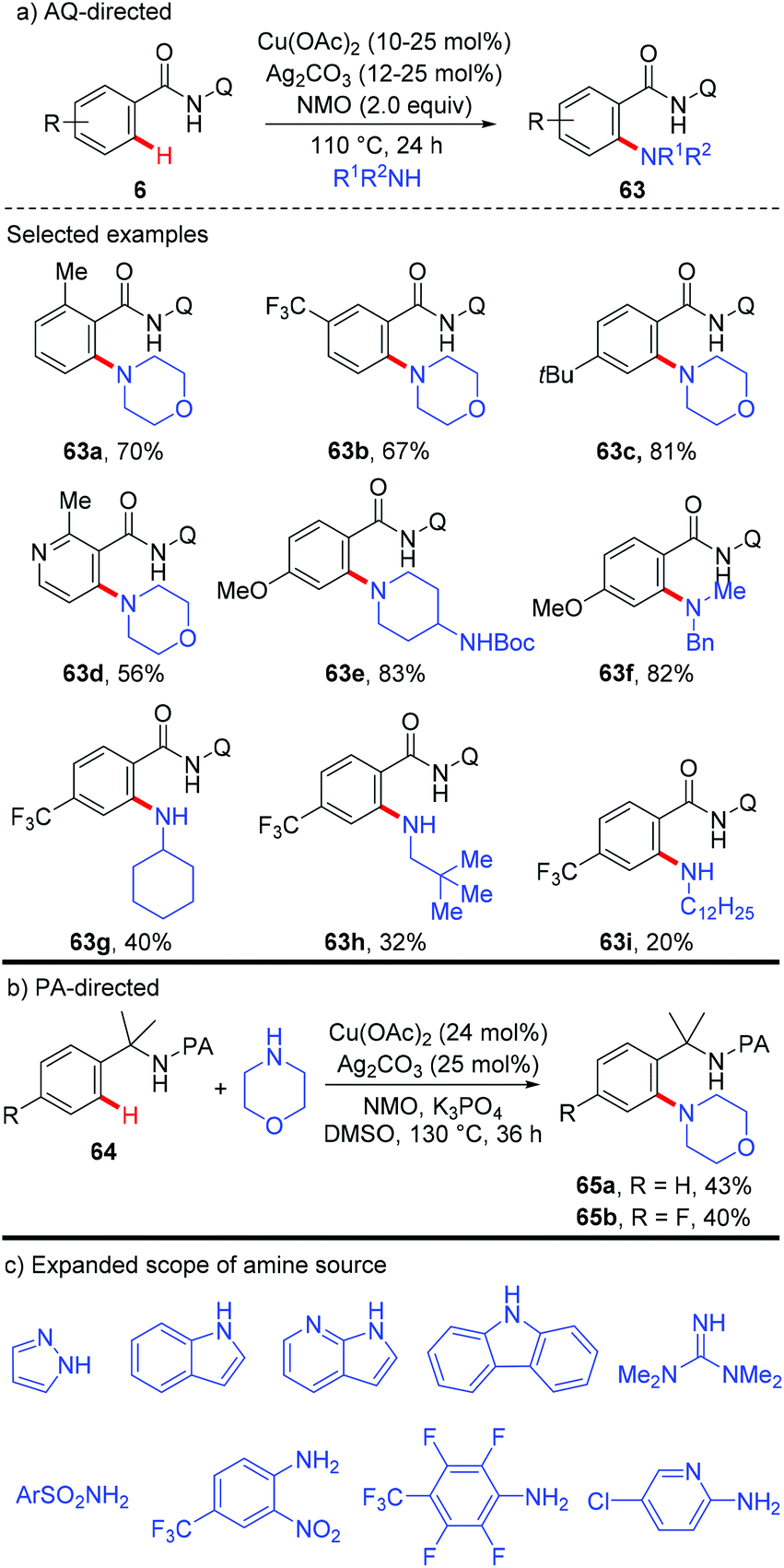
On the basis of these precedents, the Daugulis group realized the copper-catalyzed C–H amination using the removable bidentate AQ directing group (Scheme 27a).29a The amination of both electron-rich and electron-deficient benzamides proceeded smoothly to provide the corresponding products in moderate to high yields. Notably, the amination selectively delivered mono-functionalization products at the less sterically hindered position. Secondary amines possessing various functional groups, such as ester, ether, cyano, and NHBoc, were all compatible with this reaction, affording the amination products in excellent yields. Primary amines were also able to serve as the coupling partner, albeit in lower to moderate yields. They further presented that α,α-disubstituted benzylamines could also be aminated with the assistance of the PA auxiliary under the optimized conditions, albeit in moderate yields (Scheme 27b). Although this transformation showed good functional group tolerance and substrate scope, it still limited to the use of silver salt as a cocatalyst and NMO as a terminal oxidant. Moreover, only primary and secondary aliphatic amines were effective coupling partners. To overcome these limitations, a general method using Cu(OAc)2 or Cu2(OH)2CO3 as the catalyst and oxygen from air as the sole oxidant was recently reported by Daugulis and co-workers.29b Notably, primary and secondary aliphatic and aromatic amines, N-heteroarenes, such as indoles, pyrazole, and carbazole, sulphonamides, as well as electron-deficient aromatic and heteroaromatic amines were tolerated under the reaction conditions (Scheme 27c).
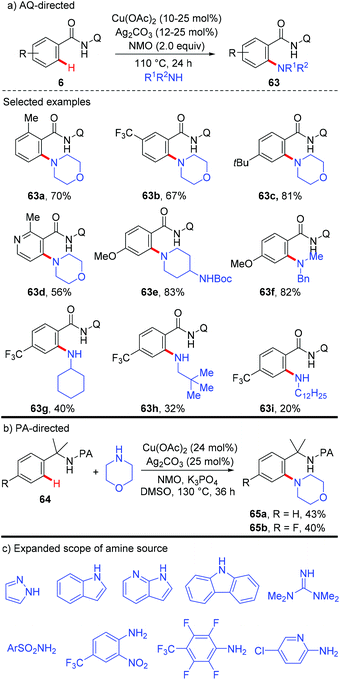 |
| | Scheme 27 Copper-catalyzed direct amination of unactivated C(sp2)–H bonds. | |
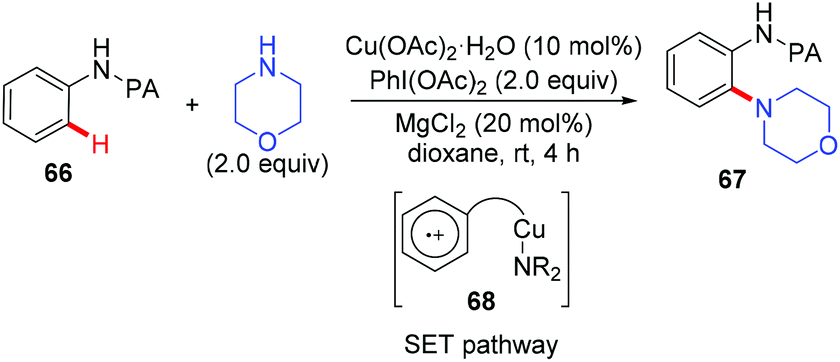
In 2014, the Carretero and Chen group independently reported a copper-catalyzed PA-directed amination of aniline derivatives at room temperature (Scheme 28).30 Although the amine partners were limited to cyclic secondary amines, a broad range of anilines bearing various functional groups were tolerated. The PA directing group could be removed under basic conditions. Mechanistic studies revealed that an aromatic substitution-type pathway mediated by SET might be involved in the reaction.
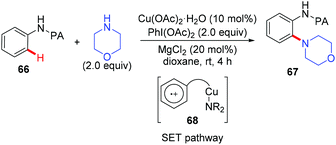 |
| | Scheme 28 Copper-catalyzed PA-directed amination of anilines via a SET pathway. | |
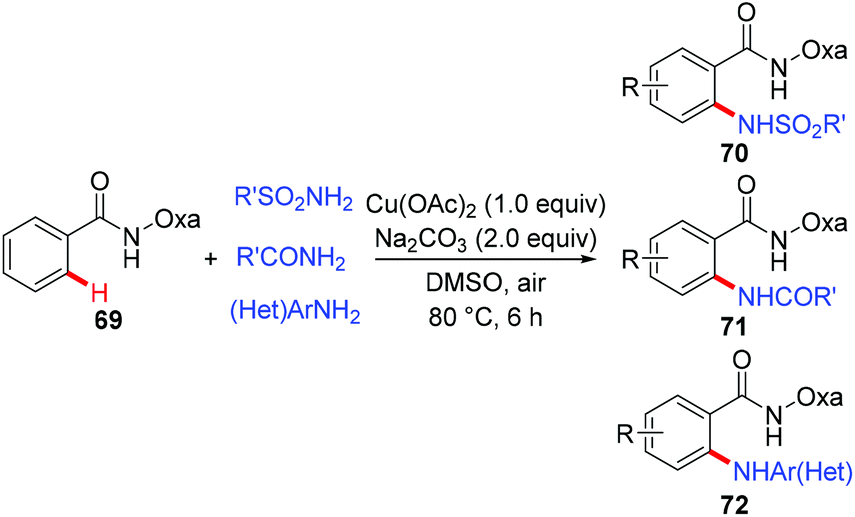
In 2014, a copper-mediated amidation and amination of (hetero)arenes with a broad range of sulfonamides, amides, and anilines was reported by Yu's group (Scheme 29).31a The amide-oxazoline was used as a removable directing group in this transformation. While the stoichiometric copper in the reaction was necessary, exceptional compatibility with heterocyclic arenes and amine donors was observed in the reaction, which should be practically attractive to medicinal chemists. A related copper-catalyzed AQ-directed amination of benzamides with pyrroles, indoles, pyrazoles and carbazoles via dehydrogenative coupling has recently been reported by Punniyamurthy.31b
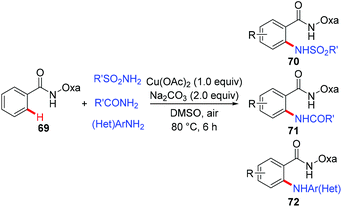 |
| | Scheme 29 Copper-mediated amidation and amination of C(sp2)–H bonds. | |
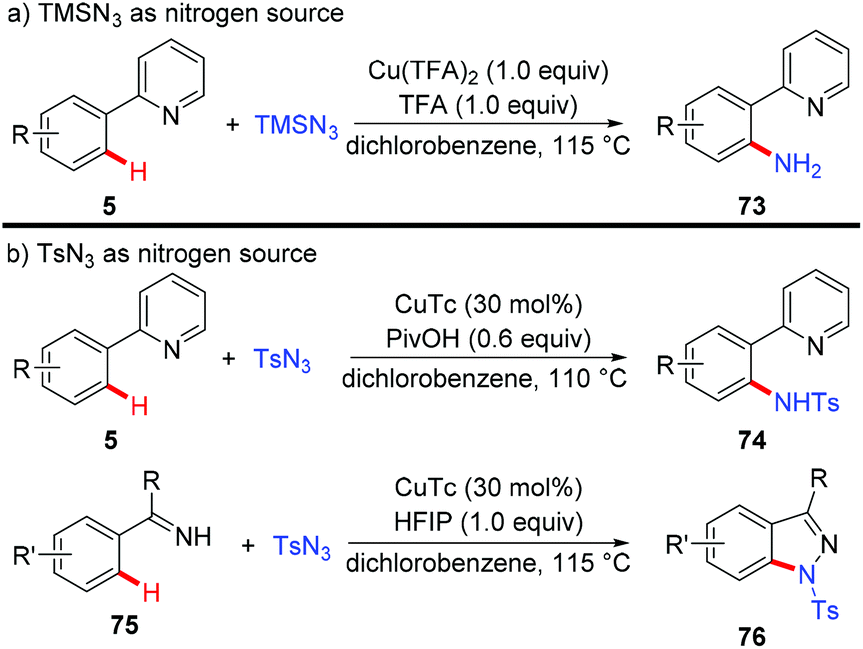
Organic azides have been proven to be an efficient and convenient amino source in transition metal-catalyzed C–H amination reactions.32 Zhu and co-workers recently described several examples of copper-mediated formation of aromatic C–N bonds with azides (Scheme 30).33 They disclosed that when TMSN3 was used as a nitrogen source, the free amino group could be directly introduced into the aromatic ring, thus providing a redox-neutral access to the primary amino group (Scheme 30a).33a Notably, a key intermediate for the synthesis of inhibitors of phosphate transport protein was prepared in 62% isolated yield with this protocol. Moreover, the copper-catalyzed amidation of C(sp2)–H bonds could be achieved, when TsN3 was used as an amino source (Scheme 30b). Indazoles were obtained with amidine or imine as the directing group via tandem C–N/N–N bond formation. Mechanistic investigations indicated that an aryl-Cu(III) intermediate rather than a copper nitrenoid intermediate via an inner-sphere pathway was involved in the process.
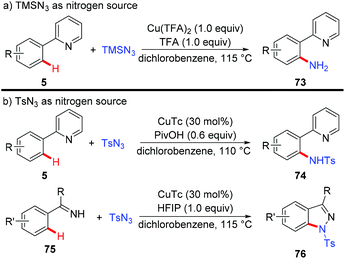 |
| | Scheme 30 Copper-mediated formation of C–N bonds with organic azides as amino donors. | |
3.2 Intramolecular amination
Inspired by the seminal work of Buchwald on benzimidazole synthesis via copper-catalyzed intramolecular C–H amination,34a a number of heterocycles were synthesized via a related SEAr pathway.34 These elegant studies have recently been reviewed and will not be discussed here.34k
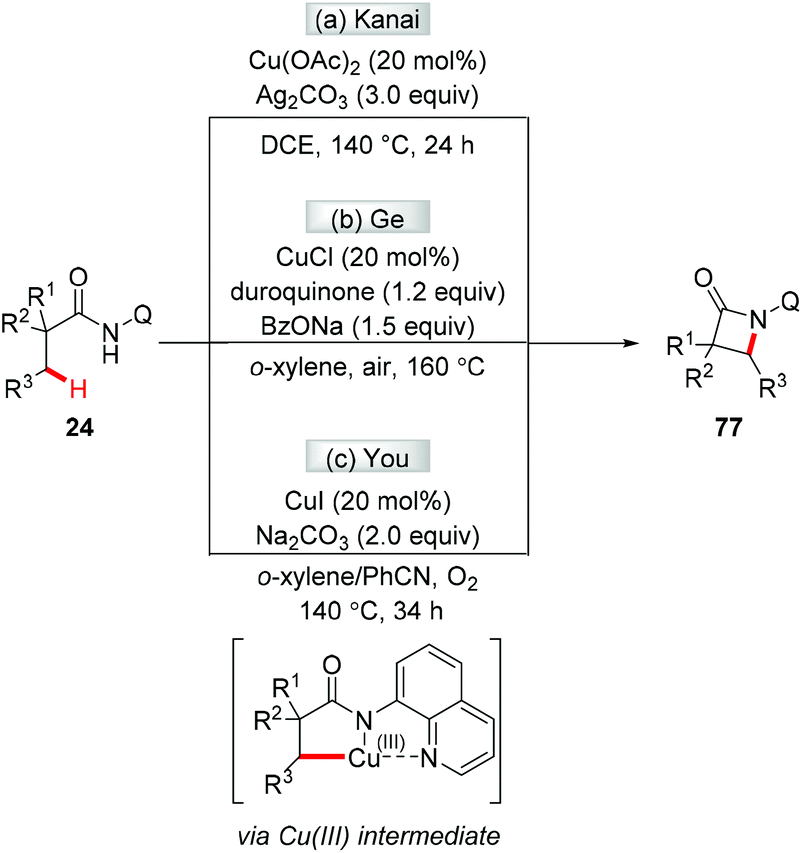
The first chelation-assisted amination of unactivated C(sp3)–H bonds was independently reported by Kanai and Ge in 2014. Biologically important β-lactams were successfully synthesized via the Cu-catalyzed AQ-directed intramolecular C(sp3)–H amination (Scheme 31a and b).35a,b,36 Shortly after, the You group reported a copper-catalyzed AQ-directed intramolecular C(sp3)–H amination using oxygen as the sole external oxidant (Scheme 31c).35c To increase the practicality of these reactions, the removal of 5-methoxy-8-aminoquinoline was demonstrated to give β-lactams with a free NH group. The reactions were proposed to proceed via reductive elimination of a Cu(III) intermediate.
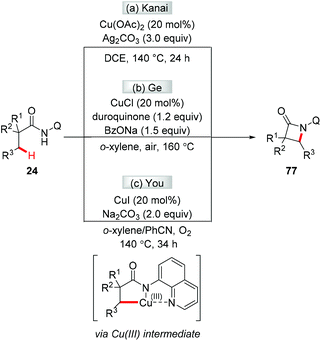 |
| | Scheme 31 The synthesis of β-lactams via copper-catalyzed intramolecular C(sp3)–H amination. | |
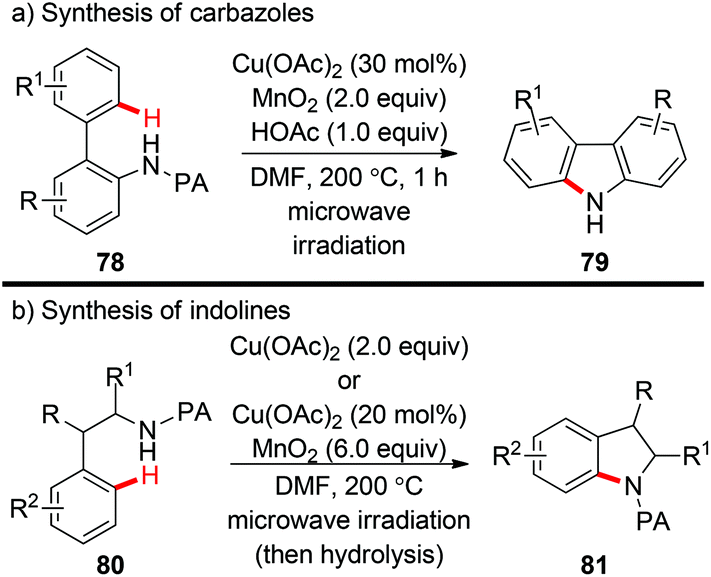
Miura et al. reported a copper-catalyzed PA-directed intramolecular amination of C(sp2)–H bonds for the synthesis of carbazoles under microwave irradiation (Scheme 32a).37a Interestingly, the PA directing group could be spontaneously detached after the formation of C–N bonds. In 2015, they successfully extended this protocol to the construction of indoline scaffolds (Scheme 32b).37b In some cases, the use of excess Cu(OAc)2 was superior to the combination of catalytic Cu(OAc)2 and excess MnO2 as the terminal oxidant.
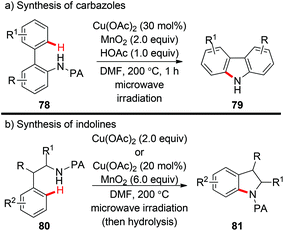 |
| | Scheme 32 The synthesis of carbazoles and indolines via copper-catalyzed intramolecular C(sp2)–H amination. | |
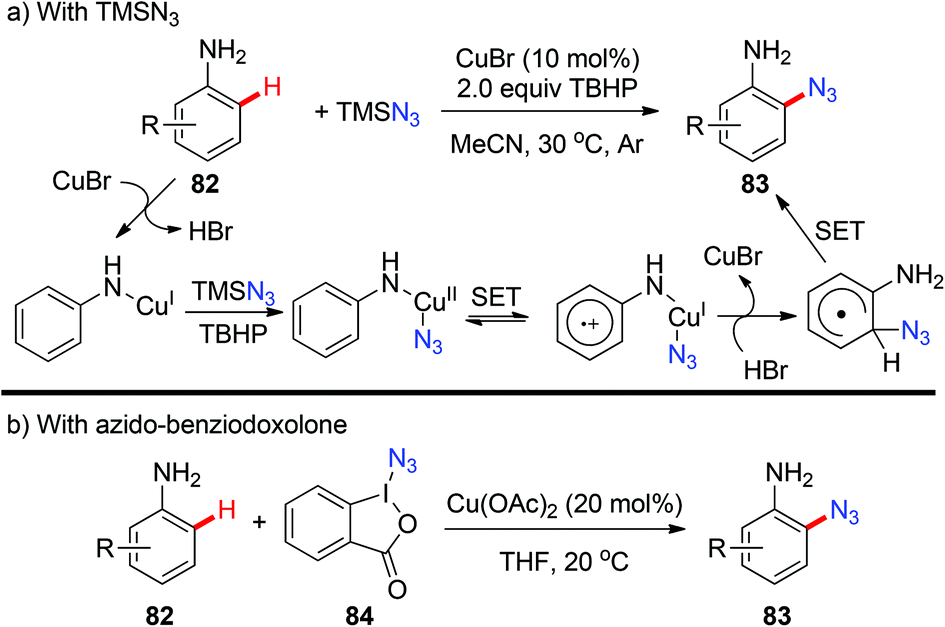
3.3 Azidation
Organic azides are an important class of compounds and have been extensively applied in organic synthesis as building blocks or as “masked amines”.38 Despite their importance, the synthesis of organic azides via copper-mediated C–H azidation has been less studied. In 2012, the Jiao group reported the first copper-catalyzed C–H azidation of anilines with TMSN3 directed by the amino group under rather mild conditions (Scheme 33a).39a The amino group can be converted into various groups, such as Cl, Br, I, CN, OH or H, by the Sandmeyer reaction. The azidation reaction was proposed to proceed via a SET pathway. In 2014, Hao reported an analogous ortho-azidation of anilines using azido-benziodoxolone 84 catalyzed by Cu(OAc)2 (Scheme 33b).39b
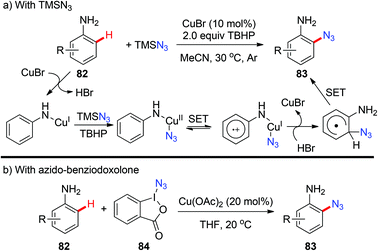 |
| | Scheme 33 Copper-catalyzed ortho-azidation of anilines. | |
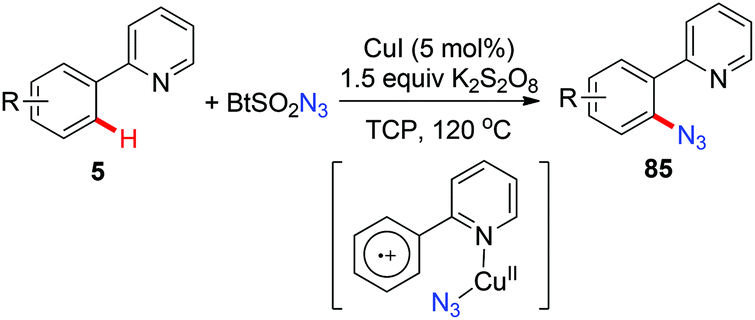
In 2015, a copper-catalyzed pyridyl-directed ortho-azidation using benzotriazole sulphonyl azide as the azidating reagent in the solvent 1,2,3-trichloropropane (TCP) was reported by Narula and co-workers.39c A SET pathway was also proposed for this azidation reaction (Scheme 34).
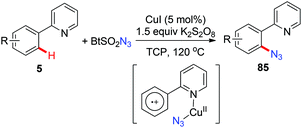 |
| | Scheme 34 Copper-catalyzed ortho-azidation of 2-arylpyridines. | |
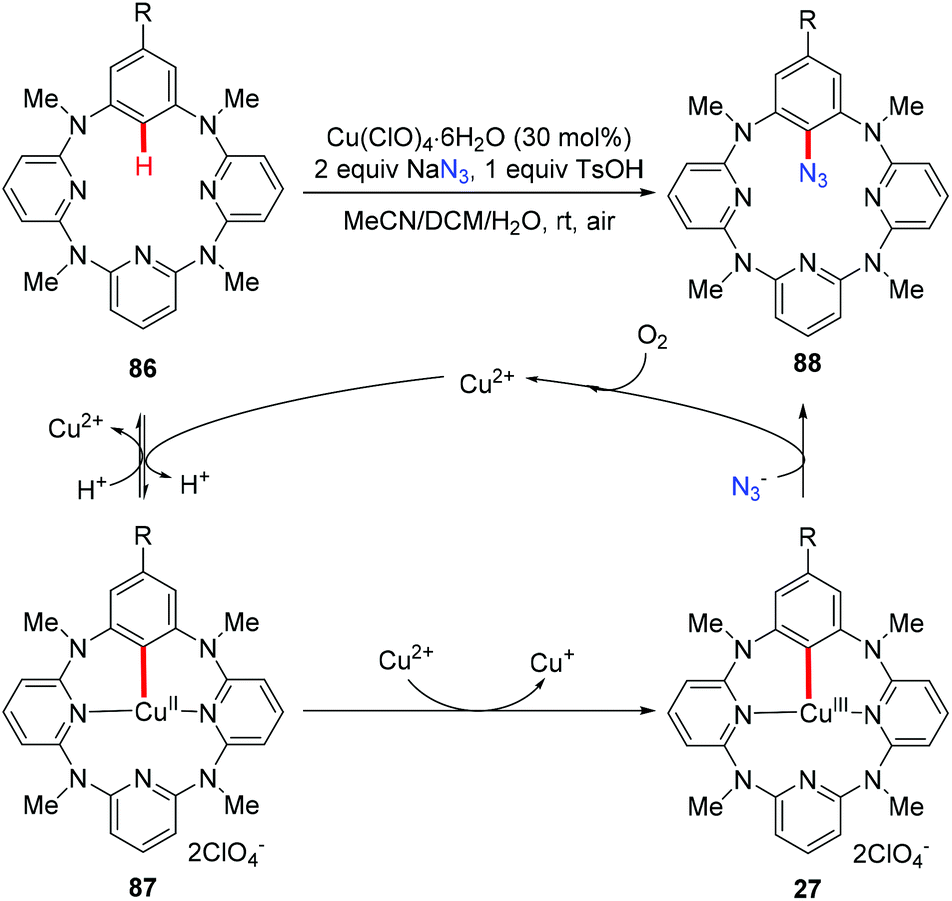
The above mentioned azidation reactions all proceeded through a SET mechanism without involving an organometallic C–H activation step. In 2014, Wang and co-workers reported the first copper-catalyzed azidation via an organometallic C–H activation pathway through an arylcopper(III) intermediate (Scheme 35).40 Stoichiometric reactions of the isolated arylcopper(II) species 87 and arylcopper(III) species 27 with sodium azide clearly indicated that the arylcopper(III) was an active species in this azidation protocol. Thus, an unprecedented Cu(II)–ArCu(II)–ArCu(III)–Cu(I) catalytic cycle was proposed for this reaction.
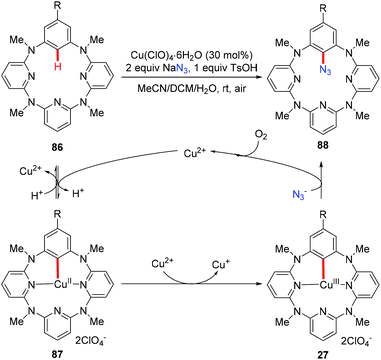 |
| | Scheme 35 Copper-catalyzed azidation of azacalix[1]arene[3]pyridines. | |
3.4 Nitration
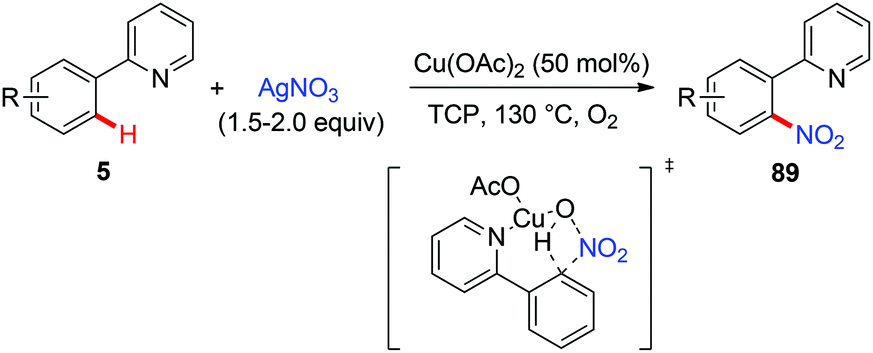
In 2011, Bi and Liu reported a copper-catalyzed ortho-nitration of 2-arylpyridines with AgNO3 using oxygen as an oxidant (Scheme 36).41 Mechanistic studies revealed that the reaction proceeded through a four-centered transition state, with simultaneous cleavage of the ortho-C–H bond and the N–O bond of the nitrate anion on the 2-arylpyridine-coordinated copper(II) intermediate.
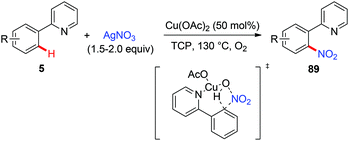 |
| | Scheme 36 Copper-mediated ortho-nitration of 2-arylpyridines. | |
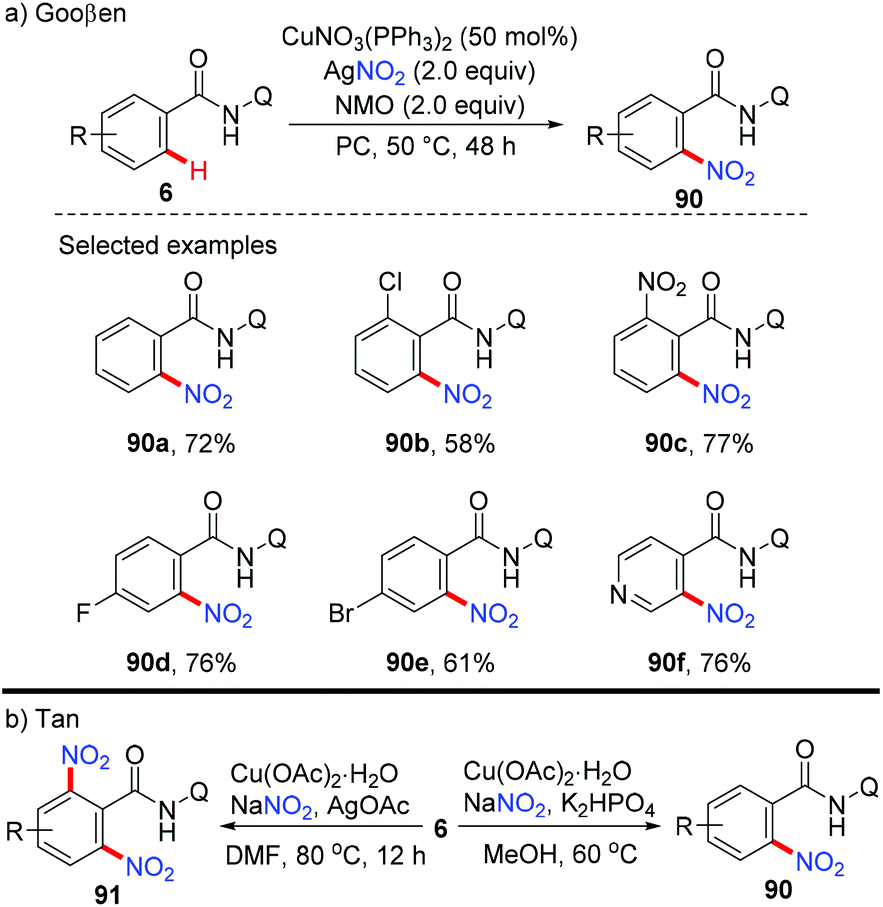
In 2014, the Gooßen group reported a copper-mediated ortho-nitration of (hetero)arenes with the assistance of the removable AQ directing group (Scheme 37a).42a The reactions of both electron-rich and electron-deficient benzamides with AgNO2 proceed smoothly in the presence of NMO as the oxidant and propylene carbonate (PC) as the solvent. In contrast to the previous pyridyl-directed nitration,41 isotope-labeling experiments and control experiments with TEMPO as the radical scavenger revealed that C–H bond activation was the rate-determining step and a radical pathway was not involved in the process. Shortly after, the Tan group reported the copper-mediated AQ-directed nitration of aryl C–H bonds using sodium nitrite as the nitration agent. Notably, good selectivity can be achieved by simply tuning the reaction conditions (Scheme 37b).42b
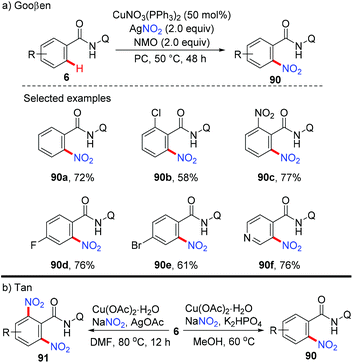 |
| | Scheme 37 Copper-mediated ortho-nitration of (hetero)arene amides. | |
4. Carbon–oxygen bond formation
4.1 Intermolecular carbon–oxygen bond formation
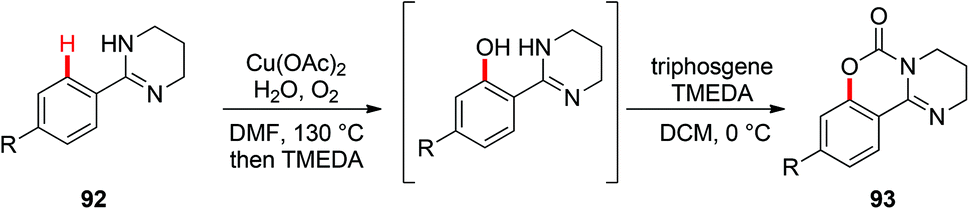
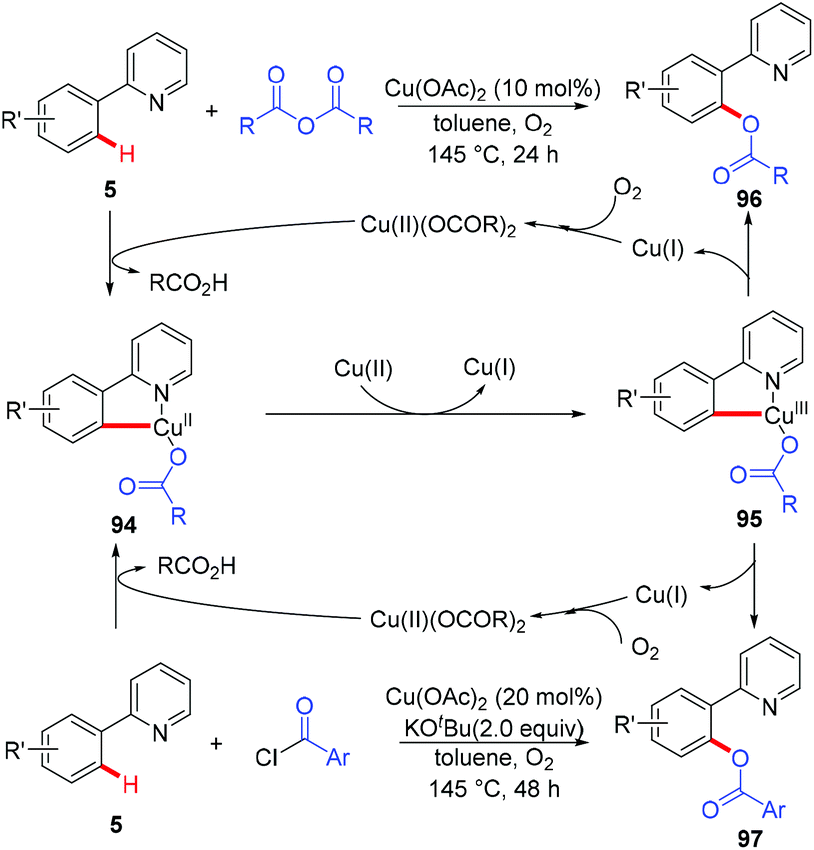
In 2006, the Yu group described the first example of Cu(OAc)2-catalyzed ortho-acetoxylation of 2-arylpyridines in HOAc/Ac2O using O2 as the terminal oxidant.5a A SET mechanism for the reaction was proposed. The direct oxygenation of a structurally well-defined aryl-Cu(III) complex 27a with various carboxylates at room temperature was reported by Wang in 2009.16b In 2009, Ohno and co-workers reported a Cu(OAc)2-mediated ortho-functionalization of C(sp2)–H bonds using tetrahydropyrimidine as the directing group, where the ortho-hydroxylation of aromatic C–H bonds followed by cyclization with triphosgene was described (Scheme 38).43 Shortly after, Cheng et al. used anhydrides44a or acyl chlorides44b as the coupling partners for copper-catalyzed acyloxylation of 2-arylpyridines (Scheme 39). In sharp contrast to Yu's SET mechanism, this reaction was considered to proceed through an organometallic C–H activation, followed by the reductive elimination of a key RCO2–Cu(III)–Ar intermediate 95. More recently, our group also reported a copper-catalyzed PIP-directed acyloxylation of the C(sp2)–H bond with sterically bulky benzoic acids.44c
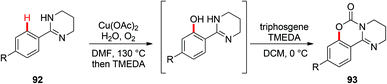 |
| | Scheme 38 Copper-catalyzed ortho-hydroxylation of aromatic C–H bonds followed by cyclization with triphosgene. | |
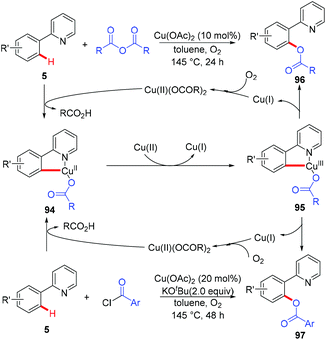 |
| | Scheme 39 Copper-catalyzed aerobic acyloxylation of C(sp2)–H bonds. | |
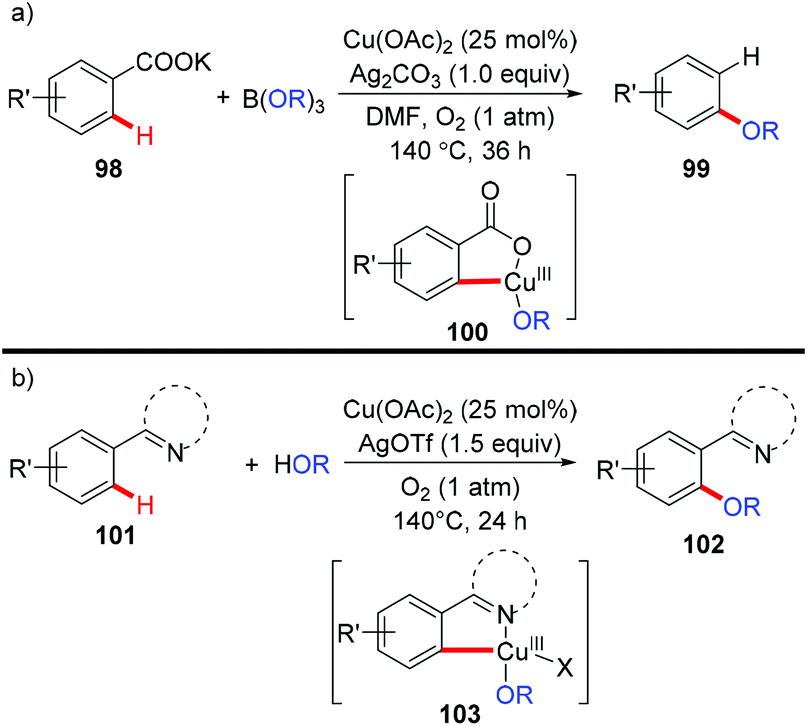
In 2013, the Gooßen group reported a copper-catalyzed carboxylate-directed ortho-alkoxylation of benzoates with B(OR)3 (Scheme 40a).45 Interestingly, concomitant decarboxylation was observed in the process. The presence of silver carbonate was crucial for the success of this reaction, not only for the decarboxylation, but also for the alkoxylation process. Inspired by the copper-mediated pyridyl-directed phenoxylation by Yu5a and the observation of facile C–O reductive elimination from the copper(III) center,16b,e,f the bimetallic copper/silver system was further extended to the regiospecific dehydrogenative cross-coupling of arenes and alcohols with the assistance of pyridine, pyrimidine or pyrazole (Scheme 40b).46 These reactions were proposed to go through Ar–Cu(III)–OR intermediates 100 and 103.
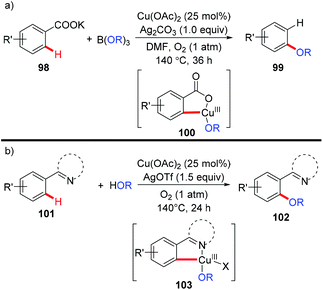 |
| | Scheme 40 (a) Copper-catalyzed carboxylate-directed alkoxylation with concomitant protodecarboxylation; (b) copper-catalyzed dehydrogenative alkoxylation with alcohols. | |
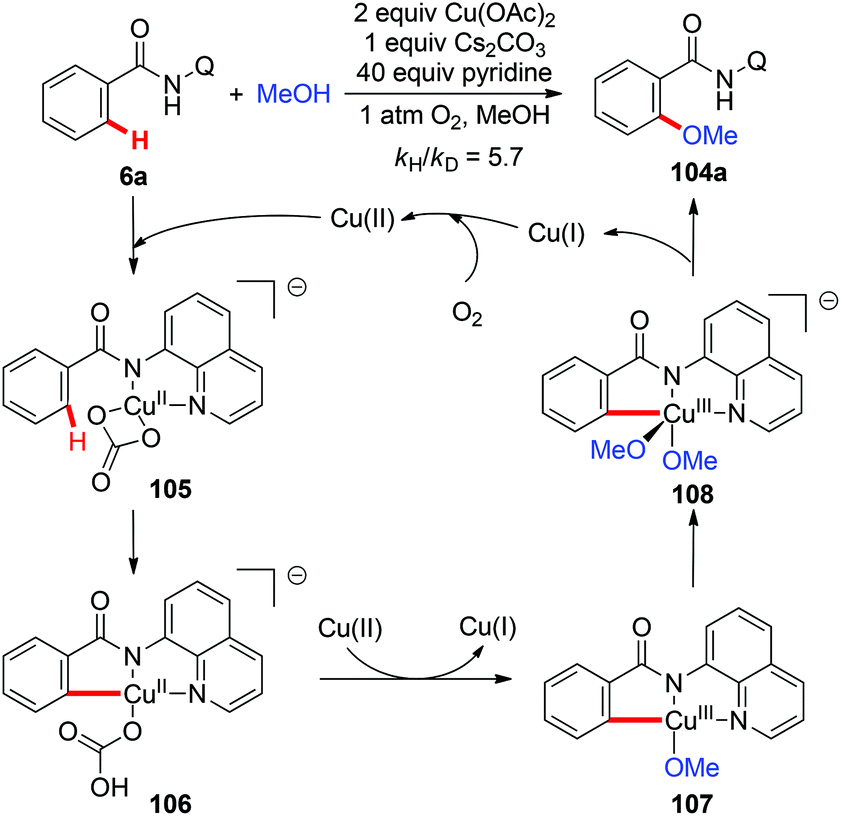
Although copper(II)-mediated C–H oxidation has been well investigated, the mechanisms of many of these reactions remain unclear. In an instructive study, Stahl and co-workers gave clear evidence for the involvement of a basic ligand induced, rate-limiting organometallic C–H activation step in the Cu(OAc)2-mediated AQ-directed methoxylation reactions based on the experimental and computational studies (Scheme 41).47
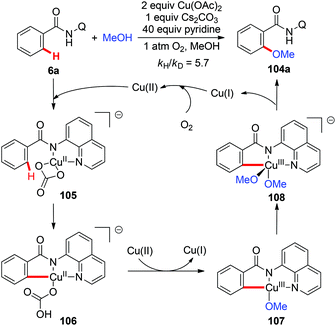 |
| | Scheme 41 Cu(OAc)2-mediated AQ-directed methoxylation and mechanistic studies. | |
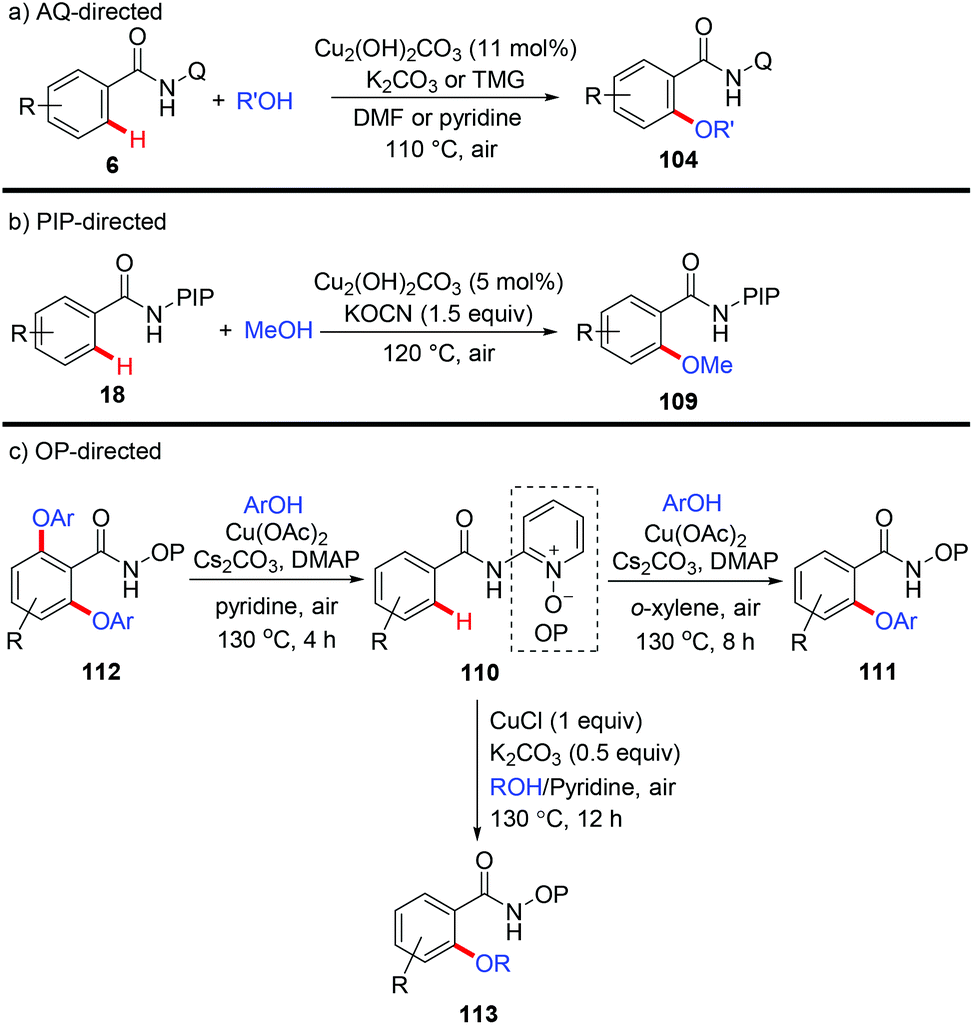
The Daugulis group reported the etherification of benzamides directed by 8-aminoquinoline with Cu2(OH)2CO3 as the catalyst and air as the oxidant (Scheme 42a).48 The aryloxylation could be achieved in the presence of K2CO3 and DMF; while C–H alkoxylation occurred when tetramethylguanidine (TMG) was used as a base and pyridine as a solvent. They found that PA is also an efficient directing group for this protocol. In 2015, a copper-catalyzed PIP-directed methoxylation of C(sp2)–H bonds with MeOH was achieved by our group (Scheme 42b).49 The reaction employed Cu2(OH)2CO3 as the catalyst, KOCN as the base and air as the oxidant. Selective mono-methoxylation of both aromatic and heteroaromatic amides has been accomplished under the optimized conditions.
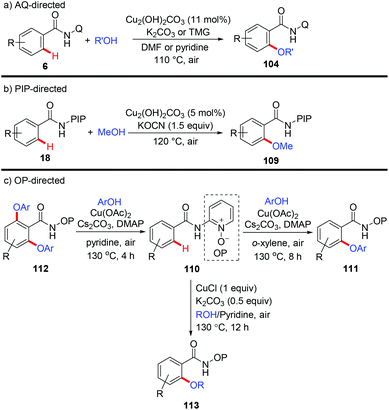 |
| | Scheme 42 Copper-catalyzed directed etherification of C(sp2)–H bonds. | |
Recently, Song et al. reported a copper-mediated mono- and diaryloxylation of benzamides using a N,O-bidentate auxiliary OP (Scheme 42c).50a Selective mono- or diaryloxylation could be tuned by simply switching the amounts of ingredients and solvents. Next, they employed the identical auxiliary for CuCl-mediated C–H alkoxylation of (hetero)arenes with alcohols (Scheme 42c).50b The use of pyridine substantially improved the reaction efficiency to give a superior result. Notably, besides primary alcohols, secondary alcohols including isopropanol and HFIP could also serve as coupling partners.
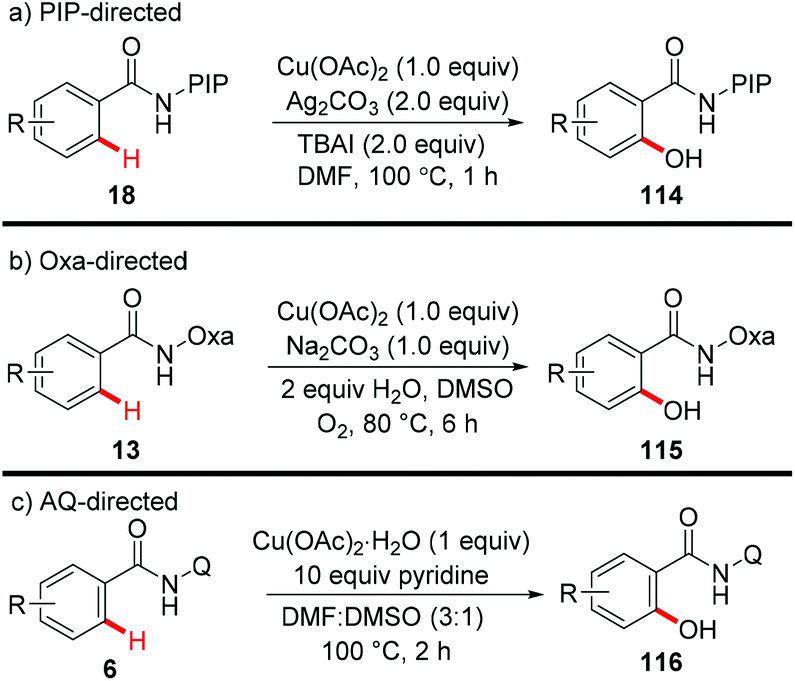
In 2014, our group reported the efficient synthesis of phenol derivatives via copper-mediated PIP-directed hydroxylation of (hetero)arenes (Scheme 43a).51 The method employed Ag2CO3 as the oxidant and tetrabutylammonium iodide (TBAI) as the additive. Notably, the procedure could also proceed smoothly at the gram scale and was compatible with a wide range of functional groups. Further mechanistic studies showed that a basic ligand-enabled, irreversible, rate-determining concerted-metalation–deprotonation (CMD) C–H cleavage was likely involved in the process. Later on, an aerobic copper-mediated hydroxylation of C(sp2)–H bonds with the assistance of the amide-oxazoline directing group was reported by Yu and Dai (Scheme 43b).52 Dioxygen was used as the sole oxidant and water was found to remarkably facilitate the reaction. Significant KIEs and the results of the reaction with TEMPO as the radical scavenger indicated that a copper-mediated C–H cleavage step rather than an SEAr or a radical pathway was involved in the process. More recently, Jana reported the hydroxylation of benzamides with copper(II) acetate monohydrate and a pyridine ligand directed by AQ (Scheme 43c).7c
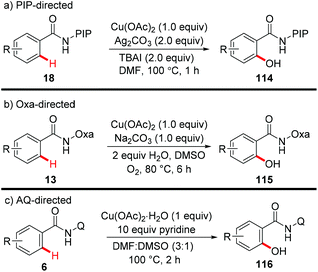 |
| | Scheme 43 Copper-mediated directed hydroxylation of C(sp2)–H bonds. | |
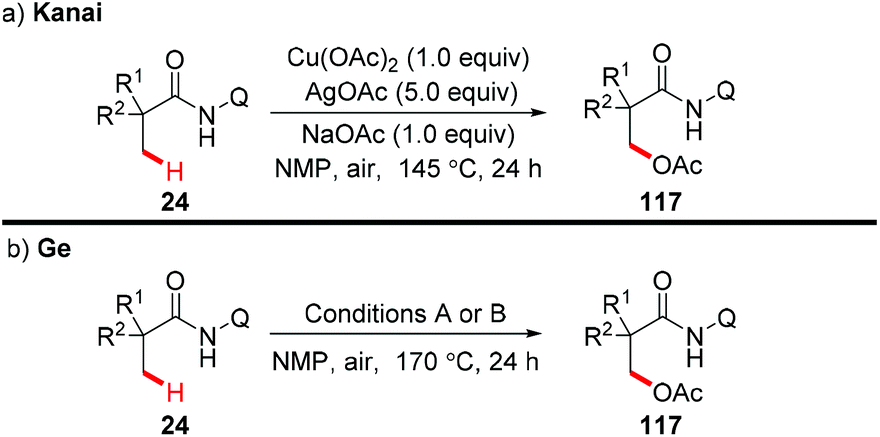
In 2014, the Kanai group and the Ge group independently reported for the first time that direct acetoxylation of methyl C(sp3)–H bonds of aliphatic amides was enabled by the AQ directing group in the presence of copper salt as the catalyst and silver salt as the oxidant (Scheme 44).53
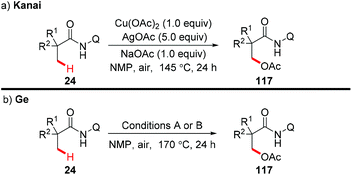 |
| | Scheme 44 Copper-mediated AQ-directed acetoxylation of methyl C(sp3)–H bonds. Conditions A: Cu(OAc)2 (50 mol%), AgOAc (3.0 equiv.), KH2PO4 (1.5 equiv.). Conditions B: CuI (40 mol%), Ag2O (4.0 equiv.), KH2PO4 (2.0 equiv.), HOAc. | |
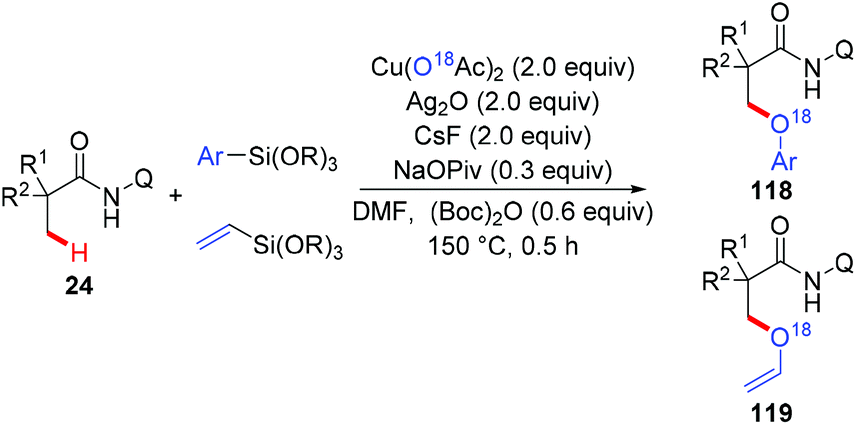
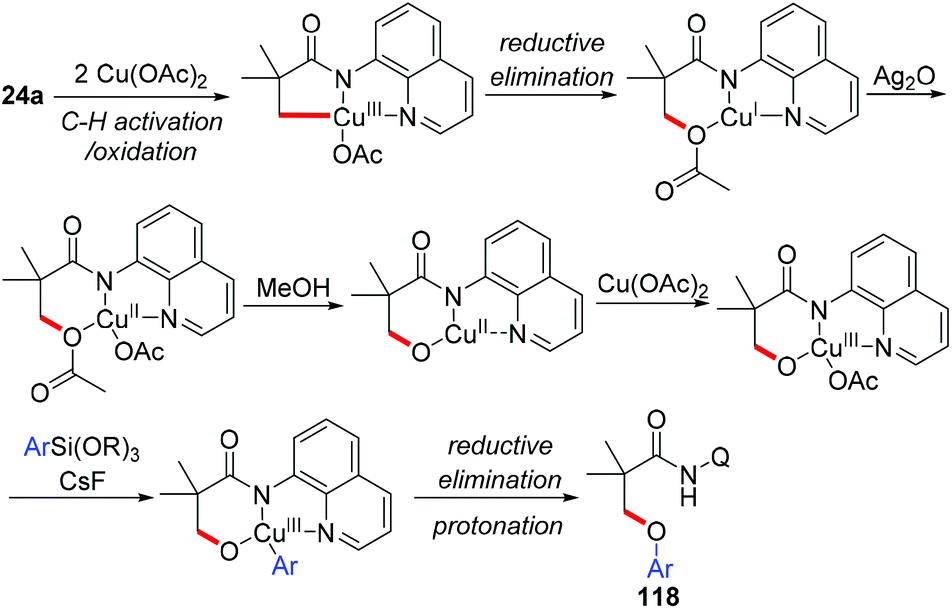
The Zhang group achieved the copper-mediated etherification of methyl C–H bonds of aliphatic amides with organosilanes employing the same AQ directing group (Scheme 45).54 The reaction proceeded in a tandem way of the first oxidation of β-C(sp3)–H bonds and subsequent arylation/vinylation to give the etherification products 118 or 119. Isotope-labeling experiments revealed that Cu(OAc)2 served as the source of oxygen in the aryloxylation products. A plausible mechanism was proposed as shown in Scheme 46.
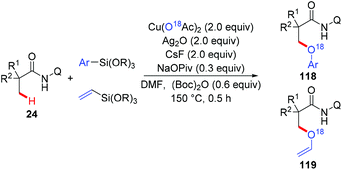 |
| | Scheme 45 Copper-mediated aryloxylation and vinyloxylation of methyl C(sp3)–H bonds with organosilanes. | |
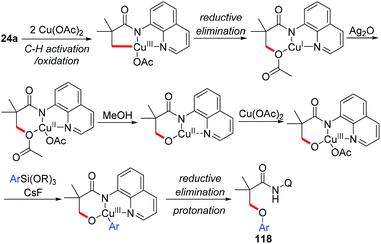 |
| | Scheme 46 Proposed mechanism. | |
Recently, the Baran group reported a scalable, high yielding, and site-specific oxidation of the steroidal C12-position with copper and molecular oxygen by the improvement of Schönecker's oxidation protocol.55a–d This protocol has been previously applied in the total synthesis of cyclopamine55e and (+)-cephalostatin 1.55f These reactions proceed through an inner-sphere way, which was mechanistically different from the topic of this review.
4.2 Intramolecular carbon–oxygen bond formation
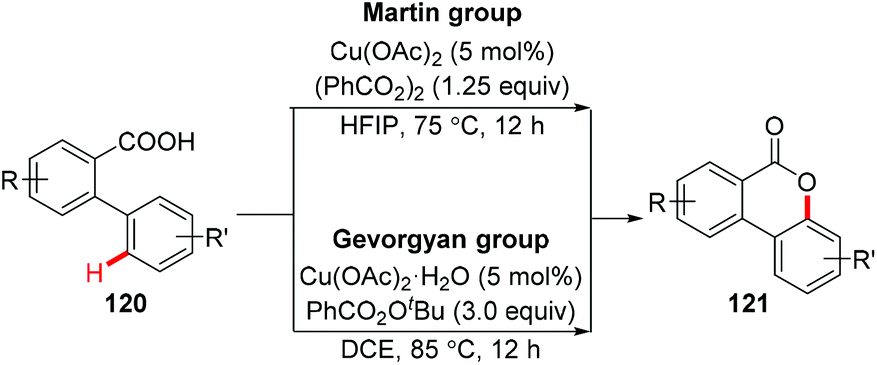
In 2008, Nagasawa et al. reported a copper-catalyzed intramolecular oxidative C–O coupling of benzanilides for the synthesis of 2-arylbenzoxazoles.56 In 2012, Zhu et al. reported the synthesis of dibenzofurans via Cu-catalyzed oxidative C(sp2)–H cycloetherification of o-arylphenols.57 However, a nitro group at the para- or meta-position was necessary to the successful cycloetherification. In 2013, Martin and Gevorgyan independently reported the copper-catalyzed intramolecular esterification of C(sp2)–H bonds (Scheme 47).58
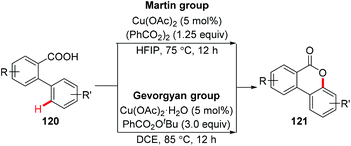 |
| | Scheme 47 Copper-catalyzed intramolecular esterification of C(sp2)–H bonds. | |
5 Carbon–sulfur bond formation
Since Yu reported the first example of copper-mediated direct thioetherification of 2-phenylpyridine,5a several methods of C–S bond formation based on the strategy of copper-mediated auxiliary-directed C–H functionalization have been developed in the following years. In 2010, the Qing group reported that widely available DMSO was able to work as a methylthiolation reagent for copper-mediated methylthiolation of 2-arylpyridines (Scheme 48).59 The presence of excess CuF2 was found to be essential to the successful transformation based on their consideration that the organic sulfur compound could deactivate copper salt through the binding effect.
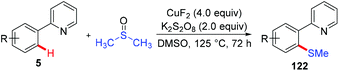 |
| | Scheme 48 Copper-mediated methylthiolation of 2-arylpyridines with DMSO. | |
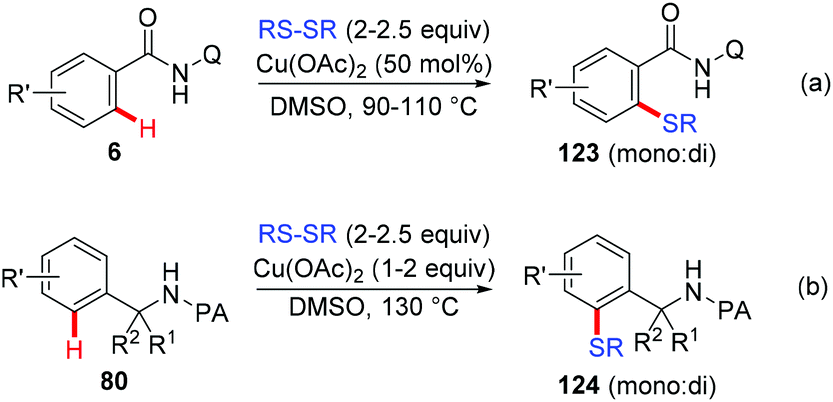
In 2012, the Daugulis group reported the copper-mediated AQ-directed sulfenylation of C(sp2)–H bonds (Scheme 49a).60 Disulfide compounds acted as both an effective thiolating reagent and oxidant. DMSO was selected as the solvent based on the consideration that it was able to in situ regenerate RSSR by oxidizing the resulting RSH. Both electron-rich and electron-deficient benzamides were compatible with this protocol, providing the thiolated products in good yields. Nevertheless, the direct sulfenylation of benzylamines bearing the PA directing group could also be accomplished under almost identical conditions (Scheme 49b).
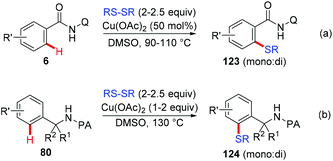 |
| | Scheme 49 Copper-mediated sulfenylation of C(sp2)–H bonds with disulfides. | |
Liang et al. described a copper-catalyzed AQ-directed thiolation of benzamides with aryl and aliphatic thiols using Ag2CO3 as the oxidant (Scheme 50).61 The protocol exhibited broad substrate generality and primary, secondary and tertiary thiols all could serve as thiolation reagents.
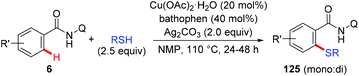 |
| | Scheme 50 Copper-catalyzed thiolation of C(sp2)–H bonds with aryl and aliphatic thiols. | |
The above mentioned methods still suffer from shortages of the use of odorous sulfur sources and the formation of undesired toxic byproducts. As such, identifying a useful and operationally convenient sulfur source to increase the practicality of this class of reactions was highly appealing. The use of elemental sulfur (S8) as a low-cost and non-odorous sulfur source in copper-mediated sulfenylation of C(sp2)–H bonds has been recently realized by our group (Scheme 51).62 TBAI as the additive was found to be crucial for the success of this transformation. Moreover, the reaction was scalable, and tolerated a variety of functional groups and even several heteroarenes to afford various benzoisothiazolones in moderate to excellent yields. Additionally, the synthetic transformation of the resulting benzoisothiazolones has been performed.
 |
| | Scheme 51 Copper-mediated C–S/N–S bond formation with elemental sulfur. | |
The copper-mediated direct sulfonylation of C(sp2)–H bonds with aryl sulfinate salts was developed independently by Tan and our group.63 As described by Tan et al., the reaction required stoichiometric Cu(OAc)2 and proceeded under air with the assistance of 8-aminoquinoline (Scheme 52).63a In contrast, we employed our newly developed PIP group as the auxiliary, and the sulfonylation of C–H bonds could take place in the presence of a catalytic amount of Cu(OAc)2. However, Ag2CO3 as the terminal oxidant was necessary in the transformation (Scheme 53).63b Both the methods specifically furnished mono-sulfonylated products, which was quite different from previous copper-mediated auxiliary-directed C–H thiolation reactions.
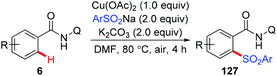 |
| | Scheme 52 Copper-mediated AQ-directed sulfonylation of C(sp2)–H bonds. | |
 |
| | Scheme 53 Copper-catalyzed PIP-directed sulfonylation of C(sp2)–H bonds. | |
6 Carbon–halogen bond formation
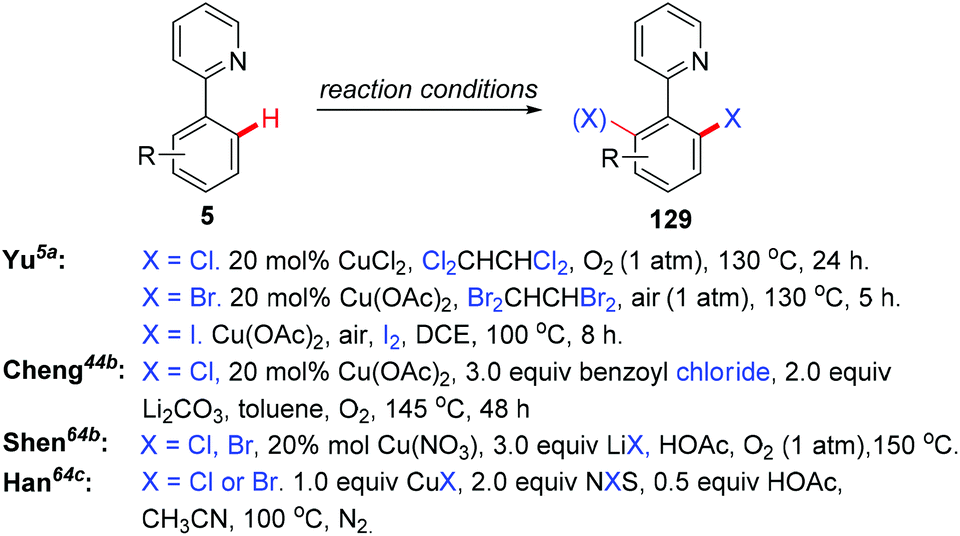
In the seminal study by Yu and co-workers, they found that the reaction of 2-arylpyridines 5 with catalytic copper acetate (20 mol%) in Cl2CHCHCl2 gave the corresponding chlorinated products 129 in good yields (Scheme 54). Analysis of the reaction mixture using 1H NMR and pH measurements revealed that the Cl− anion was provided by Cl2CHCHCl2. Bromination or iodination occurred when Br2CHCHBr2 or iodide was used as the halogen source, respectively. Inspired by this work, various halogen sources, such as benzoyl chloride, lithium halides and NXS, have been used by Cheng,44b Shen,64a,b and Han,64c respectively. In 2011, the copper-catalyzed ortho-chlorination of 2-arylpyridines with an acyl chloride was described by Cheng's group (Scheme 54).44b Shen et al. discovered that cheap and easy-to-handle LiX (X = Cl, Br) could serve as the halogen sources for copper-catalyzed ortho-halogenation of 2-phenylpyridines (Scheme 54).64a,b However, toxic CrO3 was used as the terminal oxidant. In 2014, Han developed a mild protocol for copper-mediated highly selective mono- and di-halogenation of 2-arylpyridines (Scheme 54).64c Acyl hypohalites, generated in situ from the readily available carboxylic acid and N-halosuccinimides (NXS; X = Br, Cl), were crucial for the good selectivity.
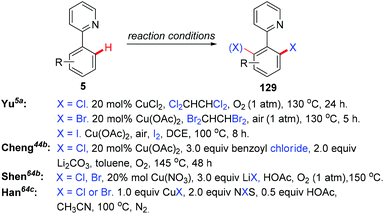 |
| | Scheme 54 Copper-mediated halogenation of 2-arylpyridines with various halogen sources. | |
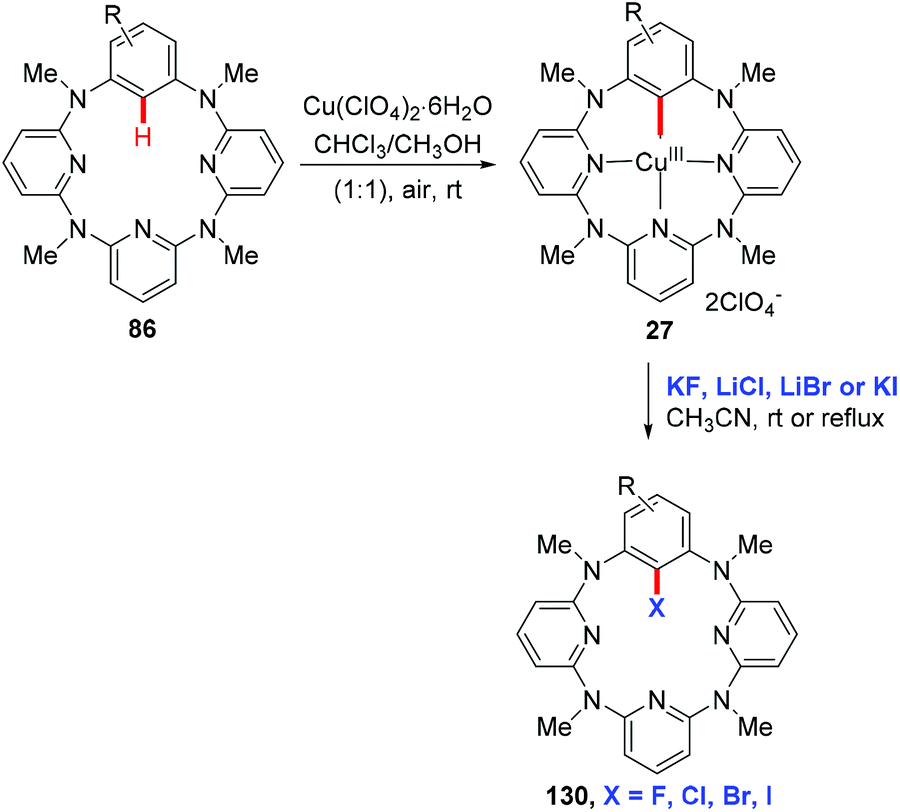
In 2012, Wang and co-workers reported the regioselective halogenation of azacalix[1]arene[3]pyridines at the lower rim position of the benzene ring with alkali metal halides as the halogen source. The reaction proceeded through the copper(II)-mediated aryl C–H activation, which gave structurally well-defined aryl-Cu(III) intermediates, followed by a subsequent carbon–halogen reductive elimination (Scheme 55).65 It is worth noting that the cost-effective and operation friendly potassium fluoride is used as the fluorine source, providing an effective method to access fluoroarenes.
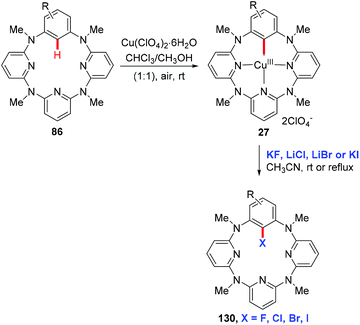 |
| | Scheme 55 Copper-mediated halogenation of azacalix[1]arene[3]pyridines. | |
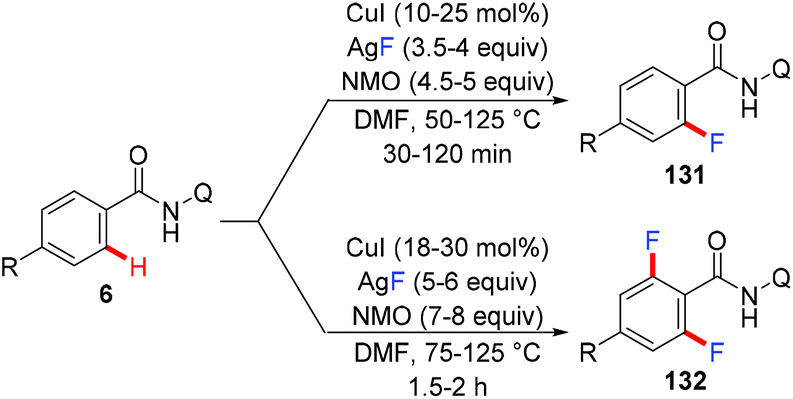
In 2013, the Daugulis group reported the copper-catalyzed fluorination of C(sp2)–H bonds directed by AQ and PA directing groups (Scheme 56).66 The reaction employed CuI as the catalyst and AgF as the nucleophilic fluoride source. Selective mono- or difluorination of benzamide substrates could be achieved by tuning the reaction temperature and the amounts of CuI catalyst, oxidant, and AgF.
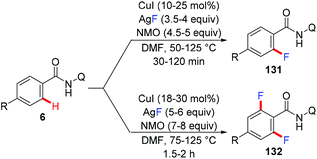 |
| | Scheme 56 Copper-catalyzed fluorination of C(sp2)–H bonds. | |
An easily removable N-(2-pyridyl)sulfonyl auxiliary for the selective halogenation of C(sp2)–H bonds of anilines under copper catalysis was developed by Carretero's group (Scheme 57).67ortho-Mono-halogenation of anilines could be achieved with the CuX2–NXS (X = Cl, Br) system. The one-step removal of the 2-pyridylsulfonyl auxiliary under mild reductive conditions afforded the free primary amine in high yield.
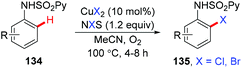 |
| | Scheme 57 Copper-catalyzed halogenation of protected anilines with NXS. | |
In 2015, the ortho-halogenation of benzamides with NXS (X = Cl, Br, I) under copper catalysis was developed by our group using the removable PIP group (Scheme 58).68 A wide range of functional groups could be tolerated. The PIP auxiliary could be easily removed to give ortho-halogenated benzoic acids.
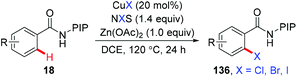 |
| | Scheme 58 Copper-catalyzed PIP-directed halogenation of C(sp2)–H bonds with NXS. | |
7 Carbon–phosphorus bond formation
The ability of 8-aminoquinoline to enable phosphorylation of benzamides under copper catalysis was demonstrated by Yu and co-workers in 2014 (Scheme 59).69 They showed that both aromatic and heteroaromatic benzamides were suitable substrates for phosphorylation with H-phosphonates. It is noteworthy that the protocol exclusively delivered the mono-phosphorylated products.
 |
| | Scheme 59 Copper-catalyzed AQ-directed phosphorylation of C(sp2)–H bonds. | |
8 Conclusions
In this review, we have summarized recent advances in copper-mediated chelation-assisted functionalization of unactivated C–H bonds. Several N-based monodentate groups and amide-based bidentate groups70 have been developed to serve as effective directing groups for copper-mediated functionalization of C–H bonds and could remarkably facilitate a variety of transformations, including the formation of carbon–carbon, carbon–nitrogen, carbon–oxygen, carbon–sulfur, carbon–halogen, and carbon–phosphorus. From a synthetic point of view, these reactions provide novel and efficient tools for constructing various synthetically useful compounds. However, the detailed mechanism of copper-mediated auxiliary-directed functionalization of C–H bonds remains to be elucidated, and the functionalization of C(sp3)–H bonds remains underdeveloped. As such, more efforts will continue to be devoted to the development of this area.
Acknowledgements
Financial support from the National Basic Research Program of China (2015CB856600), the NSFC (21572201, 21422206, and 21272206) and the Fundamental Research Funds for the Central Universities (2015XZZX004-03) is gratefully acknowledged.
Notes and references
-
(a) F. Ullmann and J. Bielecki, Ber. Dtsch. Chem. Ges., 1901, 34, 2174 CrossRef CAS;
(b) F. Ullmann, Ber., 1903, 36, 2382 CrossRef;
(c) F. Ullmann, Ber., 1904, 37, 853 CrossRef;
(d) F. Ullmann and P. Sponagel, Ber., 1905, 38, 2211 CrossRef CAS;
(e) I. Goldberg, Ber. Dtsch. Chem. Ges., 1906, 39, 1691 CrossRef.
- For selected reviews on copper-mediated cross-coupling reactions, see:
(a) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed;
(b) D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450 CrossRef CAS PubMed;
(c) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed;
(d) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed;
(e) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13 RSC.
- For selected reviews on transition metal-catalyzed direct functionalization of C–H bonds, see:
(a) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468 CrossRef CAS PubMed;
(b) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC;
(c) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC;
(d) R. K. Rit, R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450 CrossRef CAS;
(e) B. Liu, F. Hu and B.-F. Shi, ACS Catal., 2015, 5, 1864 Search PubMed;
(f) X.-F. Wu, Chem. – Eur. J., 2015, 21, 12252 CrossRef CAS PubMed;
(g) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843 RSC;
(h) C. Zhao, M. R. Crimmin, F. D. Toste and R. G. Bergman, Acc. Chem. Res., 2014, 47, 517 CrossRef CAS PubMed;
(i) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed;
(j) L. C. M. Castro and N. Chatani, Chem. Lett., 2015, 44, 410 CrossRef CAS;
(k) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed;
(l) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed;
(m) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed;
(n) D. Zhao, J. You and C. Hu, Chem. – Eur. J., 2011, 17, 5466 CrossRef CAS PubMed;
(o) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed;
(p) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed;
(q) B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949 CAS;
(r) O. Daugulis, V. G. Zaitsev, D. Shabashov, Q.-N. Pham and A. Lazareva, Synlett, 2006, 15, 3382 CrossRef.
- For selected reviews, see:
(a) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed;
(b) Y. Liang, Y.-F. Liang and N. Jiao, Org. Chem. Front., 2015, 2, 403 RSC;
(c) J. Miao and H. Ge, Eur. J. Org. Chem., 2015, 7859 CrossRef CAS;
(d) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed;
(e) K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704 RSC;
(f) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC;
(g) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC;
(h) T. Liu and Q. Shen, Eur. J. Org. Chem., 2012, 6679 CrossRef CAS;
(i) K. Hirano and M. Miura, Synlett, 2011, 294 CAS;
(j) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed;
(k) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed.
-
(a) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed;
(b) T. Uemura, S. Imoto and N. Chatani, Chem. Lett., 2006, 35, 842 CrossRef CAS.
-
(a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed;
(b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed. For representative reviews, see:
(c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed;
(d) M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896 CrossRef CAS PubMed;
(e) B. Zhang, H.-X. Guan, B. Liu and B.-F. Shi, Chin. J. Org. Chem., 2014, 34, 1487 CrossRef CAS;
(f) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1063 CrossRef PubMed;
(g) M. R. Yadav, R. K. Rit, M. Shankar and A. K. Sahoo, Asian J. Org. Chem., 2015, 4, 846 CrossRef CAS;
(h) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635 CrossRef CAS PubMed.
-
(a) X. Chen, G. Dobereiner, X.-S. Hao, R. Giri, N. Maugel and J.-Q. Yu, Tetrahedron, 2009, 65, 3085 CrossRef CAS;
(b) J. Le, Y. Gao, Y. Ding and C. Jiang, Tetrahedron Lett., 2016, 57, 1728 CrossRef CAS;
(c) B. K. Singh and R. Jana, J. Org. Chem., 2016, 81, 831 CrossRef CAS PubMed.
-
(a) M. Kitahara, N. Umeda, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2011, 133, 2160 CrossRef CAS PubMed;
(b) M. Nishino, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 4457 CrossRef CAS PubMed;
(c) R. Odani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2013, 78, 11045 CrossRef CAS PubMed;
(d) R. Odani, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2014, 53, 10784 CrossRef CAS PubMed.
-
(a) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, Org. Lett., 2014, 16, 5666 CrossRef CAS PubMed;
(b) Q. Gui, X. Chen, L. Hu, D. Wang, J. Liu and Z. Tan, Adv. Synth. Catal., 2016, 358, 4 CrossRef.
-
(a) F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187 RSC;
(b) Q. Zhang, K. Chen, W.-H. Rao, Y.-J. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588 CrossRef CAS PubMed.
-
(a) S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 4069 RSC;
(b) Y.-J. Liu, Y.-H. Liu, S.-Y. Yan and B.-F. Shi, Chem. Commun., 2015, 51, 6388 RSC;
(c) S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu, Z.-Z. Zhang and B.-F. Shi, Chem. Commun., 2015, 51, 7341 RSC;
(d) Y.-J. Liu, Z.-Z. Zhang, S.-Y. Yan, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 7899 RSC;
(e) Y.-H. Liu, Y.-J. Liu, S.-Y. Yan and B.-F. Shi, Chem. Commun., 2015, 51, 11650l RSC;
(f) B.-B. Zhan, Y.-H. Liu, F. Hu and B.-F. Shi, Chem. Commun., 2016, 52, 4934 RSC.
-
(a) S. Zhao, Y.-J. Liu, S.-Y. Yan, F.-J. Chen, Z.-Z. Zhang and B.-F. Shi, Org. Lett., 2015, 17, 3338 CrossRef CAS PubMed;
(b) S. Zhao, J. Yuan, Y.-C. Li and B.-F. Shi, Chem. Commun., 2015, 51, 12823 RSC.
- X. Wu, Y. Zhao and H. Ge, Chem. Sci., 2015, 6, 5978 RSC.
-
(a)
F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology and Material Science, Wiley-VCH, Weinheim, 2005 Search PubMed;
(b) S. Toyota, Chem. Rev., 2010, 110, 5398 CrossRef CAS PubMed.
-
(a) Z. Shao and F. Peng, Angew. Chem., Int. Ed., 2010, 49, 9566 CrossRef CAS PubMed;
(b) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2096 CrossRef CAS PubMed;
(c) S. Messaoudi, J.-D. Brion and M. Alami, Eur. J. Org. Chem., 2010, 6495 CrossRef CAS.
-
(a) Z.-L. Wang, L. Zhao and M.-X. Wang, Org. Lett., 2012, 14, 1472 CrossRef CAS PubMed;
(b) B. Yao;, D.-X. Wang, Z.-T. Huang and M.-X. Wang, Chem. Commun., 2009, 2899 RSC;
(c) L. M. Hufman and S. Stahl, J. Am. Chem. Soc., 2008, 130, 9196 CrossRef PubMed;
(d) A. E. King, L. M. Huffman, A. Casitas, M. Costas, X. Ribas and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 12068 CrossRef CAS PubMed;
(e) Z.-L. Wang, L. Zhao and M.-X. Wang, Org. Lett., 2011, 13, 6560 CrossRef CAS PubMed;
(f) L. M. Huffman, A. Casitas, M. Font, M. Canta, M. Costas, X. Ribas and S. S. Stahl, Chem. – Eur. J., 2011, 17, 10643 CrossRef CAS PubMed;
(g) A. Casitas and X. Ribas, Chem. Sci., 2013, 4, 2301 RSC;
(h) H. Zhang, B. Yao, L. Zhao, D.-X. Wang, B.-Q. Xu and M.-X. Wang, J. Am. Chem. Soc., 2014, 136, 6326 CrossRef CAS PubMed.
-
(a) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590 CrossRef CAS PubMed;
(b) Y.-J. Liu, Y.-H. Liu, X.-S. Yin, W.-J. Gu and B.-F. Shi, Chem. – Eur. J., 2015, 21, 205 CrossRef CAS PubMed.
- M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 10439 CrossRef CAS PubMed.
-
(a) J. Dong, F. Wang and J. You, Org. Lett., 2014, 16, 2884 CrossRef CAS PubMed;
(b) Y. Zhang, Q. Wang, H. Yu and Y. Huang, Org. Biomol. Chem., 2014, 12, 8844 RSC.
- W. Zhu, D. Zhang, N. Yang and H. Liu, Chem. Commun., 2014, 50, 10634 RSC.
- H.-L. Wang, M. Shang, S.-Z. Sun, Z.-L. Zhou, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Org. Lett., 2015, 17, 1228 CrossRef CAS PubMed.
-
(a) W. Miura, K. Hirano and M. Miura, Org. Lett., 2015, 17, 4034 CrossRef CAS PubMed;
(b) K. Takamatsu, K. Hirano and M. Miura, Org. Lett., 2015, 17, 4066 CrossRef CAS PubMed.
- J. Zhang, D. Li, H. Chen, B. Wang, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2016, 358, 792 CrossRef CAS.
- Q. Wen, J. Jin, L. Zhang, Y. Luo, P. Lu and Y. Wang, Tetrahedron Lett., 2014, 55, 1271 CrossRef CAS.
-
(a) J. Jin, Q. Wen, P. Lu and Y. Wang, Chem. Commun., 2012, 48, 9933 RSC;
(b) J. Kim, H. Kim and S. Chang, Org. Lett., 2012, 14, 3924 CrossRef CAS PubMed.
-
(a) J. Kim and S. Chang, J. Am. Chem. Soc., 2010, 132, 10272 CrossRef CAS PubMed;
(b) J. Kim, J. Choi, K. Shin and S. Chang, J. Am. Chem. Soc., 2012, 134, 2528 CrossRef CAS PubMed;
(c) G. Zhang, X. Ren, J. Chen, M. Hu and J. Cheng, Org. Lett., 2011, 13, 5004 CrossRef CAS PubMed;
(d) X. Ren, J. Chen, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 6725 RSC. For reviews, see:
(e) J. Kim, H. J. Kim and S. Chang, Eur. J. Org. Chem., 2013, 3201 CrossRef CAS;
(f) J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948 CrossRef CAS PubMed.
-
(a) S. Ding and N. Jiao, J. Am. Chem. Soc., 2011, 133, 12374 CrossRef CAS PubMed;
(b) Y. Yan, Y. Yuan and N. Jiao, Org. Chem. Front., 2014, 1, 1176 RSC;
(c) H. Xu, P.-T. Liu, Y.-H. Li and F.-S. Han, Org. Lett., 2013, 15, 3354 CrossRef CAS PubMed;
(d) X. Kou, M. Zhao, X. Qiao, Y. Zhu, X. Tong and Z. Shen, Chem. – Eur. J., 2013, 19, 16880 CrossRef CAS PubMed;
(e) C. Pan, H. Jin, P. Xu, X. Liu, Y. Cheng and C. Zhu, J. Org. Chem., 2013, 78, 9494 CrossRef CAS PubMed.
-
(a) A. John and K. M. Nicholas, J. Org. Chem., 2011, 76, 4158 CrossRef CAS PubMed;
(b) Q. Shuai, G. Deng, Z. Chua, D. S. Bohle and C.-J. Li, Adv. Synth. Catal., 2010, 352, 632 CrossRef CAS;
(c) H. Xu, X. Qiao, S. Yang and Z. Shen, J. Org. Chem., 2014, 79, 4414 CrossRef CAS PubMed;
(d) L. Wang, D. L. Priebbenow, W. Dong and C. Bolm, Org. Lett., 2014, 16, 2661 CrossRef CAS PubMed;
(e) G. Li, C. Jia, Q. Chen, K. Sun, F. Zhao, H. Wu, Z. Wang, Y. Lv and X. Chen, Adv. Synth. Catal., 2015, 357, 1311 CrossRef CAS.
-
(a) L. D. Tran, J. Roane and O. Daugulis, Angew. Chem., Int. Ed., 2013, 52, 6043 CrossRef CAS PubMed;
(b) J. Roane and O. Daugulis, J. Am. Chem. Soc., 2016, 138, 4601 CrossRef CAS PubMed.
-
(a) Q. Li, S.-Y. Zhang, G. He, Z. Ai, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 1764 CrossRef CAS PubMed;
(b) A. M. Martinez, N. Rodriguez, R. Gomez Arrayas and J. C. Carretero, Chem. Commun., 2014, 50, 2801 RSC.
-
(a) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354 CrossRef CAS PubMed;
(b) P. Sadhu and T. Punniyamurthy, Chem. Commun., 2016, 52, 2803 RSC.
-
(a) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040 CrossRef CAS PubMed;
(b) X. Huang and J. T. Groves, ACS Catal., 2016, 6, 75 Search PubMed.
-
(a) J. Peng, M. Chen, Z. Xie, S. Luo and Q. Zhu, Org. Chem. Front., 2014, 1, 777 RSC;
(b) J. Peng, Z. Xie, M. Chen, J. Wang and Q. Zhu, Org. Lett., 2014, 16, 4702 CrossRef CAS PubMed;
(c) Z. Xie, J. Peng and Q. Zhu, Org. Chem. Front., 2016, 3, 82 RSC.
-
(a) G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932 CrossRef CAS PubMed;
(b) H. Wang, Y. Wang, C. Peng, J. Zhang and Q. Zhu, J. Am. Chem. Soc., 2010, 132, 13217 CrossRef CAS PubMed;
(c) Y. Lv, Y. Li, T. Xiong, W. Pu, H. Zhang, K. Sun, Q. Liu and Q. Zhang, Chem. Commun., 2013, 49, 6439 RSC;
(d) X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636 CrossRef CAS PubMed;
(e) Q. Gui, Z. Yang, X. Chen, J. Liu, Z. Tan, R. Guo and W. Yu, Synlett, 2013, 24, 1016 CrossRef CAS;
(f) W. Zhou, Y. Liu, Y. Yang and G. J. Deng, Chem. Commun., 2012, 48, 10678 RSC;
(g) P.-C. Huang, K. Parthasarathy and C.-H. Cheng, Chem. – Eur. J., 2013, 19, 460 CrossRef CAS PubMed;
(h) J. Huang, C. Wan, M.-F. Xu and Q. Zhu, Eur. J. Org. Chem., 2013, 1876 CrossRef CAS;
(i) S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996 CrossRef CAS PubMed;
(j) Y. Ou and N. Jiao, Chem. Commun., 2013, 49, 3473 RSC. For a recent review, see:
(k) P. Subramanian, G. C. Rudolf and K. P. Kaliappan, Chem. – Asian J., 2016, 11, 168 CrossRef CAS PubMed.
-
(a) Z. Wang, J. Ni, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 3496 CrossRef CAS PubMed;
(b) X. Wu, Y. Zhao, G. Zhang and H. Ge, Angew. Chem., Int. Ed., 2014, 53, 3706 CrossRef CAS PubMed;
(c) C. Wang, Y. Yang, D. Qin, Z. He and J. You, J. Org. Chem., 2015, 80, 8424 CrossRef CAS PubMed.
- For a review, see: Q. Zhang, K. Chen and B.-F. Shi, Synlett, 2014, 25, 1941 CrossRef CAS.
-
(a) K. Takamatsu, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 2892 CrossRef CAS PubMed;
(b) K. Takamatsu, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2015, 80, 3242 CrossRef CAS PubMed.
-
(a) S. Brase, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef CAS PubMed;
(b) H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
-
(a) C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924 CrossRef CAS PubMed;
(b) Y. Fan, W. Wan, G. Ma, W. Gao, H. Jiang, S. Zhu and J. Hao, Chem. Commun., 2014, 50, 5733 RSC;
(c) C. S. Azad and A. K. Narula, RSC Adv., 2015, 5, 100223 RSC.
- B. Yao, Y. Liu, L. Zhao, D.-X. Wang and M.-X. Wang, J. Org. Chem., 2014, 79, 11139 CrossRef CAS PubMed.
- L. Zhang, Z. Liu, H. Li, G. Fang, B.-D. Barry, T. A. Belay, X. Bi and Q. Liu, Org. Lett., 2011, 13, 6536 CrossRef CAS PubMed.
-
(a) D. Katayev, K. F. Pfister, T. Wendling and L. J. Gooßen, Chem. – Eur. J., 2014, 20, 9902 CrossRef CAS PubMed;
(b) J. Liu, S. Zhuang, Q. Gui, X. Chen, Z. Yang and Z. Tan, Adv. Synth. Catal., 2015, 357, 732 CrossRef CAS.
- T. Mizuhara, S. Inuki, S. Oishi, N. Fujii and H. Ohno, Chem. Commun., 2009, 3413 RSC.
-
(a) W. Wang, F. Luo, S. Zhang and J. Cheng, J. Org. Chem., 2010, 75, 2415 CrossRef CAS PubMed;
(b) W. Wang, C. Pan, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 3978 RSC;
(c) S. Zhao, F.-J. Chen, B. Liu and B.-F. Shi, Sci. China: Chem., 2015, 58, 1302 CrossRef CAS.
- S. Bhadra, W. I. Dzik and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 2959 CrossRef CAS PubMed.
- S. Bhadra, C. Matheis, D. Katayev and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 9279 CrossRef CAS PubMed.
- A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed.
- J. Roane and O. Daugulis, Org. Lett., 2013, 15, 5842 CrossRef CAS PubMed.
- X.-S. Yin, Y.-C. Li, J. Yuan, W.-J. Gu and B.-F. Shi, Org. Chem. Front., 2015, 2, 119 RSC.
-
(a) X.-Q. Hao, L.-J. Chen, B. Ren, L.-Y. Li, X.-Y. Yang, J.-F. Gong, J.-L. Niu and M.-P. Song, Org. Lett., 2014, 16, 1104 CrossRef CAS PubMed;
(b) L.-B. Zhang, X.-Q. Hao, S.-K. Zhang, K. Liu, B. Ren, J.-F. Gong, J.-L. Niu and M.-P. Song, J. Org. Chem., 2014, 79, 10399 CrossRef CAS PubMed.
- X. Li, Y.-H. Liu, W.-J. Gu, B. Li, F.-J. Chen and B.-F. Shi, Org. Lett., 2014, 16, 3904 CrossRef CAS PubMed.
- S.-Z. Sun, M. Shang, H.-L. Wang, H.-X. Lin, H.-X. Dai and J.-Q. Yu, J. Org. Chem., 2015, 80, 8843 CrossRef CAS PubMed.
-
(a) Z. Wang, Y. Kuninobu and M. Kanai, Org. Lett., 2014, 16, 4790 CrossRef CAS PubMed;
(b) X. Wu, Y. Zhao and H. Ge, Chem. – Asian J., 2014, 9, 2736 CrossRef CAS PubMed.
- J. Zhang, H. Chen, B. Wang, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 2768 CrossRef CAS PubMed.
-
(a) Y. Y. See, A. T. Herrmann, Y. Aihara and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 13776 CrossRef CAS PubMed;
(b) B. Schönecker, T. Zheldakova, Y. Liu, M. Kötteritzsch, W. Günther and H. Görls, Angew. Chem., Int. Ed., 2003, 42, 3240 CrossRef PubMed;
(c) B. Schönecker, T. Zheldakova, C. Lange, W. Günther, H. Görls and M. Bohl, Chem. – Eur. J., 2004, 10, 6029 CrossRef PubMed;
(d) B. Schönecker and C. Lange, J. Organomet. Chem., 2006, 691, 2107 CrossRef;
(e) A. Giannis, P. Heretsch, V. Sarli and A. Stößel, Angew. Chem., Int. Ed., 2009, 48, 7911 CrossRef CAS PubMed;
(f) K. C. Fortner, D. Kato, Y. Tanaka and M. D. Shair, J. Am. Chem. Soc., 2010, 132, 275 CrossRef CAS PubMed.
- S. Ueda and H. Nagasawa, Angew. Chem., Int. Ed., 2008, 47, 6411 CrossRef CAS PubMed.
-
(a) J. Zhao, Y. Wang, Y. He, L. Liu and Q. Zhu, Org. Lett., 2012, 14, 1078 CrossRef CAS PubMed;
(b) J. Zhao, Q. Zhang, L. Liu, Y. He, J. Li, J. Li and Q. Zhu, Org. Lett., 2012, 14, 5362 CrossRef CAS PubMed.
-
(a) J. Gallardo-Donaire and R. Martin, J. Am. Chem. Soc., 2013, 135, 9350 CrossRef CAS PubMed;
(b) Y. Wang, A. V. Gulevich and V. Gevorgyan, Chem. – Eur. J., 2013, 19, 15836 CrossRef CAS PubMed.
- L. Chu, X. Yue and F.-L. Qing, Org. Lett., 2010, 12, 1644 CrossRef CAS PubMed.
- L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237 CrossRef CAS PubMed.
- X.-B. Yan, P. Gao, H.-B. Yang, Y.-X. Li, X.-Y. Liu and Y.-M. Liang, Tetrahedron, 2014, 70, 8730 CrossRef CAS.
- F.-J. Chen, G. Liao, X. Li, J. Wu and B.-F. Shi, Org. Lett., 2014, 16, 5644 CrossRef CAS PubMed.
-
(a) J. Liu, L. Yu, S. Zhuang, Q. Gui, X. Chen, W. Wang and Z. Tan, Chem. Commun., 2015, 51, 6418 RSC;
(b) W.-H. Rao and B.-F. Shi, Org. Lett., 2015, 17, 2784 CrossRef CAS PubMed.
-
(a) Y. Lu, R. Wang, X. Qiao and Z. Shen, Synlett, 2011, 1038 CAS;
(b) S. Mo, Y. Zhu and Z. Shen, Org. Biomol. Chem., 2013, 11, 2756 RSC;
(c) Z.-J. Du, L.-X. Gao, Y.-J. Lin and F.-S. Han, ChemCatChem, 2014, 6, 123 CrossRef CAS;
(d) Z. Du, L. Gao and Y. Lin, Chem. Res. Chin. Univ., 2015, 31, 197 Search PubMed.
- B. Yao, Z.-L. Wang, H. Zhang, D.-X. Wang, L. Zhao and M.-X. Wang, J. Org. Chem., 2012, 77, 3336 CrossRef CAS PubMed.
- T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342 CrossRef CAS PubMed.
- B. Urones, A. M. Martinez, N. Rodriguez, R. G. Arrayas and J. C. Carretero, Chem. Commun., 2013, 49, 11044 RSC.
- B. Li, B. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 5093 RSC.
- S. Wang, R. Guo, G. Wang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2014, 50, 12718 RSC.
- J. Liu, G. Chen and Z. Tan, Adv. Synth. Catal., 2016, 358, 1174 CrossRef CAS.
Footnote |
| † Current address: College of Chemistry and Chemical Engineering, Xinyang Normal University, Xinyang 464000, China. |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.