
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
“Click-fluors”: triazole-linked saccharide sensors†
Wenlei
Zhai
a,
Brette M.
Chapin
ab,
Akina
Yoshizawa
a,
Hui-Chen
Wang
a,
Stephen A.
Hodge
c,
Tony D.
James
 c,
Eric V.
Anslyn
b and
John S.
Fossey
*a
c,
Eric V.
Anslyn
b and
John S.
Fossey
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK
bDepartment of Chemistry and Biochemistry, The University of Texas at Austin, Austin, Texas 78712, USA
cDepartment of Chemistry, University of Bath, Bath, Claverton Down BA2 7AY, UK
First published on 14th June 2016
Abstract
A series of boronic acid-containing saccharide receptors was synthesised via copper catalysed azide–alkyne cycloaddition (CuAAC) reactions. Their saccharide binding capacity was studied by 1H and 11B NMR spectroscopy titrations and isothermal titration calorimetry (ITC) techniques. Fluorescent sensors were generated by linking a phenylboronic acid (PBA) receptor with fluorophores via a triazole-linker. Fluorescence titrations with fructose revealed that the substitution pattern about the PBA influences the fluorescence response to saccharides. Titrations studied by 1H NMR spectroscopy suggested that fructose binding is enhanced when the aromatic ring bearing the boronic acid has the triazole-containing substituent at the ortho position. No evidence of either a dative N–B bond or solvent insertion (between B and N) was observed by 11B NMR spectroscopy. These results demonstrate that synthetic accessible triazole receptors may allow rapid sensor synthesis, screening and discovery.
Introduction
Saccharides participate in some of the most essential chemical processes in life, particularly energy metabolism and cell recognition.1 Accurate detection of saccharides could have an impact on clinical diagnosis of several diseases. For example, certain types of glycoproteins are over-expressed on the surface of cancer cells.2 As a result, the development of sensing techniques to target these cancer biomarkers are in high demand.3In past decades progress in synthetic molecular probes (chemosensors) has shown significant promise.4 Synthetic boronic acid-based saccharide detection, which relies on the reversible formation of boronic esters through covalent bonding between diol motifs and boronic acids,5 is a well-established approach in the glyco-detection area.6 As illustrated in Scheme 1, a common strategy to assemble fluorescent molecular sensors is to connect a receptor and a fluorophore using a linker motif,7 numerous molecular chemosensors have been designed and synthesised following this strategy.8 However, chemosensor synthesis is often non-trivial, requiring multiple synthetic steps and challenging purifications.9 Strategies for rapid glyco-sensor assembly would be beneficial if chemo-diagnostics could provide new sensors at a relatively comparable rate to biomarker discovery,10 and therapeutic use.11
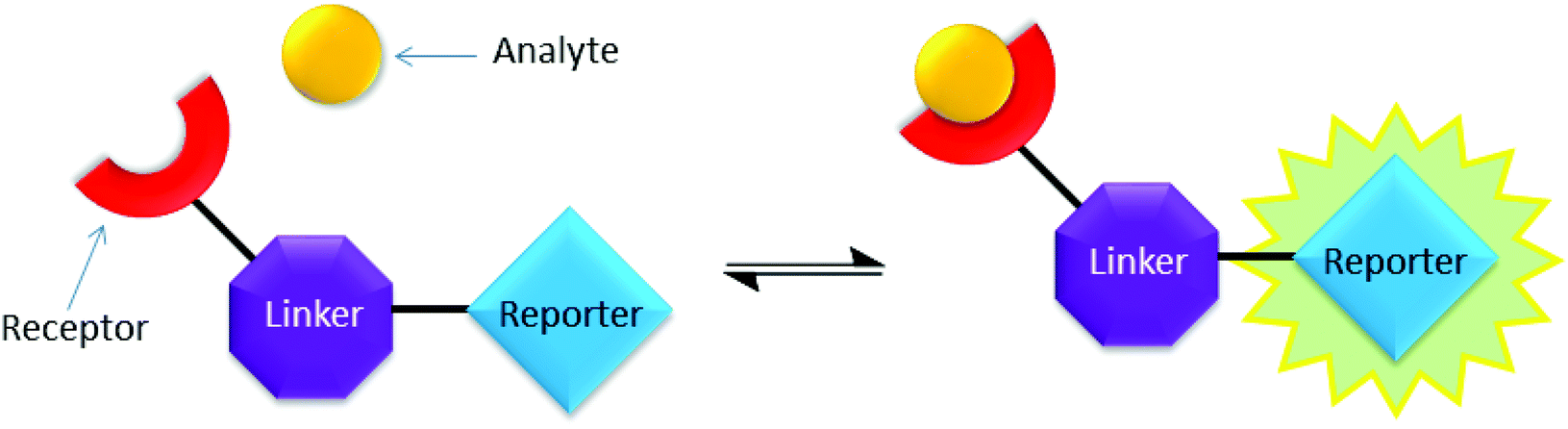 | ||
| Scheme 1 Schematic illustration of the process of sensing in a “three-component” chemosensor system.8b | ||
In order to establish a simple but effective method for boronic acid-based sensor creation, Scrafton et al. (including some co-authors of this report) employed the copper catalyzed azide–alkyne cycloaddition (CuAAC) reaction, commonly referred to as a “click” reaction.12 For such receptors, the term click-fluor was coined.13 The CuAAC reaction has been extensively exploited across the chemical sciences due to well-known merits, such as high yields, operational simplicity, and wide-ranging reaction regimes.14 In fact, the synthetic advantages of creating molecular chemosensors via CuAAC reactions have also been demonstrated in other recent studies,15 and more boronate ester analogues have also been synthesised as modular building blocks for Suzuki coupling.16
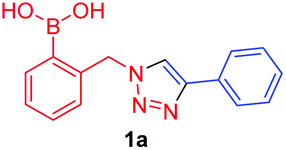
In the Scrafton study, compound 1a (Fig. 1a) showed fluorescence enhancements upon addition of fructose and mannose. This was assumed to be due to fluorescence emission recovery from the conjugated triazole after the binding of diols, however this assumption has proved to be incorrect and is discussed in this manuscript.
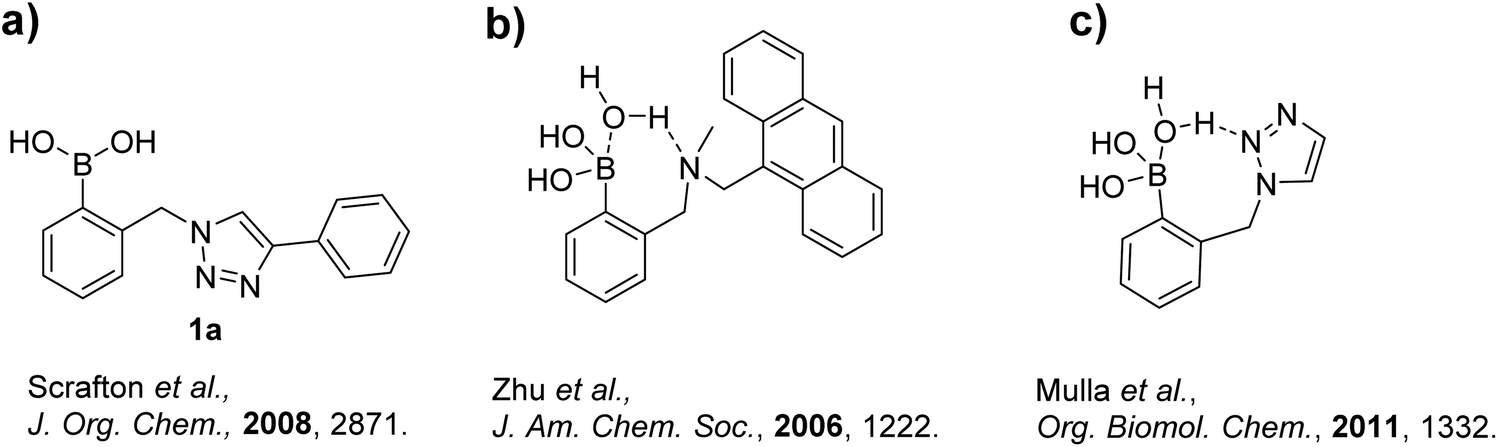 | ||
| Fig. 1 (a) Chemical structure of the first “click-fluor”; (b) the model for amine-based boronic acid interactions in James and Shinkai's system; (c) proposed model of triazole nitrogen–boronic acid interaction in aqueous media. | ||
Additionally, an interesting possibility exists with a triazole linker in a boron-based chemosensor. It was previously proposed that one or more of the triazole's nitrogen atoms could interact with an adjacent boronic acid, similar to the ortho-aminomethyl phenylboronic acid-based systems reported by James et al.17 and carefully studied by Zhu et al.18 (Fig. 1b). This interaction could either involve a direct N–B dative bond, or as Zhu et al. found, a solvent insertion. The postulate of a triazole to boronic acid interaction was further investigated in a separate study by Mulla et al. with supporting computational analysis (Fig. 1c).19 Their computational study revealed a solvent insertion using the central nitrogen of the triazole.
Herein, the so-called click chemistry strategy is used to prepare a series of boronic acids with varying regiochemistry of substitution on both the phenylboronic acid and triazole rings, and their performance as saccharide sensors is probed. Importantly, the earlier click-fluor observations are revised following investigations into weak background fluorescence witnessed across multiple analyte samples. Further, we set out to investigate whether the computationally proposed structure 1c would have experimental support, and report that the 11B NMR and fluorescence titrations are best interpreted such that the triazole has no interaction, neither N–B bond nor solvent insertion, with the boronic acid, but rather simply plays the role of a ready synthesised linker. The reasons for this are explained based upon physical organic chemistry principles.
Results and discussion
First generation “click-fluors”
Against a backdrop of our previous report of just a single boronic acid derivative of a triazole,13 the assembled team wished to study structural effects on boron–saccharide interactions and prepared six regioisomers of compound 1a (Scheme 2). Starting from commercially available boronic acid compounds, pinacol protection was performed to minimize side reactions. Moreover, free boronic acids have strong interactions with silica gel, but carrying pinacol esters through the synthetic sequence minimized problems for flash chromatography. Bromomethyl phenyl boronic acid (3a–c) and terminal alkyne (6a–c) functionalised intermediates were obtained after bromination and TMS deprotection of 2a–c and 5a–c, respectively. Azide derivatives for the CuAAC reaction were prepared in situ, and triazoles 4a–c and 7a–c were formed in 33–65% isolated yield.20 Deprotection of pinacol esters was carried out by conversion to a potassium trifluoroborate salt, which was subsequently hydrolyzed to deliver the corresponding boronic acids 1a–c and 8a–c in good yield.21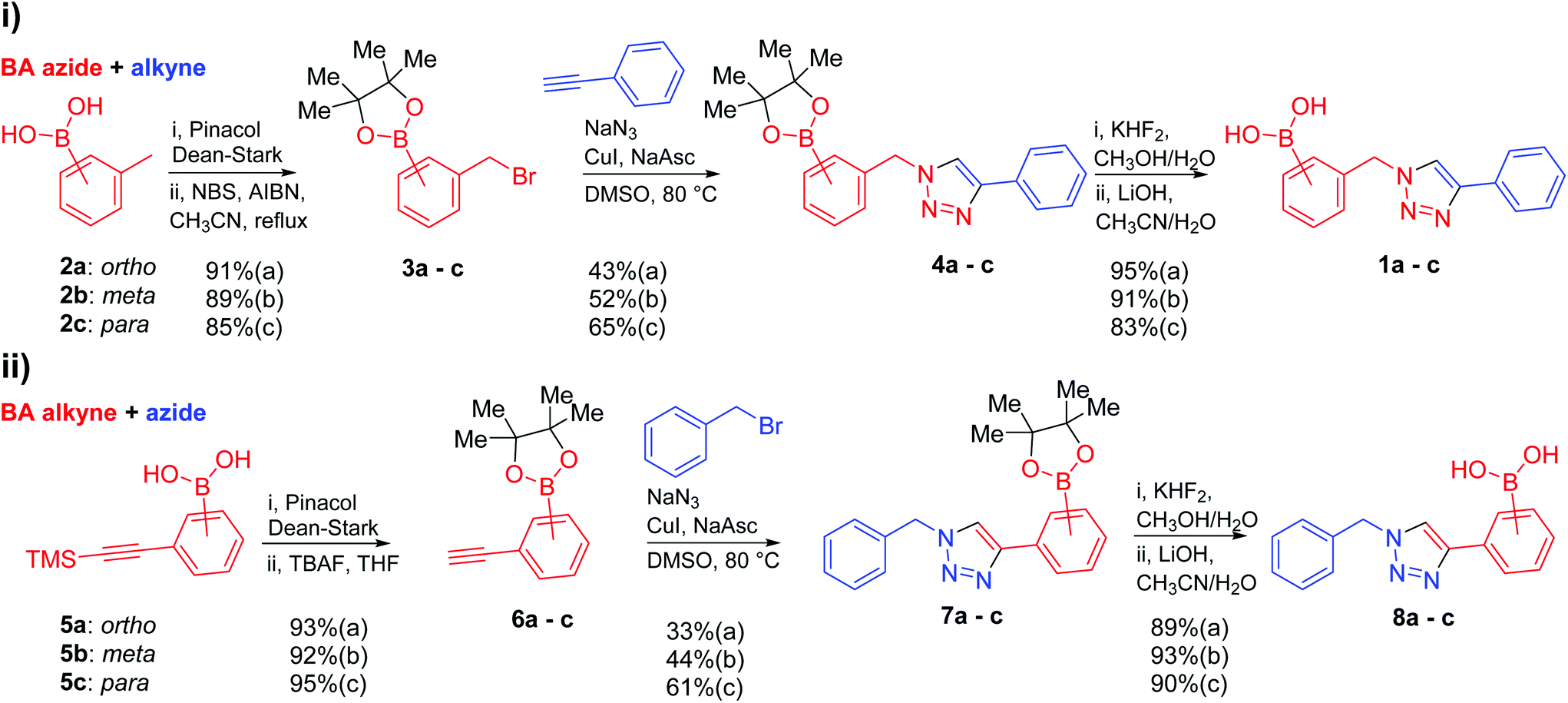 | ||
| Scheme 2 Synthetic routes to the six borono-regioisomers of the first generation “click-fluor”. (i) Starting from tolylboronic acids (a: ortho, b: meta, c: para), followed by pinacol protection, bromination, CuAAC reaction and pinacol deprotection to furnish 1a–c; (ii) starting from [(trimethylsilyl)ethynyl]phenylboronic acids (a: ortho, b: meta, c: para), followed by pinacol protection, TMS deprotection, CuAAC reaction and pinacol deprotection delivering 8a–c. | ||
Crystals, suitable for single crystal X-ray diffraction structure determination, of compound 8a were obtained from a methanol solution (Fig. 2). The X-ray structure revealed incorporation of one methoxy group (from methanol). An intramolecular O–H⋯N hydrogen bond with the proximal nitrogen of the triazole ring ((N(33)–H(37) = 1.74(3) Å)) was observed in a close to linear arrangement (O(36)–H(37)–N(33) = 161(2)°). It is worth noting that the torsion angle between the boronic acid group and phenyl ring is −19.4(3)° (O(36)–B(1)–C(2)–C(11)), which suggests the boronic acid group was twisted in the system to facilitate the formation of the intramolecular hydrogen bond. This crystal structure shows that in the solid state it is possible for compound 8a to form a B–OH⋯N bond interaction, which is akin to solvent insertion prior to sugar binding. It can be further noted that the basicity of the N-atom acting as a hydrogen bond acceptor is enhanced in a triazole ring due to the fact that the nitrogen 2-atoms away in the ring donates electrons via resonance (a fact we will return to in regards to a discussion of Fig. 5).
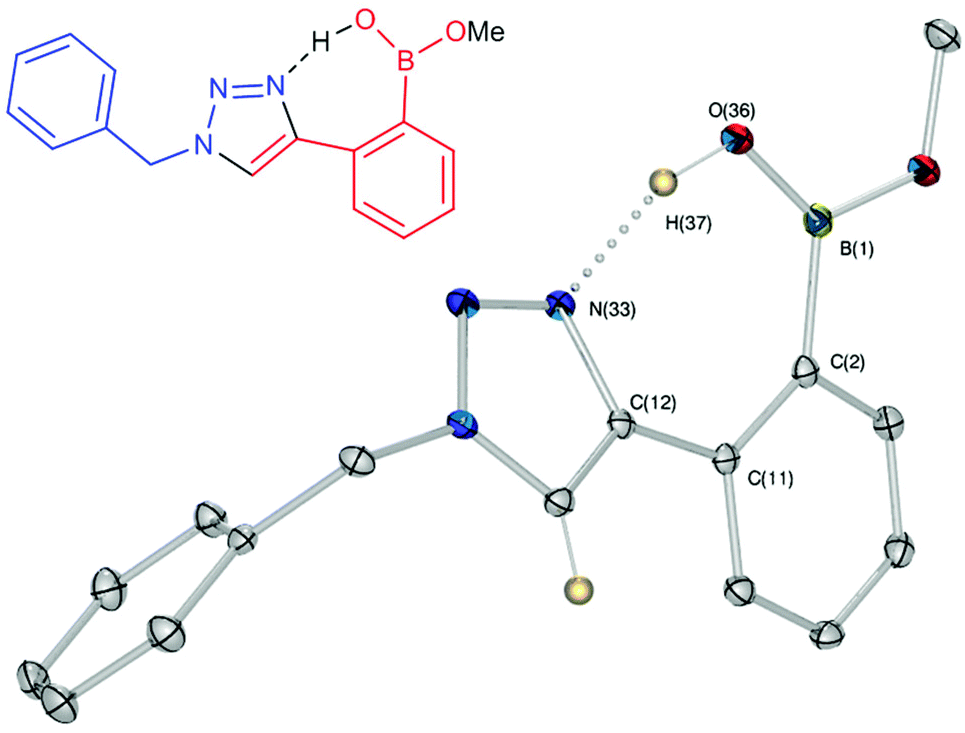 | ||
| Fig. 2 Molecular and X-ray structure of compound 8a crystallised from methanol. ORTEP ellipsoids set at 30%, selected bond length (Å), angle (°) and torsions (°): N(33)–H(37) 1.74(3); C(11)–C(2)–B(1) 129.06(14); N(33)–C(12)–C(11)–C(2) 23.20; C(12)–C(11)–C(2)–B(1) 7.65. | ||
Compounds 1a–c were tested as fluorescent saccharide sensors following previously reported procedures.13 Solid D-fructose was added to a solution of given concentration boronic acid (1a–c) in pH 8.21 methanolic buffer. Whilst the fluorescence responses closely matched that of previously reported,13 further study of control conditions revealed samples of fructose were contributing to the weak fluorescence observed (see ESI, Fig. S1 and S2†). In other words, samples of fructose appear to elicit a weak fluorescence response in the absence of any boronic acid. Indeed, in order to verify the origin of the observed signal, the experiments were repeated using different fluorimeters at more than one institution and numerous sources of fructose, including that prepared in situ by cleavage of sucrose. Fructose samples were examined by NMR spectroscopy and elemental analysis, and no fluorescent impurity was successfully identified from any analytical technique. Possible origins of the fluorescence include contamination by small amounts of highly fluorescent aromatic species, or n-to-π* transitions of the open-chain form of the sugars22 (see ESI, Fig. S3 and S4, Table S1† for other probed saccharides). Whilst the origin of the weak fluorescent signal of the samples of fructose is not resolved in this study, it remains an intriguing issue warranting further investigation.
Because the fluorescence responses of compounds 1a–c to saccharides were too weak to be useful (once the background signal from saccharide samples was accounted for), an alternative approach to determine saccharide binding capability was needed. As such, isothermal titration calorimetry (ITC) was employed to determine the saccharide binding constants. ITC is an effective method for studying the binding of small molecules to large biomolecules like proteins and DNA.23 Furthermore, it has also been used to determine the binding strength between boronic acid derivatives and diol-containing molecules.24
Fig. 3 shows the results from titrating fructose into solutions of each of the six synthesised boronic acid compounds (1a–c and 8a–c), respectively. The ITC measurements were carried out at pH 8.21 in methanolic buffer. Both the binding site information and binding constants are shown in Table 1. All boronic acid triazole receptors tested in an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with fructose, which agrees with the widely accepted PBA–fructose binding model.25
:1 ratio with fructose, which agrees with the widely accepted PBA–fructose binding model.25
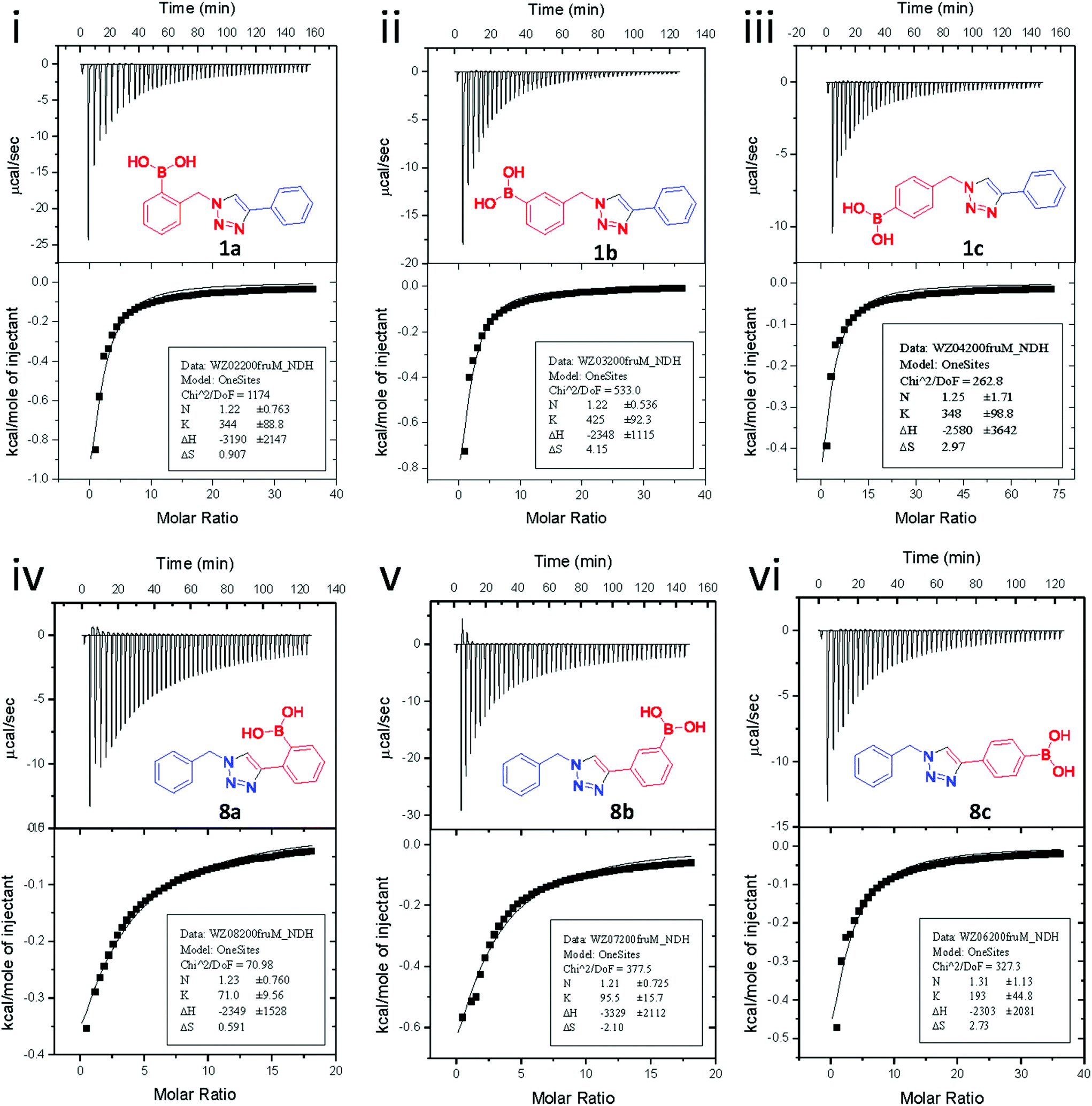 | ||
| Fig. 3 (i–iii) ITC results of compounds 1a–c, respectively, binding with D-fructose in pH 8.21 methanolic buffer; (iv–vi) ITC results of compounds 8a–c, respectively, binding with D-fructose in pH 8.21 methanolic buffer. | ||
| Entry | Compound | Binding site | Binding constants (M−1) |
|---|---|---|---|
| 1 |
|
1.2 | 3.4 × 102 |
| 2 |
|
1.2 | 4.2 × 102 |
| 3 |
|
1.3 | 3.5 × 102 |
| 4 |
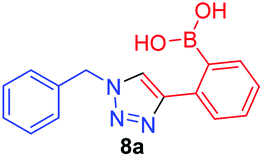
|
1.2 | 7.1 × 101 |
| 5 |
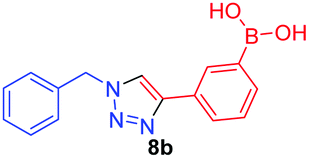
|
1.2 | 9.5 × 101 |
| 6 |
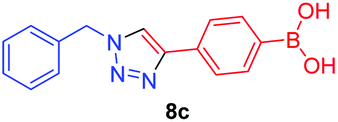
|
1.3 | 1.9 × 102 |
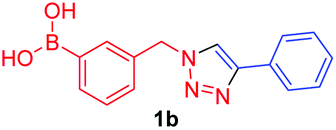
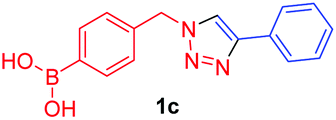
A few trends in this data are worth noting. First, in terms of binding constants, boronic acids–triazoles derived azido boronic esters, ortho (1a), meta (1b) and para (1c) (entries 1 to 3, respectively) consistently gave higher binding constants than boronic acids–triazoles derived alkyne boronic esters (entries 4 to 6), by factors between 2 and 4. Apart from that, compound 1a–c also showed stronger binding strength towards fructose comparing with PBA (binding constant = 1.6 × 102 M−1, see ESI Fig. S7†). We postulate this is due to an electronic factor, where the triazoles in entries 4–6 deactivate the boron to complexation via resonance donation from the triazole, which would reduce the electrophilicity of the boron. Second, there is little to no difference in the binding affinities between the regioisomers 1a–c in entries 1, 2, and 3. It makes sense that 1b–c would bind similarly, but until verified we postulated that 1a could possess some form of a N–B interaction, so a different affinity may have been expected. Similarly, there is little difference in entries 4, 5, and 6, although 8c does bind somewhat more strongly than 8a–b.
Titrations probed by proton NMR spectroscopy have been previously utilised in the study of supramolecular interactions.26 Herein, titrations of 1a and 1c with fructose were studied by 1H NMR spectroscopy, following the protocol of Mulla et al.19 These two compounds were selected in order to compare the effect of a potential triazole–boronic acid interaction on fructose binding. In the studies of Mulla et al., it was proposed that the second triazolyl nitrogen might interact with boron through solvent insertion, which stabilises the boronic acid–diol complex. As such, compounds 1a and 1c were dissolved in DMSO-d6, and a fructose solution (phosphate buffer prepared in D2O, pD 8.21) was titrated into the boronic acid solutions. As it is shown in Fig. 4i, upon addition of fructose solution in D2O to 1a (ortho), the signal of the exchangeable boronic acid OH protons (Hb) disappear due to deuterium exchange. However, the signal of the triazole C–H proton (Ha) shifts more than 0.2 ppm downfield, indicating a deshielding effect after fructose binding.
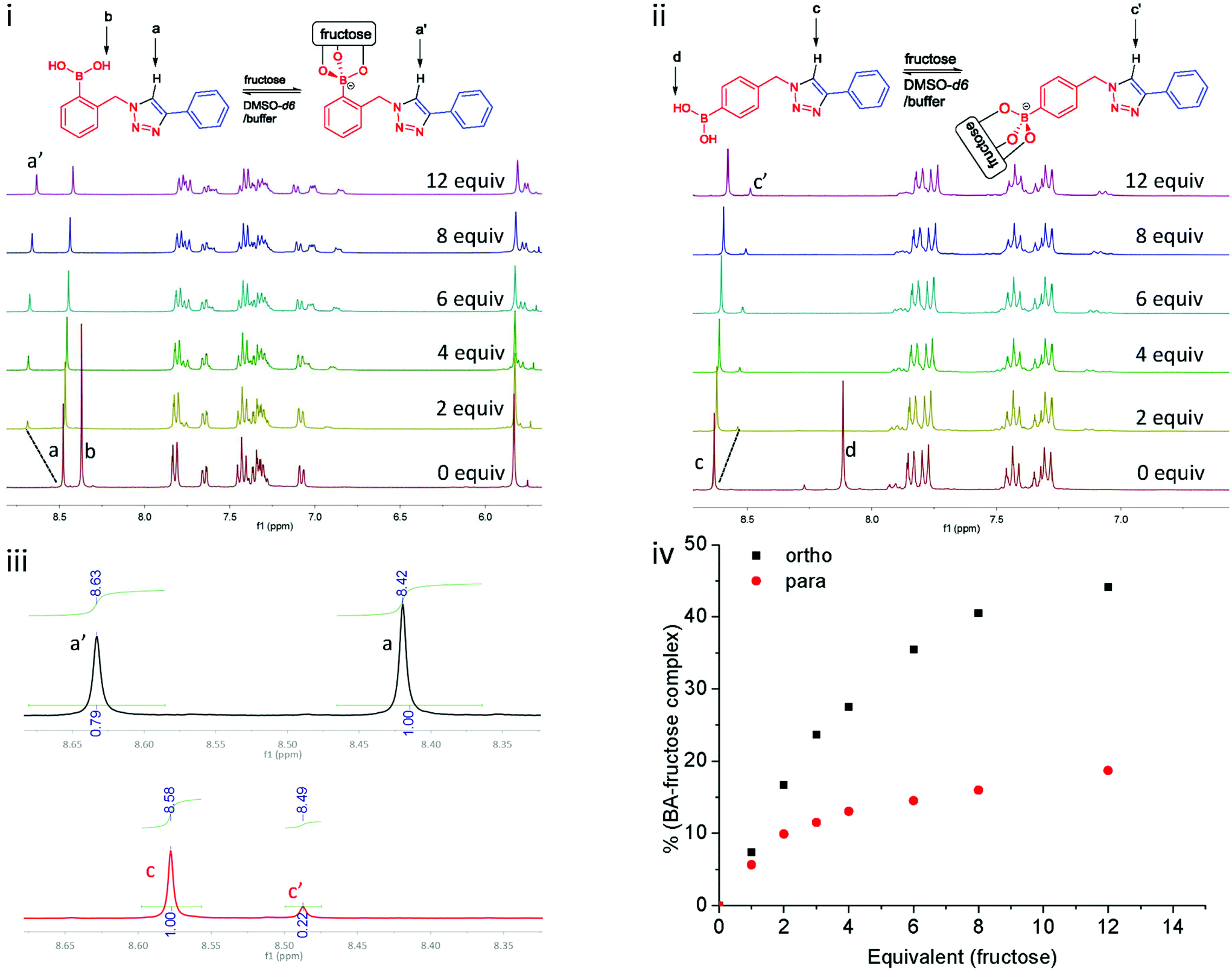 | ||
| Fig. 4 (i) 1H NMR titration of 1a with D-fructose in DMSO-d6 and phosphate buffer solution (pD 8.21); (ii) 1H NMR titration of 1c with D-fructose in DMSO-d6 and phosphate buffer solution (pD 8.21), 1H NMR drift slightly (<0.1 ppm) due to the change of solvent composition during titration; (iii) integration of triazolyl protons for compound 1a (up) and 1c (bottom); (iv) the equivalent of fructose plotting against the percentage of BA–fructose complex in the system. | ||
For the experiment with 1c (para) under the same condition, the spectral region of interest is given in Fig. 4ii. The triazole proton of para1c (Hc) shifts 0.1 ppm to upfield, in contrast to the 0.2 ppm downfield shift noted for ortho1a. This suggests that binding of fructose has a different influence on the triazole.27 A downfield shift is indicative of deshielding, such as an inductive effect when the boronic acid binding an electron withdrawing saccharide. The ratio of free boronic acid to fructose bound boronic ester were determined by comparison of the integration of the triazole C–H protons in both cases (Ha versus Ha′ and Hc versus Hc′, Fig. 4iv). All the proton NMR spectra of this titration experiment are presented in ESI, see Fig. S9 and S10.†28
Next, 11B NMR spectroscopy was used to determine whether or not an interaction between a triazole nitrogen atom and the boron atom could be modulating the binding with fructose. If a triazole nitrogen atom were to interact with the boronic acid, it could do so directly, through a N–B bond, or indirectly by inserting a solvent molecule between the nitrogen and boron atoms, or via an intramolecular hydrogen bond (as shown in Fig. 1). In aprotic solvent, such as CD3CN, the N–B interaction produces a 11B NMR signal at approximately 15 ppm. In protic solvent, such as CD3OD, a solvent could insert, which would produce a signal at approximately 10 ppm. This same chemical shift would occur for a sugar that binds via three hydroxyl groups, as does fructose.29 In comparison, a trigonal boron produces a signal at approximately 30 ppm.
The 11B NMR spectra of compounds 1b–c and 8a–c in CD3CN all showed one 11B signal at 28–29 ppm, indicating no N–B dative bonding with the triazole nitrogen atoms takes place and that the boron atom remains trigonal (see ESI, Fig. S12.† Unfortunately, compound 1a was not soluble enough in CD3CN to obtain comparative data). In CD3OD, all six compounds showed one signal, again at 28–29 ppm, indicating that no solvent insertion takes place between the triazole nitrogen atom and the boron atom as found when more basic nitrogen atoms are placed in the ortho position (see ESI, Fig. S13†). When fructose was added to the solutions in CD3OD, the signal at 28–29 ppm was replaced by a new signal at 11 ppm, indicative of fructose binding and tetrahedral boron (see ESI, Fig. S14†). This signal can be attributed to the tridentate binding of fructose to the pyramidalised boron atom.18 From these experiments, we concluded that there is no N–B interaction, and hence a change in such an interaction cannot be responsible for the change in fluorescence.
These results call into question the assignment of solvent insertion, as found by Mulla (Fig. 1c). Yet, this experimental data makes sense based upon pKa reasoning. The most basic nitrogen of a triazole ring is indicated in Fig. 5, indicating where one would expect hydrogen bond acceptance. This site is basic due to resonance, as in imidazole, leading to hydrogen bond acceptance behaviour, as depicted in Fig. 2. However, the pKa of the conjugate acid is still only 1 to 2. Thus, neither of the triazole nitrogens would be expected to make a strong hydrogen bond accepting interaction with an inserted solvent (except in a solid state, Fig. 2), nor a reasonably strong dative N–B interaction. Indeed, such interactions are not found with this most basic nitrogen. Hence, using the nitrogen with an even lower basicity seems unlikely.
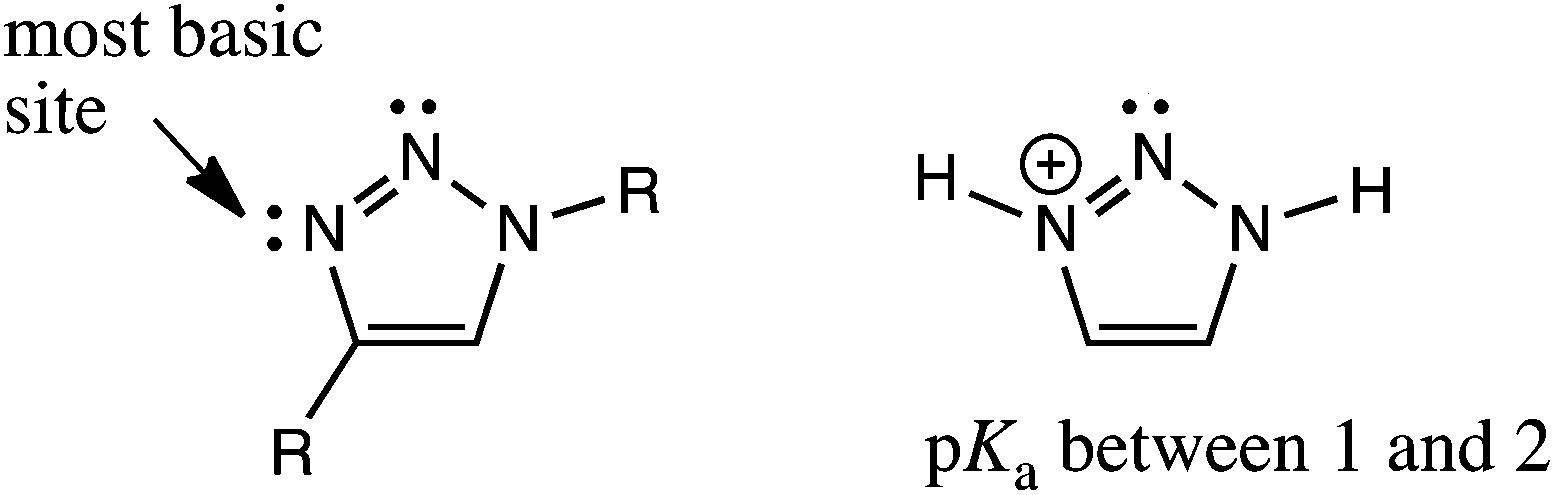 | ||
| Fig. 5 pKa of the third nitrogen of triazole (left) and imidazole (right). | ||
Second generation “click-fluors”
To rapidly assemble boronic acid receptors suitable for fluorescent sensing via triazole formation, with fluorescent responses significantly stronger than any residual background fluorescence from the saccharide samples noted earlier, building blocks that contain fluorophores were used. Considering that boronic acid–triazoles derived from bromomethyl benzenes (1a–c) showed greater saccharide binding potential by ITC (Table 1, entries 1 to 3) than the correspond methylacetylene benzenes (Table 1, entries 4 to 6), it was reasoned that superior fluorescent sensors would be derived from azides formed from methyl bromobenzenes. Noting that binding regimes of ortho1a and para1c were demonstrated to be divergent (Fig. 4), fluorophore-containing versions were selected as suitable structures for further investigation. Terminal alkyne-functionalised fluorophores were readily incorporated into boronic acid-containing triazole derivatives through CuAAC reactions. The alkyne-functionalised fluorophores were either commercially available or readily prepared via Sonogashira cross-coupling reactions.30 Among them, 4-ethynyl-1,8-naphthalimide and 7-ethynylcoumarin have been reported as fluorescent tags in the area of bioorthogonal chemistry.31 In this study, they were selected together with diphenylaniline and pyrene based on their electronic properties and scope of fluorescent signaling mechanisms accessible.The fluorescence responses upon saccharide binding, of both electronically divergent as well as ortho and para substituted isomers, could provide further information to understand the role of the triazoles in the boronic acid-based saccharide sensors. Therefore, eight fluorophore-containing boronic acid triazole deriviatives (14–17, a and b respectively) were prepared, and are named “second generation click-fluors” in this report (Scheme 3).
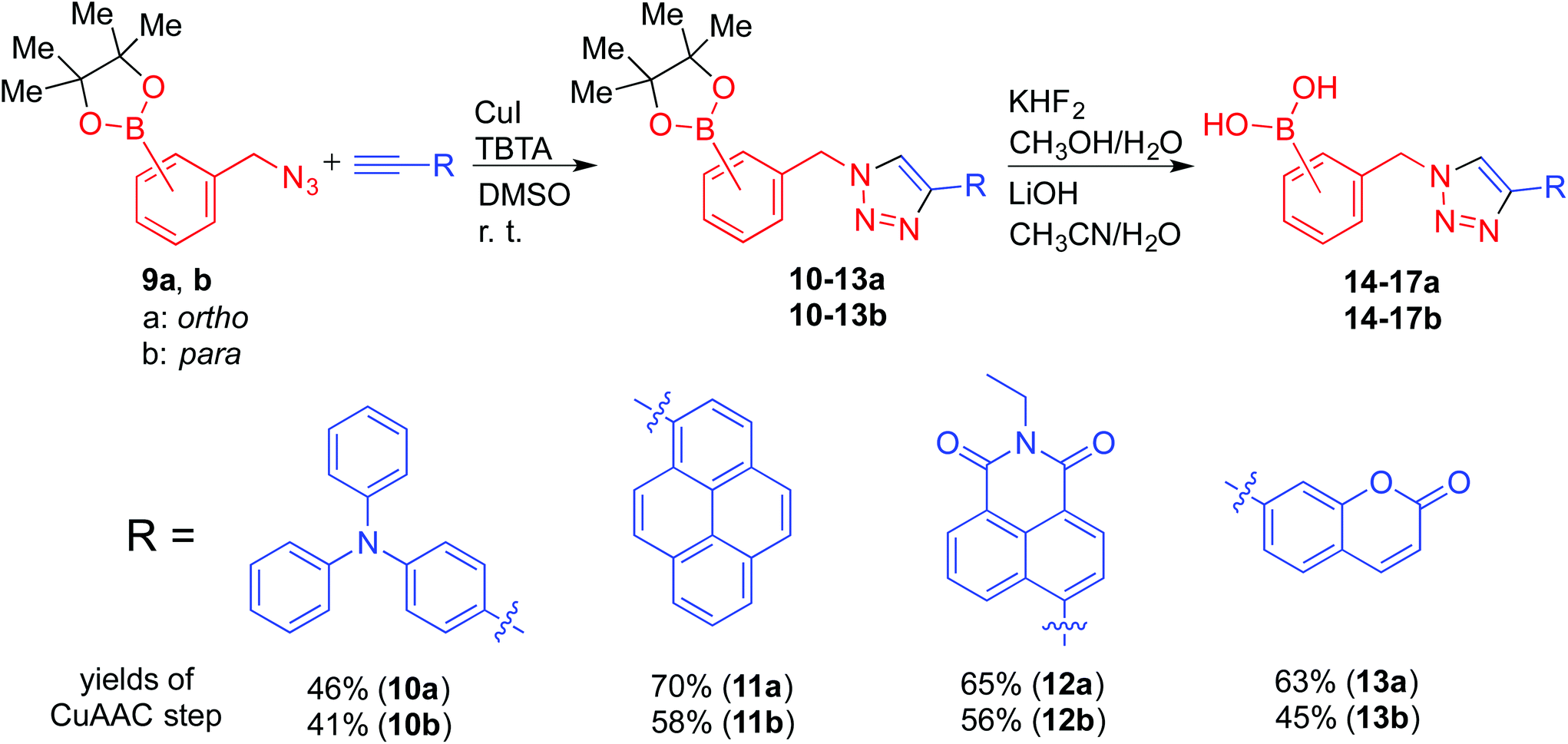 | ||
| Scheme 3 Synthetic route of second generation “click-fluors” with modified CuAAC reaction condition. Starting from alkyne functionalised fluorophores and (azidomethyl)phenylboronic acid pinacol esters (a: ortho, b: para), the designed “click-fluors” were synthesised by CuAAC reactions and pinacol deprotections in 34 to 63% overall yields. | ||
Copper-mediated side-reactions plagued the preliminary synthetic efforts due to undesired deborylation and the formation of oxidation byproducts. Alternative synthetic methods have been developed to avoid the undesired outcomes of side reactions.16,32 For example, Wang et al. reported a method to protect the boronic acid group by adding fluoride.33 Maison et al. also suggested the addition of tris(benzyltriazolylmethyl)amine (TBTA) as a ligand for copper.34 In these studies, boronic esters with pyrene and coumarin fluorophores were synthesised with modest yields.15b,35 In the present work, optimisation of the reaction conditions revealed that isolation of boronic acid azide derivatives (rather than in situ preparation) offered significant improvements in formation of the desired products. Copper-mediated deborylation and oxidation were overcome by reducing the copper catalyst loading and adding TBTA to accelerate the CuAAC reactions.36 As a result, complete conversion of the alkynes was achieved and yields of the CuAAC step were improved from 32 to 70% (11a), 34 to 65% (12a) and 29 to 63% (13a). Further work is currently underway to understand how to improve the isolated yields of this class of click reaction products.32 Material suitable for single crystal X-ray diffraction structure determination of key intermediate 13a was obtained (Fig. 6) by crystallisation from a mixture of hexane and ethyl acetate. No interaction between triazole ring nitrogen atoms and boronic ester was observed in the material crystallised from aprotic solvent. However, the distance between O(3) and H(26) is measured as 2.655(1) Å, which could be indicative of a weak hydrogen bond with triazole C–H being the hydrogen bond donor.37
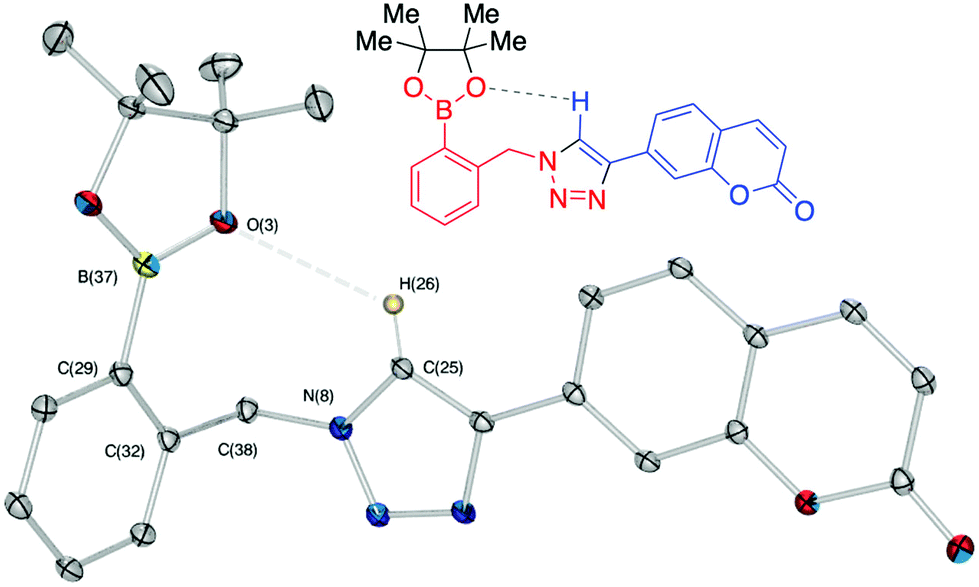 | ||
| Fig. 6 Chemical and X-ray crystal structure of compound 13a. ORTEP, ellipsoids plot at 30%, selected angle: O(3)–H(26) 2.655(1) Å; selected torsion: O(3)–B(37)–C(29)–C(32) 6.833°. | ||
ortho- and para-regioisomers of 14–17 (a and b respectively) were synthesised from each of the four selected alkyne-appended fluorophores. The synthesised compounds were tested as fluorescent probes for fructose binding using the same protocol as described by Scrafton et al. The ortho and para congeners (14a and 14b) gave strikingly different fluorescence responses to fructose, with ortho14a showing a stronger fluorescence recovery (Fig. 7i, ii). A similar trend of fluorescence responses was observed with compound 17a and 17b under the same experimental conditions (Fig. 7iii, iv). These results agree with the 1H NMR titration study, which showed divergent responses between ortho and para triazole isomers of 1a and 1c to fructose. Because there is no evidence of a hypothetical N–B interaction facilitated by saccharide binding from the 11B NMR spectroscopic studies earlier, it is possible that the stronger binding strength is caused by the different pKa between ortho and para boronic acids. With the triazole group at the ortho position, the electron-withdrawing capacity through induction is greater than that of the para isomers. As a result, the ortho boronic acids could have a lower pKa, which means stronger binding strength under the experimental pH (8.21). The same fluorescence recovery was not recorded in the cases of compounds 15a,b and 16a,b (see ESI, Fig. S5†). This result suggests that the properties of the fluorophore also have an impact on fluorescence modulation, possibly due to the overall effect on the pKa of the boronic acids. Differences in fluorescence quantum yields can be difficult to decipher, and thus the reason for greater recovery with the ortho-derivatives is unclear. However, the most common deactivation process for fluorescence is internal conversion from loose rotors, and with a triazole in the ortho position hydrogen bonds as observed Fig. 2 and 6 with a bound tridentate fructose would restrict bond rotations within the complexes possibly leading to higher extents of emission turn-on than for other regioisomers. Clearly, more direct evidence is needed in future studies.
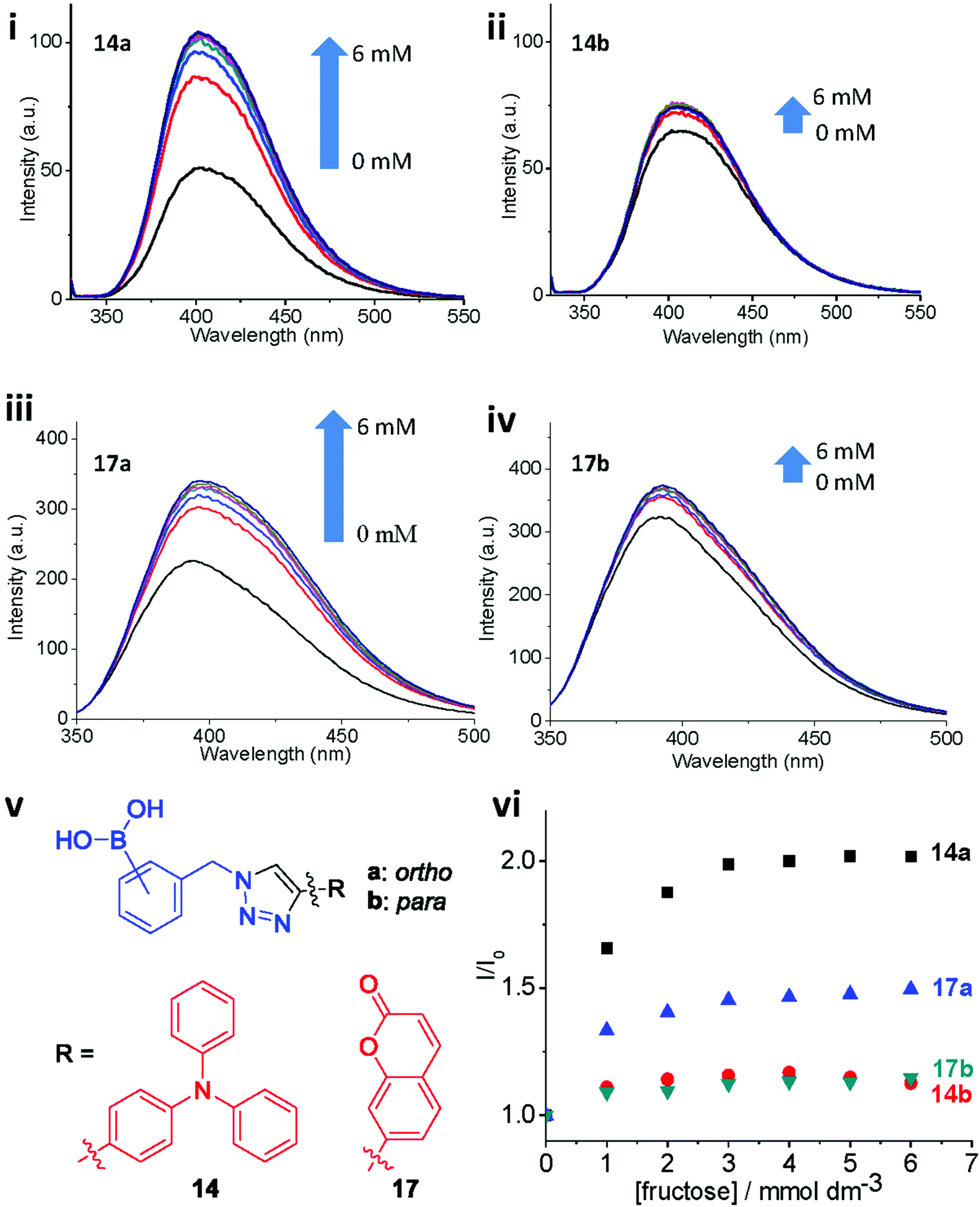 | ||
| Fig. 7 (i) Fluorescence spectra of 14a in the presence of increasing concentration of D-fructose (0–6 mM); (ii) fluorescence spectra of 14b in the presence of increasing concentration of D-fructose (0–6 mM); (iii) fluorescence spectra of 17a in the presence of increasing concentration of D-fructose (0–6 mM); (iv) fluorescence spectra of 17b in the presence of increasing concentration of D-fructose (0–6 mM); (v) chemical structures of the tested compounds; (vi) plotting fluorescence enhancement of the tested compounds against the concentrations of D-fructose. Excitation wavelength: 290 nm (14a,b); 330 nm (17a,b). | ||
Furthermore, fluorescence titrations were carried out with the synthesised compounds and glucose. The anticipated low binding affinity for mono-boronic acids with glucose5a was also apparent in the present system as only weak responses were recorded (see ESI, Fig. S6†).
Conclusion
In summary, six regioisomers of previously reported 1a (including 1a) were synthesised as “click-fluors” in this work. 1H, 11B NMR titrations and ITC were employed to study the effect of triazole–boronic acid distance upon saccharide binding capacity. The result of 1H NMR spectroscopy titrations suggests that the ortho-triazole compound has stronger fructose binding strength than the para-triazole isomer under the experimental conditions. However, no indication of direct N–B interaction was observed by comparing 11B NMR signals of the synthesised “click-fluors” before and after binding with fructose. As a result, a possible hypothesis of different pKa of the boronic acids as a result of differing triazole substitution was suggested. In the case of ortho-triazole compound 1a, the adjacent triazole ring could have a stronger inductive electron-withdrawing effect comparing with the para-triazole isomer 1c.A fluorescence study was conducted following the procedure of Scrafton et al. For the first generation “click-fluors,” the result suggested that the fluorescence of these molecules needed to be improved for saccharide sensing. Therefore, four alkyne-functionalised fluorophores with different electronic properties were selected for the synthesis of second generation “click-fluors.” Specifically, triphenylamine, coumarin, 1,8-naphthalimide and pyrene were attached on different positions of PBA through CuAAC reaction. By comparing the fluorescence response of ortho and para regioisomers upon addition of fructose, it is found that ortho-triazole sensors generate stronger fluorescence enhancement after binding with fructose, which could be due to the difference between the pKa of ortho (14a, 17a) and para (14b, 17b) boronic acids and difference in internal conversion.
Considering the wide range and easy access of alkyne-functionalised fluorophores, it is reasonable to expect more sensor molecules with larger fluorescence responses could be created using the same strategy. Moreover, the improvement on the efficiency of CuAAC reaction with boron moieties could be beneficial to develop boronic acid functionalised materials via click chemistry.38 Ongoing efforts are focused on synthesising and studying multi-boronic acid compounds using the same strategy.
Contributions
WZ participated in all aspects, performed the synthesis, fluorescence studies and ITC experiments and co-wrote the manuscript. JSF conceived the project, oversaw all aspects and co-wrote the manuscript. BMC designed and carried out the NMR titration experiments with EVA, who both contributed to writing aspects of the manuscript. SAH synthesised one compound of the report prior to the main work being carried out. AY carried out XRD experiments, Louise Male is thanked for assistance with XRD experiments. JSF, EVA and TDJ discussed the manuscript and contributed to the interpretation of the results. All co-authors commented on the manuscript.Acknowledgements
David Scrafton and Mathew Wright are acknowledged for carrying out related preliminary work. Dr Cécile Le Duff, the University of Birmingham, is acknowledged for assistance with aspects of 11B NMR spectroscopy. WZ, TDJ, EVA and JSF thank The Catalysis and Sensing for our Environment (CASE) group for networking opportunities.39 China Scholarship Council (CSC) and the University of Birmingham are also thanked for providing studentship support (WZ). JSF thanks the University of Birmingham for support, the Royal Society for an Industrial Fellowship and the EPSRC for funding (EP/J003220/1). JSF and TDJ are grateful for past collaborative support (DT/F00267X/1). TDJ, JSF and SAH thank the University of Bath for past support. WZ, BMC, JSF and EVA thank the Royal Society International Joint Project Scheme for support. EVA also thanks the Welch Regents Chair for support (F-0046).References
- J. F. Robyt, in Essentials of Carbohydrate Chemistry, Springer, New York, NY, 1998, pp. 157–227. DOI:10.1007/978-1-4612-1622-3_6.
- E. F. Petricoin, C. Belluco, R. P. Araujo and L. A. Liotta, Nat. Rev. Cancer, 2006, 6, 961–967 CrossRef CAS PubMed.
- A. Stephenson-Brown, S. Yong, M. H. Mansor, Z. Hussein, N.-C. Yip, P. M. Mendes, J. S. Fossey and F. J. Rawson, Chem. Commun., 2015, 51, 17213–17216 RSC.
- (a) W. Zhai, X. Sun, T. D. James and J. S. Fossey, Chem. – Asian J., 2015, 10, 1836–1848 CrossRef CAS PubMed; (b) J. S. Fossey, F. D'Hooge, J. M. H. van den Elsen, M. P. P. Morais, S. I. Pascu, S. D. Bull, F. Marken, A. T. A. Jenkins, Y.-B. Jiang and T. D. James, Chem. Rec., 2012, 12, 464–478 CrossRef CAS PubMed.
- (a) J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 CrossRef CAS; (b) X. Wu, X.-X. Chen, M. Zhang, Z. Li, P. A. Gale and Y.-B. Jiang, Chem. Commun., 2016, 52, 6981–6984 RSC.
- X. Sun, J. S. Fossey, W. Zhai and T. D. James, Chem. Commun., 2016, 52, 3456–3469 RSC.
- R. A. Bissell, A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, G. E. M. Maguire and K. R. A. S. Sandanayake, Chem. Soc. Rev., 1992, 21, 187–195 RSC.
- (a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; (b) J. S. Fossey and T. D. James, in Reviews in Fluorescence 2007, Springer, New York, 2009, ch. 5, vol. 2007, pp. 103–118 Search PubMed.
- X. Sun and T. D. James, Chem. Rev., 2015, 115, 8001–8037 CrossRef CAS PubMed.
- (a) X. Wu, Z. Li, X.-X. Chen, J. S. Fossey, T. D. James and Y.-B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 RSC; (b) Z.-J. Chen, Z. Tian, K. Kallio, A. L. Oleson, A. Ji, D. Borchardt, D.-E. Jiang, S. J. Remington and H.-W. Ai, J. Am. Chem. Soc., 2016, 138, 4900–4907 CrossRef CAS PubMed; (c) K. A. Andersen, T. P. Smith, J. E. Lomax and R. T. Raines, ACS Chem. Biol., 2016, 11, 319–323 CrossRef CAS PubMed.
- K. Okuro, M. Sasaki and T. Aida, J. Am. Chem. Soc., 2016, 138, 5527–5530 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- D. K. Scrafton, J. E. Taylor, M. F. Mahon, J. S. Fossey and T. D. James, J. Org. Chem., 2008, 73, 2871–2874 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC; (c) C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- (a) C. Dai, Y. Cheng, J. Cui and B. Wang, Molecules, 2010, 15, 5768–5781 CrossRef CAS PubMed; (b) L. Du, N. Ni, M. Li and B. Wang, Tetrahedron Lett., 2010, 51, 1152–1154 CrossRef CAS PubMed; (c) S.-L. Zheng, S. Reid, N. Lin and B. Wang, Tetrahedron Lett., 2006, 47, 2331–2335 CrossRef CAS; (d) S. Jin, C. Zhu, Y. Cheng, M. Li and B. Wang, Bioorg. Med. Chem., 2010, 18, 1449–1455 CrossRef CAS PubMed.
- J. R. White, G. J. Price, S. Schiffers, P. R. Raithby, P. K. Plucinski and C. G. Frost, Tetrahedron Lett., 2010, 51, 3913–3917 CrossRef CAS.
- T. D. James, K. R. A. S. Sandanayake, R. Iguchi and S. Shinkai, J. Am. Chem. Soc., 1995, 117, 8982–8987 CrossRef CAS.
- L. Zhu, S. H. Shabbir, M. Gray, V. M. Lynch, S. Sorey and E. V. Anslyn, J. Am. Chem. Soc., 2006, 128, 1222–1232 CrossRef CAS PubMed.
- K. Mulla, P. Dongare, N. Zhou, G. Chen, D. W. Thompson and Y. Zhao, Org. Biomol. Chem., 2011, 9, 1332–1336 CAS.
- X. Sun, S.-Y. Xu, S. E. Flower, J. S. Fossey, X. Qian and T. D. James, Chem. Commun., 2013, 49, 8311–8313 RSC.
- A. K. L. Yuen and C. A. Hutton, Tetrahedron Lett., 2005, 46, 7899–7903 CrossRef CAS.
- M. P. O'Sullivan and A. C. Testa, J. Am. Chem. Soc., 1970, 92, 5842–5844 CrossRef.
- T. K. Dam and C. F. Brewer, Chem. Rev., 2002, 102, 387–430 CrossRef CAS PubMed.
- (a) M. S. Melicher, J. Chu, A. S. Walker, S. J. Miller, R. H. G. Baxter and A. Schepartz, Org. Lett., 2013, 15, 5048–5051 CrossRef CAS PubMed; (b) Ö. Torun, F. C. Dudak, D. Baş, U. Tamer and İ. H. Boyacı, Sens. Actuators, B, 2009, 140, 597–602 CrossRef.
- J. E. Jeffery, F. Kerrigan, T. K. Miller, G. J. Smith and G. B. Tometzki, J. Chem. Soc., Perkin Trans. 1, 1996, 2583–2589, 10.1039/p19960002583.
- M. Lisbjerg, H. Valkenier, B. M. Jessen, H. Al-Kerdi, A. P. Davis and M. Pittelkow, J. Am. Chem. Soc., 2015, 137, 4948–4951 CrossRef CAS PubMed.
- (a) B. E. Collins, P. Metola and E. V. Anslyn, Supramol. Chem., 2013, 25, 79–86 CrossRef CAS PubMed; (b) J. D. Larkin, J. S. Fossey, T. D. James, B. R. Brooks and C. W. Bock, J. Phys. Chem. A, 2010, 114, 12531–12539 CrossRef CAS PubMed.
- Mulla et al. that the “Wulff-type” a nitrogen–boron interaction is important in saccharide binding. Our experiments from this point were planned, to exam nearby nitrogen effect so ortho and para isomers are compared, in respect of saccharide binding capabilities.
- B. E. Collins, S. Sorey, A. E. Hargrove, S. H. Shabbir, V. M. Lynch and E. V. Anslyn, J. Org. Chem., 2009, 74, 4055–4060 CrossRef CAS PubMed.
- (a) M. Chtchigrovsky, A. Primo, P. Gonzalez, K. Molvinger, M. Robitzer, F. Quignard and F. Taran, Angew. Chem., Int. Ed., 2009, 48, 5916–5920 CrossRef CAS PubMed; (b) G.-C. Kuang, H. A. Michaels, J. T. Simmons, R. J. Clark and L. Zhu, J. Org. Chem., 2010, 75, 6540–6548 CrossRef CAS PubMed; (c) L. Yuan, Z. Zhang, X. Xu and X. Zhou, Synth. Commun., 2014, 44, 1007–1011 CrossRef CAS; (d) L. Zhang, L. Zou, J. Xiao, P. Zhou, C. Zhong, X. Chen, J. Qin, I. F. A. Mariz and E. Macoas, J. Mater. Chem., 2012, 22, 16781–16790 RSC.
- (a) C. Le Droumaguet, C. Wang and Q. Wang, Chem. Soc. Rev., 2010, 39, 1233–1239 RSC; (b) M. Sawa, T.-L. Hsu, T. Itoh, M. Sugiyama, S. R. Hanson, P. K. Vogt and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12371–12376 CrossRef CAS PubMed; (c) S. A. Ingale and F. Seela, J. Org. Chem., 2013, 78, 3394–3399 CrossRef CAS PubMed; (d) P. Kumar, K. I. Shaikh, A. S. Jørgensen, S. Kumar and P. Nielsen, J. Org. Chem., 2012, 77, 9562–9573 CrossRef CAS PubMed.
- G. A. Molander and J. Ham, Org. Lett., 2006, 8, 2767–2770 CrossRef CAS PubMed.
- S. Jin, G. Choudhary, Y. Cheng, C. Dai, M. Li and B. Wang, Chem. Commun., 2009, 5251–5253, 10.1039/b909575f.
- F. Wienhold, D. Claes, K. Graczyk and W. Maison, Synthesis, 2011, 4059–4067 CAS.
- D. Luvino, C. Amalric, M. Smietana and J.-J. Vasseur, Synlett, 2007, 3037–3041 CAS.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed.
- (a) Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC; (b) R. O. Ramabhadran, Y. Liu, Y. Hua, M. Ciardi, A. H. Flood and K. Raghavachari, J. Am. Chem. Soc., 2014, 136, 5078–5089 CrossRef CAS PubMed.
- A. Stephenson-Brown, A. L. Acton, J. A. Preece, J. S. Fossey and P. M. Mendes, Chem. Sci., 2015, 6, 5114–5119 RSC.
- (a) J. S. Fossey and W. D. G. Brittain, Org. Chem. Front., 2015, 2, 101–105 RSC; (b) D. T. Payne, J. S. Fossey and R. B. P. Elmes, Supramol. Chem., 2016 DOI:10.1080/10610278.2016.1150595.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1475996 and 1475987. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00171h |
| This journal is © the Partner Organisations 2016 |