
C–C bond migration in the cycloisomerization of 1,6-enynes†‡
Susan M.
Stevenson
a,
Eric T.
Newcomb
b and
Eric M.
Ferreira
*a
aDepartment of Chemistry, University of Georgia, Athens, GA 30602, USA. E-mail: emferr@uga.edu
bArcus Biosciences, Hayward, CA 94545, USA
First published on 30th June 2016
Abstract
A full account of our investigation of C–C bond migration in the cycloisomerization of oxygen-tethered 1,6-enynes is described. Under Pt(II) and/or Ir(I) catalysis, cyclic and acylic alkyl groups were found to undergo 1,2-shifts into metal carbenoid intermediates. Interestingly, this process does not appear to be driven by the release of ring strain, and thus provides access to large carbocyclic frameworks. The beneficial effect of CO on the Pt(II) and Ir(I) catalytic systems is also evaluated.
Introduction
Enyne cycloisomerization through metal-catalyzed alkyne activation is a widely studied strategy for attaining structural complexity from simple starting materials.1 In the cycloisomerization of 1,6-enynes, a variety of skeletally diverse products can be accessed depending on the substitution of the enyne, the enyne tether, and the chosen reaction conditions. Catalysts that have been shown to induce enyne cycloisomerization include π-acidic metal complexes of Au, Pt, Ir, and several others. Our group has been specifically interested in the cycloisomerization of oxygen-tethered 1,6-enynes to give bicyclo[4.1.0]heptene derivatives.2–4 Related to these studies, both the Fensterbank/Malacria group5 and our group6 recently reported catalytic examples of similar enyne cycloisomerizations (using Au and Pt/Ir, respectively), these specific cases involving a C–C bond migration. The migration presumably occurs via the metal carbenoid intermediate of the proposed reaction mechanism (2 → 4, Scheme 1). This process exploits the reactive capacity of metal carbenoids, which have been of specific interest for our group for some time.7 These reactive species have been described to undergo a number of transformations,8 such as cyclopropanation,9 oxidation,10 C–H bond insertion,11 and bond migration (H, C, Si, S, etc.).12 Although C–C bond migration into metal carbenoids has been reported via alkyne activation,13 we had found that alkyl migrations were relatively unexplored when paired with the cycloisomerization of oxygen-tethered 1,6-enynes to bicyclo[4.1.0]heptene derivatives. Herein, we provide a full account of our efforts in this area, where we demonstrate that both cyclic (up to a 10-membered ring) and acyclic C–C bonds are able to migrate into Pt(II)- and Ir(I)-generated carbenoids, providing access to large carbocyclic frameworks and macrolactones. Additionally, we describe our full catalytic optimization studies of this transformation, ultimately finding that both the Pt(II) and Ir(I) catalytic systems are significantly enhanced in the presence of CO.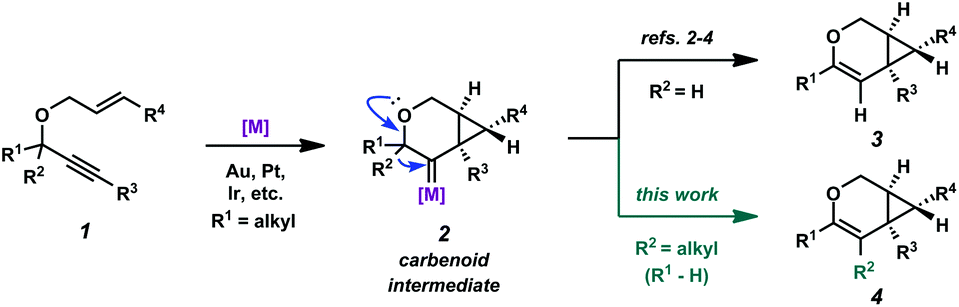 | ||
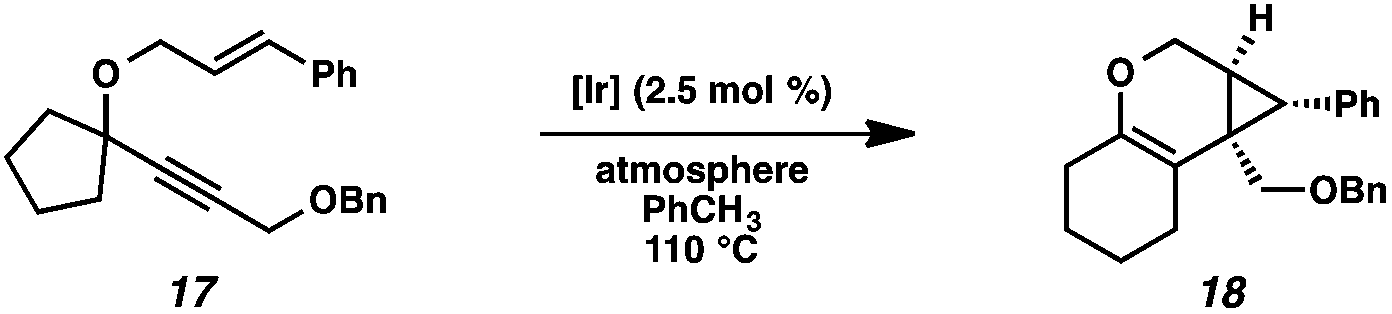
| Scheme 1 Proposed 1,2-alkyl migration into metal carbenoid. | ||
Results and discussion
Preliminary results
In order to test the propensity for alkyl migration in the cycloisomerization of oxygen-tethered 1,6-enynes, enyne 7 containing a fully substituted propargylic carbon was synthesized in two steps in straightforward fashion (Scheme 2). The alkene and alkyne substitution patterns of this enyne had proven effective in our previous cycloisomerization studies.2a Gratifyingly, when this substrate was exposed to catalytic PtCl2 in toluene at 60 °C, the desired ring-expanded product (8) was formed, indicating that C–C bond migration was a feasible outcome of this cycloisomerization. The structure of product 8 was confirmed by X-ray crystallography. The observed expansion to the six-membered ring confirmed our initial hypothesis, that alkyl groups would be capable of migration in this process if hydrogen migration was not available (2 → 4, Scheme 1).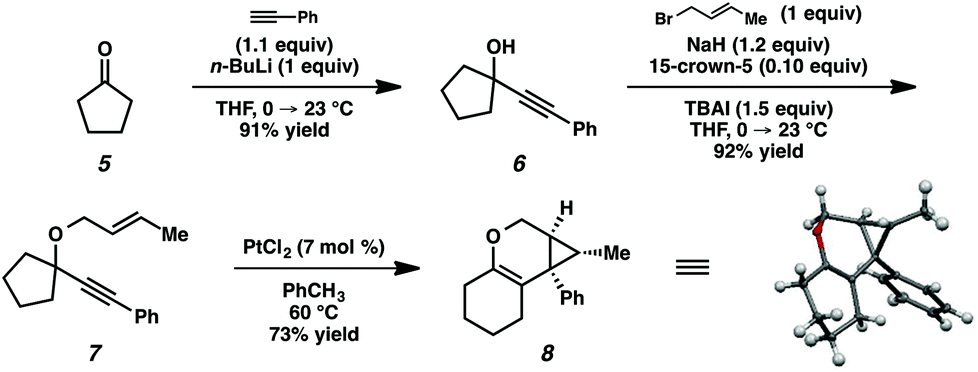 | ||
| Scheme 2 Preliminary cycloisomerization/alkyl migration result. | ||
Reaction optimization
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Temperature (°C) | Time (h) | GC yielda (%) |
| a GC yields measured using 4,4′-di-tert-butylbiphenyl as internal standard b Complex mixture obtained | ||||
| 1 | PtCl2 (7) | 60 | 2.5 | 73 |
| 2 | PtCl4 (7) | 60 | 3 | 78 |
| 3 | [(C2H4)PtCl2]2 (2.5) | 23 | 3.5 | 86 |
| 4 | (PhCN)2PtCl2 (2.5) | 23 | 2 | <5 |
| 5 | (Ph3P)2PtCl2 (2.5) | 80 | 20 | <5 |
| 6 | (Ph3P)Au(NTf2) (3) | −78→23 | 12 | 63 |
| 7 | Rh(CO)2Cl]2 (2.5) | 80 | 23 | —b |
Next a solvent screen was performed with Zeise's dimer (Table 2). Both polar and nonpolar solvents provided the product (8) in good yields, but none gave better reactivity than the original solvent toluene (entry 1). Overall, Zeise's dimer in toluene at ambient temperature gave us the optimal results for this substrate.
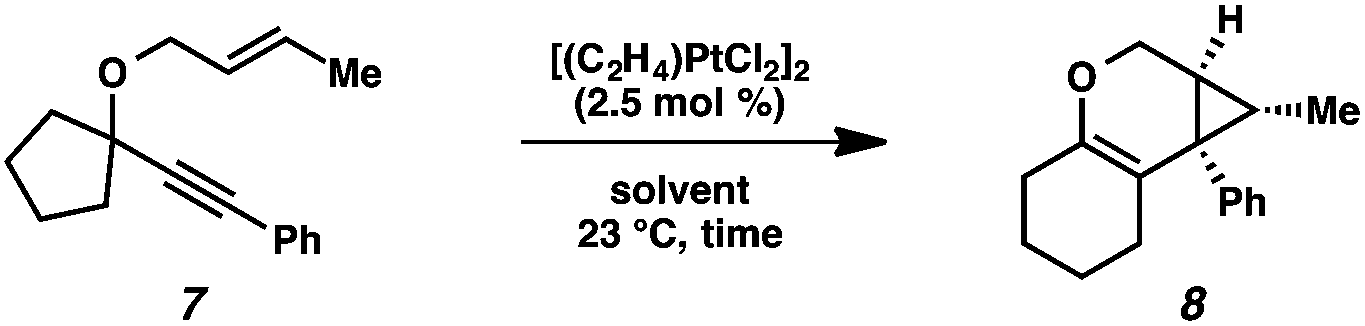
Moving forward, a variety of enyne starting materials were synthesized by nucleophilic addition of an alkyne into a cyclic ketone, followed by etherification with an allylic halide (Scheme 3).
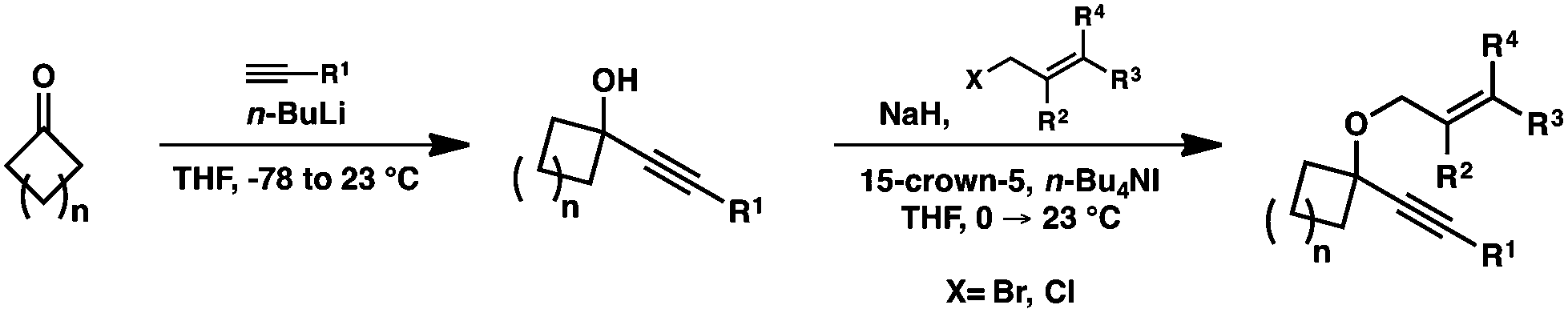 | ||
| Scheme 3 Enyne synthesis. | ||
When the optimized reaction conditions for enyne 7 (Zeise's dimer, toluene, 23 °C) were applied to other substrates, however, results varied (Table 3). For example, disubstituted alkene substrate 9 underwent the cycloisomerization with Zeise's dimer at ambient temperature in 70% isolated yield, which was comparable to what was observed under these reaction conditions with enyne 7 (Table 1, entry 3). In contrast, increased substitution on the alkene (enynes 10 and 11) required elevated temperatures in order for full conversion to be achieved, and even then product yields were poor. Clearly, further optimization was needed in order to encompass a broader scope of substrates.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Enyne | R1 | R2 | R3 | R4 | Temperature (°C) | Product | Isol. yield (%) |
| 1 | 9 | Ph | H | Ph | H | 23 | 12 | 70 |
| 2 | 10 | n-Bu | Me | Me | H | 60 | 13 | 26 |
| 3 | 11 | n-Bu | H | Me | Me | 60 | 14 | 11 |
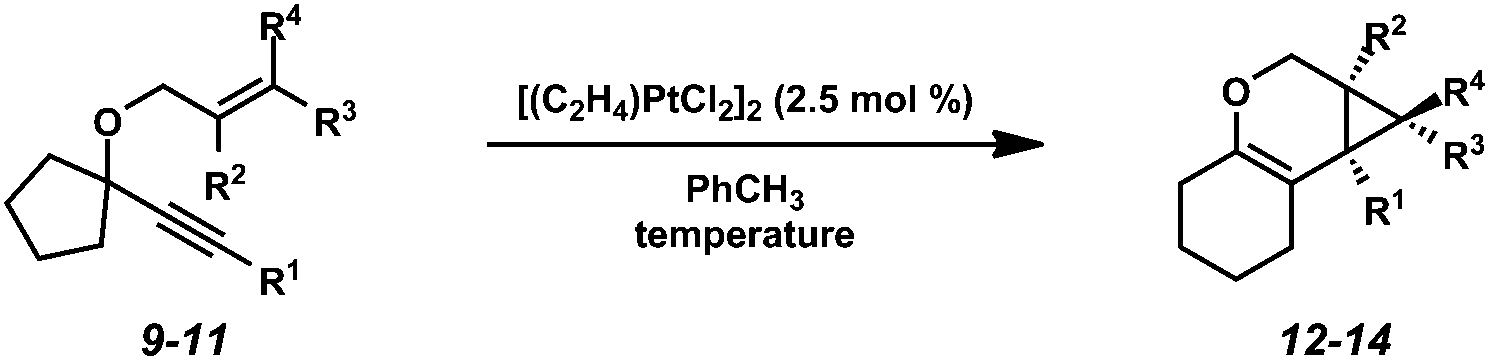
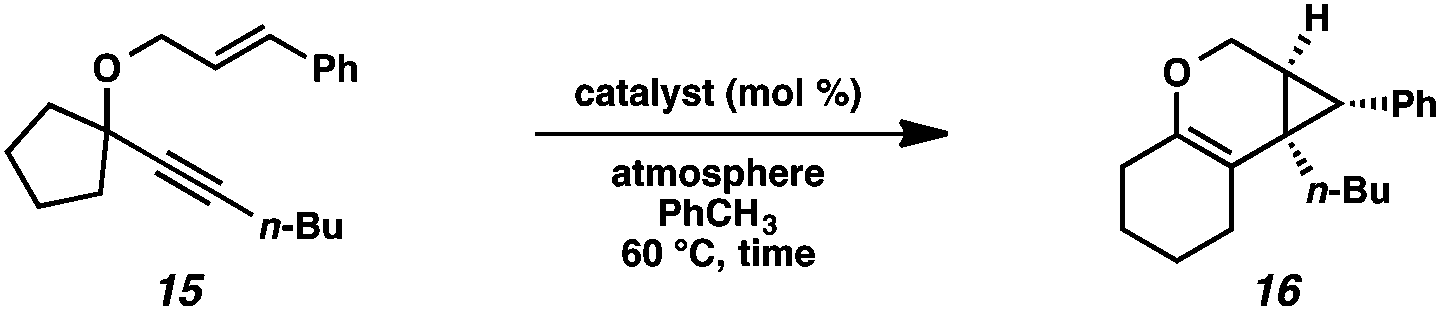
Hoping to see a similar increase in reaction rate and yield, we evaluated the effect of CO on the cycloisomerization of enyne 15, which had occurred in only moderate yield under Ar with PtCl2 (Table 4, entry 1). Encouragingly, the PtCl2-catalyzed cycloisomerization run under CO proceeded in 75% yield and in a shorter reaction time (entry 2). Likewise, Zeise's dimer in the presence of CO gave product 16 in 88% yield, compared to 63% yield under Ar (entries 3 and 4). Thus, our final optimized Zeise's dimer conditions were concluded to be 2.5 mol% catalyst in toluene at 60 °C under CO.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Atmosphere | Time (h) | Isol. yield (%) |
| 1 | PtCl2 (7) | Ar | 43 | 54 |
| 2 | PtCl2 (7) | CO (1 atm) | 25 | 75 |
| 3 | [(C2H4)PtCl2]2 (2.5) | Ar | 43 | 63 |
| 4 | [(C2H4)PtCl2]2 (2.5) | CO (1 atm) | 30 | 88 |
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Catalyst (mol%) | Atmosphere | Time (h) | Resulta |
| a Conversions are approximated by 1H NMR; yields are isolated. | |||||
| 1 | CH2OBn | IrCl(CO)(PPh3)2 (10) | Ar | 16 | <5% conversion |
| 2 | CH2OBn | IrCl(CO)(PPh3)2 (5) | CO (1 atm) | 16 | <5% conversion |
| 3 | n-Bu | Ir4(CO)12 (3) | CO, then Ar | 40 | ∼50% conversion |
| 4 | n-Bu | Ir(CO)2(acac) (5) | CO, then Ar | 40 | <10% conversion |
| 5 | n-Bu | [Ir(cod)Cl]2 (2.5) | Ar | 60 | 24% yield |
| 6 | n-Bu | [Ir(cod)Cl]2 (2.5) | CO, then Ar | 16 | 37% yield |
| 7 | n-Bu | [Ir(dbcot)Cl]2 (2.5) | CO, then Ar | 3 | 68% yield |
|
|
|||
|---|---|---|---|
| Entry | [Ir] | Isolated yield (%) | |
| CO (1 atm) | CO, then Ar | ||
| 1 | [Ir(dbcot)Cl]2 | 72 | 87 |
| 2 | [Ir(cod)Cl]2 | 63 | 81 |
| 3 | [Ir(coe)2Cl]2 | 47 | 50 |
If this theory is correct and the active Ir catalyst is being formed in situ through reaction with CO, then it would seem as if the starting Ir complex should not matter. In the cycloisomerization of enyne 17, however, we observed a slight variance in yield between [Ir(dbcot)Cl]2 and [Ir(cod)Cl]2, and a more significant difference between these two and [Ir(coe)2Cl]2 (Table 6).
We considered that the differential reactivity between the three Ir(I) catalysts may simply be attributed to the ease of [Ir(CO)2Cl]n formation from [Ir(dbcot)Cl]2 and [Ir(cod)Cl]2 compared to [Ir(coe)2Cl]2. When comparing the stability of each complex and their metal–ligand bond strengths, however, this explanation is insufficient. Of the three ligands, the bidentate chelation of dbcot and cod should render the [Ir(dbcot)Cl]2 and [Ir(cod)Cl]2 complexes more stable than [Ir(coe)2Cl]2 due to the chelate effect.22 Further, comparing the dbcot and cod ligands, dbcot should bind more strongly to the metal due to its greater π-accepting character.23 If [Ir(CO)2Cl]n complex formation is dependent solely on ligand displacement, we would expect it to form most efficiently from [Ir(coe)2Cl]2 and least efficiently from [Ir(dbcot)Cl]2 (Fig. 1). Optimal reactivity is obtained, however, using [Ir(dbcot)Cl]2 as the iridium precursor, prompting us to wonder whether the displaced ligand may be playing a role in the reactivity of the metal.
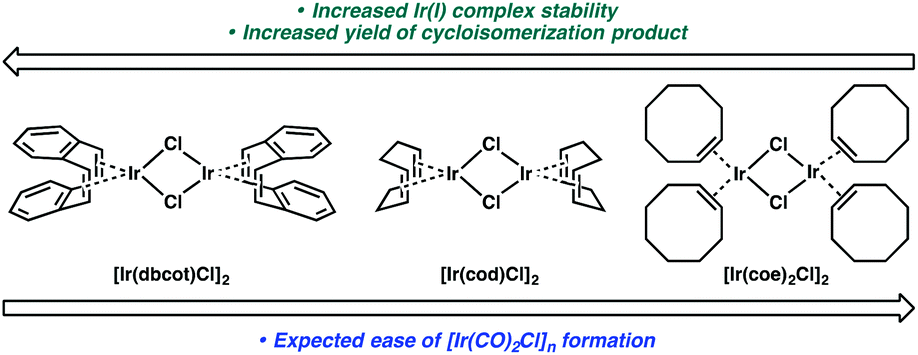 | ||
| Fig. 1 Structures and relative stabilities of three Ir(I) catalysts. | ||
To better understand the catalyst system and to probe whether or not the displaced ligand could be influencing the reactivity of the metal, experiments were performed where the displaced ligand was removed from the reaction mixture prior to addition of the enyne. For these experiments, the Ir(I) catalyst was taken up in toluene and CO was bubbled through the solution, causing the dark blue [Ir(CO)2Cl]n precipitate to form. The toluene, which contained the displaced ligand (confirmed by 1H NMR), was then removed from the flask as thoroughly as possible, leaving the solid behind. The dark blue solid was then taken up in fresh toluene, enyne 15 was added, and the reaction was performed as usual. The yields of product 16 with the displaced ligand still present and with the ligand removed were compared (Table 7). A small decrease in yield from 68% to 64% was observed when dbcot was removed from reaction mixture (entry 1), but almost no change in yield was observed for the [Ir(cod)Cl]2 with and without excess ligand present (entry 2). In the case of [Ir(coe)2Cl]2, a slightly larger decrease in yield was observed when the excess coe was removed from the reaction mixture (entry 3). Ultimately, the yield changes observed when the excess ligand was removed from the reaction mixture were not substantial enough to conclusively indicate whether or not the displaced ligand has an effect on reactivity. It remains possible, however, that when the displaced ligand is still present, some sort of beneficial competitive binding between the ligand and the enyne substrate with the [Ir(CO)2Cl]n complex may be operative. Alternatively, there could simply be more subtle differences in the physical nature of the [Ir(CO)2Cl]n polymeric complex based on the iridium precursor used, and these differences are impacting net reactivity.
|
|
|||
|---|---|---|---|
| Entry | [Ir] | Isolated yield (%) | |
| CO then Ar | CO, then Ar (excess lig. removed) | ||
| 1 | [Ir(dbcot)Cl]2 | 68 | 64 |
| 2 | [Ir(cod)Cl]2 | 37 | 38 |
| 3 | [Ir(coe)2Cl]2 | 49 | 39 |
Substrate scope
In the interest of exploring new reaction conditions and comparing the Pt(II) and Ir(I) systems for different substrates, we investigated the substrate scope of this transformation using both our optimized Zeise's dimer and [Ir(dbcot)Cl]2 conditions. PtCl2 was also employed, specifically for the different ring-size substrates.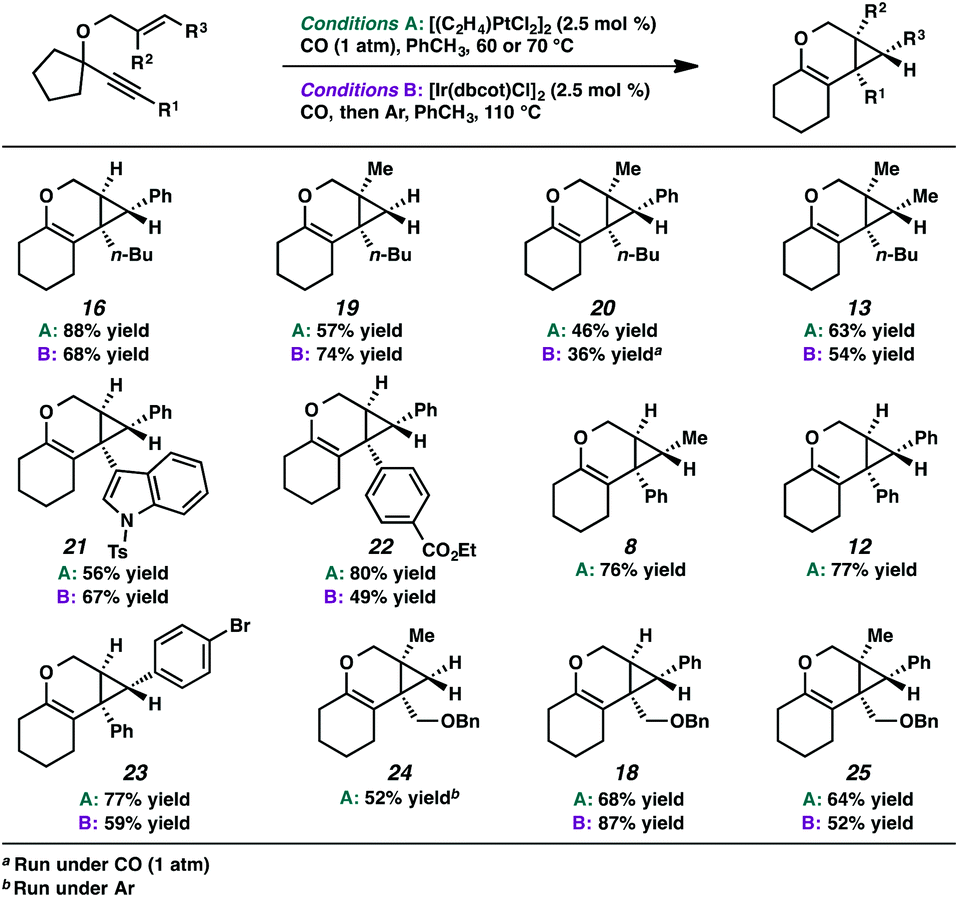 | ||
| Scheme 4 Scope of alkene and alkyne variation. | ||
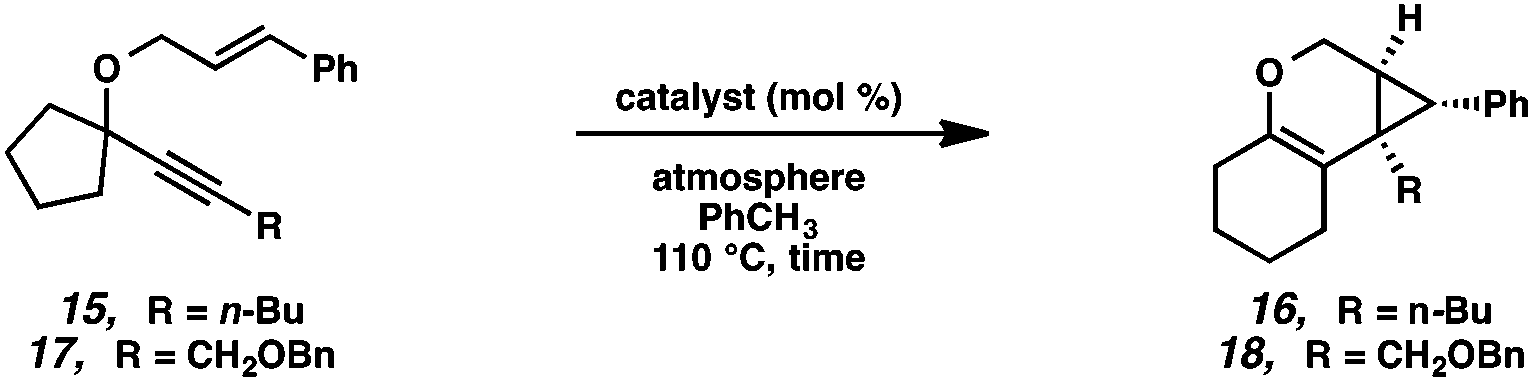
In terms of the Pt(II) vs. Ir(I) conditions, we were not able to draw any conclusions about why one set of conditions seemed to work better than the other; patterns in yields were hard to discern. For example, the Pt(II) conditions worked better for the formation of product 16 (88% yield) vs. 68% yield with the Ir(I) conditions. For the formation of product 18, however, originating from an enyne with the same alkene component and also an alkyl-substituted alkyne, essentially the opposite reactivity trend was observed: Pt(II) catalysis gave the product in 68% yield, while Ir(I) catalysis gave 87% yield. Ultimately, optimal reaction conditions appeared to be fairly substrate-dependent.
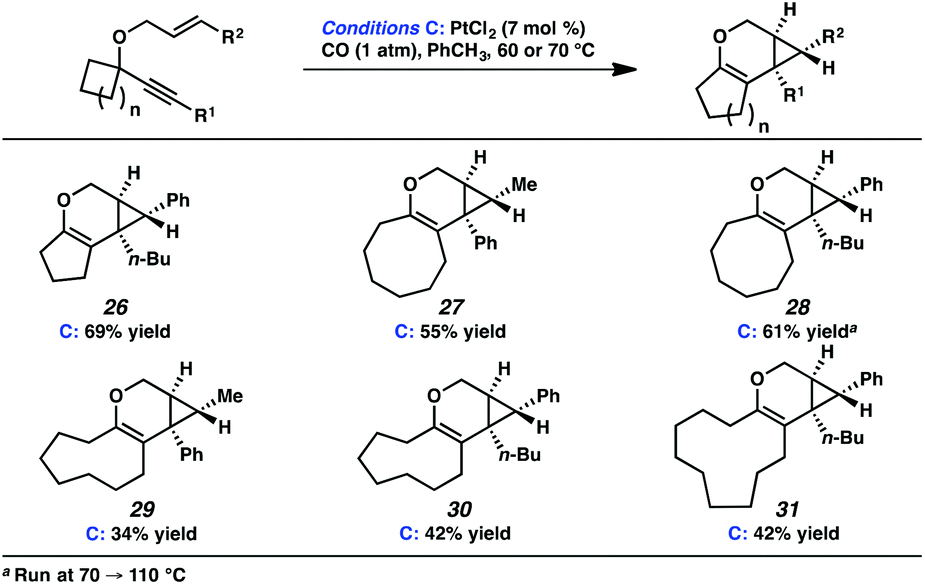 | ||
| Scheme 5 Scope of ring size variation. | ||
These results are in contrast to previous reports by Toste and coworkers where, in the Au-catalyzed cycloisomerization of carbon-tethered 1,5-enynes, 4- and 5-membered rings underwent a ring expansion into the Au-carbenoid, while 6- and 7-membered rings underwent a C–H insertion.24 Though this variance in reactivity may simply be catalyst- or substrate-dependent, both transformations are thought to proceed through similar carbenoid intermediates. Perhaps, in the oxygen-tethered cases here, stabilization by the adjacent oxygen atom through oxocarbenium formation promotes the alkyl migration for the larger ring substrates. Ring strain may also be playing a role; 4- and 5-membered rings have higher strain energies than 6- and 7-membered rings (although 5- and 7-membered ring strains are very close),25 thus the smaller rings will more readily undergo ring expansion. This rationalization would imply that in Toste's Au-catalyzed 1,5-enyne cycloisomerization, product formation is driven by release of ring strain, but in our 1,6-enyne Pt(II)-catalyzed system, release of ring strain may not be playing a significant role.
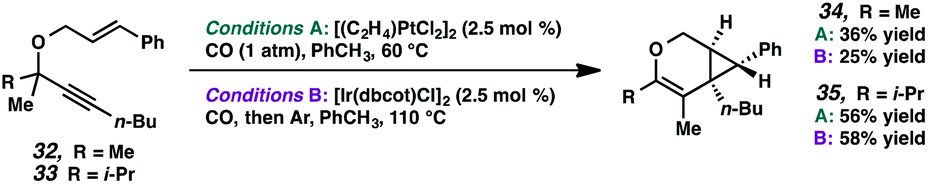 | ||
| Scheme 6 Acyclic C–C bond migrations. | ||
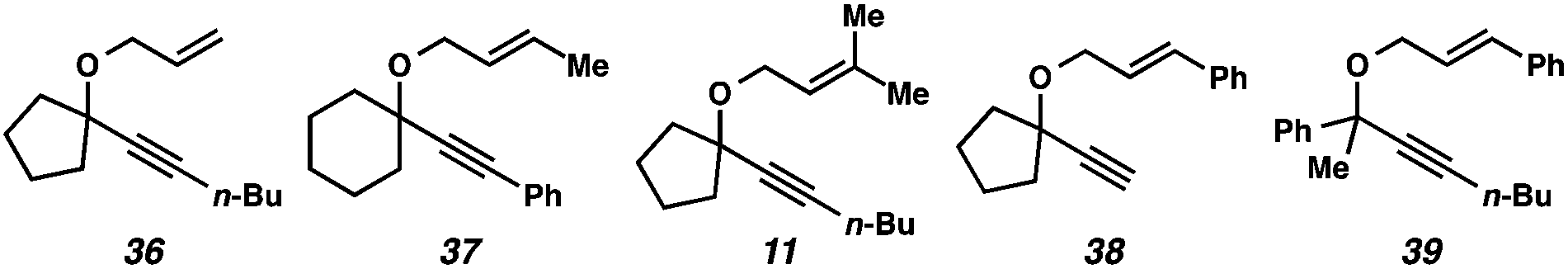 | ||
| Fig. 2 Substrates that did not efficiently undergo the cycloisomerization. | ||
In the case of enyne 11, we observed that the enyne was being consumed under the cycloisomerization conditions, yet the yield of the desired product was very low (11% yield, see Table 3). This result was somewhat surprising considering the carbocation intermediate that would result from nucleophilic attack of the geminal dimethyl alkene should be relatively stable. To try to explain this consumption of starting material but low yield, we wondered if product decomposition may be occurring. Probing this hypothesis, we exposed cycloisomerization product 14 to the reaction conditions (Zeise's dimer, PhCH3, 70–105 °C, ∼18 h), but no product decomposition was observed by TLC or 1H NMR.
We also considered that some kind of catalyst inhibition could be occurring, where product generated during the reaction could be inhibiting the formation of more product through complexation of the catalyst with the enol ether moiety. To test this hypothesis, the cycloisomerization of prenyl substrate 11 was performed with 0.5 equivalents of the product (14) also included in the initial reaction mixture. Substrate 11 was completely consumed despite the presence of the extra product, so product inhibition was disproved as well.
Lastly, the cycloisomerization of enyne 11 was performed in an NMR tube to ascertain side products in the reaction mixture that could have been removed through the workup procedure. The 1H NMR experiment on the cycloisomerization of prenyl substrate 11 revealed diagnostic signals corresponding to 3-methyl-2-butenal (40). Aldehyde 40 could arise from ionization of enyne 11 to give the propargyl cation and oxygen anion, then oxidation of the resulting alcohol (Scheme 7a). The formation of this aldehyde may explain why the starting material was consumed, but a low yield of product 14 was obtained. In addition, we believe this ether ionization/aldehyde formation is also likely occurring in some of our other, more modest-yielding cycloisomerizations.27
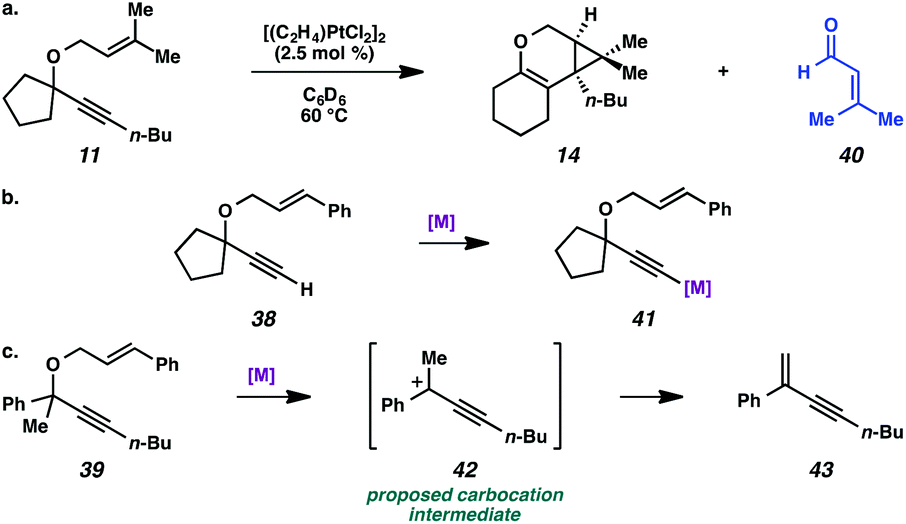 | ||
| Scheme 7 (a) Decomposition of enyne 11 to aldehyde 40. (b) Proposed deactivation of metal by terminal alkyne. (c) Proposed ionization of ether 39. | ||
Terminal alkyne substrate 38 also did not undergo the cycloisomerization. This outcome is not uncommon in the enyne cycloisomerization literature, although certain approaches to terminal alkyne substrates have seen success.3g,28 A possible explanation for this lack of reactivity could be deprotonation or C–H insertion of the metal at the terminus of the alkyne (41), rendering the catalyst incapable of promoting the cycloisomerization (Scheme 7b).
Lastly, acyclic substrate 39 with phenyl substitution at the propargylic position also did not undergo the cycloisomerization, perhaps due to the stability of benzylic carbocation (42) upon ionization of the tethered ether (Scheme 7c). Only enyne 43 was observed.29
Synthesis of macrolactones from cycloisomerization products
Macrocycle synthesis is of increasing interest in the natural product community due to the prevalence of this motif in biologically active molecules.30 We envisioned that our cycloisomerization/ring expansion process could provide access to macrolactones via oxidative cleavage of the cyclic enol ether of the products. With this in mind, a two-step method to convert the tricyclic products into macrolactones was devised (Scheme 8). Reaction of tricycles 16 and 18 with catalytic OsO4 gave diols 44 and 45 in 76% and 58% yield, respectively.31 The resulting diols were then cleaved with Pb(OAc)4 to give macrolactones 46 and 47.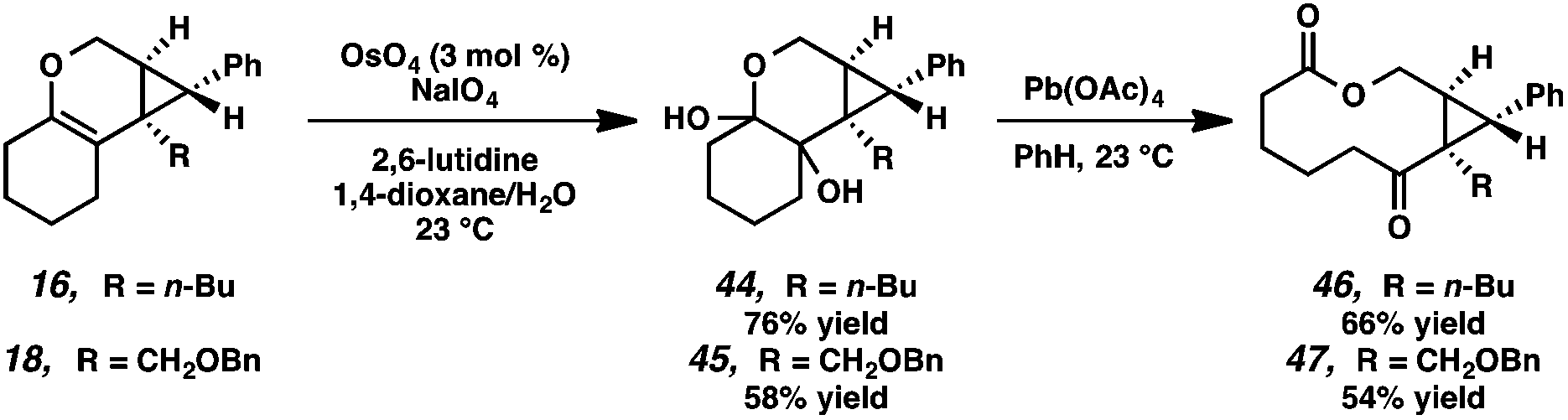 | ||
| Scheme 8 Oxidative cleavage to access macrolactones. | ||
Conclusions
We have developed Pt(II)- and Ir(I)-catalyzed enyne cycloisomerization/C–C bond migration reactions that provide access to tricyclic enol ethers. A variety of substituted enyne substrates undergo the cycloisomerization, and both cyclic and acyclic alkyl groups are able to migrate into the carbenoid intermediates. The influence of CO on both the Pt(II)- and Ir(I)-catalyst systems was explored. The cycloisomerization products can also be cleaved to generate macrolactones. Ultimately, this ring expansion process can provide access to large carbocycles, which are traditionally synthetically challenging.Experimental section
General procedure for the cycloisomerization of oxygen-tethered 1,6-enynes catalyzed by ([(C2H4)PtCl2]2)
To a solution of the 1,6-enyne (1 equiv.) in toluene (0.06 M) in a 2-dram vial under argon at 23 °C was quickly added Zeise's dimer (2.5 mol%). CO was bubbled through the solution using a balloon and needle outlet (ca. 30 s). The balloon and outlet were then removed, and the solution was stirred at the described temperature until all of the starting material was consumed, as determined by TLC. The reaction mixture was allowed to cool to ambient temperature and diluted with an approximately equal amount of hexanes. The mixture was stirred for 15 min, then passed through a small plug of Al2O3 (hexanes → 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexanes/EtOAc eluent). The volatile materials were removed by rotary evaporation, and the resulting residue was purified by flash chromatography to afford the product enol ether.
:1 hexanes/EtOAc eluent). The volatile materials were removed by rotary evaporation, and the resulting residue was purified by flash chromatography to afford the product enol ether.
General procedure for the cycloisomerization of oxygen-tethered 1,6-enynes catalyzed by [Ir(dbcot)Cl]2
To a solution of the 1,6-enyne (1 equiv.) in toluene (0.06 M) in a 16 × 125 mm glass culture tube under argon at 23 °C was quickly added [Ir(dbcot)Cl]2 (2.5 mol%). CO was bubbled through the solution using a balloon and needle outlet (ca. 30 s), during which time the reaction mixture turned a dark-blue/black color. The balloon was removed, and argon was bubbled through the reaction mixture in the same manner. The septum was quickly replaced with a Teflon cap, and the reaction mixture was stirred at 110 °C until all of the starting material was consumed, as determined by TLC. The reaction mixture was allowed to cool to ambient temperature and diluted with an approximately equal amount of hexanes. 1,3-Bis(diphenylphosphino)propane (dppp) (0.25 equiv.) was also added to the mixture. The mixture was stirred for 15 min, then passed through a small plug of Al2O3 (hexanes → 1:1 hexanes/EtOAc eluent). The volatile materials were removed by rotary evaporation, and the resulting residue was purified by flash chromatography to afford the product enol ether.
General procedure for the cycloisomerization of oxygen-tethered 1,6-enynes catalyzed by PtCl2
To a solution of the 1,6-enyne (1 equiv.) in toluene (0.06 M) in a 2-dram vial under argon at 23 °C was quickly added PtCl2 (7 mol%). CO was bubbled through the solution using a balloon and needle outlet (ca. 30 s). The balloon and outlet were then removed, and the solution was stirred at the described temperature until all of the starting material was consumed, as determined by TLC. The reaction mixture was allowed to cool to ambient temperature and diluted with an approximately equal amount of hexanes. The mixture was stirred for 15 min, then passed through a small plug of Al2O3 (hexanes → 1:1 hexanes/EtOAc eluent). The volatile materials were removed by rotary evaporation, and the resulting residue was purified by flash chromatography to afford the product enol ether.
Acknowledgements
The National Institutes of Health (NIGMS, R01GM110560) is gratefully acknowledged. Dr Brian Newell is acknowledged for X-ray crystallographic expertise. Prof. Louis Fensterbank is acknowledged for helpful discussions.Notes and references
- (a) B. M. Trost and M. J. Krische, Synlett, 1998, 1–16 CrossRef; (b) C. Nieto-Oberhuber, S. López, E. Jiménez-Núñez and A. M. Echavarren, Chem. – Eur. J., 2006, 12, 5916–5923 CrossRef CAS PubMed; (c) L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271–2296 CrossRef CAS; (d) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed; (e) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed; (f) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS PubMed; (g) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed; (h) S. I. Lee and N. Chatani, Chem. Commun., 2009, 371–384 RSC; (i) H. Schmidbaur and A. Schier, Organometallics, 2010, 29, 2–23 CrossRef CAS; (j) A. Marinetti, H. Jullien and A. Voituriez, Chem. Soc. Rev., 2012, 41, 4884–4908 RSC; (k) L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953–965 CrossRef CAS PubMed; (l) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed.
- (a) E. T. Newcomb and E. M. Ferreira, Org. Lett., 2013, 15, 1772–1775 CrossRef CAS PubMed; (b) E. T. Newcomb, P. C. Knutson, B. A. Pedersen and E. M. Ferreira, J. Am. Chem. Soc., 2016, 138, 108–111 CrossRef CAS PubMed.
- For select examples of related cycloisomerizations to form oxabicyclo[4.1.0]heptene compounds, see: (a) J. Blum, H. Beer-Kraft and Y. Badrieh, J. Org. Chem., 1995, 60, 5567–5569 CrossRef CAS; (b) A. Fürstner, F. Stelzer and H. Szillat, J. Am. Chem. Soc., 2001, 123, 11863–11869 CrossRef; (c) C. Nevado, C. Ferrer and A. M. Echavarren, Org. Lett., 2004, 6, 3191–3194 CrossRef CAS PubMed; (d) C. Ferrer, M. Raducan, C. Nevado, C. K. Claverie and A. M. Echavarren, Tetrahedron, 2007, 63, 6306–6316 CrossRef CAS; (e) A. Tenaglia and S. Gaillard, Angew. Chem., Int. Ed., 2008, 47, 2454–2457 CrossRef CAS PubMed; (f) C.-M. Chao, D. Beltrami, P. Y. Toullec and V. Michelet, Chem. Commun., 2009, 6988–6990 RSC; (g) S. H. Sim, S. I. Lee, J. H. Park and Y. K. Chung, Adv. Synth. Catal., 2010, 352, 317–322 CrossRef CAS; (h) S. Y. Kim, Y. Park and Y. K. Chung, Angew. Chem., Int. Ed., 2010, 49, 415–418 CrossRef CAS PubMed; (i) Z. Chen, Y.-X. Zhang, Y.-H. Wang, L.-L. Zhu, H. Liu, X.-X. Li and L. Guo, Org. Lett., 2010, 12, 3468–3471 CrossRef CAS PubMed; (j) A. Pradal, C.-M. Chao, P. Y. Toullec and V. Michelet, Beilstein J. Org. Chem., 2011, 7, 1021–1029 CrossRef CAS PubMed; (k) S. Y. Kim, Y. Park, S. Son and Y. K. Chung, Adv. Synth. Catal., 2012, 354, 179–186 CrossRef CAS; (l) H. Teller, M. Corbet, L. Mantilli, G. Gopakumar, R. Goddard, W. Thiel and A. Fürstner, J. Am. Chem. Soc., 2012, 134, 15331–15342 CrossRef CAS PubMed; (m) T. Nishimura, Y. Takiguchi, Y. Maeda and T. Hayashi, Adv. Synth. Catal., 2013, 355, 1374–1382 CrossRef CAS; (n) E. M. Barreiro, E. V. Boltukhina, A. J. P. White and K. K. Hii, Chem. – Eur. J., 2015, 21, 2686–2690 CrossRef CAS PubMed.
- For seminal examples of cycloisomerizations of oxygen-tethered enynes based on catalytic alkyne π-activation, see: (a) A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS; (b) B. Martín-Matute, D. J. Cárdenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2001, 40, 4754–4757 CrossRef.
- A. Simonneau, Y. Harrak, L. Jeanne-Julien, G. Lemière, V. Mouriès-Mansuy, J.-P. Goddard, M. Malacria and L. Fensterbank, ChemCatChem, 2013, 5, 1096–1099 CrossRef CAS.
- S. M. Stevenson, E. T. Newcomb and E. M. Ferreira, Chem. Commun., 2014, 50, 5239–5241 RSC.
- (a) P. A. Allegretti and E. M. Ferreira, Org. Lett., 2011, 13, 5924–5927 CrossRef CAS PubMed; (b) P. A. Allegretti and E. M. Ferreira, Chem. Sci., 2013, 4, 1053–1058 RSC; (c) P. A. Allegretti and E. M. Ferreira, J. Am. Chem. Soc., 2013, 135, 17266–17269 CrossRef CAS PubMed; (d) P. A. Allegretti, K. Huynh, T. J. Ozumerzifon and E. M. Ferreira, Org. Lett., 2016, 18, 64–67 CrossRef CAS PubMed.
- K. Ohe, Bull. Korean Chem. Soc., 2007, 28, 2153–2161 CrossRef CAS.
- For select examples, see: (a) M. J. Johansson, D. J. Gorin, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 18002–18003 CrossRef CAS PubMed; (b) E. Mainetti, V. Mouriés, L. Fensterbank, M. Malacria and J. Marco-Contelles, Angew. Chem., Int. Ed., 2002, 41, 2132–2135 CrossRef CAS; (c) K. Miki, K. Ohe and S. Uemura, Tetrahedron, 2003, 44, 2019–2022 CrossRef CAS.
- J. Xiao and X. Li, Angew. Chem., Int. Ed., 2011, 50, 7226–7236 CrossRef CAS PubMed.
- T. de Haro and C. Nevado, Synthesis, 2011, 2530–2539 CAS.
- (a) R. K. Shiroodi and V. Gevorgyan, Chem. Soc. Rev., 2013, 42, 4991–5001 RSC; (b) B. Crone and S. F. Kirsch, Chem. – Eur. J., 2008, 14, 3514–3522 CrossRef CAS PubMed; (c) A. S. Dudnik, Y. Xia, Y. Li and V. Gevorgyan, J. Am. Chem. Soc., 2010, 132, 7645–7655 CrossRef CAS PubMed; (d) Y. Xia, A. S. Dudnik, Y. Li and V. Gevorgyan, Org. Lett., 2010, 12, 5538–5541 CrossRef CAS PubMed; (e) D. Garayalde and C. Nevado, Beilstein J. Org. Chem., 2011, 7, 767–780 CrossRef CAS PubMed; (f) D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272–286 CrossRef CAS.
- For select examples of alkyl migrations into metal carbenoids in enyne cycloisomerization, see: (a) D. J. Gorin, N. R. Davis and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260–11261 CrossRef CAS PubMed; (b) H. Kusama, Y. Miyashita, J. Takaya and N. Iwasawa, Org. Lett., 2006, 8, 289–292 CrossRef CAS PubMed; (c) A. S. K. Hashmi, M. Wieteck, I. Braun, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 10633–10637 CrossRef CAS PubMed; (d) A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644–661 CrossRef CAS; (e) T. Lauterbach, S. Gatzweiler, P. Nösel, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2013, 355, 2481–2487 CrossRef CAS; (f) P. Nösel, L. Nunes dos Santos Comprido, T. Lauterbach, M. Rudolph, F. Rominger and A. S. K. Hashmi, J. Am. Chem. Soc., 2013, 135, 15662–15666 CrossRef PubMed; (g) M. V. Vita, P. Mieville and J. Waser, Org. Lett., 2014, 16, 5768–5771 CrossRef CAS PubMed.
- A. Fürstner, P. W. Davies and T. Gress, J. Am. Chem. Soc., 2005, 127, 8244–8245 CrossRef PubMed.
- (a) N. Chatani, T. Morimoto, T. Muto and S. Murai, J. Am. Chem. Soc., 1994, 116, 6049–6050 CrossRef CAS; (b) A. Fürstner and P. W. Davies, J. Am. Chem. Soc., 2005, 127, 15024–15025 CrossRef PubMed; (c) A. Fürstner and C. Aïssa, J. Am. Chem. Soc., 2006, 128, 6306–6307 CrossRef PubMed.
- Y. Gimbert, L. Fensterbank, V. Gandon, J.-P. Goddard and D. Lesage, Organometallics, 2013, 32, 374–376 CrossRef CAS.
- T. Shibata, Y. Kobayashi, S. Maekawa, N. Toshida and K. Takagi, Tetrahedron, 2005, 61, 9018–9024 CrossRef CAS.
- L. Vaska and J. W. DiLuzio, J. Am. Chem. Soc., 1961, 83, 2784–2785 CrossRef CAS.
- No migrations were observed with N-tethered 1,6-enynes under any Ir or Pt conditions we evaluated.
- dbcot: dibenzo[a,e]cyclooctatetraene.
- D. Roberto, E. Cariati, R. Psaro and R. Ugo, Organometallics, 1994, 13, 4227–4231 CrossRef CAS.
- J. F. Hartwig, Organotransition Metal Chemistry, University Science Books, Mill Valley, California, 2010, p. 48 Search PubMed.
- (a) D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 621–627 CrossRef CAS; (b) A. Singh and P. R. Sharp, Organometallics, 2006, 25, 678–683 CrossRef CAS.
- (a) M. R. Luzung, J. P. Markham and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 10858–10859 CrossRef CAS PubMed; (b) Y. Horino, T. Yamamoto, K. Uedo, S. Kuroda and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 2809–2811 CrossRef CAS PubMed.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, New York, 1994, pp. 675–678 Search PubMed.
- No product derived from migration of the i-Pr group was observed in the crude reaction mixture (determined by 1H NMR).
- Substrates with substituents cis to the ethereal linker (e.g., Z-alkenes) were generally ineffective in this process. The methyl substituent in enyne 11 is also reflective of this observation.
- E. Benedetti, A. Simonneau, A. Hours, H. Amouri, A. Penoni, G. Palmisano, M. Malacria, J.-P. Goddard and L. Fensterbank, Adv. Synth. Catal., 2011, 353, 1908–1912 CrossRef CAS.
- Curiously, we did not observe cinnamaldehyde in the crude reaction mixture.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- The use of NaIO4 in the first step did not result in diol cleavage.
Footnotes |
| † Dedicated to Professor Barry Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, characterization data, and spectra. CCDC 963880. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00224b |
| This journal is © the Partner Organisations 2016 |