
Direct and metal-free oxidative amination of sp3 C–H bonds for the construction of 2-hetarylquinazolin-4(3H)-ones†
Huanhuan
Liu
a,
Tianran
Zhai
a,
Shiteng
Ding
a,
Yalei
Hou
a,
Xiangyu
Zhang
a,
Lei
Feng
a and
Chen
Ma
*ab
aSchool of Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, P. R. China
bState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing, 100191, P. R. China. E-mail: chenma@sdu.edu.cn
First published on 6th July 2016
Abstract
A new method for the synthesis of 2-hetarylquinazolin-4(3H)-ones from 2-aminobenzamides and (2-azaaryl)methanes under transition metal-free conditions is described. This protocol features a wide substrate scope with a broad range of functional group tolerance under mild conditions. The oxidation of (2-azaaryl)methanes to (2-azaaryl)methanals is proposed as the key step in this transformation.
Quinazolin-4(3H)-ones and their derivatives have attracted considerable attention because of their important role in medicinal chemistry.1 Many derivatives exhibit biological activities, including anticancer,2 antiviral,3 anti-inflammatory,4 antibacterial,5 antifungal,6 anticonvulsant,7 and antimalarial8 properties. Furthermore, the 2-heterocycle-substituted quinazolin-4(3H)-one skeleton is present in various biologically active agents.9 For example, luotonin A has cytotoxicity toward the murine leukemia P388 cell line,10 while luotonin B has cytotoxicity toward leukemia P388 cells,10b and bouchardatine11 is present in the natural product Bouchardatia neurococca.
Although numerous methods for the synthesis of quinazolin-4(3H)-ones have been developed,9d,12 reports about construction of 2-hetarylquinazolin-4(3H)-ones are still rare. Traditionally, 2-hetarylquinazolin-4(3H)-ones are prepared by the dehydration condensation of 2-aminobenzamides with (2-azaaryl)methanals or (2-azaaryl)methanoic acids.13 However, the corresponding (2-azaaryl)methanals are usually expensive and are easily oxidized to their acid derivatives, thus limiting their further direct use, and the vast majority of (2-azaaryl)methanals even couldn't be synthesized until now. On the other hand, harsh conditions are usually needed for the corresponding (2-azaaryl)methanoic acids due to their low reactivity. Moreover, multistep synthesis leads to low atom economy in the above cases. To overcome these drawbacks, it is highly desirable to develop a more straightforward, efficient and convenient approach for the synthesis of 2-hetarylquinazolin-4(3H)-ones.
Recent research has revealed that a suitable Lewis or Brønsted acid can convert the 2-methylazaarene to its enamine counterpart,14 which makes it a powerful strategy for C–H functionalization. Recently, Zhou, Yin and coworkers reported a Cu-catalyzed strategy from 2-aminobenzamides and (2-azaaryl)methanes to assemble 2-hetarylquinazolin-4(3H)-ones (Scheme 1).15 The copper catalyst was required in this transformation and excessive (2-azaaryl)methane was needed. In addition, this reaction was also completed with a relatively high temperature. In order to develop a direct and metal-free method for the construction of these heterocyclic compounds, we report an efficient and convenient reaction of 2-aminobenzamides and (2-azaaryl)methanes for the construction of 2-hetarylquinazolin-4(3H)-ones under mild conditions.
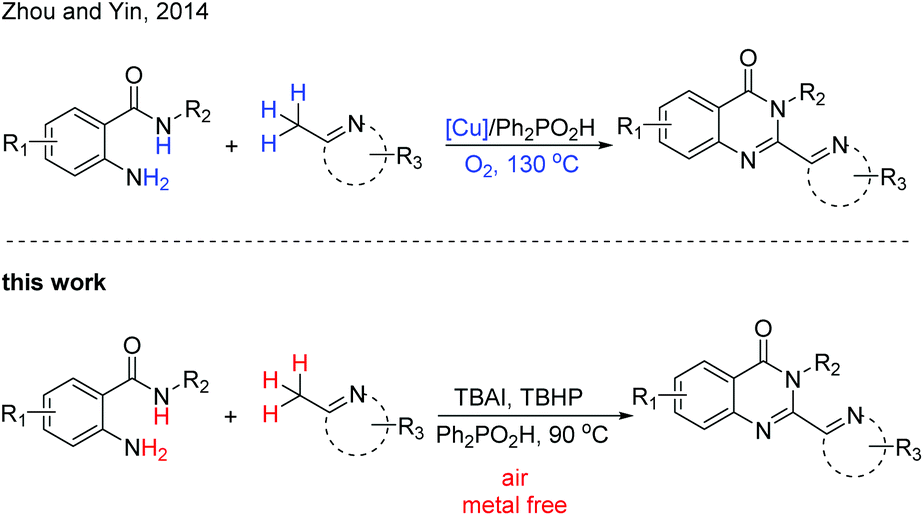 | ||
| Scheme 1 Different protocols for the synthesis of 2-hetarylquinazolin-4(3H)-ones. | ||
At the outset of this study, we employed 2-aminobenzamide 1a and 2-methylquinoline 2a as the model substrates to optimize the reaction conditions (Table 1). We found that an acid, iodine source and oxidant are all essential for the reaction (entries 1–3). Different acids were screened (entries 4–8), and Ph2PO2H showed the best activity. When the reaction was mediated by other iodine sources instead of TBAI (entries 9–13), the yield of 3a was decreased. Then, we changed the iodine source to TBAB with no corresponding product observed (entry 14), which indicated that TBAI played an important role in this reaction rather than serving as a phase transfer catalyst. Different oxidants were screened (entries 15–18), and TBPB gave a comparable yield of 3a compared with TBHP (entry 16). The effect of solvents was also investigated (entries 19–21) and DMSO was found to be the best choice.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Acid | [I] | [O] | Solvent | Yieldb, % |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.3 mmol), acid (0.3 mmol), [I] (0.3 mmol), [O] (1.2 mmol) and solvent (5 mL), 90 °C, under air, 10 h. TBHP (70% in water). b Isolated yield. c Reaction with TBAB. | |||||
| 1 | — | TBAI | TBHP | DMSO | Trace |
| 2 | Ph2PO2H | — | TBHP | DMSO | None |
| 3 | Ph2PO2H | TBAI | — | DMSO | Trace |
| 4 | Ph 2 PO 2 H | TBAI | TBHP | DMSO | 93 |
| 5 | CH3CO2H | TBAI | TBHP | DMSO | 53 |
| 6 | CF3CO2H | TBAI | TBHP | DMSO | 86 |
| 7 | TsOH·H2O | TBAI | TBHP | DMSO | 52 |
| 8 | CH3SO3H | TBAI | TBHP | DMSO | 87 |
| 9 | Ph2PO2H | KI | TBHP | DMSO | 55 |
| 10 | Ph2PO2H | ZnI2 | TBHP | DMSO | 37 |
| 11 | Ph2PO2H | I2 | TBHP | DMSO | 16 |
| 12 | Ph2PO2H | NIS | TBHP | DMSO | 12 |
| 13 | Ph2PO2H | NH4I | TBHP | DMSO | 22 |
| 14c | Ph2PO2H | — | TBHP | DMSO | None |
| 15 | Ph2PO2H | TBAI | DTBP | DMSO | 29 |
| 16 | Ph2PO2H | TBAI | TBPB | DMSO | 90 |
| 17 | Ph2PO2H | TBAI | K2S2O8 | DMSO | Trace |
| 18 | Ph2PO2H | TBAI | BPO | DMSO | 15 |
| 19 | Ph2PO2H | TBAI | TBHP | Toluene | 22 |
| 20 | Ph2PO2H | TBAI | TBHP | Dioxane | 13 |
| 21 | Ph2PO2H | TBAI | TBHP | PhCl | 31 |
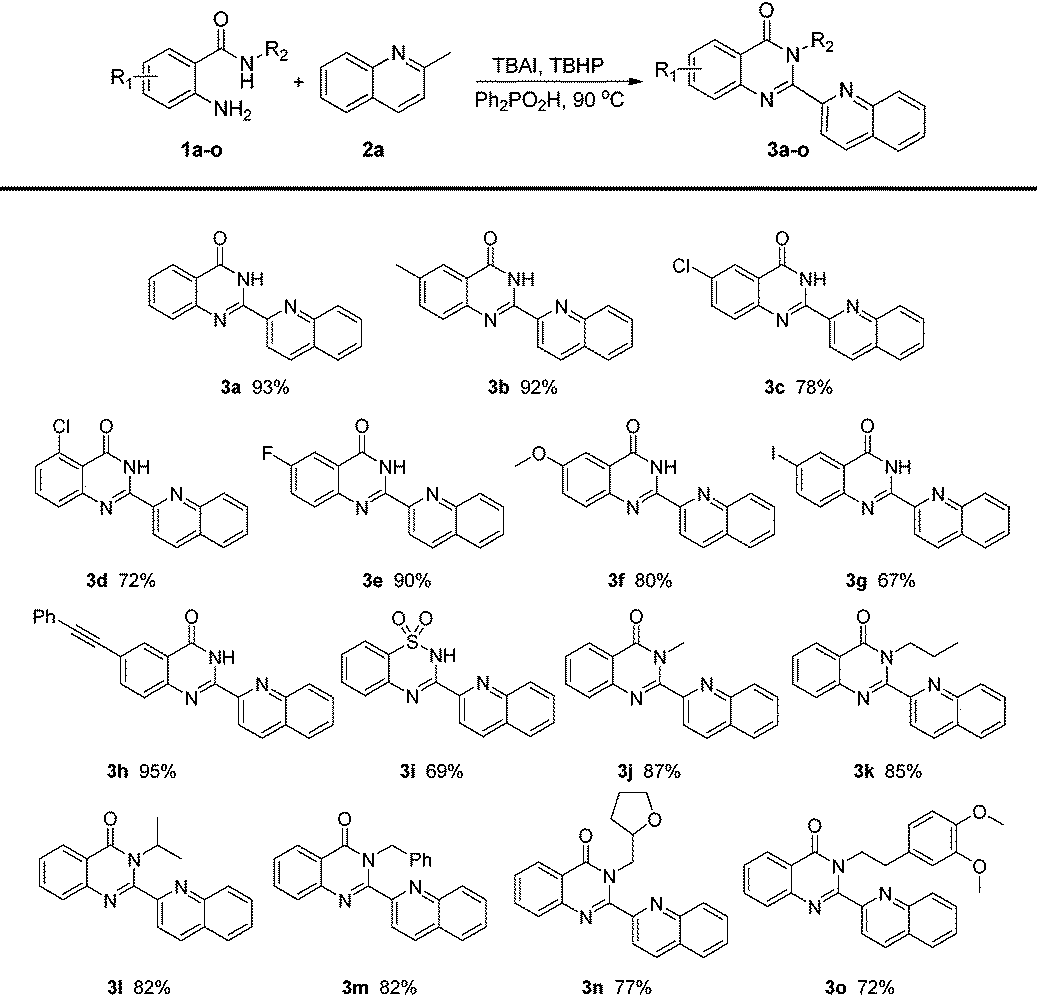
With the optimized conditions in hand, the scope of 2-aminobenzamides was firstly investigated. As shown in Table 2, substrates with electron-withdrawing groups or electron-donating groups can react smoothly to afford the corresponding products in good to excellent yields (3b–h). The electronic nature of substituents showed no significant impact on the yields of 3. To our delight, product 3g could be obtained in 67% yield and its iodine group could be further transformed into other functional groups easily, while 1h bearing a phenylethynyl group was also tolerated to give 3h in 95% yield. Substrates with different R2 groups also worked well to give the corresponding products (3j–o). Product 3j with a methyl group could be obtained in 87% yield while the yields of the other products with more hindered R2 groups decreased modestly. Notably, 2-aminobenzenesulfonamide was also tolerated to give the corresponding product 3i in 69% yield. It is worth mentioning that the product 3a acted as a valuable intermediate for luotonin A, luotonin B and luotonin E which were a series of biologically active compounds.13a
| a Reaction conditions: 1 (0.3 mmol), 2a (0.3 mmol), Ph2PO2H (0.3 mmol), TBAI (0.3 mmol), TBHP (1.2 mmol) and DMSO (5 mL), 90 °C, under air, 10 h. |
|---|
|
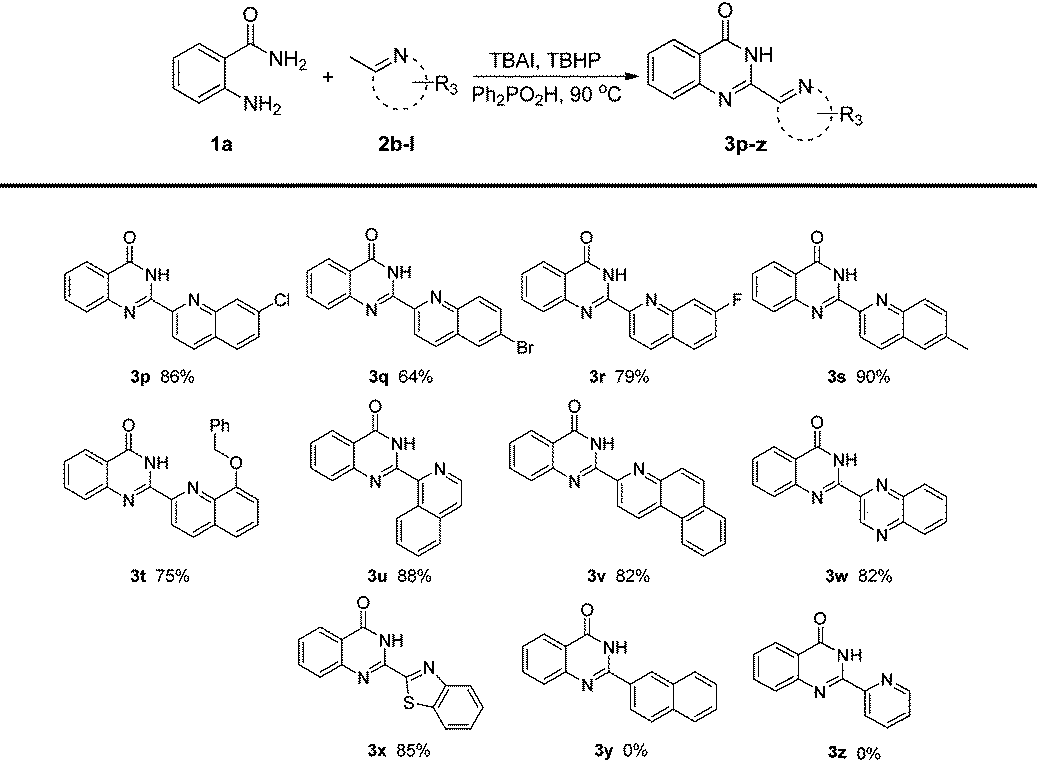
Encouraged by the above promising results, we next examined the substituted (2-azaaryl)methanes as shown in Table 3. 2-Methylquinolines with electron-withdrawing groups (fluoro, chloro and bromo) or electron-donating groups (methyl, benzyloxy) were tolerated under the optimal reaction conditions (3p–t). Besides 2-methylquinolines, the reaction system could also be applied to other (2-azaaryl)methanes. For example, 1-methylisoquinoline could give a relatively high yield of 88%. 3-Methylbenzo[f]quinolone could be employed to afford 82% 3v. 2-Methylquinoxaline which contains another nitrogen atom was suitable for the protocol to obtain 3w in 82% yield. In addition, 2-methylbenzo[d]thiazole which consists of a five-membered thiazole and a benzene ring could afford 85% 3x under the standard reaction conditions. It should be noted that the substrate 2-methylnaphthalene could not give the desired product 3y. This result may indicate that the nitrogen atom was essential for our protocol. In addition, we have examined pyridine derivatives such as 2-methylpyridine, 2,6-dimethylpyridine, 2-iodo-6-methylpyridine, etc. The reactants were recovered and no desired product generated, even up to 130 °C . We speculate that it's more difficult for the pyridine derivatives to perform the dearomatization process than the quinolines.
| a Reaction conditions: 1a (0.3 mmol), 2 (0.3 mmol), Ph2PO2H (0.3 mmol), TBAI (0.3 mmol), TBHP (1.2 mmol) and DMSO (5 mL), 90 °C, under air, 10 h. |
|---|
|
To demonstrate the practicability of this newly established protocol, a gram-scale experiment was carried out under the standard reaction conditions (Scheme 2). The reaction solution was poured into water after the reaction was completed, and 3a precipitated out. Chemically pure 3a was collected by filtration rather than column chromatography. To our delight, product 3a was obtained with 1.35 g in 90% yield, which made it more convenient for its further applications.
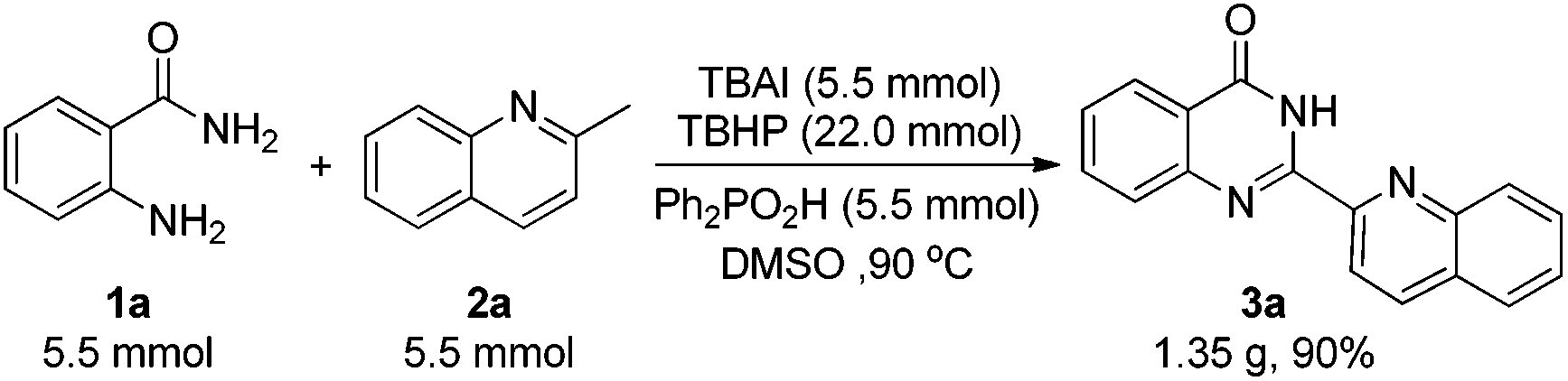 | ||
| Scheme 2 Gram-scale synthesis of 3a. | ||
To gain some insights into the reaction mechanism, several control experiments were carried out. First, a trace amount of 3a was observed when TEMPO was added under the standard conditions (Scheme 3a). This indicated that the reaction might include a radical process. To our surprise, we obtained quinoline-2-carbaldehyde in 68% yield when the reaction was performed without 1a (Scheme 3b). To the best of our knowledge, quinoline-2-carbaldehyde was usually synthesized by using poisonous reagents such as SeO2 or H3SeO3, as there was no such direct and mild method.16 Subsequently, we obtained 3a in 95% yield when 2a was replaced by quinoline-2-carbaldehyde in the reaction with 1a under the standard conditions (Scheme 3c). The oxidation of 2-methylquinoline to quinoline-2-carbaldehyde was proposed as a key step in this transformation. In 2014, Li's group reported a radical method with 2-aminobenzamides and methylarenes to construct quinazolinones.12l When we carried out the reaction of 1a and 2a under the conditions in Li's report (Scheme 3d), only a trace amount of 3a was detected, which suggested that their reaction conditions were not suitable for the preparation of 2-hetarylquinazolin-4(3H)-ones.
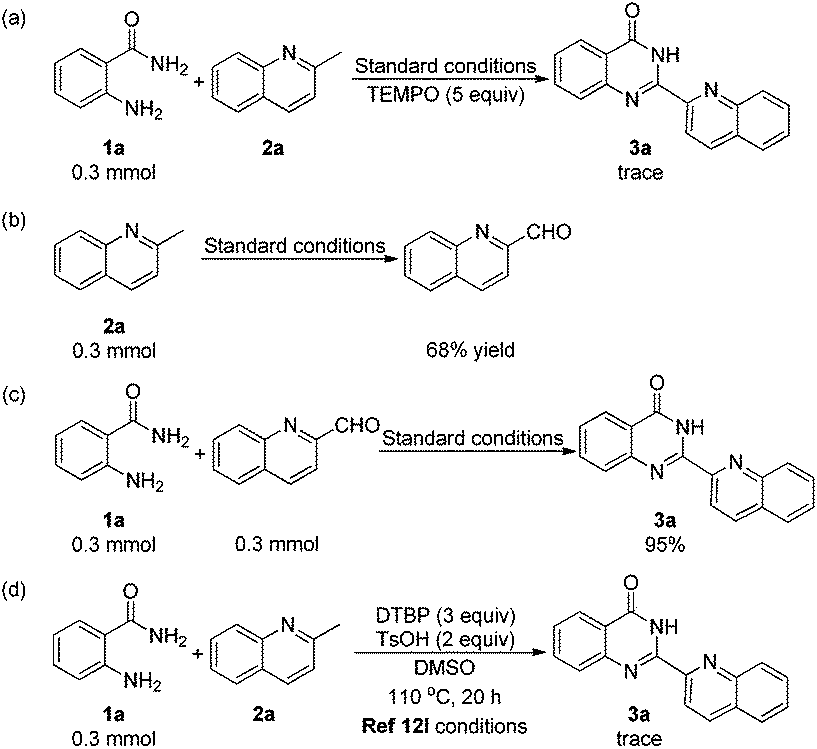 | ||
| Scheme 3 Control experiments. | ||
An assumed pathway for the formation of 2-hetarylquinazolin-4(3H)-one derivatives is illustrated in Scheme 4 according to the results above and previous research.17,18 The tert-butoxyl and tert-butylperoxy radicals were generated from the TBAI–TBHP system. The addition of a RO radical with 2a′, which was generated from 2a by the acid-promoted isomerization, afforded intermediate 4. Then, 4 could be further oxidized by a RO radical to give 5 and ROH. After that, another acid-promoted isomerization afforded 5′, and then addition of a RO radical gave 6 which was oxidized to 7, next hydrolysis of 7 led to quinoline-2-carbaldehyde. Dehydration condensation of quinoline-2-carbaldehyde with 1a afforded an imine intermediate which could undergo intramolecular addition and oxidation to afford the final product 3a.
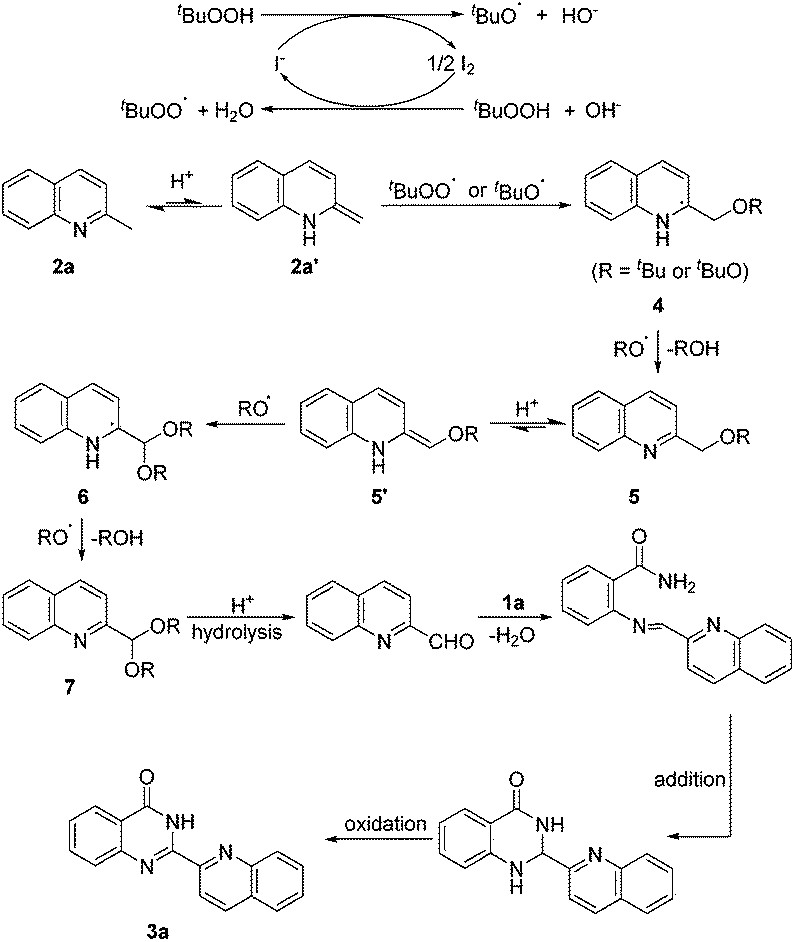 | ||
| Scheme 4 Proposed reaction mechanism. | ||
In summary, we have developed a direct and metal-free oxidative amination of sp3 C–H bonds for the construction of 2-hetarylquinazolin-4(3H)-ones. A wide range of substrates are tolerated in this reaction, and the products are obtained in high yields under mild conditions. A proposed pathway for the formation of 2-hetarylquinazolin-4(3H)-one derivatives is illustrated. In addition, we also report a significant method for preparing quinoline-2-carbaldehyde, which may be of great importance for the synthesis of N-containing heterocycles.
Acknowledgements
We are grateful to the National Science Foundation of China (No. 21572117) and the State Key Laboratory of Natural and Biomimetic Drugs of Peking University (No. K20140208) for the financial support of this research.Notes and references
- I. Khan, A. Ibrar, N. Abbas and A. Saeed, Eur. J. Med. Chem., 2014, 76, 193 CrossRef CAS PubMed.
- S.-L. Cao, Y.-P. Feng, Y.-Y. Jiang, S.-Y. Liu, G.-Y. Ding and R.-T. Li, Bioorg. Med. Chem. Lett., 2005, 15, 1915 CrossRef CAS PubMed.
- Z. Wang, M. Wang, X. Yao, Y. Li, J. Tan, L. Wang, W. Qiao, Y. Geng, Y. Liu and Q. Wang, Eur. J. Med. Chem., 2012, 53, 275 CrossRef CAS PubMed.
- S. E. de Laszlo, C. S. Quagliato, W. J. Greenlee, A. A. Patchett, R. S. L. Chang, V. J. Lotti, T. B. Chen, S. A. Scheck and K. A. Faust, et al. , J. Med. Chem., 1993, 36, 3207 CrossRef CAS PubMed.
- P.-P. Kung, M. D. Casper, K. L. Cook, L. Wilson-Lingardo, L. M. Risen, T. A. Vickers, R. Ranken, L. B. Blyn, J. R. Wyatt, P. D. Cook and D. J. Ecker, J. Med. Chem., 1999, 42, 4705 CrossRef CAS PubMed.
- N. J. Liverton, D. J. Armstrong, D. A. Claremon, D. C. Remy, J. J. Baldwin, R. J. Lynch, G. Zhang and R. J. Gould, Bioorg. Med. Chem. Lett., 1998, 8, 483 CrossRef CAS PubMed.
- M. M. Aly, Y. A. Mohamed, K. A. M. El-Bayouki, W. M. Basyouni and S. Y. Abbas, Eur. J. Med. Chem., 2010, 45, 3365 CrossRef CAS PubMed.
- S. Kobayashi, M. Ueno, R. Suzuki and H. Ishitani, Tetrahedron Lett., 1999, 40, 2175 CrossRef CAS.
- (a) A. L. D'Yakonov and M. V. Telezhenetskaya, Chem. Nat. Compd., 1997, 33, 221 CrossRef; (b) J. P. Michael, Nat. Prod. Rep., 2004, 21, 650 RSC; (c) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787 CrossRef CAS; (d) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed.
- (a) Z.-Z. Ma, Y. Hano, T. Nomura and Y.-J. Chen, Heterocycles, 1999, 51, 1883 CrossRef CAS; (b) Z.-Z. Ma, Y. Hano, T. Nomura and Y.-J. Chen, Heterocycles, 1997, 46, 541 CrossRef CAS; (c) Z.-Z. Ma, Y. Hano, T. Nomura and Y.-J. Chen, Phytochemistry, 2000, 53, 1075 CrossRef CAS PubMed.
- C. Wattanapiromsakul, P. I. Forster and P. G. Waterman, Phytochemistry, 2003, 64, 609 CrossRef CAS PubMed.
- For selected examples, see: (a) D. J. Connolly, D. Cusack, T. P. O'Sullivan and P. J. Guiry, Tetrahedron, 2005, 61, 10153 CrossRef CAS; (b) D. A. Patil, P. O. Patil, G. B. Patil and S. J. Surana, Mini-Rev. Med. Chem., 2011, 11, 633 CrossRef CAS PubMed; (c) T. M. Potewar, R. N. Nadaf, T. Daniel, R. J. Lahoti and K. V. Srinivasan, Synth. Commun., 2005, 35, 231 CrossRef CAS; (d) Y. Mitobe, S. Ito, T. Mizutani, T. Nagase, N. Sato and S. Tokita, Bioorg. Med. Chem. Lett., 2009, 19, 4075 CrossRef CAS PubMed; (e) X. Liu, H. Fu, Y. Jiang and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 348 CrossRef CAS PubMed; (f) B. Ma, Y. Wang, J. Peng and Q. Zhu, J. Org. Chem., 2011, 76, 6362 CrossRef CAS PubMed; (g) W. Xu and H. Fu, J. Org. Chem., 2011, 76, 3846 CrossRef CAS PubMed; (h) L. Xu, Y. Jiang and D. Ma, Org. Lett., 2012, 14, 1150 CrossRef CAS PubMed; (i) Y.-F. Wang, F.-L. Zhang and S. Chiba, Org. Lett., 2013, 15, 2842 CrossRef CAS PubMed; (j) X. Jiang, T. Tang, J.-M. Wang, Z. Chen, Y.-M. Zhu and S.-J. Ji, J. Org. Chem., 2014, 79, 5082 CrossRef CAS PubMed; (k) X. Yang, G. Cheng, J. Shen, C. Kuai and X. Cui, Org. Chem. Front., 2015, 2, 366 RSC; (l) D. Zhao, T. Wang and J.-X. Li, Chem. Commun., 2014, 50, 6471 RSC.
- (a) S. B. Mhaske and N. P. Argade, J. Org. Chem., 2004, 69, 4563 CrossRef CAS PubMed; (b) M. Viji and R. Nagarajan, J. Chem. Sci., 2014, 126, 1075 CrossRef CAS; (c) N. Y. Kim and C.-H. Cheon, Tetrahedron Lett., 2014, 55, 2340 CrossRef CAS.
- (a) F.-F. Wang, C.-P. Luo, G. Deng and L. Yang, Green Chem., 2014, 16, 2428 RSC; (b) F. Xiao, S. Chen, Y. Chen, H. Huang and G.-J. Deng, Chem. Commun., 2015, 51, 652 RSC; (c) Z. Jamal and Y.-C. Teo, RSC Adv., 2015, 5, 26949 RSC; (d) G.-W. Wang, S.-X. Li, Q.-X. Wu and S.-D. Yang, Org. Chem. Front., 2015, 2, 569 RSC.
- Q. Li, Y. Huang, T. Chen, Y. Zhou, Q. Xu, S.-F. Yin and L.-B. Han, Org. Lett., 2014, 16, 3672 CrossRef CAS PubMed.
- For selected examples, see: (a) C. Perez-Melero, A. B. S. Maya, B. Del Rey, R. Pelaez, E. Caballero and M. Medarde, Bioorg. Med. Chem. Lett., 2004, 14, 3771 CrossRef CAS PubMed; (b) L. Achremowicz, Synth. Commun., 1996, 26, 1681 CrossRef CAS; (c) Y. Tagawa, K. Yamashita, Y. Higuchi and Y. Goto, Heterocycles, 2003, 60, 953 CrossRef CAS; (d) V. S. Gopinath, J. Pinjari, R. T. Dere, A. Verma, P. Vishwakarma, R. Shivahare, M. Moger, P. S. Kumar Goud, V. Ramanathan, P. Bose, M. V. S. Rao, S. Gupta, S. K. Puri, D. Launay and D. Martin, Eur. J. Med. Chem., 2013, 69, 527 CrossRef CAS PubMed.
- For reviews on the TBAI–TBHP oxidation system, see: X.-F. Wu, J.-L. Gong and X. Qi, Org. Biomol. Chem., 2014, 12, 5807 CAS.
- For selected examples, see: (a) J. Zhang, Y. Shao, H. Wang, Q. Luo, J. Chen, D. Xu and X. Wan, Org. Lett., 2014, 16, 3312 CrossRef CAS PubMed; (b) H. Yu and J. Shen, Org. Lett., 2014, 16, 3204 CrossRef CAS PubMed; (c) Z. Liu, J. Zhang, S. Chen, E. Shi, Y. Xu and X. Wan, Angew. Chem., Int. Ed., 2012, 51, 3231 CrossRef CAS PubMed; (d) K. K. Sharma, D. I. Patel and R. Jain, Chem. Commun., 2015, 51, 15129 RSC; (e) L. Gao, H. Tang and Z. Wang, Chem. Commun., 2014, 50, 4085 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data for products. See DOI: 10.1039/c6qo00231e |
| This journal is © the Partner Organisations 2016 |