
MeOTf-catalyzed annulation of aldehydes and arylalkynes leading to 2,3-disubstituted indanones†
Yu
Liu
a,
Peng
Zhao
a,
Bo
Zhang
a and
Chanjuan
Xi
*ab
aKey Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China. E-mail: cjxi@tsinghua.edu.cn; Tel: +86-10-62782695
First published on 12th July 2016
Abstract
The MeOTf-catalyzed construction of indanones via arylalkyne–aldehyde metathesis and subsequent Nazarov cyclization has been reported. This reaction represents a sustainable and atom-economical approach for the preparation of indanones from readily available substrates with high stereoselectivity.
The indanone skeleton is found in a wealth of natural products and many indanone structures have demonstrated various pharmacological and biological activities in cancer and Alzheimer's types of diseases, for example by acting as endothelin antagonists,1 antiproliferatives,2 antibiotics,3 or an acetylcholinesterase inhibitor as a therapeutic in Alzheimer's disease.4 Additionally, hundreds of indanone natural products are known,5 such as dilemmaones,5b mukagolactone and monachosorin A.5c,5d Consequently, various synthetic methodologies for the synthesis of indanones have been reported. For example, the intramolecular Friedel–Crafts cyclization reaction of arylpropionic acids or 3-chloro-1-(substituted phenyl)propan-1-one is one of the most common methods for the preparation of 1-indanones.6 Nevertheless, direct cyclization usually requires an excess of protic acids such as sulfuric acid, hydrogen fluoride,7 methanesulfonic acid,8 and polyphosphoric acid,9 or Lewis acids such as AlCl3 and SnCl4.7 Recently, transition-metal-catalyzed ortho-bifunctionalized arenes were used as a significant strategy for the synthesis of 1-indanones.10 However, many polysubstituted ortho-bifunctionalized arenes are not readily available, and the transformation usually requires the use of expensive catalysts (e.g. [Rh(C2H4)2Cl]2, Pd(OAc)2, or Cp*RuCl(COD)). In 2008, Hanzawa, Saito, and co-workers pioneered an SbF5-catalyzed reaction of phenylalkyne derivatives with aldehydes in the presence of EtOH to form indanones through a tandem alkyne–aldehyde metathesis/Nazarov cyclization.11 More recently, Luo and co-workers developed an In(OTf)3/PhCO2H dual acid-catalyzed tandem [2 + 2] cycloaddition and Nazarov reaction between alkynes and acetals to afford indanones.12 Although the previous studies provided atom-economical approaches to indanones using a catalytic amount of metal salt, an efficient route for the synthesis of diverse indanones with readily available starting materials and a simple reaction process under metal-free conditions is still highly desirable. Especially, metal-free reactions in the drug scanning process are of great interest since removing potentially toxic trace metals from the end products is omitted. As a part of our ongoing projects on alkyltriflate-triggered annulation,13 herein, we report a new one-pot procedure for the stereoselective synthesis of 2,3-disubstituted 1-indanones via a methyltriflate (MeOTf)-catalyzed coupling reaction of arylalkynes and aldehydes under metal-free conditions.
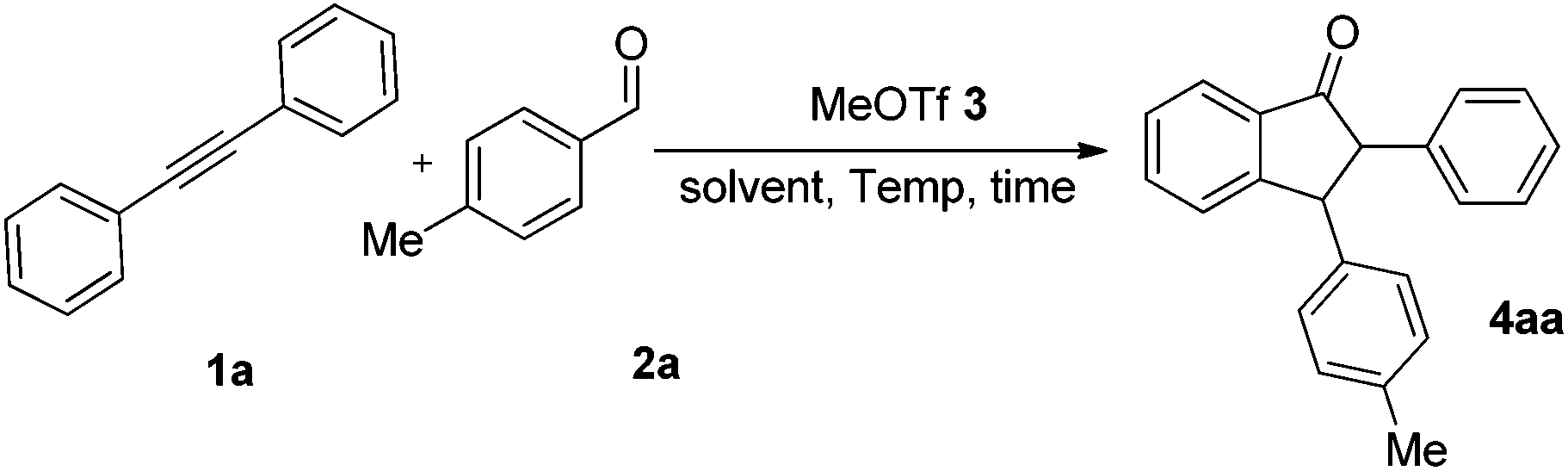
In the preliminary experiment, we chose 1,2-diphenylethyne 1a and 4-methylbenzaldehyde 2a as model substrates. Although it was previously reported that dichloroethane (DCE) is a satisfactory solvent in MeOTf-triggered annulation,13 we again screened solvents such as DCE, dichloromethane (CH2Cl2), chloroform (CHCl3), tetrachloromethane (CCl4), and n-hexane for this reaction (Table 1, entries 1–5). Indeed, DCE is confirmed as the superior solvent in this reaction (entry 1). Then we tried using different amounts of MeOTf (entries 1 and 6–8). To our delight, when 20 mol% MeOTf was employed, the reaction proceeded in satisfactory yield (entry 7). When the ratio of 1a and 2a was adjusted (entries 9 and 10), the optimal ratio of diphenylacetylene 1a, 4-methylbenzaldehyde 2a and MeOTf 3 was 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0:0.2, in which the product 4aa was afforded in 58% yield (entry 10). The effect of reaction time was also examined, and the yield of the product was increased with an extended reaction time (entries 10–13). Furthermore, temperature screening experiments (entries 13–15) revealed that the best reaction temperature was 50 °C. On the basis of the above results, the optimal conditions are shown in entry 12.
:1.0:0.2, in which the product 4aa was afforded in 58% yield (entry 10). The effect of reaction time was also examined, and the yield of the product was increased with an extended reaction time (entries 10–13). Furthermore, temperature screening experiments (entries 13–15) revealed that the best reaction temperature was 50 °C. On the basis of the above results, the optimal conditions are shown in entry 12.
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Temp. [°C] | Time [h] | Solvent | Ratio [1a:2a:3] |
Yieldb [%] |
| a Reaction conditions: a sealed tube under nitrogen. b The yield was evaluated by 1H NMR with CH2Br2 as the internal standard. | |||||
| 1 | 50 | 12 | DCE | 1:1:1 |
48 |
| 2 | 50 | 12 | CH2Cl2 | 1:1:1 |
NR |
| 3 | 50 | 12 | CHCl3 | 1:1:1 |
10 |
| 4 | 50 | 12 | CCl4 | 1:1:1 |
5 |
| 5 | 50 | 12 | n-Hexane | 1:1:1 |
Trace |
| 6 | 50 | 12 | DCE | 1:1:0.1 |
16 |
| 7 | 50 | 12 | DCE | 1:1:0.2 |
49 |
| 8 | 50 | 12 | DCE | 1:1:0.4 |
47 |
| 9 | 50 | 12 | DCE | 1:1.1:0.2 |
43 |
| 10 | 50 | 12 | DCE | 1.1:1:0.2 |
58 |
| 11 | 50 | 18 | DCE | 1.1:1:0.2 |
68 |
| 12 | 50 | 24 | DCE | 1.1:1:0.2 |
72 |
| 13 | 50 | 36 | DCE | 1.1:1:0.2 |
73 |
| 14 | 40 | 24 | DCE | 1.1:1:0.2 |
44 |
| 15 | 60 | 24 | DCE | 1.1:1:0.2 |
46 |
Under the optimized conditions, a study on the substrate scope was carried out. First, diphenylacetylene 1a reacted with a range of substituents of aromatic aldehydes 2, including 4-methyl, 4-methoxyl, 4-isopropyl, 4-bromo, 4-chloro, 4-fluoro, 4-trifluoromethyl, and 3-methyl groups. Both electron-withdrawing and electron-donating groups can be accommodated in this reaction. The representative results are summarized in Fig. 1. When 3,4-dimethylbenzaldehyde 2j was used in this reaction, the product 4aj was obtained in 62% yield. When 2-naphthaldehyde 2k was employed, the product 4ak was formed in 42% yield. In each case only one diastereomer was obtained, which was assigned with a trans relationship between the substituents at the 2- and 3-position. This stereochemical assignment was corroborated by an X-ray crystallographic study of a representative compound 4be and its structure is shown in Fig. 2. Unfortunately, when alkylaldehydes such as propionaldehyde, isobutyraldehyde, cyclohexanecarbaldehyde and pivalaldehyde were used, the reaction did not proceed.
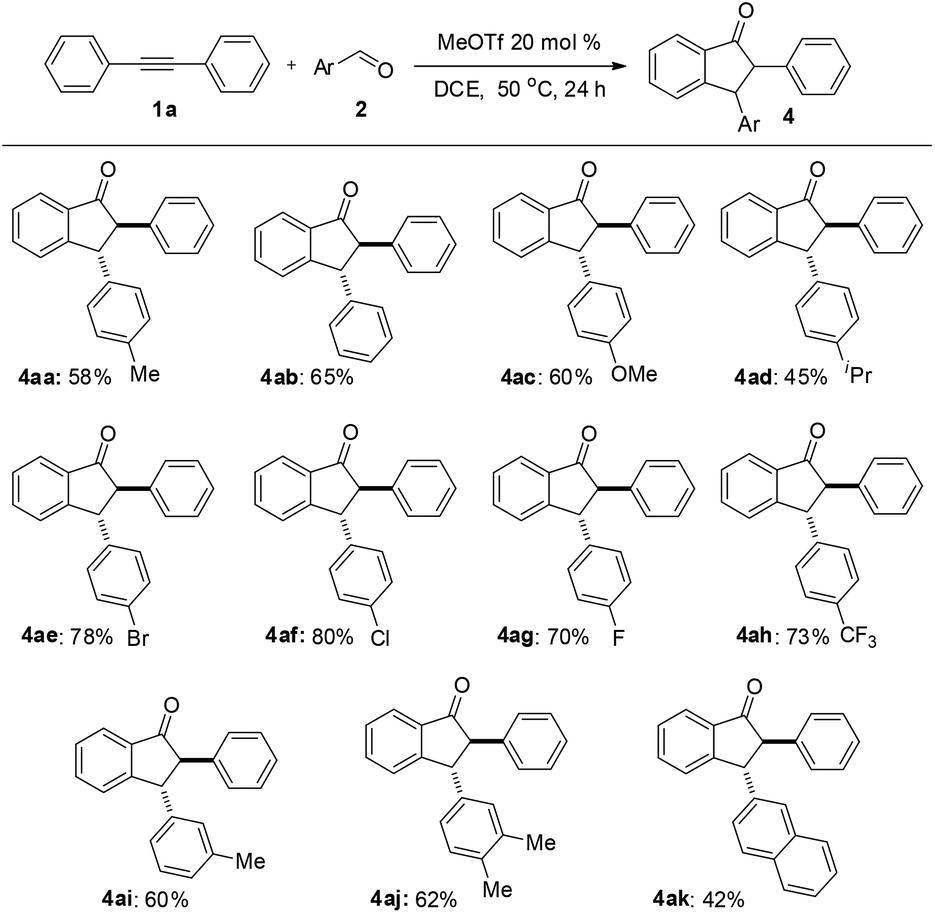 | ||
| Fig. 1 Substrate scope of aldehydes. Reaction conditions: di-phenylacetylene 1a (0.22 mmol), aromatic aldehydes 2 (0.2 mmol), MeOTf (0.04 mmol), and 0.5 mL of DCE, N2, 50 °C, sealed, 24 h. Isolated yield is based on aromatic aldehydes 2. | ||
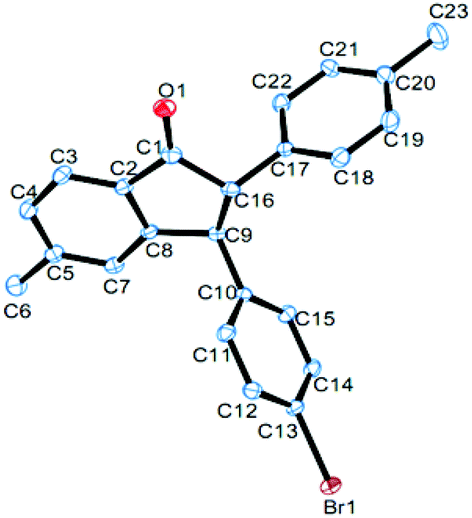 | ||
| Fig. 2 The X-ray crystal structure of 4be. Thermal ellipsoids are shown at the 30% probability level; hydrogen atoms have been omitted for clarity. CCDC 1483374. | ||
The substrate scope and limitation of alkynes for annulation were then investigated. The representative results are summarized in Fig. 3. First, we tested some symmetrical diarylacetylenes. Diarylacetylenes with electron-donating groups such as methyl and methoxyl at the para-position of the benzene ring reacted smoothly to give indanone 4ba, 4be, and 4ce in 75%, 94%, and 70% yields, respectively. When diarylacetylenes bearing electron-withdrawing groups such as chloro, fluoro, and triflourmethyl at the para-position were used, the reaction did not proceed (4de, 4ee, and 4fe). Next, asymmetric diarylalkynes reacted regioselectively to form desired 1-indanones such as 4gf, 4hf, and 4if as a single isomer. Annulation occurred selectively in the electron-rich benzene ring. Furthermore, the reaction could also proceed with aryl alkyl alkynes. The reaction of 1-phenyl-1-propyne 1j, 1-phenyl-1-hexyne 1k, and 1-phenyl-1-(5-chloro)butyne 1l afforded 1-indanones 4jf–4lf, 4jb, 4ja, and 4kb in moderate to good yields with an alkyl group at the 2-position. In this case a long reaction time is required. The reaction of terminal alkynes such as 1-phenylacetylene 1m failed to afford the desired product even at 90 °C for 48 h but enone 5ma was obtained in 62% isolated yield.
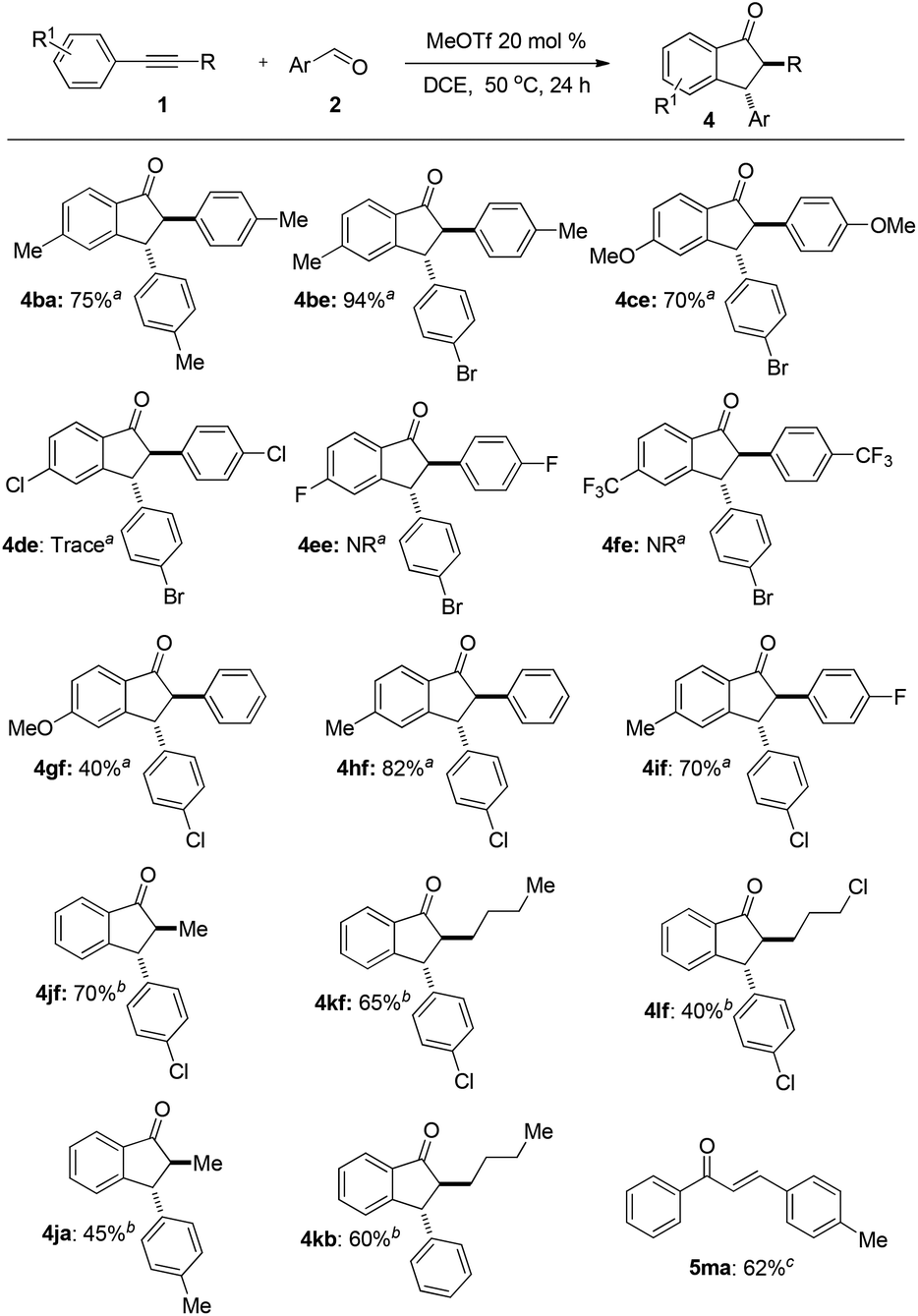 | ||
| Fig. 3 Substrate scope. Reaction conditions: alkynes 1 (0.22 mmol), aromatic aldehydes 2 (0.2 mmol), MeOTf 3 (0.04 mmol), and 0.5 mL of DCE, N2, sealed. a50 °C, 24 h. b50 °C, 36 h. c90 °C, 48 h. Isolated yield is based on aromatic aldehydes 2. | ||
The reaction of 1-phenyl-1-propyne 1j with 4-methylbenzaldehyde 2a at 40 °C for 12 h in the presence of 20 mol% MeOTf afforded enone 5ja in 59% isolated yield (eqn (1)). The compound 5ja could be converted into the corresponding 1-indanone 4ja at 50 °C within 36 h in 80% yield with excellent selectivity (eqn (2)). Therefore, we suspected that the one-pot formation of 4ja from 1-phenyl-1-propyne 1j and aldehyde 2a would be composed of (i) the formal alkyne-carbonyl metathesis between the two substrates and (ii) Nazarov cyclization of the phenyl alkenyl ketone intermediate 5ja.
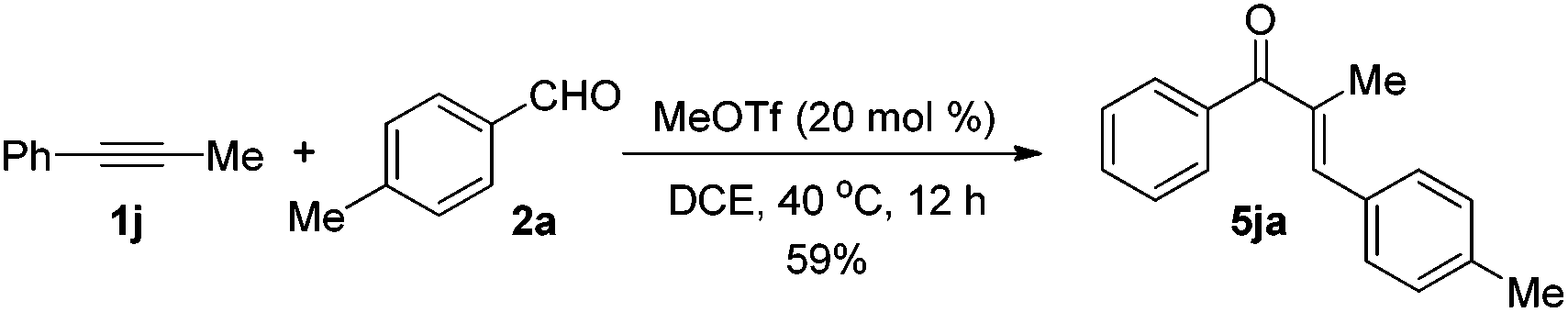 | (1) |
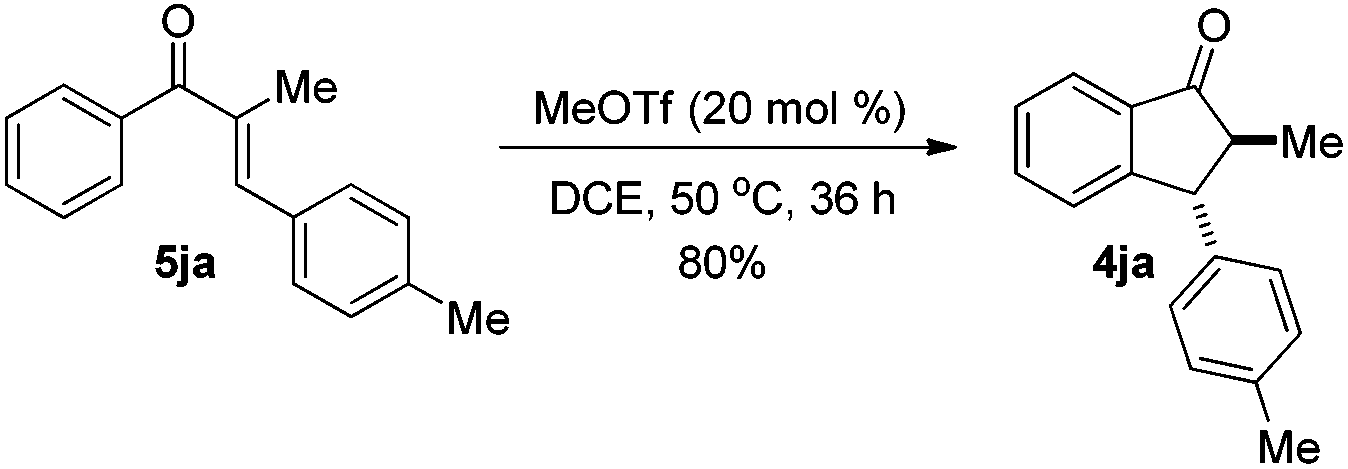 | (2) |
On the basis of the experimental results as well as reported literature,11,12,14 a plausible mechanism is proposed (Scheme 1). First, MeOTf as an electrophile reacts with aldehyde to give the oxonium A, which couples with alkyne to form the highly active oxetenium intermediate Bvia [2 + 2] cycloaddition. Then, the intermediate B undergoes spontaneous isomerization to form the 4π-Nazarov intermediate C, followed by Nazarov cyclization to afford 1-indanone 4 and the regeneration of MeOTf. When 1-phenyl-1-propyne 1j was used in the reaction, the alkyne-carbonyl metathesis product 5ja was isolated under the present conditions at 40 °C, which further supports the reaction mechanism.
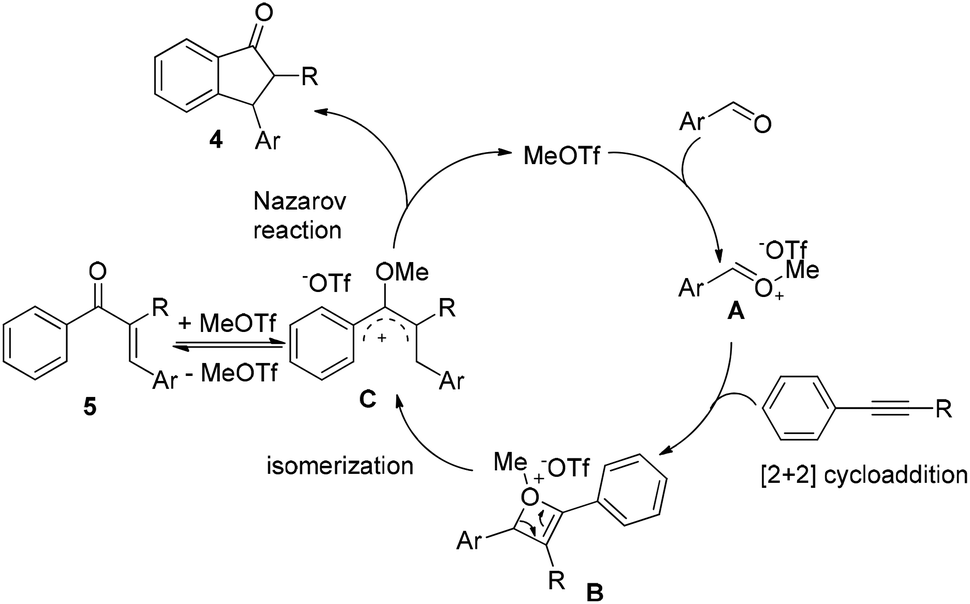 | ||
| Scheme 1 Proposed reaction mechanism. | ||
In conclusion, a series of 2,3-disubstituted 1-indanones were synthesized in good yields via MeOTf-catalyzed tandem metathesis and Nazarov cyclization of arylalkyne and aldehydes under mild conditions. This route is a practical and convenient method for the synthesis of 2,3-disubstituted 1-indanones from readily available starting materials in a one-pot reaction under metal-free conditions.
Notes and references
- M. A. Lago, J. I. Luengo, C. E. Peishoff and J. D. Elliott, in Ann. Rep. Med. Chem, ed. A. B. James, Academic Press, 1996, vol. 31, p. 81 Search PubMed.
- (a) H. O. Saxena, U. Faridi, S. Srivastava, J. K. Kumar, M. P. Darokar, S. Luqman, C. S. Chanotiya, V. Krishna, A. S. Negi and S. P. S. Khanuja, Bioorg. Med. Chem. Lett., 2008, 18, 3914 CrossRef CAS PubMed; (b) A. Morrell, S. Antony, G. Kohlhagen, Y. Pommier and M. Cushman, Bioorg. Med. Chem. Lett., 2004, 14, 3659 CrossRef CAS PubMed; (c) A. Morrell, S. Antony, G. Kohlhagen, Y. Pommier and M. Cushman, Bioorg. Med. Chem. Lett., 2006, 16, 1846 CrossRef CAS PubMed; (d) W. Kemnitzer, N. Sirisoma, B. Nguyen, S. Jiang, S. Kasibhatla, C. Crogan-Grundy, B. Tseng, J. Drewe and S. X. Cai, Bioorg. Med. Chem. Lett., 2009, 19, 3045 CrossRef CAS PubMed; (e) C. Tseng, Y. Chen, K. Chung, C. Cheng, C. Wang and C. Tzeng, Bioorg. Med. Chem., 2009, 17, 7465 CrossRef CAS PubMed; (f) B. Insuasty, F. Orozco, C. Lizarazo, J. Quiroga, R. Abonia, M. Hursthouse, M. Nogueras and J. Cobo, Bioorg. Med. Chem., 2008, 16, 8492 CrossRef CAS PubMed.
- E. A. Anderson, E. J. Alexanian and E. J. Sorensen, Angew. Chem., Int. Ed., 2004, 43, 1998 CrossRef CAS PubMed.
- (a) C. H. Rojas-Fernandez, Ann. Pharmacother., 2001, 35, 202 CAS; (b) W. Moraes, D. Poyares, L. Sukys-Claudino, C. Guilleminault and S. Tufik, Chest, 2008, 133, 677 CrossRef CAS PubMed.
- (a) B. Fugmann, S. Lang-Fugmann and W. Steglich, Rompp Encyclopedia of Natural Products, Thieme, New York, 2000 Search PubMed; (b) D. R. Beukes, M. T. Davies-Coleman, M. Kelly-Borges, M. K. Harper and D. J. Faulkner, J. Nat. Prod., 1998, 61, 699 CrossRef CAS PubMed; (c) T. Satake, T. Murakami, N. Yokote, Y. Saike and C. Chen, Chem. Pharm. Bull., 1985, 33, 4175 CrossRef CAS; (d) M. Saito, M. Umeda, M. Enomoto, Y. Hatanaka, S. Natori, K. Yoshihira, M. Fukuoka and M. Kuroyanagi, Experientia, 1975, 31, 829 CrossRef CAS PubMed.
- (a) A. Bhattacharya, B. Segmuller and A. Ybarra, Synth. Commun., 1996, 26, 1775 CrossRef CAS; (b) C. De Castro, J. Primo and A. Corma, J. Mol. Catal. A: Chem., 1998, 134, 215 CrossRef CAS; (c) V. Gevorgyan, L. G. Quan and Y. Yamamoto, Tetrahedron Lett., 1999, 40, 4089 CrossRef CAS; (d) H. Rudler and B. Denise, J. Mol. Catal. A: Chem., 2000, 154, 277 CrossRef CAS; (e) G. K. S. Prakash, P. Yan, B. Török and G. A. Olah, Catal. Lett., 2003, 87, 109 CrossRef; (f) S. V. Gagnier and R. C. Larock, J. Am. Chem. Soc., 2003, 125, 4804 CrossRef CAS PubMed; (g) E. Fillion and D. Fishlock, Org. Lett., 2003, 5, 4653 CrossRef CAS PubMed.
- W. S. Johnson, Org. React., 1949, 2, 114 Search PubMed.
- R. B. Miller and J. M. Frincke, J. Org. Chem., 1981, 46, 2972 CrossRef CAS.
- F. D. Popp and W. E. McEwen, Chem. Rev., 1958, 58, 321 CrossRef CAS.
- (a) Y. Yu and M. Xu, J. Org. Chem., 2013, 78, 2736 CrossRef CAS PubMed; (b) S. V. Gagnier and R. C. Larock, J. Am. Chem. Soc., 2003, 125, 4804 CrossRef CAS PubMed; (c) W. Zi and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 12600 CrossRef CAS PubMed; (d) A. Arefalk, M. Larhed and A. Hallberg, J. Org. Chem., 2005, 70, 938 CrossRef CAS PubMed; (e) R. S. Senaiar, J. A. Teske, D. D. Young and A. Deiters, J. Org. Chem., 2007, 72, 7801 CrossRef CAS PubMed.
- A. Saito, M. Umakoshi, N. Yagyu and Y. Hanzawa, Org. Lett., 2008, 10, 1783 CrossRef CAS PubMed.
- L. Zhu, Z. Xi, J. Lv and S. Luo, Org. Lett., 2013, 15, 4496 CrossRef CAS PubMed.
- (a) X. Yan, S. Zou, P. Zhao and C. Xi, Chem. Commun., 2014, 50, 2775 RSC; (b) X. Yan, X. Yi and C. Xi, Org. Chem. Front., 2014, 1, 657 RSC; (c) P. Zhao, X. Yan, H. Yin and C. Xi, Org. Lett., 2014, 16, 1120 CrossRef CAS PubMed; (d) P. Zhao, Y. Liu and C. Xi, Org. Lett., 2015, 17, 4388 CrossRef CAS PubMed.
- W. Zhao, Z. Li and J. Sun, J. Am. Chem. Soc., 2013, 135, 4680 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and full characterization including 1H NMR and 13C NMR data and spectra for all compounds. CCDC 1483374. For ESI and crystallographic data in CIF or other electronic formats see DOI: 10.1039/c6qo00253f |
| This journal is © the Partner Organisations 2016 |