
Selective Pd-catalyzed α- and β-arylations of the furan rings of (ortho-bromophenyl)furan-2-yl-methanones: C(CO)–C bond cleavage with a furan ring as a leaving group and synthesis of furan-derived fluorenones†
Xiaoting
Zhang‡
,
Jianchao
Liu‡
,
Yongjie
Yang
,
Furong
Wang
,
Huanfeng
Jiang
and
Biaolin
Yin
*
School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, P.R. China. E-mail: blyin@scut.edu.cn; Fax: +86-20-8711-3735; Tel: +86-20-8711-3735
First published on 13th July 2016
Abstract
Selective palladium diacetate-catalyzed α- and β-arylations of the furan rings of (ortho-bromophenyl)furan-2-yl-methanones 1 under two different conditions are reported. In the presence of potassium tert-butoxide as a strong base and triphenylphosphine as a ligand, methanones 1 undergo α-arylation accompanied by C(CO)–C bond cleavage. In contrast, in the presence of potassium carbonate as the base and tricyclohexylphosphonium tetrafluoroborate as the ligand, methanones 1 undergo intramolecular β-arylation to afford furan-derived fluorenones in high yields from a wide variety of substrates. In addition, a one-pot protocol for the successive direct intramolecular β-arylation and intermolecular α-arylation of the furan rings of 1 has been achieved.
Furans are excellent building blocks, not only because they can be readily prepared from biomass-derived synthetic platforms such as furfural and 5-hydroxymethylfurfural but also because they have a unique reactivity both as electron-rich heteroaromatic compounds and as equivalents of alkenes, 1,3-dienes, alkynes, 1,4-diketones, and enol ethers.1 Although the synthetic applications of furans have been greatly expanded, the elucidation of new chemical properties and the development of a novel transformation of furans are still highly desirable goals,2 given the current interest in sustainable chemistry.
The Pd-catalyzed arylation of furans is a powerful tool for the construction of arylfurans,3 some of which display interesting bioactivities and physical properties.4 Among the methods used to accomplish this transformation, the direct arylation of furyl C–H bonds with aryl halides is of special interest because it does not require preactivation of the substrates. This reaction usually occurs regioselectively at the more reactive α-position of the furan ring.5 Direct arylation at the less reactive β-position is more challenging, and there are only a few reports of this reaction. Specifically, β-arylation of furans occurs only when both of the α-positions are substituted6 or when a furan 2-carboxamide is used as the substrate; in the latter case, chelation-assisted regioselective functionalization of the ortho-C–H bonds occurs with the amide as the directing group.7 In addition, a protocol involving Pd-catalyzed α-arylation of 2-furoic acid substrates with aryl halides accompanied by C–C(O)OH bond cleavage (decarboxylation) has also received much attention because of the ready availability of 2-furoic acid.8 To our knowledge, direct β-arylation of furan rings substituted with an electron-withdrawing benzoyl group and α-arylation of α-substituted furan rings accompanied by C(CO)–C bond cleavage have never been reported in the literature.
Owing to our ongoing interest in the synthetic applications of furans9 and given that fluorenones have important biological activities and are useful synthetic intermediates,10 we wished to develop a route to unexplored furan-derived fluorenones 3 starting from simple (ortho-bromophenyl)furan-2-yl-methanones 1via palladium-catalyzed direct β-arylation of the furan ring.11,12 Substrates 1 were prepared easily either by the reactions of lithium furyl compounds with 2-bromobenzaldehydes followed by MnO2 oxidation of the resultant alcohols or by Friedel–Crafts reactions of furans with acyl chlorides (Scheme 1).
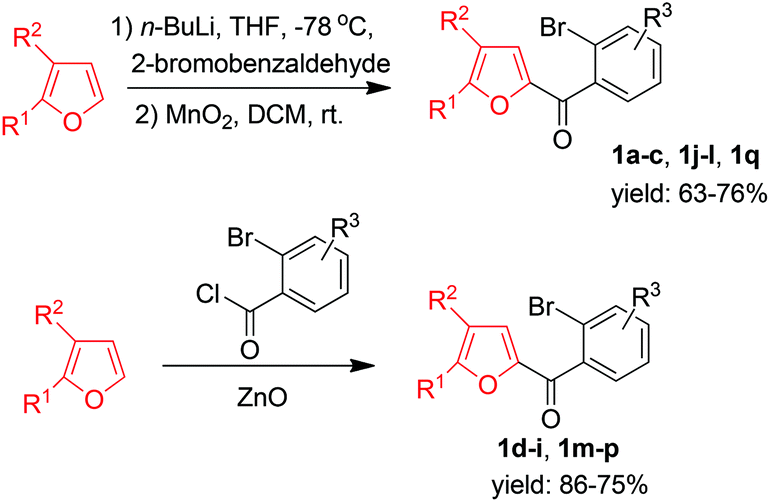 | ||
| Scheme 1 Syntheses of 1. | ||
Initially, we attempted to produce 3a by treatment of 1a with NaOtBu in the presence of Pd(OAc)2 as a catalyst, PPh3 as a ligand, and KI as an additive to activate the aryl bromide. However, only a trace of 3a was obtained, along with a substantial amount of protonated product 2a (Scheme 2). Interestingly, an unexpected product, [2-(5-methyl-furan-2-yl)phenyl]methanone (4a), was also obtained, albeit in a low yield (16%). The presence of the newly introduced furyl group at the ortho-position of the phenyl ring of 4a clearly indicated the occurrence of a novel α-arylation reaction of the α-substituted furan and an interesting palladium-catalyzed C(CO)–C bond cleavage with a furan ring as a leaving group. To improve the yield of 4a and gain insight into the mechanism of this reaction, we directed our efforts at optimizing the reaction parameters (additive, base, ligand, and solvent).
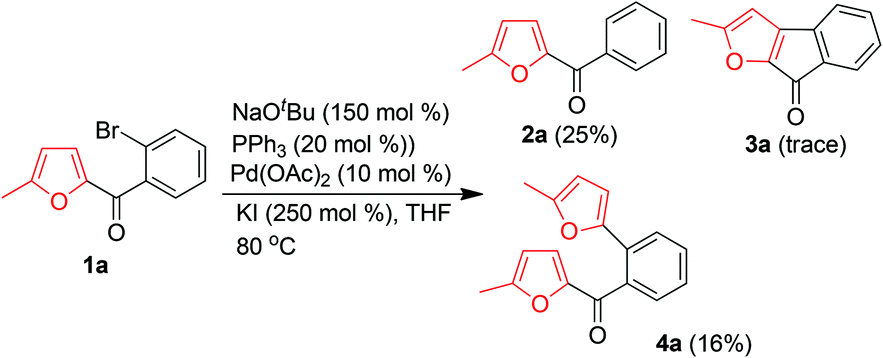 | ||
| Scheme 2 Palladium-catalyzed transformations of 1a in the presence of NaOtBu. | ||
As shown in Table 1, when 1a was subjected to the above-mentioned conditions in the absence of Pd(OAc)2, no 4a formed (entry 1), which indicated that Pd(OAc)2 was essential for C–C bond cleavage. Screening of various bases revealed that KOtBu was optimal (entries 2–7). The presence of additives clearly influenced the reaction. Several potassium salts were screened, and KCl gave the highest yield (43%; entries 2 and 8–11). The use of various chloride salts including NaCl, LiCl, and tetrabutylammonium chloride did not further improve the yield (entries 12–14). Various other ligands were examined (L1–L6), but none were found to be better than PPh3 (entries 15–20). Of the various solvents that were evaluated, the aprotic solvent toluene was the best, providing 4a in 66% yield (entry 22).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions | Yieldb (%) | |||
| Additive | Base | L | Solvent | ||
| a Reaction conditions, unless otherwise noted: 1a (0.5 mmol), Pd(OAc)2 (0.05 mmol), additive (1.25 mmol), base (0.75 mmol), ligand (0.1 mmol), solvent (5 mL), 80 °C under N2 for 16 h. TBAC = tetrabutylammonium chloride; L1 = tri-tert-butylphosphine; L2 = tricyclohexylphosphine; L3 = tri-p-tolylphosphine; L4 = tri(2-furyl)phosphine; L5 = (±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthalene; L6 = 1,1′-bis(diphenylphosphino)ferrocene; ND = not detected; NR = no reaction. b Isolated yield (4a was produced from 2 equiv. of 1a). c No Pd(OAc)2 was used. | |||||
| 1c | KI | NaOtBu | PPh3 | THF | ND |
| 2 | KI | KOtBu | PPh3 | THF | 25 |
| 3 | KI | NaOtBu | PPh3 | THF | 16 |
| 4 | KI | LiOtBu | PPh3 | THF | ND |
| 5 | KI | K2CO3 | PPh3 | THF | ND |
| 6 | KI | Cs2CO3 | PPh3 | THF | ND |
| 7 | KI | Et3N | PPh3 | THF | NR |
| 8 | KF | KOtBu | PPh3 | THF | ND |
| 9 | KBr | KOtBu | PPh3 | THF | 26 |
| 10 | KCl | KOtBu | PPh3 | THF | 43 |
| 11 | K3PO4 | KOtBu | PPh3 | THF | 27 |
| 12 | NaCl | KOtBu | PPh3 | THF | 40 |
| 13 | LiCl | KOtBu | PPh3 | THF | ND |
| 14 | TBAC | KOtBu | PPh3 | THF | Trace |
| 15 | KCl | KOtBu | L 1 | THF | ND |
| 16 | KCl | KOtBu | L 2 | THF | ND |
| 17 | KCl | KOtBu | L 3 | THF | 40 |
| 18 | KCl | KOtBu | L 4 | THF | Trace |
| 19 | KCl | KOtBu | L 5 | THF | 13 |
| 20 | KCl | KOtBu | L 6 | THF | 8 |
| 21 | KCl | KOtBu | PPh3 | DMF | ND |
| 22 | KCl | KO t Bu | PPh 3 | PhMe | 66 |
| 23 | KCl | KOtBu | PPh3 | DMSO | ND |
| 24 | KCl | KOtBu | PPh3 | 1,4-Dioxane | 40 |
Next we investigated the reaction scope by subjecting numerous furfuryl phenyl ketones 1 to the optimized conditions (Table 2). The electron density of the furan ring markedly influenced the reaction. When R3 = R2 = H, R1 could be a H, an alkyl group (Me, Et, or Bn), an unsubstituted phenyl group, or a phenyl group with an electron-donating or electron-withdrawing substituent; the reactions of these substrates afforded 4a–4h in moderate to good yields. However, the reaction of a substrate in which R1 was an electron-withdrawing ester group did not give the expected product (4i). A substrate with two electron-donating groups (R1 = Me, R2 = SMe) also did not give the expected product (4j), and a benzofuran-type substrate did not give the product of C–C bond cleavage (4k). When R1 was a methyl group, the reactions gave the corresponding products (4l–4p) in moderate yields.
| a 1 (0.5 mmol), Pd(OAc)2 (0.05 mmol), PPh3 (0.05 mmol), KOtBu (0.75 mmol), KCl (1.25 mmol), and PhMe (5 mL) at 80 °C under N2 for 18 h. All yields are isolated yields. |
|---|
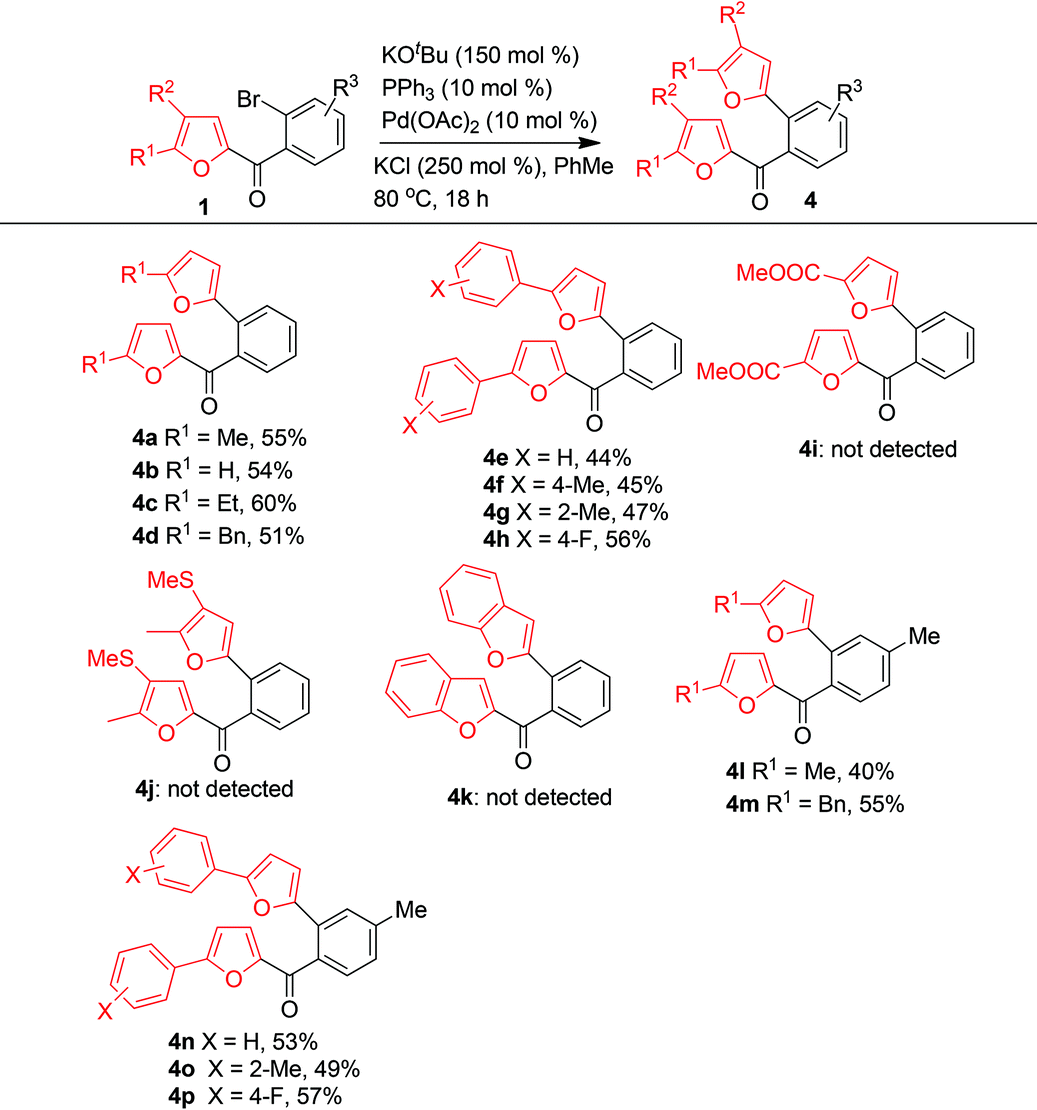
|
The mechanism of this transformation was explored by means of four control experiments (Scheme 3). Ketone 2a did not react under the standard conditions, indicating that the aryl bromide moiety was essential for C–C bond cleavage (Scheme 3a). The fact that reaction of 1a with p-methylphenyl bromide gave 5a clearly demonstrated that the cleaved furan ring could be trapped by a phenyl bromide (Scheme 3b). This was confirmed by the results of a cross reaction between 1a and 1e under the standard reaction conditions, which afforded cross products 4s and 4t, as indicated by ESI/MS (Scheme 3c). When the radical scavenger 2,2,6,6-tetramethyl piperidinyloxy was added under the standard conditions, 4a was obtained in 25% isolated yield, suggesting that the reaction did not proceed by a radical-mediated mechanism (Scheme 3d).
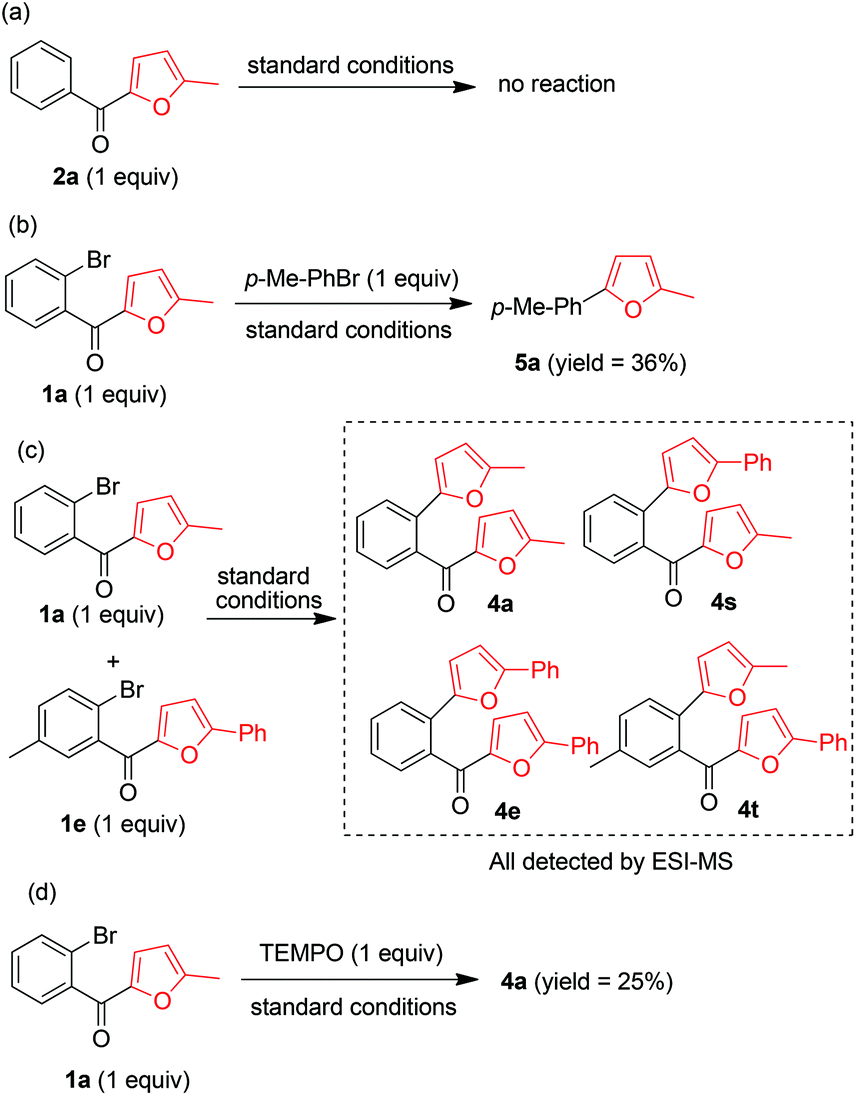 | ||
| Scheme 3 Control experiments. | ||
On the basis of the above-described results, we propose the mechanism outlined in Scheme 4. Oxidative addition of PdLn to 1 affords complex 6, which undergoes nucleophilic addition by KOtBu to afford 7. Complex 7 is then cleaved to produce furyl anion 8 and cation 9. Reaction of 8 with 6 produces complex 10, which undergoes reductive elimination to generate 4 and reaction of 9 with KOtBu affords 11, detected by ESI/MS.
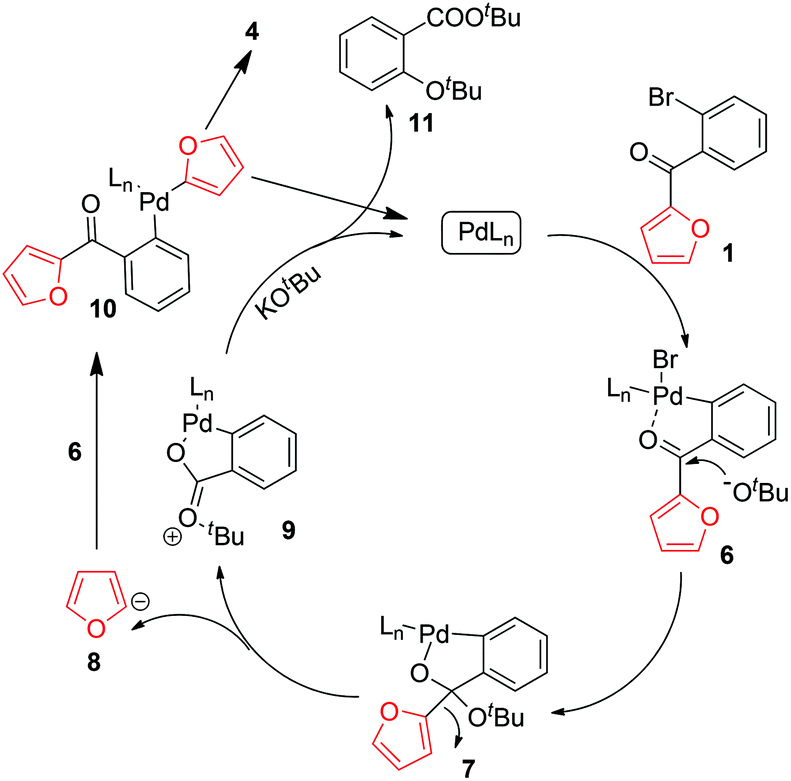 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Having investigated α-arylation accompanied by C–C bond cleavage, we next searched for suitable conditions for achieving intramolecular β-arylation to synthesize 3via suppression of C–C bond cleavage and protonation. Considering that cleavage of the C–C bond of 1 may have resulted from the nucleophilicity of KOtBu or from the presence of KCl, we hypothesized that using a weakly nucleophilic base and omitting KCl might minimize C–C bond cleavage. In addition, because the formation of a protonated product might have resulted from the weak acidity of β-H of the furan ring, we suspected that rescreening of various ligands and reaction temperatures would be helpful. We began by screening a series of bases in reactions at 80 °C with Pd(OAc)2 as the catalyst, PPh3 as the ligand, and THF as the solvent in the absence of an additive (Table 3). Among the relatively weak bases (entries 1–4), K2CO3 provided 3a in the best yield (20%), and 4a was not detected. Of the various ligands that were evaluated (entries 5–10), PCy3·HBF4 was the best, providing 3a in 68% yield (entry 10). We suggest that BF4− may have coordinated with the carbonyl group, thus increasing the acidity of β-H. Changing the solvent from THF to mesitylene, which is less polar, increased the yield to 77% (entry 13), and increasing the reaction temperature to 150 °C resulted in a 95% yield (entry 16).
a
|
|
||||
|---|---|---|---|---|
| Entry | L | Base | Solvent | Yieldb (%) |
| a Reaction conditions, unless otherwise noted: 1 (0.5 mmol), Pd(OAc)2 (0.01 mmol), ligand (0.04 mmol), base (1.0 mmol), solvent (5 mL), T = 80 °C. dppf = bis(diphenylphosphino)ferrocene; BINAP = (±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthalene; L7, tris(4-methylphenyl)phosphine; L8, PCy3·HBF4; Tol = toluene; Mes = mesitylene. b All yields are isolated yields. NR = no reaction; ND = not detected. c T = 110 °C. d T = 130 °C. e T = 150 °C. | ||||
| 1 | PPh3 | K2CO3 | THF | 20 |
| 2 | PPh3 | Cs2CO3 | THF | 15 |
| 3 | PPh3 | Na2CO3 | THF | 8 |
| 4 | PPh3 | K3PO4 | THF | Trace |
| 5 | L 7 | K2CO3 | THF | 32 |
| 6 | PCy3 | K2CO3 | THF | 35 |
| 7 | L 1 | K2CO3 | THF | NR |
| 8 | dppf | K2CO3 | THF | 48 |
| 9 | BINAP | K2CO3 | THF | 54 |
| 10 | L 8 | K2CO3 | THF | 68 |
| 11 | L 8 | K2CO3 | DMF | ND |
| 12 | L 8 | K2CO3 | Tol | 65 |
| 13 | L 8 | K2CO3 | Mes | 77 |
| 14c | L 8 | K2CO3 | Mes | 84 |
| 15d | L 8 | K2CO3 | Mes | 88 |
| 16e | L 8 | K2CO3 | Mes | 95 |
A library of furan-derived fluorenones 3 was synthesized from various substrates 1 under the optimized reaction conditions (Table 4). Despite the high reaction temperature, the reaction tolerated a wide variety of functional groups, and the electron density of the furan ring had only a slight influence on the outcome of the reaction. When R3 = R2 = H, R1 could be a H, an alkyl group (Me, Et, or Bn), or a phenyl group, either unsubstituted or with an electron-donating or electron-withdrawing substituent and 3a–3h were produced in good to excellent yields. Substrates with a phenyl R1 group generally gave lower yields than substrates with an alkyl R1 group. A substrate with an electron-withdrawing ester group as R1 also gave the expected product (3i) in a good yield (72%). A substrate with two electron-donating groups (R1 = Me, R2 = SMe) on the furan ring afforded 3j in 88% yield, and the benzofuran-type product 3k was obtained in 90% yield. R3 could be an electron-neutral H atom (3a–3k), an electron-donating methyl group (3l–3p), or an electron-withdrawing fluorine atom (3r). In contrast, a substrate with a strongly electron-donating 4-methoxyl group did not give the desired product (3q).
a
| a All yields are isolated yields. |
|---|
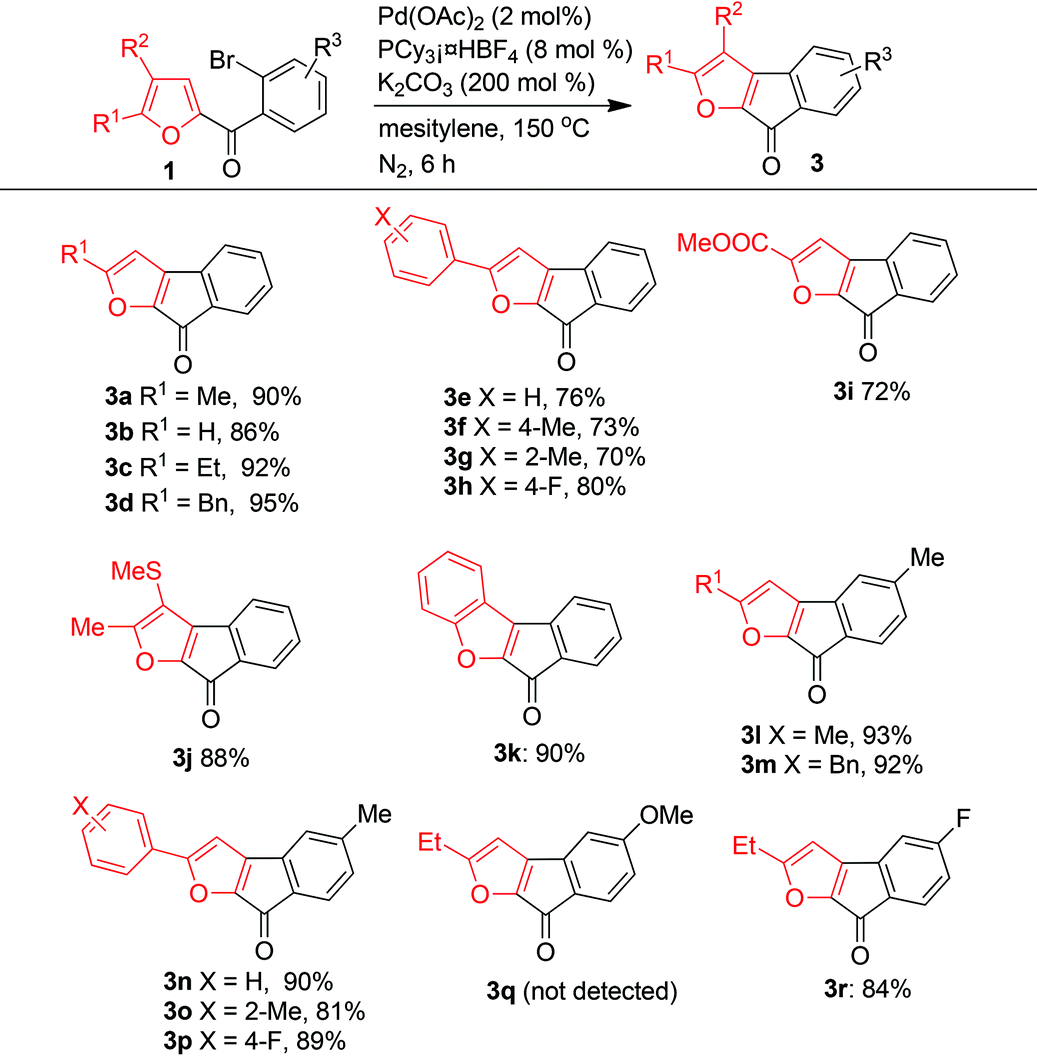
|
Encouraged by the above-described results, we next investigated a one-pot procedure for successive direct intramolecular β-arylation and intermolecular α-arylation of the furan ring of 1b by means of double C–H activation of the ring (Scheme 5). After the completion of the β-arylation reaction was detected by TLC, 1.2 equiv. of an aryl bromide was added to the reaction mixture under a nitrogen atmosphere, and the reaction was continued for 6 h at 150 °C; this protocol led to the formation of di-arylated products 3e, 3f, 3h, and 3s in moderate to good isolated yields.
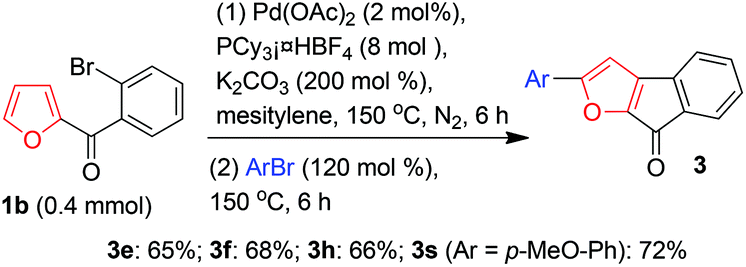 | ||
| Scheme 5 One-pot synthesis of 3e, 3f, 3h, and 3s from 1b. | ||
In summary, we have developed a protocol for selective α- and β-arylations of the furan rings of (ortho-bromophenyl)furan-2-yl-methanones. The α-arylation involved a novel palladium-catalyzed C(CO)–C bond cleavage with a furan ring as a leaving group in the presence of KOtBu and the subsequent coupling of the cleaved furan ring with an aryl bromide. This transformation might be useful for the development of new reactions involving C–C bond cleavage. We achieved intramolecular β-arylation of the furan ring by suppressing the C(CO)–C bond cleavage using K2CO3 as the base and PCy3·HBF4 as the ligand, and these reaction conditions were used to synthesize furan-derived fluorenones in high yields from a wide variety of substrates. In addition, a one-pot protocol for successive direct intramolecular β-arylation and intermolecular α-arylation of the furan ring was developed. This protocol may be useful for the synthesis of biologically interesting fluorenone derivatives.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (no. 21272078 and 21572068), the Science and Technology Planning Project of Guangdong Province, China (no. 2014A020221035), and the Program for New Century Excellent Talents in Universities (no. NCET-12-0189).References
- For reviews on the conversion of furans, see:
(a) B. H. Lipshutz, Chem. Rev., 1986, 86, 795 CrossRef CAS
; (b) F. W. Lichtenthaler, Acc. Chem. Res., 2002, 35, 728 CrossRef CAS PubMed
- For selected recent examples, see:
(a) L. I. Palmer and J. Read de Alaniz, Angew. Chem., Int. Ed., 2011, 50, 7167 CrossRef CAS PubMed
- For selected reviews on C–H arylations of furans, see:
(a) D. A. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed
- For selected examples of biological activities and physical properties of C-3-arylated furan derivatives, see:
(a) S. Jiang, S. R. Tala, H. Lu, N. E. Abo-Dya, I. Avan, K. Gyanda, L. Lu, A. R. Katritzky and A. K. Debnath, J. Med. Chem., 2011, 54, 572 CrossRef CAS PubMed
- For selected recent examples, see:
(a) E. Ö. Karaca, N. Gürbüz, İ. Özdemir, H. Doucet, O. Şahin, O. Büyükgüngör and B. Çetinkaya, Organometallics, 2015, 34, 2487 CrossRef CAS
-
(a) T. Nishikata, Y. Yamane, Y. Yamaguchi and S. Ishikawa, Asian J. Org. Chem., 2016, 5, 466 CrossRef CAS
-
(a) K. S. Larbi, H. Y. Fu, N. Laidaoui, K. Beydoun, A. Miloudi, D. E. Abed, S. Djabbar and H. Doucet, ChemCatChem, 2012, 4, 815 CrossRef
-
(a) F. Bilodeau, M.-C. Brochu, N. Guimond, K. H. Thesen and P. Forgione, J. Org. Chem., 2010, 75, 1550 CrossRef CAS PubMed
-
(a) B.-L. Yin, J.-Q. Lai, Z.-R. Zhang and H.-F. Jiang, Adv. Synth. Catal., 2011, 353, 1961 CrossRef CAS
- For the bioactivities of fluorenones and their derivatives, see:
(a) M. L. Greenlee, J. B. Laub, G. P. Rouen, F. DiNinno, M. L. Hammond, J. L. Huber, J. G. Sundelof and G. G. Hammond, Bioorg. Med. Chem. Lett., 1999, 9, 3225 CrossRef CAS PubMed
- For examples of the synthesis of fluorenones and their derivatives via direct palladium-catalyzed intramolecular arylation of 2-bromoarylketones, see:
(a) G. Qabaja and G. B. Jones, Tetrahedron Lett., 2000, 41, 5317 CrossRef CAS
- For selected examples of C(CO)–C bond cleavage caused by the nucleophilic attack of –OtBu at the ketone group, see:
(a) H. Serizawa, K. Aikawa and K. Mikami, Chem. – Eur. J., 2013, 19, 17692 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. See DOI: 10.1039/c6qo00277c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |