
Silver(I)-promoted C5–H phosphonation of 8-aminoquinoline amides with H-phosphonates†
Mengmeng
Sun
a,
Suyan
Sun
a,
Huijie
Qiao
a,
Fan
Yang
*a,
Yu
Zhu
a,
Jianxun
Kang
a,
Yusheng
Wu
*bc and
Yangjie
Wu
*a
aThe College of Chemistry and Molecular Engineering, Henan Key Laboratory of Chemical Biology and Organic Chemistry, Key Laboratory of Applied Chemistry of Henan Universities, Zhengzhou University, Zhengzhou 450052, China. E-mail: yangf@zzu.edu.cn; wyj@zzu.edu.cn
bTetranov Biopharm, LLC., 75 Daxue Road, Zhengzhou, 450052, China. E-mail: yusheng.wu@tetranovglobal.com
cCollaborative Innovation Center of New Drug Research and Safety Evaluation, Henan Province, China. E-mail: yusheng.wu@tetranovglobal.com
First published on 19th September 2016
Abstract
A simple and efficient protocol for silver(I)-promoted phosphonation of 8-aminoquinoline amides with dialkyl H-phosphonates has been developed, affording the desired products in moderate yields. This method features C5–H regioselectivity of 8-aminoquinoline amides, a broad substrate scope and general functional group compatibility.
Phosphorous compounds have been widely applied to many fields, such as organic synthesis, medicinal chemistry, and materials chemistry.1 Since the Hirao group first introduced a Pd-catalyzed version for C–P bond formation in 1981,2 thereafter many efforts have been devoted to developing new methodologies for the construction of C–P bonds.3 However, these synthetic routes to the C–P bond typically require prefunctionalization of the aryl C–H bond.
Over the past few decades, the direct functionalization of C–H bonds has become a reliable and robust tool in organic synthesis due to the elimination of the prefunctionalization step.4,5 As a result, a series of transformations from C–H to C–C4 and C–heteroatom5 bonds have been demonstrated. Particularly, phosphonation of C–H bonds can be realized with or without the help of a directing group under transition metal catalysis or even transition metal-free conditions (Scheme 1).6,7 For the phosphonation without a directing group, a number of catalytic systems have been successfully developed by several groups such as Zhang,6k Ishii,6l Bosch.6m and Kottmann.6n However, these catalytic protocols suffer from poor and indefinite regioselectivity (Scheme 1a).6 On the other hand, the directing group-assisted phosphonation could take place exclusively at the ortho-position of the directing groups, and great progress has been achieved (Scheme 1b).7 For example, Yu's group described a pyridine-directed ortho-C–H phosphonation by slowly adding the phosphonating reagent using a syringe pump;7i the group of Hong7a and Murakami7j successfully realized ortho-C–H phosphonation using oxime and pyridine as the directing group, respectively, employing an α-hydroxylalkyl phosphonate as a phosphonating agent to prevent the catalyst from deactivation; Yu's group7f presented ortho-C–H phosphonation using amide and copper(II) acetate as the directing group and catalyst, respectively.
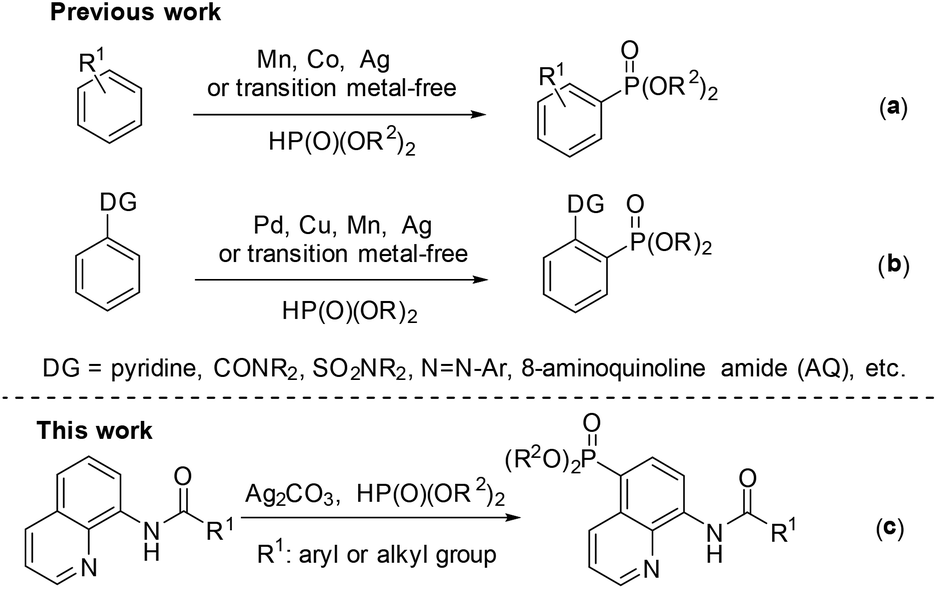 | ||
| Scheme 1 Direct phosphonation of the aryl C–H bond. | ||
In 2014, Yu and co-workers described a copper(II)-catalyzed ortho-C–H phosphonation with the help of a 8-aminoquinoline moiety as a bidentate directing group, and indicated that this reaction did not involve the radical process, but proceeded via Cu(III) intermediates.7f Just recently, we demonstrated an efficient copper-catalyzed C5–H sulfonylation of aminoquinolines with sulfonyl chlorides, which proceeded via a single electron transfer (SET) process.8 The difference in the mechanism between these two reactions may lead to a great difference in the reactive site. To date, reports on C5–H phosphonation of 8-aminoquinoline amides remain rare. As a continuation of this work, we envisioned developing a general protocol for the C5–H phosphonation of 8-aminoquinoline amides with dialkyl H-phosphonates (Scheme 1c).
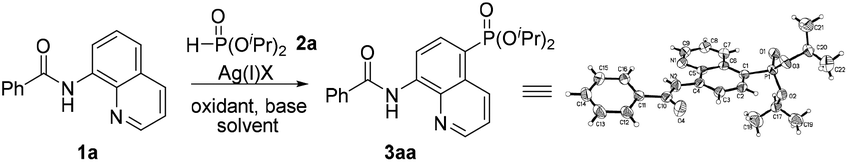
Initially, the phosphonation of N-(quinolin-8-yl)benzamide (1a) with diisopropyl H-phosphonate (2a) was selected as a model reaction to optimize the reaction parameters, and the results are displayed in Table 1. After various silver salts and oxidants were checked, Ag2CO3 and K2S2O8 showed the best reactivity, affording the desired product 3aa in 41% yield in the presence of a base K2CO3 under a nitrogen atmosphere (Table 1, entries 1–4, and ESI†). The desired product 3aa was hardly observed in the absence of a silver salt, indicating that the silver(I) species plays an irreplaceable role (Table 1, entry 5). Next, among the bases tested, NaOH exhibited the highest reactivity; however, the reaction did not occur under the base-free conditions (entry 1 vs. entries 6–9 and ESI†). Then, other solvents were also examined, and dioxane gave a higher yield (Table 1, entry 1 vs. entries 10 and 11 and ESI†). The molecular structure of 3aa was unambiguously confirmed by the single crystal X-ray diffraction study.9 Finally, some additional observations are to be noted: by changing the amount of Ag2CO3 to 2.5 equiv. and 3.0 equiv., the phosphonated products were obtained in a higher yield of 68% and a lower yield of 60%, respectively, suggesting that 2.5 equiv. should be the appropriate loading of the silver salt (Table 1, entry 10 vs. entries 12 and 13); performing the reaction in air led to the target product in a decreased yield of 39% (Table 1, entry 12 vs. entry 14); when the temperature was increased to 130 °C or decreased to 110 °C, no better result was obtained (Table 1, entry 12 vs. entries 15 and 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | AgX | Oxidant | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), silver salt (0.2 mmol), oxidant (0.2 mmol), base (0.2 mmol), solvent (1 ml) at 120 °C under N2 for 24 h. b Isolated yield. c Silver salt (0.25 mmol). d Silver salt (0.3 mmol). e Silver salt (0.25 mmol) in air. f Silver salt (0.25 mmol) at 130 °C. g Silver salt (0.25 mmol) at 110 °C. | |||||
| 1 | Ag2CO3 | K2S2O8 | K2CO3 | Toluene | 41 |
| 2 | AgNO3 | K2S2O8 | K2CO3 | Toluene | 24 |
| 3 | Ag2O | K2S2O8 | K2CO3 | Toluene | 18 |
| 4 | Ag2SO4 | K2S2O8 | K2CO3 | Toluene | <5 |
| 5 | — | K2S2O8 | K2CO3 | Toluene | <5 |
| 6 | Ag2CO3 | K2S2O8 | KOH | Toluene | 37 |
| 7 | Ag2CO3 | K2S2O8 | NaOH | Toluene | 46 |
| 8 | Ag2CO3 | K2S2O8 | t BuOLi | Toluene | 36 |
| 9 | Ag2CO3 | K2S2O8 | — | Toluene | <5 |
| 10 | Ag2CO3 | K2S2O8 | NaOH | Dioxane | 51 |
| 11 | Ag2CO3 | K2S2O8 | NaOH | t AmOH | 48 |
| 12 | Ag 2 CO 3 | K 2 S 2 O 8 | NaOH | Dioxane | 68 |
| 13d | Ag2CO3 | K2S2O8 | NaOH | Dioxane | 60 |
| 14e | Ag2CO3 | K2S2O8 | NaOH | Dioxane | 39 |
| 15f | Ag2CO3 | K2S2O8 | NaOH | Dioxane | 53 |
| 16g | Ag2CO3 | K2S2O8 | NaOH | Dioxane | 60 |
With the optimized reaction conditions in hand, the substrate scope of the phosphonation of various 8-aminoquinoline amides with diisopropyl H-phosphonate was examined (Scheme 2). The results demonstrate that benzamides bearing electron-donating, -neutral, or -withdrawing groups on the benzene ring are all well tolerated in this reaction, and the desired products were obtained in moderate yields (3aa–3ga). However, the substrate containing strong electron-withdrawing groups on the benzene ring (NO2) did not afford the desired product probably due to its inhibiting electrophilic phosphonyl radical addition process (3ha). Note that aliphatic amides could afford the phosphonated product in good yields (3ia–3ka). Nevertheless, heterocyclic amides could also generate the products in relatively low yields (3la and 3ma). Moreover, the substrates bearing electron-donating groups (e.g., Me and MeO) at the C-2 or C-6 position could afford the products in 56% and 61% yields, respectively (3na and 3oa). These results demonstrated that the steric effect at the C-2 or C-6 position was not of crucial importance for the regioselectivity of this reaction. However, in the case of the substrate bearing an electron-withdrawing group such as the diphenylphosphinyl or nitro group at the C-7 position, the reaction did not occur at all (3pa and 3qa). In addition, N-benzylquinolin-8-amine could also be smoothly tolerated under the standard reaction conditions, although it leads to a slightly lower yield of 41% of the product (3ra).
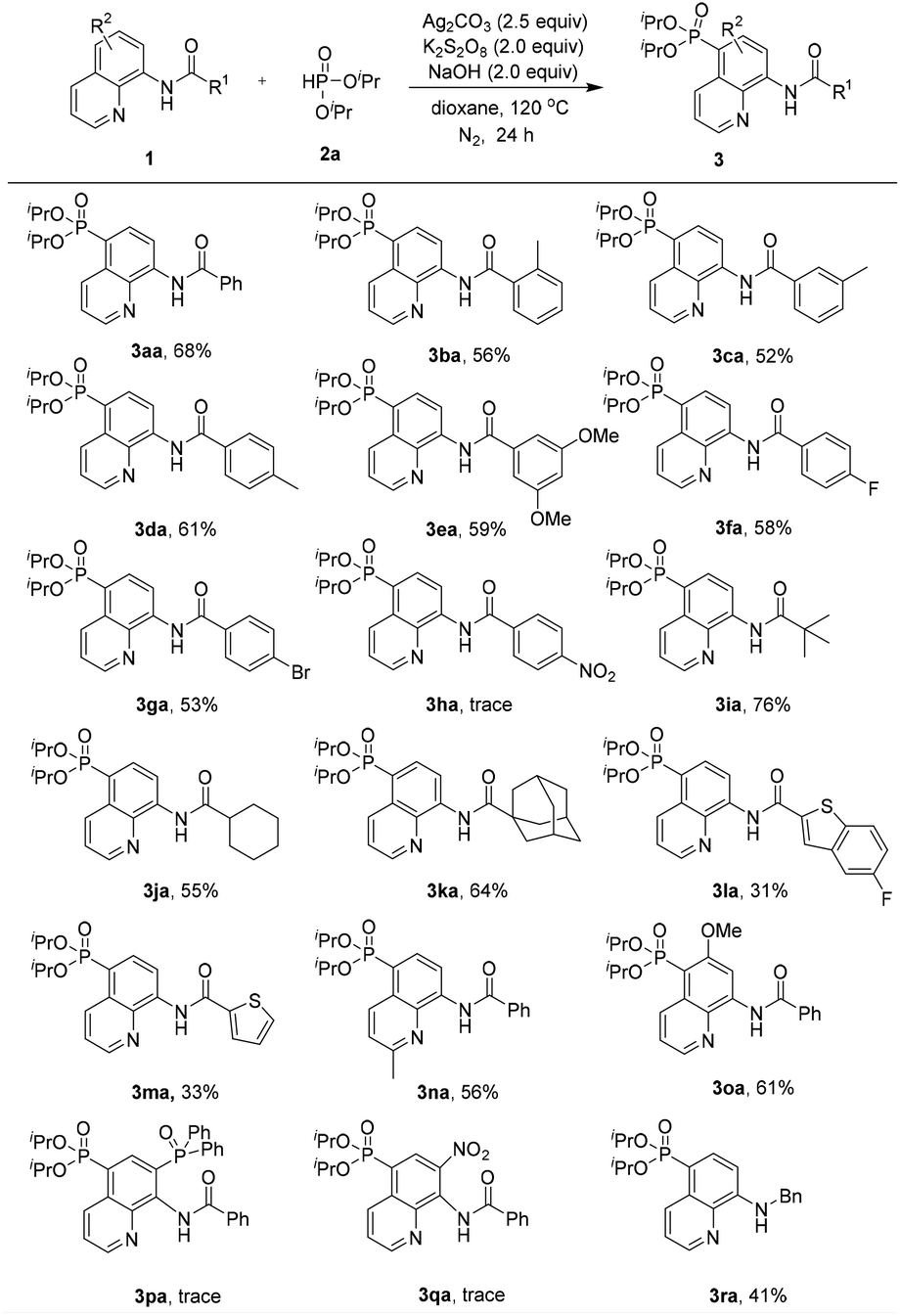 | ||
| Scheme 2 Substrate scope of 8-aminoquinoline amides. | ||
Subsequently, the effect of different H-phosphonates on the reaction was also investigated (Scheme 3). The phosphonation of N-(quinolin-8-yl)pivalamide with different dialkyl H-phosphonates under the optimized conditions was explored, the desired products were obtained in mostly moderate to good yields and the diisopropyl H-phosphonate was found to be the best coupling partner in this C5–H phosphonation (3iavs.3ib–3if).
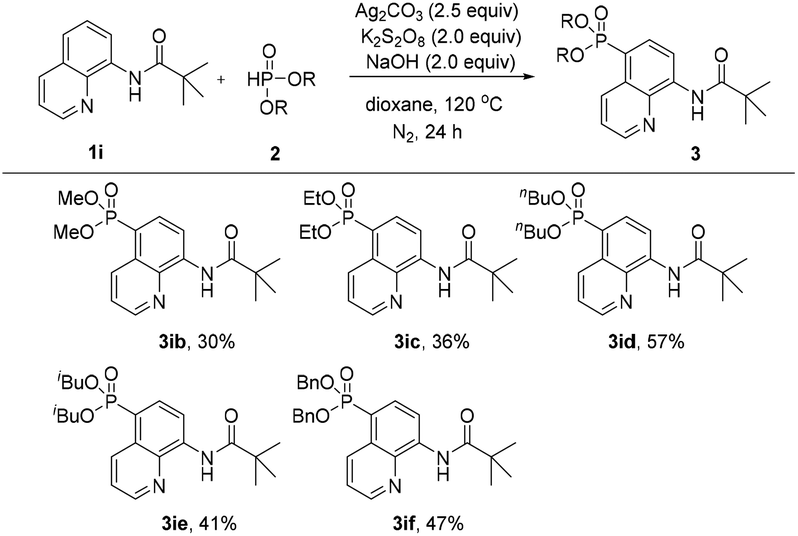 | ||
| Scheme 3 Substrate scope of different phosphonation reagents. | ||
To reveal the actual role of this bidentate directing group and obtain insight into the mechanism, control experiments were performed (Scheme 4). When some structurally similar directing groups were taken instead of the N,N-bidentate directing group (e.g., 4a, 5a, and 6a) in their phosphonation with diisopropyl H-phosphonate, only a trace amount of the products were generated, suggesting that the N,N-bidentate directing group and especially the relatively acidic NH moiety in 1a are crucial for the successful reaction (Scheme 4a). The addition of a radical scavenger such as tetramethylpiperidinyloxy (TEMPO) or 2,6-diisopropyl-4-methylphenol (BHT) inhibited the generation of product 3aa, indicating that the reaction might involve a radical process (Scheme 4b). If the C-5 position of an amide was blocked by a chlorine atom (1s), the reaction did not occur at all (Scheme 4c). Our previous report has pointed out that the C5 position of the quinoline ring shows the largest pz orbital occupancies, which implies that the C5 position might be the most likely electrophilic reactive site.8
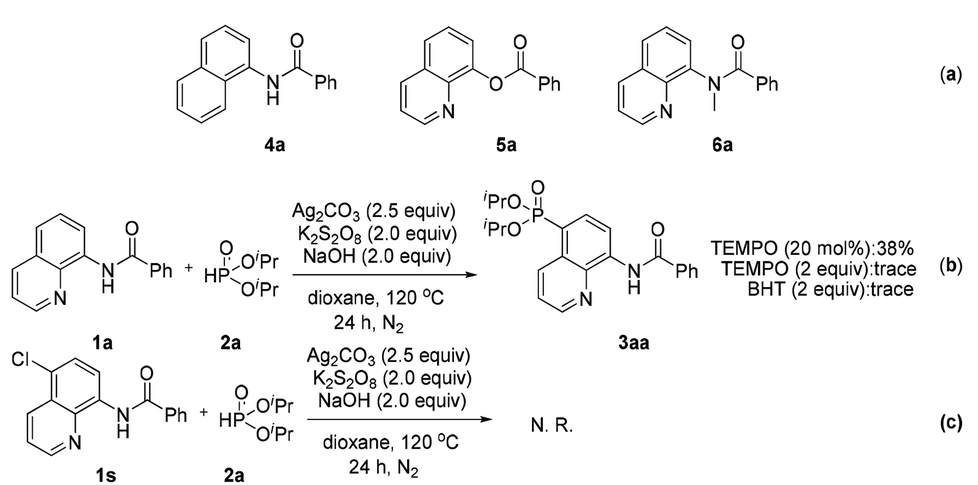 | ||
| Scheme 4 Control experiments. | ||
To demonstrate the potential application, the transformation of the product was also investigated. For example, the AQ directing group of 3aa could be easily removed by simple base hydrolysis, affording the corresponding diisopropyl(8-aminoquinolin-5-yl)phosphonate 7a in 68% yield (Scheme 5).
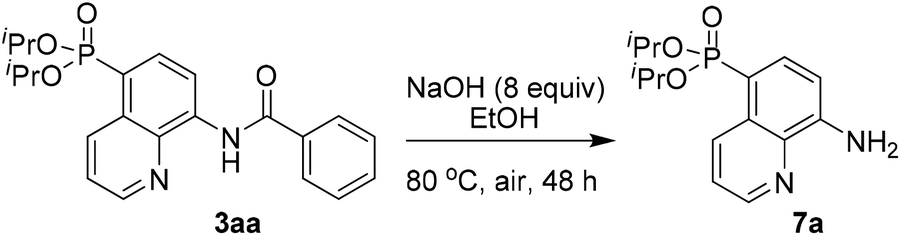 | ||
| Scheme 5 Removal of the directing group. | ||
On the basis of these experimental results and previous reports, a plausible mechanism for direct phosphonation of N-(quinolin-8-yl)benzamide 1a with diisopropyl H-phosphonate 2a is illustrated in Scheme 6. Firstly, the AgI is oxidized to unstable AgII in the presence of the persulfate anion,10 which oxidizes diisopropyl H-phosphonate 2a to afford the intermediate cation radical A. The intermediate A would be transformed into a free phosphonyl radical B after releasing a proton in the presence of a base.7c,h Then, the coordination of 1a with AgI affords the chelated intermediate C, and the radical addition of the phosphonyl radical to the intermediate C occurs at the C5 position to form an intermediate radical D, Subsequently, D is oxidized into an intermediate E by an sulfate radical via a single electron transfer process (SET), Finally, decomposition of the intermediate E successfully affords the desired product 3aa, along with the generation of the Ag2SO4 species. However, Ag2SO4 exhibits no catalytic activity (Table 1, entry 4).
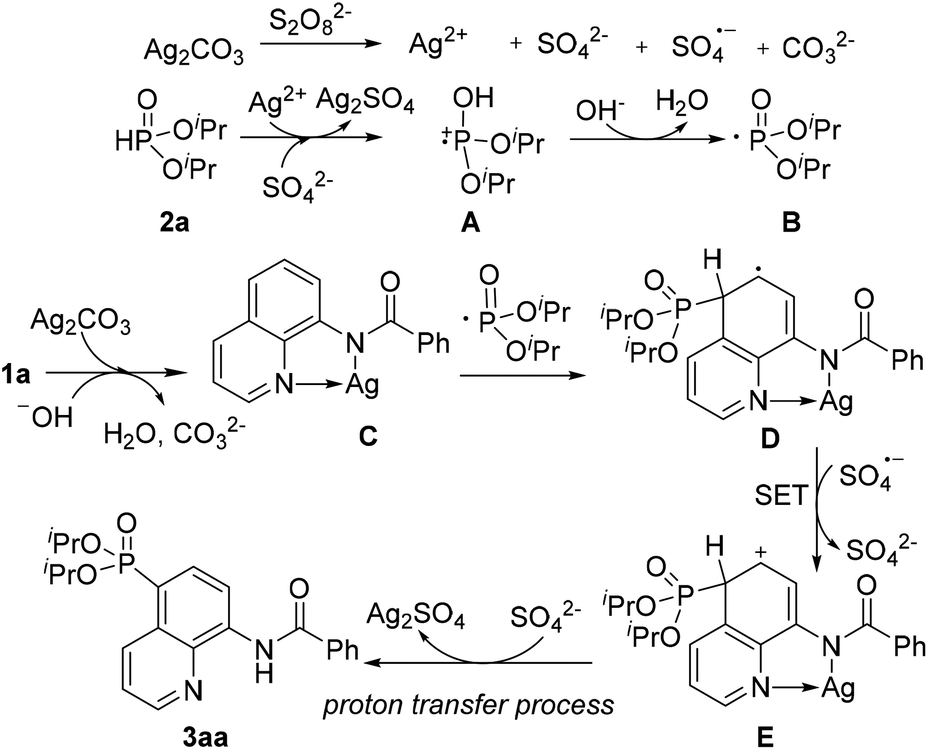 | ||
| Scheme 6 Proposed mechanism for the phosphonation of N-(quinolin-8-yl)benzamide. | ||
Conclusions
In conclusion, we have successfully developed a silver-promoted direct C5–H phosphonation of 8-aminoquinoline derivatives with H-phosphonates through a single electron transfer process, thus providing a facile and convenient route to C5 phosphonated 8-aminoquinoline derivatives. A series of C5 phosphonated amides were obtained in moderate yields with high regioselectivity.Acknowledgements
We are grateful to the National Natural Science Foundation of China (no. 21102134, 21172200) for financial support.Notes and references
- (a) J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC; (b) L. H. Zhou, H. W. Zhang, S. J. Tao, L. Bassit, T. Whitaker, T. R. McBrayer, M. Ehteshami, S. Amiralaei, U. Pradere, J. HyunCho, F. Amblard, D. Bobeck, M. Detorio, S. J. Coats and R. F. Schinazi, J. Med. Chem., 2015, 58, 3445–3458 CrossRef CAS PubMed; (c) J. L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef CAS PubMed; (d) X. Chen, D. J. Kopecky, J. Mihalic, S. Jeffries, X. Min, J. Heath, J. Deignan, S. Lai, Z. Fu, C. Guimaraes, S. Shen, S. Li, S. Johnstone, S. Thibault, H. Xu, M. Cardozo, W. Shen, N. Walker, F. Kayser and Z. Wang, J. Med. Chem., 2012, 55, 3837–3851 CrossRef CAS PubMed; (e) Y. Li, L. Q. Lu, S. Das, S. Pisiewicz, K. Junge and M. Beller, J. Am. Chem. Soc., 2012, 134, 18325–18329 CrossRef CAS PubMed; (f) C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed; (g) L. Kollár and G. Keglevich, Chem. Rev., 2010, 110, 4257–4302 CrossRef PubMed; (h) M. N. Birkholz, Z. Freixa and P. W. N. M. Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC; (i) A. George and A. Veis, Chem. Rev., 2008, 108, 4670–4693 CrossRef CAS PubMed; (j) R. F. Roush, E. M. Nolan, F. Löhr and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 3603–3609 CrossRef CAS PubMed; (k) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed.
- T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Synthesis, 1981, 56–57 CrossRef CAS.
- (a) T. Fu, H. Qiao, Z. Peng, G. Hu, X. Wu, Y. Z. Gao and Y. F. Zhao, Org. Biomol. Chem., 2014, 12, 2895–2902 RSC; (b) J. Xu, P. B. Zhang, Y. Z. Gao, Y. Y. Chen, G. Tang and Y. F. Zhao, J. Org. Chem., 2013, 78, 8176–8183 CrossRef CAS PubMed; (c) M. Li, M. Sun, H. L. Wang, Q. P. Tan and S. D. Yang, Angew. Chem., Int. Ed., 2013, 52, 3972–3976 CrossRef PubMed; (d) B. Yang, T. T. Yang, X. A. Li, J. J. Wang and S. D. Yang, Org. Lett., 2013, 15, 5024–5027 CrossRef CAS PubMed; (e) C. D. Hou, Y. L. Ren, R. Lang, X. X. Hu, C. G. Xia and F. W. Li, Chem. Commun., 2012, 48, 5181–5183 RSC; (f) Y. L. Zhao, G. J. Wu, Y. Li, L. X. Gao and F. S. Han, Chem. – Eur. J., 2012, 18, 9622–9627 CrossRef CAS PubMed; (g) J. Hu, N. Zhao, B. Yang, G. Wang, L. N. Guo, Y. M. Liang and S. D. Yang, Chem. – Eur. J., 2011, 17, 5516–5521 CrossRef CAS PubMed; (h) M. Sun, H. Y. Zhang, Q. Han, K. Yang and S. D. Yang, Chem. – Eur. J., 2011, 17, 9566–9570 CrossRef CAS PubMed; (i) D. Prasad and B. Konig, Org. Lett., 2011, 13, 3852–3855 CrossRef PubMed; (j) C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS PubMed; (k) X. Q. Pan, J. P. Zou, G. L. Zhang and W. Zhang, Chem. Commun., 2010, 46, 1721–1723 RSC; (l) S. V. Jeught and C. V. Stevens, Chem. Rev., 2009, 109, 2672–2702 CrossRef PubMed; (m) O. Basle and C. J. Li, Chem. Commun., 2009, 4124–4126 RSC.
- For transition-metal-catalyzed C–C bond formation via C–H activation, see: (a) L. Yang and H. M. Huang, Chem. Rev., 2015, 115, 3468–3517 CrossRef CAS PubMed; (b) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed; (c) C. Zhang, C. H. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464–3484 RSC; (d) A. S. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef PubMed; (e) K. M. Engle, T. S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (f) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed; (g) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (h) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC; (i) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (j) C. J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- For transition-metal-catalyzed C–heteroatom bond formation via C–H activation, see: (a) C. Shen, P. F. Zhang, Q. Sun, S. Q. Bai, T. S. A. Hor and X. G. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC; (b) X. X. Guo, D. W. Gu, Z. X. Wu and W. B. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed; (c) C. Liu, J. W. Yuan, M. Gao, S. Tang, W. Li, R. Y. Shi and A. W. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (d) W. Liu and J. Groves, Acc. Chem. Rec., 2015, 48, 1727–1735 CrossRef CAS PubMed; (e) D. G. Yu, B. J. Li and Z. J. Shi, Acc. Chem. Rec., 2010, 43, 1486–1495 CrossRef CAS PubMed; (f) C. I. Herrerías, X. Q. Yao, Z. P. Li and C. J. Li, Chem. Rev., 2007, 107, 2546–2562 CrossRef PubMed.
- For the phosphonation of C–H bonds without a directing group, see: (a) K. Luo, Y. Z. Chen, W. C. Yang, J. Zhu and L. Wu, Org. Lett., 2016, 18, 452–455 CrossRef CAS PubMed; (b) G. B. Hu, W. Z. Chen, D. M. Ma, Y. Zhang, P. X. Xu, Y. X. Gao and Y. F. Zhao, J. Org. Chem., 2016, 81, 1704–1711 CrossRef CAS PubMed; (c) K. Luo, Y. Z. Chen, L. X. Chen and L. Wu, J. Org. Chem., 2016, 81, 4682–4689 CrossRef CAS PubMed; (d) H. J. Zhang, W. D. Lin, Z. J. Wu, W. Q. Ruan and T. B. Wen, Chem. Commun., 2015, 51, 3450–3453 RSC; (e) K. Luo, Y. Z. Chen, W. C. Yang, J. Zhu and L. Wu, Org. Lett., 2015, 18, 452–455 CrossRef PubMed; (f) X. F. Huang, Q. L. Wu, J. S. He and Z. Z. Huang, Org. Biomol. Chem., 2015, 13, 4466–4472 RSC; (g) M. Yadav, S. Dara, V. Saikam, M. Kumar, S. K. Aithagani, S. Paul, R. A. Vishwakarma and P. P. Singh, Eur. J. Org. Chem., 2015, 6526–6533 CrossRef CAS; (h) O. Berger and J. L. Montchamp, Chem. – Eur. J., 2014, 20, 12385–12388 CrossRef CAS PubMed; (i) C. B. Xiang, Y. J. Bian, X. R. Mao and Z. Z. Huang, J. Org. Chem., 2012, 77, 7706–7710 CrossRef CAS PubMed; (j) H. L. Wang, X. C. Li, F. Wu and B. Wan, Synthesis, 2012, 941–945 Search PubMed; (k) W. Xu, J. P. Zou and W. Zhang, Tetrahedron Lett., 2010, 51, 2639–2643 CrossRef CAS; (l) T. Kagayama, A. Nakano, S. Sakaguchi and Y. Ishii, Org. Lett., 2006, 8, 407–409 CrossRef CAS PubMed; (m) R. Lavilla, A. Spada and J. Bosch, Org. Lett., 2000, 2, 1533–1535 CrossRef CAS PubMed; (n) F. Effenberger and H. Kottmann, Tetrahedron, 1985, 41, 4171–4182 CrossRef CAS.
- For the directing group-assisted phosphonation of ortho-C–H bonds, see: (a) M. Min, D. Kang, S. Jung and S. Hong, Adv. Synth. Catal., 2016, 358, 1296–1301 CrossRef CAS; (b) M. Gao, Y. Li, L. J. Xie, R. Chauvin and X. L. Cui, Chem. Commun., 2016, 52, 2846–2849 RSC; (c) W. B. Sun, J. F. Xue, G. Y. Zhang, R. S. Zeng, L. T. An, P. Z. Zhang and J. P. Zou, Adv. Synth. Catal., 2016, 358, 1753–1758 CrossRef CAS; (d) G. Y. Song and X. W. Li, Acc. Chem. Rec., 2015, 48, 1007–1020 CrossRef CAS PubMed; (e) J. W. Yuan, Y. Z. Li, L. R. Yang, W. P. Mai, P. Mao, Y. M. Xiao and L. B. Qu, Tetrahedron, 2015, 71, 8178–8186 CrossRef CAS; (f) S. Wang, R. Guo, G. Wang, S. Y. Chen and X. Q. Yu, Chem. Commun., 2014, 50, 12718–12721 RSC; (g) H. Wang, X. L. Cui, Y. Pei, Q. Q. Zhang, J. Bai, D. H. Wei and Y. J. Wu, Chem. Commun., 2014, 50, 14409–14411 RSC; (h) G. Hong, D. Mao, S. Y. Wu and L. M. Wang, J. Org. Chem., 2014, 79, 10629–10635 CrossRef CAS PubMed; (i) C. G. Feng, M. Ye, K. J. Xiao, S. H. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 9322–9325 CrossRef CAS PubMed; (j) C. Li, T. Yano, N. Ishida and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9801–9804 CrossRef CAS PubMed; (k) X. Mi, M. M. Huang, J. Y. Zhang, C. Y. Wang and Y. J. Wu, Org. Lett., 2013, 15, 6266–6269 CrossRef CAS PubMed.
- H. J. Qiao, S. Y. Sun, F. Yang, Y. Zhu, W. G. Zhu, Y. X. Dong, Y. S. Wu, X. T. Kong, L. Jiang and Y. J. Wu, Org. Lett., 2015, 17, 6086–6089 CrossRef CAS PubMed.
- CCDC 1474469 (3aa).† Crystal data for compound 3aa: C22H25N2O4P, M = 412.41, triclinic, a = 8.9115(7) Å, α = 83.858(6)°, b = 11.1099(7) Å, β = 86.228(6)°, c = 11.8272(9) Å, γ = 67.922(6)°, V = 1078.45(14) Å3, T = 291.15 K, space group = P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, number of reflections = 8712, independent reflections = 4383, [R(int) = 0.0373, R(sigma) = 0.0592], final R indices [I ≥ 2σ(I)] R1 = 0.0776, wR2 = 0.2075, R indices (all data) R1 = 0.1204, wR2 = 0.2482.
, Z = 2, number of reflections = 8712, independent reflections = 4383, [R(int) = 0.0373, R(sigma) = 0.0592], final R indices [I ≥ 2σ(I)] R1 = 0.0776, wR2 = 0.2075, R indices (all data) R1 = 0.1204, wR2 = 0.2482. - X. R. Mao, X. Ma, S. W. Zhang, H. W. Hu, C. J. Zhu and Y. X. Cheng, Eur. J. Org. Chem., 2013, 4245–4248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1474469 and 1474793. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00379f |
| This journal is © the Partner Organisations 2016 |