
Iodosobenzene-mediated direct and efficient oxidation of β-dicarbonyls to vicinal tricarbonyls catalyzed by iron(III) salts†
Jian
Cui
,
Ya-Nan
Duan
,
Jun
Yu
and
Chi
Zhang
*
State Key Laboratory of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, P. R. of China. E-mail: zhangchi@nankai.edu.cn
First published on 27th September 2016
Abstract
Iodosobenzene was found to be effective in the direct oxidation of β-dicarbonyls to vicinal tricarbonyl compounds catalyzed by Fe(NO3)·9H2O under mild and environmentally friendly conditions. Also, the present protocol was applied to the one-pot synthesis of heterocyclic compounds via vicinal tricarbonyls as the reactive intermediates. As for the reaction pathway, a plausible mechanism involving iodonium ylide, reactive carbene species and an α-hydroxylated intermediate was proposed.
Introduction
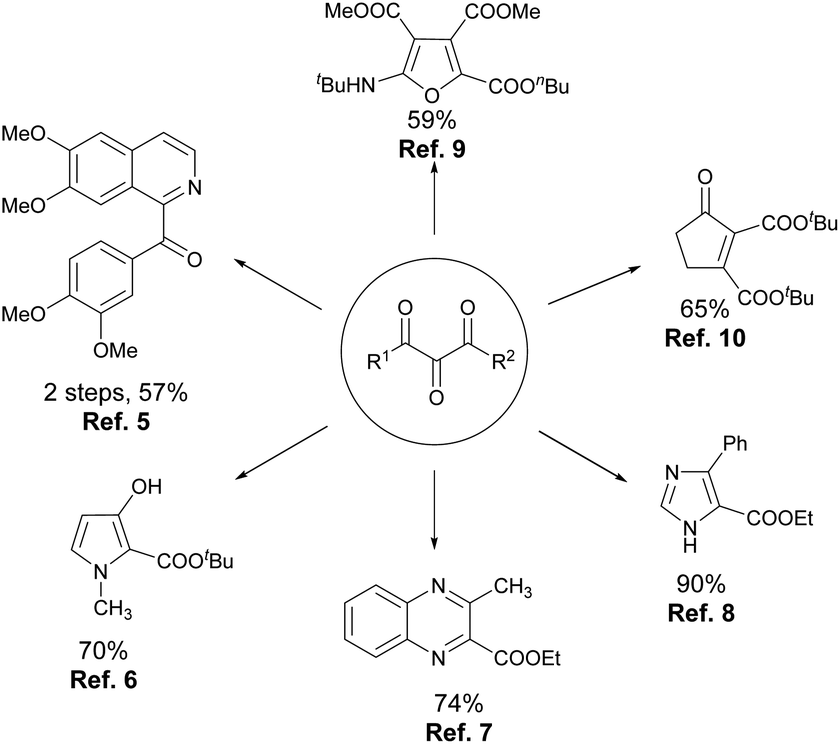
A vicinal tricarbonyl unit, together with its hemiketal form, is the key structural unit present in many biologically active natural products, such as YM47141 and YM47142,1 FK-506 and FR-900525,2 and rapamycin and 29-demethoxyrapamycin (Fig. 1).3 Besides, vicinal tricarbonyl compounds serve as important building blocks in synthetic chemistry due to their highly electrophilic nature, particularly the central one (Scheme 1).4 For example, the reactions of vicinal tricarbonyl compounds with different nitrogen nucleophiles produce isoquinoline,5 pyrrole,6 quinoxaline,7 and imidazole8 frameworks. Moreover, the corresponding furan derivatives9 and α,β-unsaturated cyclic ketones10 are provided when oxygen nucleophiles and carbon nucleophiles are respectively employed. Interestingly, vicinal tricarbonyl groups when being incorporated into polymer chains also play significant roles in the establishment of reversible cross-linking systems, which are always utilized in chemical recycling as well as self-healing materials.11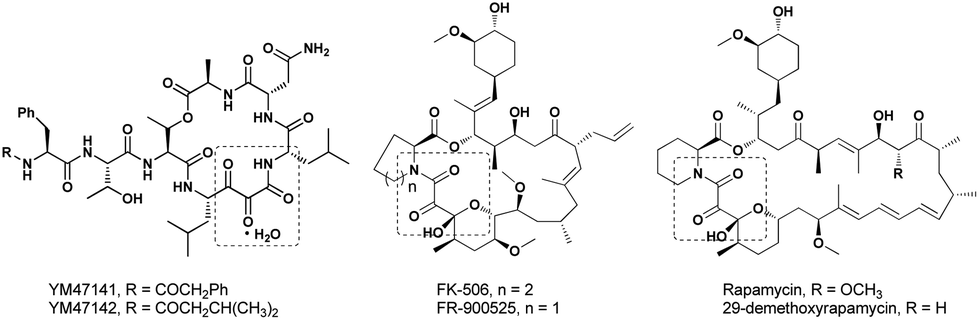 | ||
| Fig. 1 Natural products containing a vicinal tricarbonyl unit and its hemiketal forms. | ||
 | ||
| Scheme 1 Synthetic applications of vicinal tricarbonyl compounds. | ||
Usually, vicinal tricarbonyl compounds are prepared via two separate steps, that is, α-functionalization of β-dicarbonyls like bromination, and diazotization, followed by oxidation with suitable reagents such as dimethyldioxirane (DMDO), tBuOCl etc.12 The direct oxidation of the readily available β-dicarbonyl compounds is obviously the most convenient method for the synthesis of vicinal tricarbonyl compounds, which has, however, received less attention. Few examples include the use of oxidants like selenium dioxide,13 singlet oxygen,14 and hypervalent iodine(V) reagents such as DMP,15 and IBX.16 However, these methods suffer from several disadvantages such as the use of toxic reagents, harsh reaction conditions, a limited substrate scope and unsatisfactory yields. Very recently, cerium(IV) ammonium nitrate (CAN) and DDQ/TEMPO were reported as the oxidants in the synthesis of vicinal tricarbonyl compounds.17 To our knowledge, there is no protocol for direct oxidation of β-dicarbonyl compounds to vicinal tricarbonyls using iodine(III) reagents.
Iron is one of the most inexpensive and environmentally friendly metals on earth. The last few years have witnessed advances in iron-catalyzed C–H transformations,18 including site-selective C–H bond oxidation,19 and directed C–H bond activation.20 Herein, as part of our continuing research on the new synthetic applications of a commercially available hypervalent iodine(III) reagent, iodosobenzene (PhIO),21 we disclosed a straightforward strategy to vicinal tricarbonyl compounds from β-dicarbonyls by using PhIO as the oxidant and Fe(NO3)3·9H2O as the catalyst.
At the beginning, the model substrate ethyl benzoylacetate (1a) was treated with 1.5 equiv. of PhIO and 1.5 equiv. of Fe(NO3)3·9H2O in dichloromethane at room temperature. After 2 hours, it was found that the desired vicinal tricarbonyl product, ethyl 2,3-dioxo-3-phenylpropanoate (2a), could be obtained in 80% yield (Table 1, entry 1). When Fe(NO3)3·9H2O was replaced by other iron salts such as Fe(ClO4)3·9H2O, Fe2(SO4)3·6H2O, FePO4·2H2O, and FeF3, no superior yields were achieved (entries 2–5). The reaction was also conducted using PhIO alone; it was found that only 40% yield of 2a was produced after 24 h (entry 6). The use of ClCH2CH2Cl as the solvent led to 80% yield of 2a (entry 7). Ether solvents like THF, 1,2-dimethoxyethane (DME), and 1,4-dioxane resulted in lower yields of 2a compared to that in CH2Cl2 (entries 9–11). The use of EtOAc as the solvent gave the best yield of 2a, being 82% (entry 12). When polar aprotic solvents like CH3CN and CH3NO2 were employed, slightly lower yields of 2a were obtained (entries 13 and 14). Polar protic solvents like tBuOH and hexafluoroisopropanol (HFIP) gave 56% and 70% yield of 2a respectively (entries 16 and 17). Hence, EtOAc was the solvent of choice in the following investigation. When the loading of Fe(NO3)3·9H2O was reduced, the yield of 2a increased (entry 12 vs. entries 18 and 19).22 The use of 0.2 equiv. of Fe(NO3)3·9H2O led to the best yield of 2a, being 91% within 3 h (entry 19). The further reduction of the loading to 0.15 or 0.1 equiv. resulted in slightly lower yields of 2a (entries 20 and 21). Hence, the best loading of Fe(NO3)3·9H2O was 0.2 equiv. When the reaction was conducted under a N2 atmosphere, only 55% yield of the tricarbonyl was obtained even with the reaction time being prolonged from 3 h to 24 h (entry 22). When two other commonly used iodine(III) reagents (diacetoxy)iodobenzene (PIDA) and [hydroxy(tosyloxy)iodo]benzene (Koser's reagent) were employed as oxidants instead of PhIO, inferior results were obtained (entries 23 and 24).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (equiv.) | Solvent | Time (h) | Yieldb (%) |
| a The reaction was conducted using 0.5 mmol of 1a. b Isolated yield. c Data in the parentheses is the conversion of 1a. d The reaction was carried out under N2. e PIDA was utilized instead of iodosobenzene. f Koser's reagent was utilized instead of iodosobenzene. | ||||
| 1 | Fe(NO3)3·9H2O (1.5) | CH2Cl2 | 2 | 80 |
| 2 | Fe(ClO4)3·9H2O (1.5) | CH2Cl2 | 1 | 61 |
| 3 | Fe2(SO4)3·6H2O (1.5) | CH2Cl2 | 43 | 18 (72)c |
| 4 | FePO4·2H2O (1.5) | CH2Cl2 | 70 | 54 (73)c |
| 5 | FeF3 (1.5) | CH2Cl2 | 52 | 43 (89)c |
| 6 | None | CH2Cl2 | 24 | 40 (83)c |
| 7 | Fe(NO3)3·9H2O (1.5) | DCE | 17 | 80 |
| 8 | Fe(NO3)3·9H2O (1.5) | Toluene | 6 | 74 |
| 9 | Fe(NO3)3·9H2O (1.5) | THF | 24 | 21 |
| 10 | Fe(NO3)3·9H2O (1.5) | DME | 1 | 74 |
| 11 | Fe(NO3)3·9H2O (1.5) | 1,4-dioxane | 1 | 65 |
| 12 | Fe(NO3)3·9H2O (1.5) | EtOAc | 40 min | 82 |
| 13 | Fe(NO3)3·9H2O (1.5) | CH3CN | 20 min | 79 |
| 14 | Fe(NO3)3·9H2O (1.5) | CH3NO2 | 10 min | 73 |
| 15 | Fe(NO3)3·9H2O (1.5) | DMF | 3 d | 12 (92)c |
| 16 | Fe(NO3)3·9H2O (1.5) | t BuOH | 3 | 56 |
| 17 | Fe(NO3)3·9H2O (1.5) | HFIP | 4 | 70 |
| 18 | Fe(NO3)3·9H2O (0.5) | EtOAc | 1 | 85 |
| 19 | Fe(NO3)3·9H2O (0.2) | EtOAc | 3 | 91 |
| 20 | Fe(NO3)3·9H2O (0.15) | EtOAc | 8 | 82 (93)c |
| 21 | Fe(NO3)3·9H2O (0.1) | EtOAc | 8 | 80 (93)c |
| 22d | Fe(NO3)3·9H2O (0.2) | EtOAc | 24 | 55 (65)c |
| 23e | Fe(NO3)3·9H2O (0.2) | EtOAc | 24 | 70 (82)c |
| 24f | Fe(NO3)3·9H2O (0.2) | EtOAc | 5 | 61 |
After having optimized the reaction conditions, the substrate scope was investigated (Scheme 2). Both methyl and tert-butylbenzoylacetate were efficiently transformed to their corresponding vicinal tricarbonyl products 2b–2c in high yields. The successful transformation of benzyl benzoylacetate to the desired product 2d implied that the benzyl group was compatible under the present reaction conditions. Substrates bearing either electron-donating or electron-withdrawing substituents at the para or meta positions of the phenyl ring of the benzoyl moiety were also smoothly converted to the expected vicinal tricarbonyl products (2e–2i) in high yields. Other aromatic ring systems such as naphthalene, thiophene and furan were all well-tolerated under the reaction conditions as indicated by the successful formation of products 2j–2l. As for aliphatic β-keto esters, their corresponding vicinal tricarbonyl products 2m–2o were obtained in moderate to good yields. The β-diketone substrate 1,3-diphenylpropane-1,3-dione was transformed to the desired product 2p in 50% yield. Two β-ketoamide substrates were also examined, and the corresponding vicinal tricarbonyl products 2q and 2r were obtained in 67% and 68% yields, respectively.
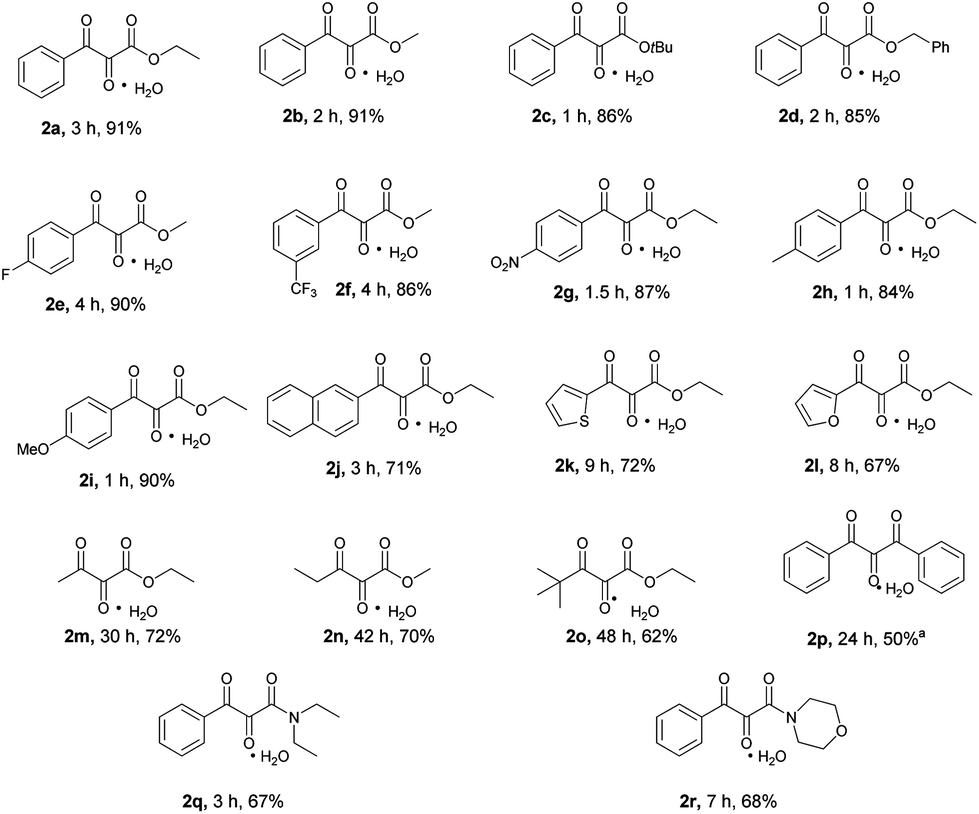 | ||
Scheme 2 Substrate scope for the synthesis of vicinal tricarbonyl compounds. All the reactions were conducted on 0.5 mmol scale and the yields shown here are isolated ones. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reaction was carried out at −20 °C. The reaction was carried out at −20 °C. | ||
The success of the vicinal tricarbonyl compound synthesis inspired us to explore the synthetic utility of the present method in the synthesis of heterocyclic compounds through a one pot procedure directly from 1,3-dicarbonyl compounds. The substrate 1a was treated under the standard conditions for 4 h, followed by the addition of benzene-1,2-diamine. It was found that a quinoxaline derivative 3a was obtained in 71% yield in one pot (Scheme 3, eqn (1)). When 2-amino-3-methylphenol was used instead of benzene-1,2-diamine, [1,4]oxazine derivative 3b was provided in 68% yield (Scheme 3, eqn (2)).
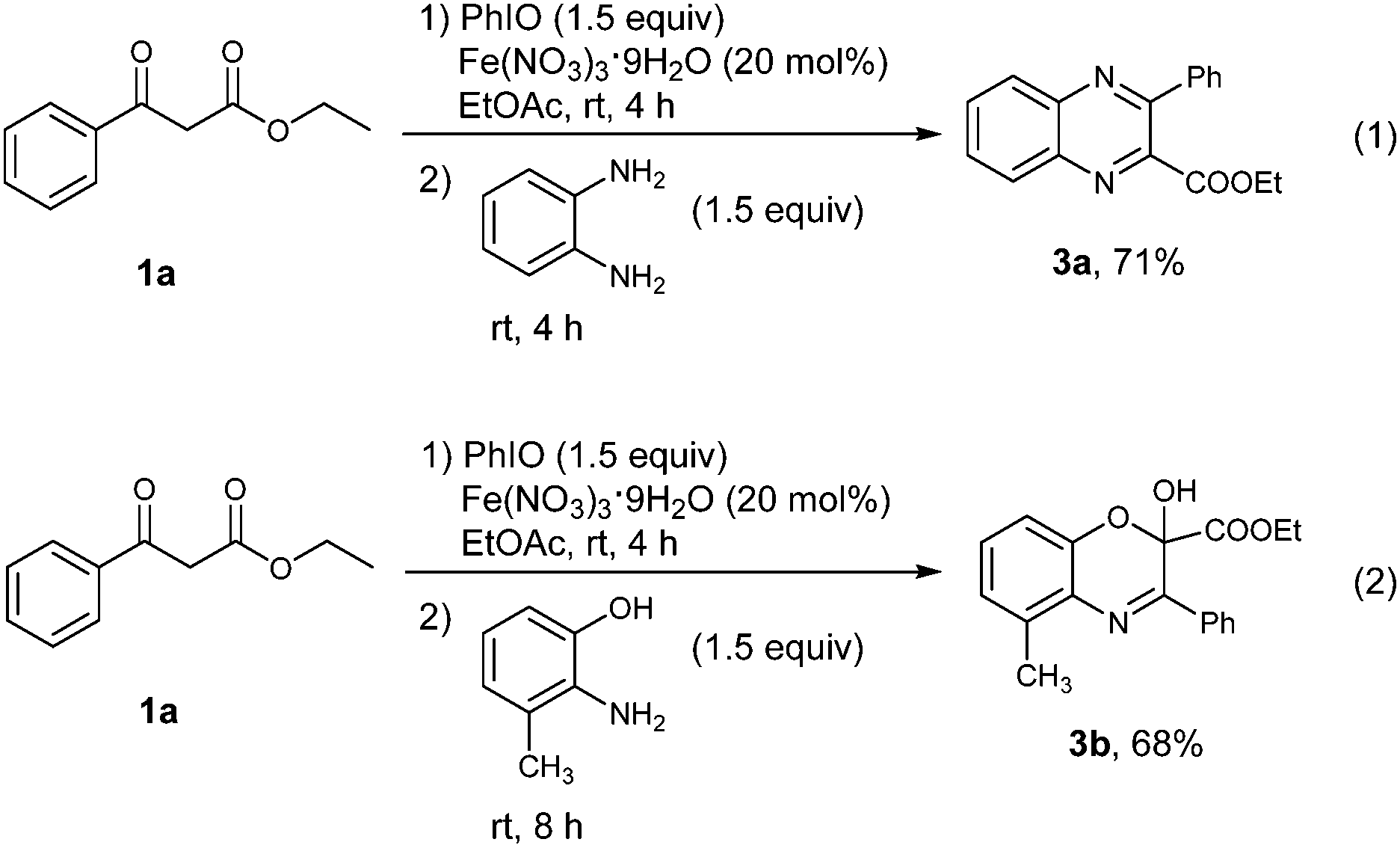 | ||
| Scheme 3 One pot procedure for the construction of two heterocyclic compounds. | ||
To explore the reaction mechanism, several control experiments were carried out (Scheme 4). There are reports to describe that PhIO can react with 1,3-dicarbonyl compounds to form iodonium ylides.23 To check whether an iodonium ylide was a possible intermediate, the iodonium ylide A was prepared according to the method reported in the literature,12m and then treated with Fe(NO3)3·9H2O in EtOAc for 10 min. It was found that the desired product 2a could be obtained in 90% yield. This fact implied the intermediacy of iodonium ylide A in the present reaction (Scheme 4, eqn (1)). There was a possibility that the central carbonyl group of the product was formed by the oxidation of a hydroxyl group. To check whether the α-hydroxylated dicarbonyl compound 4a was a possible intermediate, 4a was prepared separately,12n and then subjected to Fe(NO3)3·9H2O in EtOAc. It was found that the reaction was complete within 10 min, and the yield of the desired product 2a was 95%, indicating the intermediacy of 4a in the present reaction (Scheme 4, eqn (2)). To further investigate the source of oxygen incorporated at the central carbon of 2a, an experiment using 18O-labeled PhIO was conducted (Scheme 4, eqn (3)).24 Mass spectrometry showed that an 18O-labeled vicinal tricarbonyl product was formed, indicating that the I(III) oxidant PhIO can act as the oxygen donor in the present transformation. In addition, when 18O-labeled H2O was employed as the reaction additive under the standard conditions (Scheme 4, eqn (4)), the 18O-labeled vicinal tricarbonyl product was also formed, which shows that the central oxygen of 2a also comes from water.
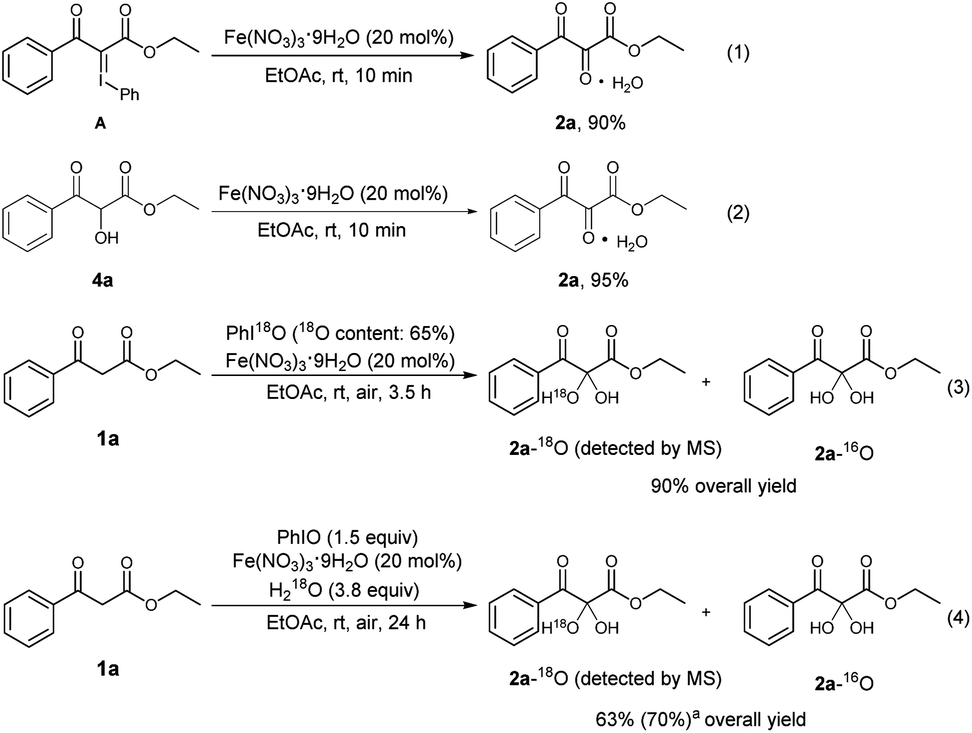 | ||
| Scheme 4 Control experiments. aThe data in the parentheses is the conversion of 1a. | ||
Based on the above experiments and previous literature reports, a plausible mechanism was proposed and is shown in Scheme 5. Firstly, 1a reacted with PhIO to form iodonium ylide A with the release of water. Then, the iodonium ylide A tautomerized to a zwitterionic intermediate B, which was stabilized by an iron(III) salt to generate the intermediate C.25 Subsequently, iodobenzene was eliminated to form carbene D, which then inserted into the O–H bond of water to give hydroxylated intermediate 4a. After further oxidation of the hydroxyl group (cf.Scheme 4, eqn (2)) and water addition, the hydrate form of the vicinal tricarbonyl product 2a was thus yielded.
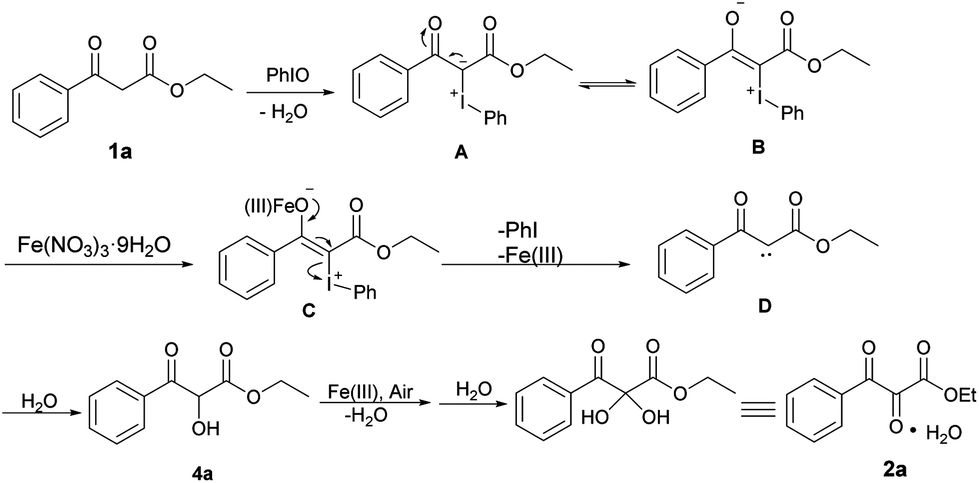 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In summary, we have developed a mild, user-friendly and efficient method for the synthesis of vicinal tricarbonyl compounds directly from β-dicarbonyls employing commercially available PhIO as the oxidant and Fe(NO3)3·9H2O as the catalyst. A plausible mechanism was also proposed involving iodonium ylide, reactive carbene, and α-hydroxylated intermediates. From a synthetic point of view, this protocol would be useful in the preparation of vicinal tricarbonyl compounds and many heterocyclic compounds through a one-pot procedure.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21172110 21472094, and 21421062).Notes and references
- (a) K. Yasumuro, Y. Suzuki, M. Shibazaki, K. Teramura, K. Abe and M. Orita, J. Antibiot., 1995, 48, 1425–1429 CrossRef CAS PubMed; (b) M. Orita, K. Yasumuro, K. Kokubo, M. Shimizu, K. Abe, T. Tokunaga and H. Kaniwa, J. Antibiot., 1995, 48, 1430–1434 CrossRef CAS PubMed; (c) H. H. Wasserman, J.-H. Chen and M. Xia, J. Am. Chem. Soc., 1999, 121, 1401–1402 CrossRef CAS.
- (a) H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto and T. Taga, J. Am. Chem. Soc., 1987, 109, 5031–5033 CrossRef CAS; (b) T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki and H. Imanaka, J. Antibiot., 1987, 40, 1249–1255 CrossRef CAS PubMed; (c) H. Hatanaka, T. Kino, M. Asano, T. Goto, H. Tanaka and M. Okuhara, J. Antibiot., 1989, 42, 620–622 CrossRef CAS PubMed; (d) T. K. Jones, S. G. Mills, R. A. Reamer, D. Askin, R. Desmond, R. P. Volante and I. Shinkai, J. Am. Chem. Soc., 1989, 111, 1157–1159 CrossRef CAS.
- (a) C. Vezina, A. Kudelski and S. N. Sehgal, J. Antibiot., 1975, 28, 721–726 CrossRef CAS PubMed; (b) D. C. N. Swindells, P. S. White and J. A. Findlay, Can. J. Chem., 1978, 56, 2491–2492 CrossRef CAS; (c) J. A. Findlay, J.-S. Liu and D. J. Burnell, Can. J. Chem., 1982, 60, 2046–2047 CrossRef CAS.
- (a) H. H. Wasserman and J. Parr, Acc. Chem. Res., 2004, 37, 687–701 CrossRef CAS PubMed; (b) M. B. Rubin and R. Gleiter, Chem. Rev., 2000, 100, 1121–1164 CrossRef CAS PubMed; (c) M. B. Rubin, Chem. Rev., 1975, 75, 177–202 CrossRef CAS.
- H. H. Wasserman, R. Amici, R. Frechette and J. H. Van Duzer, Tetrahedron Lett., 1989, 30, 869–872 CrossRef CAS.
- (a) H. H. Wasserman, J. D. Cook, J. M. Fukuyama and V. M. Rotello, Tetrahedron Lett., 1989, 30, 1721–1724 CrossRef CAS; (b) H. H. Wasserman and L. J. Lombardo, Tetrahedron Lett., 1989, 30, 1725–1728 CrossRef CAS.
- (a) J. A. Barltrop, C. G. Richards, D. M. Russell and G. Ryback, J. Chem. Soc., 1959, 1132–1142 RSC; (b) R. V. Hoffman, H.-O. Kim and A. L. Wilson, J. Org. Chem., 1990, 55, 2820–2822 CrossRef CAS; (c) R. M. Adlington, J. E. Baldwin, D. Catterick and G. J. Pritchard, J. Chem. Soc., Perkin Trans. 1, 2001, 668–679 RSC.
- M. F. Brackeen, J. A. Stafford, P. L. Feldman and D. S. Karanewsky, Tetrahedron Lett., 1994, 35, 1635–1638 CrossRef CAS.
- (a) H. H. Wasserman and G. M. Lee, Tetrahedron Lett., 1994, 35, 9783–9786 CrossRef CAS; (b) V. Nair and A. Deepthi, Tetrahedron Lett., 2006, 47, 2037–2039 CrossRef CAS.
- H. H. Wasserman, M. Shiraishi, S. J. Coats and J. D. Cook, Tetrahedron Lett., 1995, 36, 6785–6788 CrossRef CAS.
- (a) M. Yonekawa, Y. Furusho and T. Endo, Macromolecules, 2012, 45, 6640–6647 CrossRef CAS; (b) K. Morino, A. Sudo and T. Endo, Macromolecules, 2012, 45, 4494–4499 CrossRef CAS; (c) T. Yuki, M. Y. K. Matsumoto, I. Tomita and T. Endo, Polym. Bull., 2016, 73, 345–356 CrossRef CAS.
- (a) S. Wolfe, J. E. Berry and M. R. Peterson, Can. J. Chem., 1976, 54, 210–217 CrossRef CAS; (b) M. R. Mahran, W. M. Abdou, M. M. Sidky and H. Wamhoff, Synthesis, 1987, 506–508 CrossRef CAS; (c) S. J. Coats and H. H. Wasserman, Tetrahedron Lett., 1995, 36, 7735–7738 CrossRef CAS; (d) R. Gleiter and P. Schang, Angew. Chem., Int. Ed. Engl., 1980, 19, 715–716 CrossRef; (e) J. Detering and H.-D. Martin, Angew. Chem., Int. Ed. Engl., 1988, 27, 695–698 CrossRef; (f) C. M. Gasparski, A. Ghosh and M. J. Miller, J. Org. Chem., 1992, 57, 3546–3550 CrossRef CAS; (g) I. Ryu, H. Kuriyama, H. Miyazato, S. Minakata, M. Komatsu, J.-Y. Yoon and S. Kim, Bull. Chem. Soc. Jpn., 2004, 77, 1407–1408 CrossRef CAS; (h) H. H. Wasserman and W. T. Han, Tetrahedron Lett., 1984, 25, 3743–3746 CrossRef CAS; (i) H. J. Bestmann and W. Kloeters, Tetrahedron Lett., 1978, 19, 3343–3344 CrossRef; (j) H. H. Wasserman and C. B. Vu, Tetrahedron Lett., 1990, 31, 5205–5208 CrossRef CAS; (k) H. H. Wasserman, C. M. Baldino and S. J. Coats, J. Org. Chem., 1995, 60, 8231–8235 CrossRef CAS; (l) K. Schank and C. Schuhknecht, Chem. Ber., 1982, 115, 3032–3041 CrossRef CAS; (m) K. Schank and C. Lick, Synthesis, 1983, 392–395 CrossRef CAS; (n) A.-M. Lluch, M. Gibert, F. Sánchez-Baeza and A. Messeguer, Tetrahedron, 1996, 52, 3973–3982 CrossRef CAS; (o) M. S. Santos and F. Coelho, RSC Adv., 2012, 2, 3237–3241 RSC.
- F. Dayer, H. L. Dao, H. Gold, H. Rodé-Gowal and H. Dahn, Helv. Chim. Acta, 1974, 57, 2201–2209 CrossRef CAS.
- H. H. Wasserman and J. E. Pickett, J. Am. Chem. Soc., 1982, 104, 4695–4696 CrossRef CAS.
- M. J. Batchelor, R. J. Gillespie, J. M. C. Golec and C. J. R. Hedgecock, Tetrahedron Lett., 1993, 34, 167–170 CrossRef CAS.
- A. Duschek and S. F. Kirsch, Chem. – Eur. J., 2009, 15, 10713–10717 CrossRef CAS PubMed.
- (a) A. Sivan and A. Deepthi, Tetrahedron Lett., 2014, 55, 1890–1893 CrossRef CAS; (b) Z.-L. Wang, X.-L. An, L.-S. Ge, J.-H. Jin, X. Luo and W.-P. Deng, Tetrahedron, 2014, 70, 3788–3792 CrossRef CAS.
- (a) C. Bolm, J. Legros, J. L. Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed; (b) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed.
- (a) A. Company, L. Gómez, M. Güell, X. Ribas, J. M. Luis, L. Que Jr. and M. Costas, J. Am. Chem. Soc., 2007, 129, 15766–15767 CrossRef CAS PubMed; (b) M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS PubMed; (c) P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052–14055 CrossRef CAS PubMed.
- (a) R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030–6032 CrossRef CAS PubMed; (b) L. Ilies, T. Matsubara, S. Ichikawa, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 13126–13129 CrossRef CAS PubMed; (c) E. R. Fruchey, B. M. Monks and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 13130–13133 CrossRef CAS PubMed; (d) Q. Gu, H. H. Al Mamari, K. Graczyk, E. Diers and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 3868–3871 CrossRef CAS PubMed.
- (a) J. Cui, Q. Jia, R.-Z. Feng, S.-S. Liu, T. He and C. Zhang, Org. Lett., 2014, 16, 1442–1445 CrossRef CAS PubMed; (b) J. Yu, S.-S. Liu, J. Cui, X.-S. Hou and C. Zhang, Org. Lett., 2012, 14, 832–835 CrossRef CAS PubMed.
- J. Mecinović, R. B. Hamed and C. J. Schofield, Angew. Chem., Int. Ed., 2009, 48, 2796–2800 CrossRef PubMed.
- (a) P. Müller and A. Ghanem, Org. Lett., 2004, 6, 4347–4350 CrossRef PubMed; (b) H. Pellissier, Tetrahedron, 2008, 64, 7041–7095 CrossRef CAS; (c) S. R. Goudreau, D. Marcoux and A. B. Charette, J. Org. Chem., 2009, 74, 470–473 CrossRef CAS PubMed.
- B. C. Schardt and C. L. Hill, Inorg. Chem., 1983, 22, 1563–1565 CrossRef CAS.
- (a) A. S. Ivanov, I. A. Popov, A. I. Boldyrev and V. V. Zhdankin, Angew. Chem., Int. Ed., 2014, 53, 9617–9621 CrossRef CAS PubMed; (b) Y.-D. Yang, A. Azuma, E. Tokunaga, M. Yamasaki, M. Shiro and N. Shibata, J. Am. Chem. Soc., 2013, 135, 8782–8785 CrossRef CAS PubMed; (c) J.-P. Battioni, I. Artaud, D. Dupre, P. Leduc, I. Akhrem, D. Mansuy, J. Fischer, R. Weiss and I. Morgenstern-Badarau, J. Am. Chem. Soc., 1986, 108, 5598–5607 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00405a |
| This journal is © the Partner Organisations 2016 |