
Iron-catalyzed decarbonylation initiated [2 + 2 + m] annulation of benzene-linked 1,n-enynes with aliphatic aldehydes†
Leiyang
Lv
,
Xiaohui
Bai
,
Xiaoyu
Yan
 * and
Zhiping
Li
*
* and
Zhiping
Li
*
Department of Chemistry, Renmin University of China, Beijing 100872, China. E-mail: yanxy@ruc.edu.cn; zhipingli@ruc.edu.cn; Web: http://chem.ruc.edu.cn/ligroup/index.html
First published on 30th August 2016
Abstract
An iron-catalyzed decarbonylation initiated [2 + 2 + m] annulation of benzene-linked 1,n-enynes with aliphatic aldehydes has been developed. These divergent annulations allow the one-step and efficient synthesis of various fused- or spiro-polycyclic frameworks. Tertiary aldehydes undergo decarbonylation/β-C(sp3)–H cleavage to afford [2 + 2 + 2] cyclization products, whereas secondary aldehydes undergo decarbonylation/α-C(sp3)–H cleavage to generate [2 + 2 + 1] cyclization products.
Introduction
Recently, transition metal catalyzed cyclization of 1,n-enynes has attracted considerable attention from organic chemists due to the fact that it allows direct, rapid and efficient access to assemble a wide range of highly functionalized polycyclic molecules.1–3,5–7 Although a little progress has been achieved in this field, the development of unique 1,n-enyne cyclization routes to construct distinctive, unconventional architectures remains a challenge. Iron catalysts are widely used in chemical syntheses and pharmaceutical industries, probably due to the extensive abundance of iron in nature, low-lost, and particular low-toxicity in living systems.4 The development of an efficient, selective and environmentally benign 1,n-enynes cyclization catalyzed by iron would be a far-reaching advance in this field.In 2015, Tu and Li et al. realized an unprecedented iron catalyzed domino spirocyclization of benzene-linked 1,7-enynes with simple cycloalkanes by α,α-C(sp3)–H abstraction/insertion activation strategy.5 In addition, Li et al. also reported an elegant metal-free radical [2 + 2 + 1] carbocyclization of benzene-linked 1,n-enynes (n = 6,7) through dual C(sp3)–H functionalization adjacent to a heteroatom (Scheme 1, eqn (a)).6 These two alkyl radical trigged cascade spirocyclizations of 1,n-enynes are groundbreaking and pioneering and present a new synthetic method to fabricate fused [6.6.5] polycyclic architecture. Thereafter, Li et al. also realized visible light initiated or copper catalyzed annulation of 1,6-enynes with α-carbonyl alkyl bromides as the alkyl radical sources (Scheme 1b).7 Mechanistically, an intramolecular 1,5-hydride radical shift of the vinyl radical species to generate new alkyl radical intermediates was involved.8 Very recently, we reported an iron-catalyzed, radical-mediated [2 + 2 + 2] annulation of benzene linked 1,7-enynes with aromatic aldehydes (Scheme 1c).9 This aromatic aldehydic radical exhibits dual roles, which triggers and terminates the domino cyclization.
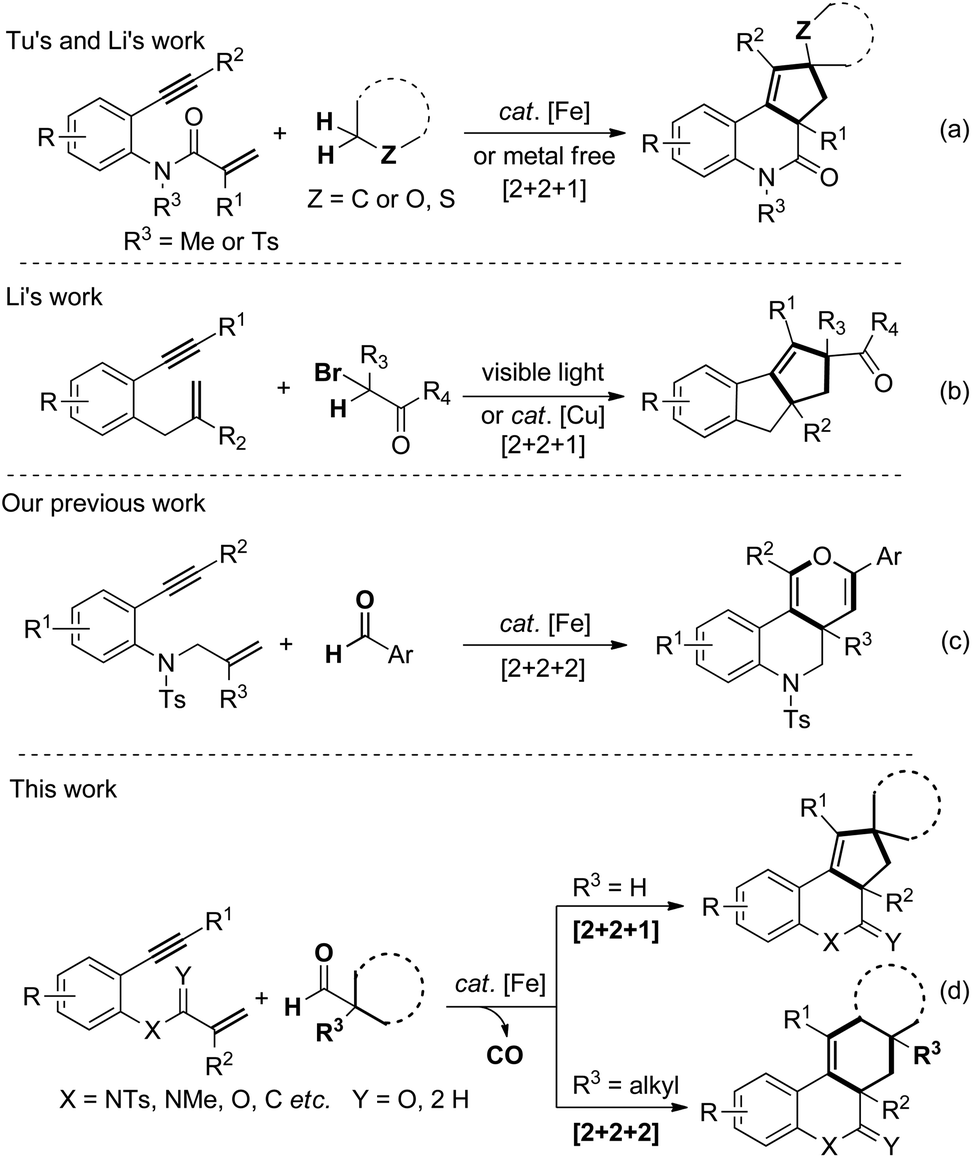 | ||
| Scheme 1 [2 + 2 + m] annulations of benzene-linked 1,n-enynes. | ||
The ordinary secondary and tertiary alkyl radicals generated from alkyl halides are extremely challenging species due to the high steric hindrance and electron-rich nature. Moreover, the alkyl radicals are really unstable and quickly undergo isomerization, disproportionation or undesired β-hydride elimination. In our hypothesis, if decarbonylation of alkyl aldehyde can be controlled, it may be utilized as an efficient precursor of alkyl radical.10 Towards this idea, herein, we present a novel decarbonylation initiated [2 + 2 + m] annulations of benzene-linked 1,n-enynes with aliphatic aldehydes (Scheme 1d).11 Tertiary aldehydes undergo decarbonylation/β-C(sp3)–H cleavage to afford [2 + 2 + 2] cyclization products, whereas secondary aldehydes undergo decarbonylation/α-C(sp3)–H cleavage to generate [2 + 2 + 1] cyclization products.
Results and discussion
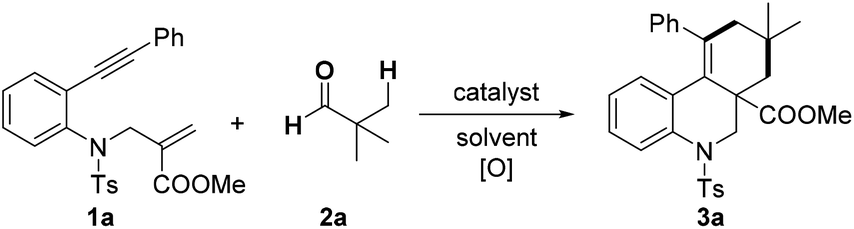
In order to optimize the reaction conditions, we commenced our studies by exploring the reactions of benzene-linked 1,7-enyne 1a with pivalaldehyde 2a (Table 1). Solvent screening showed that treatment of 1,7-enyne 1a with 2a, FeCl2 and di-tert-butyl peroxide (DTBP) in chlorobenzene led to a tricyclic product 3a in 65% yield (entries 1–4). Furthermore, catalyst screening revealed that other iron salts worked tolerably (entries 5–8), whereas other tested metal catalyst (Cu, Co, Mn, Ni and Pd) had no positive effect on the transformation (entries 9–13). Other oxidant such as tert-butyl hydroperoxide (TBHP) and tert-butyl peroxybenzoate (TBPB) reduced the efficiency of the reaction (entries 14 and 15). Notably, the reaction could also furnish product 3a in 24% yield in the absence of FeCl2 (entry 16).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | [O] | Solvent | 3a (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (1.5 mmol), catalyst (2.5 mol%), [O] (0.75 mmol), solvent (1.0 mL), 120 °C, 2 h, under N2. b Reported yields were based on 1a and determined by 1H NMR using an internal standard. | ||||
| 1 | FeCl2 | (t-BuO)2 | MeCN | 51 |
| 2 | FeCl2 | (t-BuO)2 | DCE | 57 |
| 3 | FeCl2 | (t-BuO)2 | EtOAc | 58 |
| 4 | FeCl 2 | (t-BuO) 2 | PhCl | 65 |
| 5 | FeCl3 | (t-BuO)2 | PhCl | 55 |
| 6 | FeBr2 | (t-BuO)2 | PhCl | 61 |
| 7 | Fe(OAc)2 | (t-BuO)2 | PhCl | 62 |
| 8 | Fe(acac)2 | (t-BuO)2 | PhCl | 51 |
| 9 | CuCl2 | (t-BuO)2 | PhCl | 50 |
| 10 | CoCl2 | (t-BuO)2 | PhCl | 39 |
| 11 | MnCl2 | (t-BuO)2 | PhCl | 42 |
| 12 | NiCl2 | (t-BuO)2 | PhCl | 30 |
| 13 | PdCl2 | (t-BuO)2 | PhCl | 32 |
| 14 | FeCl2 | t-BuOOH | PhCl | 32 |
| 15 | FeCl2 | PhCOOOtBu | PhCl | 42 |
| 16 | — | (t-BuO)2 | PhCl | 24 |
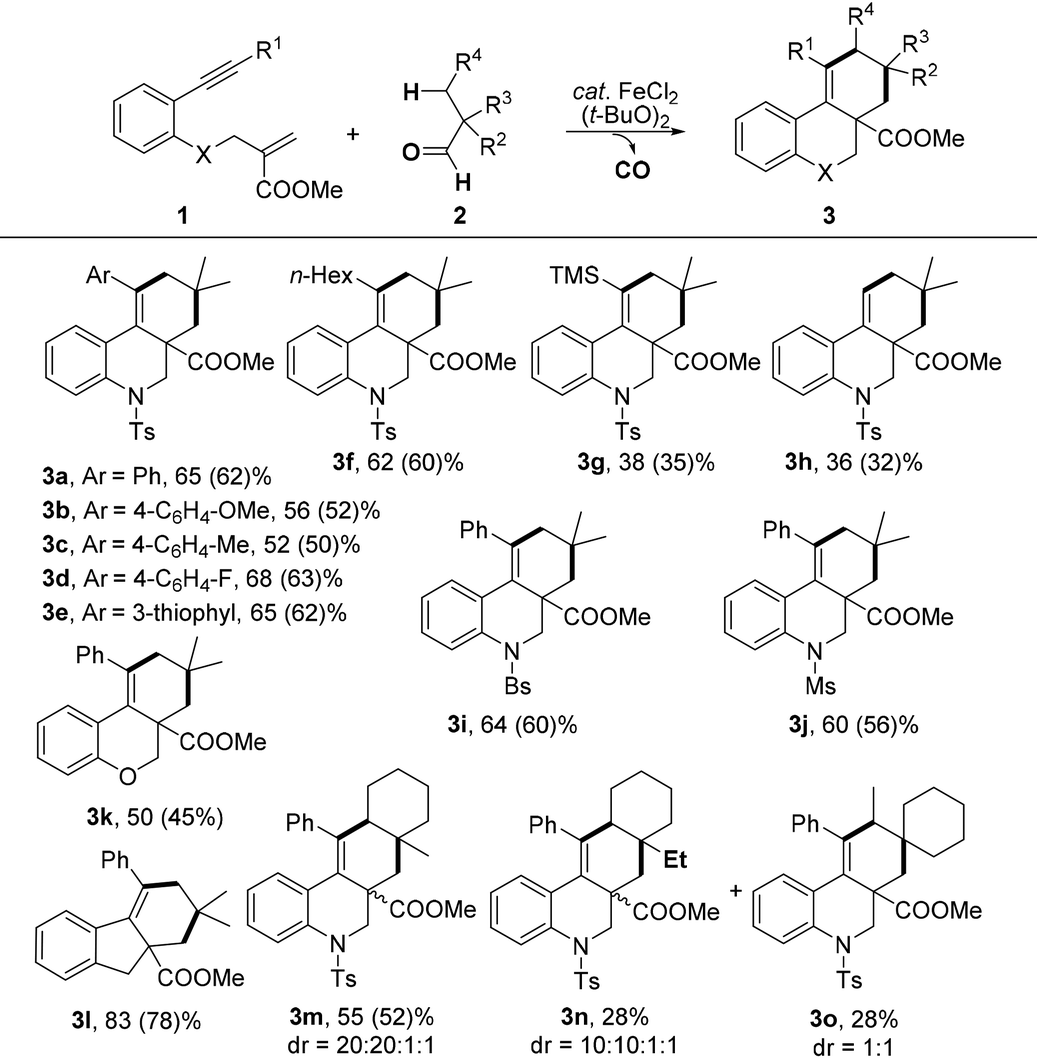
With the optimal reaction conditions in hand, we turned our attention to investigate the scope of this decarbonylation-initiated cyclization of tertiary aldehydes 2 with 1,n-enynes 1 (Table 2). Variations on the aromatic moiety of alkynes did not reduce the efficiency of this reaction (3a–e). The aliphatic alkyne 1f was also applicable for this transformation to afford the desired 3f in 62% yield. It can be noted that 3g and 3h were generated, albeit in lower yields, when trimethylsilyl alkyne 2g and terminal alkyne 2h were applied, respectively. Moreover, N-brosyl and mesyl substituted anilines also reacted smoothly to afford the desired products 3i and 3j. It is worth noting that the fused [6.6.6] oxygen-containing skeletons 3k and tetrahydro-1H-fluorene (3l) were constructed successfully. When 1-methylcyclohexane-1-carbaldehyde 2b was applied, cyclization occurred exclusively on methylene to deliver tetracyclic hydrobenzo-phenanthridine 3m in a regiospecific manner. The structure of one major isomer was confirmed by X-ray diffraction (Fig. 1).12 In contrast, 3n and 3o were produced in comparative yields when 1-ethylcyclohexane-1-carbaldehyde 2c was investigated. These results indicated that the selectivity of C–H bond functionalization of this method most likely depends on the stability of new generated alkyl radical intermediate (see mechanism in Scheme 3).
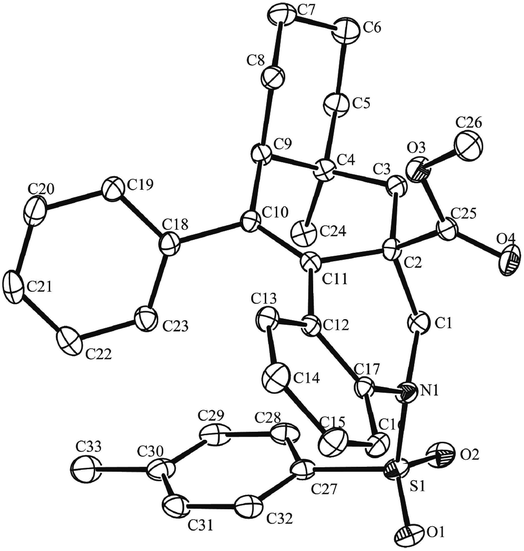 | ||
| Fig. 1 X-ray diffraction of one major isomer of 3m. | ||
| a Reaction conditions:1 (0.3 mmol), 2 (1.5 mmol), FeCl2 (2.5 mol%), (t-BuO)2 (0.75 mmol), PhCl (1.0 mL), 120 °C, 2 h, under N2; reported yields were based on 1 and determined by 1H NMR using an internal standard; the isolated yields are given in parentheses. |
|---|
|
We next investigated the scope of the transformation using secondary aldehydes 4 under the optimized conditions (Table 3). The selective C(sp3)–H bond [2 + 2 + 1] cyclization instead of [2 + 2 + 2] cyclization exclusively took place. For example, the reaction of 1a with isobutyraldehyde 4a and 2-ethyl butyraldehyde 4b afforded products 5a and 5b in good yields. When 2-methyl butyraldehyde 4c was applied, the desired 5c could be obtained in a 70% yield with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr ratio. Certainly, this method was appropriate for the [2 + 2 + 1] cyclization of 1,n-enynes with cyclic secondary aldehydes, and spiro products 5d–f were obtained in good yields. The carbonyl and alkene moieties on the ring were well tolerated to afford the desired 5g and 5h in good yields. In addition, a variety of 1,7-enynes could also be efficiently converted into the corresponding products (5i–n). It should be noted that the original cyclization strategy of cycloalkanes or oxa-cycloalkanes with enynes was reported by Li and Tu.5,6 However, the selectivity of these transformations depends mainly on the electronic characteristics of different C–H bonds. In our study, we employed carbonyls of aldehydes as traceless activation groups that could accomplish functionalization of a C(sp3)–H bond in a selective manner.
:1 dr ratio. Certainly, this method was appropriate for the [2 + 2 + 1] cyclization of 1,n-enynes with cyclic secondary aldehydes, and spiro products 5d–f were obtained in good yields. The carbonyl and alkene moieties on the ring were well tolerated to afford the desired 5g and 5h in good yields. In addition, a variety of 1,7-enynes could also be efficiently converted into the corresponding products (5i–n). It should be noted that the original cyclization strategy of cycloalkanes or oxa-cycloalkanes with enynes was reported by Li and Tu.5,6 However, the selectivity of these transformations depends mainly on the electronic characteristics of different C–H bonds. In our study, we employed carbonyls of aldehydes as traceless activation groups that could accomplish functionalization of a C(sp3)–H bond in a selective manner.
| a Reaction conditions: 1 (0.3 mmol), 4 (1.5 mmol), FeCl2 (2.5 mol%), (t-BuO)2 (0.75 mmol), PhCl (1.0 mL), 120 °C, 2 h, under N2; reported yields were based on 1 and determined by 1H NMR using an internal standard; the isolated yields are given in parentheses. |
|---|
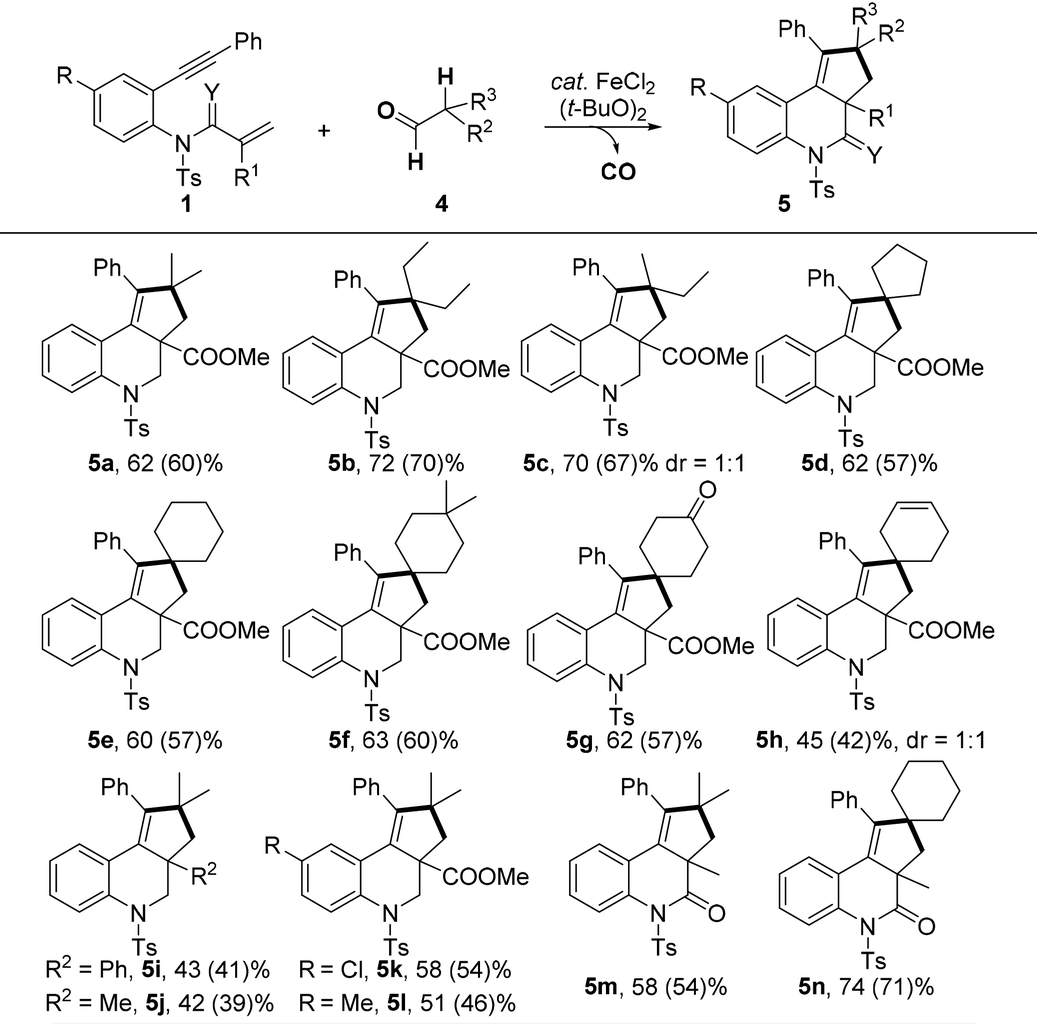
|
Furthermore, the reaction of 1a with aldehyde 6, a metabolite of cholesterol, was investigated under the standard reaction conditions (Scheme 2). The expected spiro product 7 was obtained in 48% yield. This result indicated that the method could be a potential synthetic tool for the late-stage modification of complex molecules.
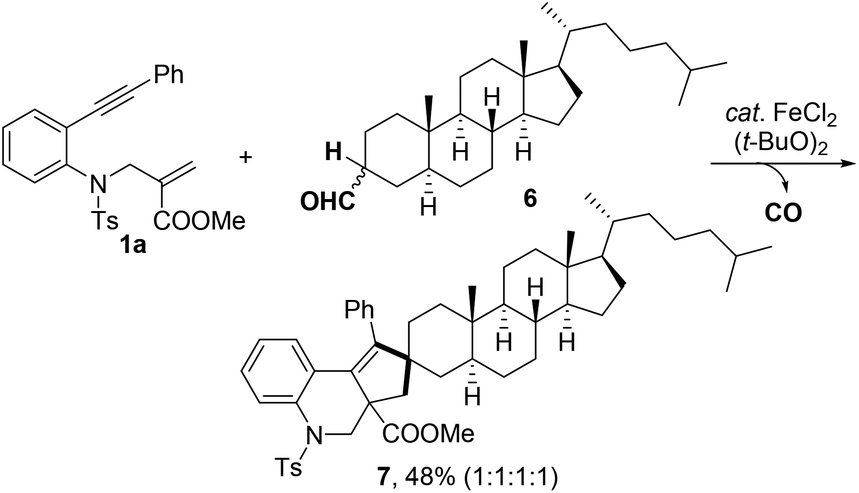 | ||
| Scheme 2 Late stage modification of cholesterol metabolite. | ||
When a primary aldehyde, such as propionaldehyde 8, was applied, the product 10 was obtained in 33% yield, accompanied by similar amount of cyclopentenone 11 (Scheme 3a). Considering the low decarbonylative constant of primary aldehydes,13 we rationalized that the ethyl radical, which was generated by the dealkylation of intermediate 9, reacted with 1a to afford 10.14 In order to gain further insight into the selectivity of H-abstraction, iso-valeraldehyde 12 was submitted to react with 1a (Scheme 3b). The generation of 13 significantly exceeded than that of 14, supporting that (1) intramolecular 1,5-H transfer is much faster than 1,6-H transfer;8,15 (2) the reactivity of C(sp3)–H bonds is related to the stability of the carbon radical generated by H-abstraction of 15.
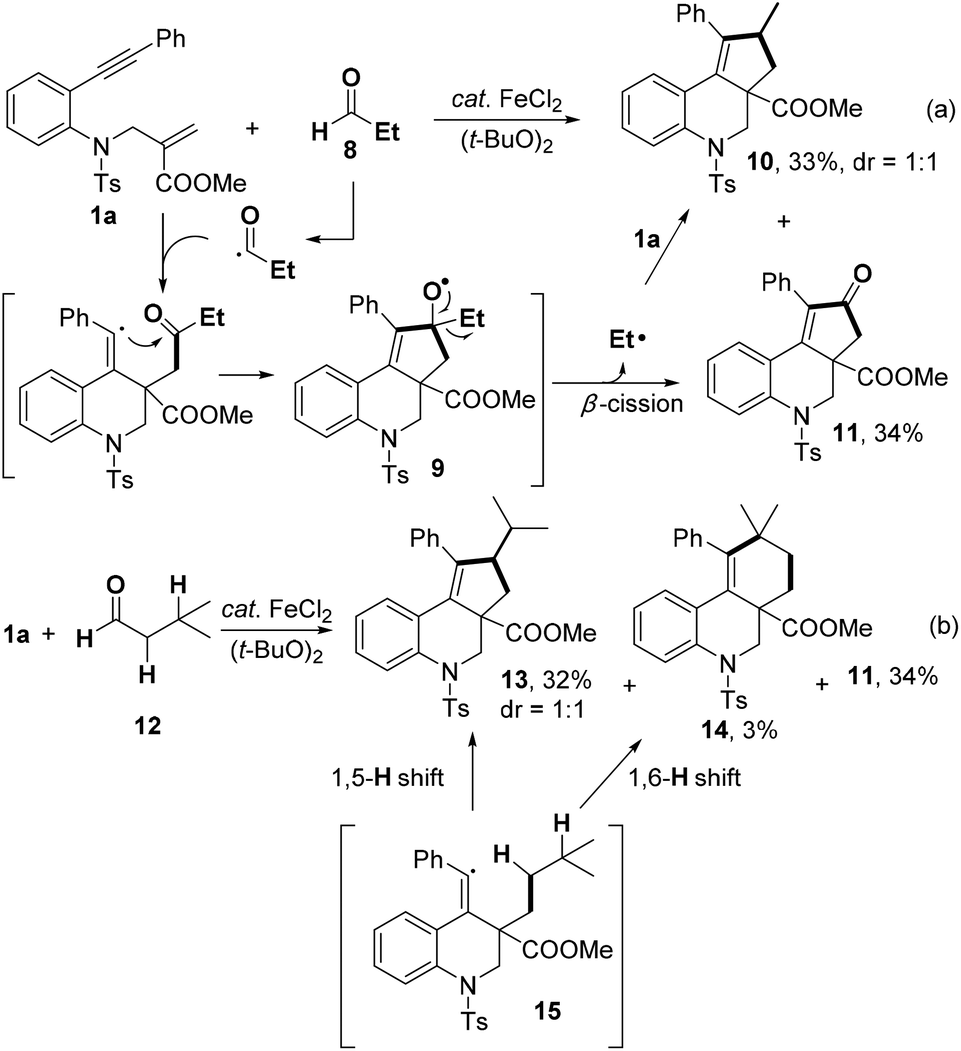 | ||
| Scheme 3 Reactions of 1a with primary aldehydes. | ||
The reaction of 1a with 4a was completely suppressed when radical inhibitors, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or butylated hydroxytoluene (BHT), was added under the standard reaction conditions (Scheme 4). The TEMPO-isopropyl adduct 16 was detected by GC-MS, which suggested that a radical pathway might be involved in this process.
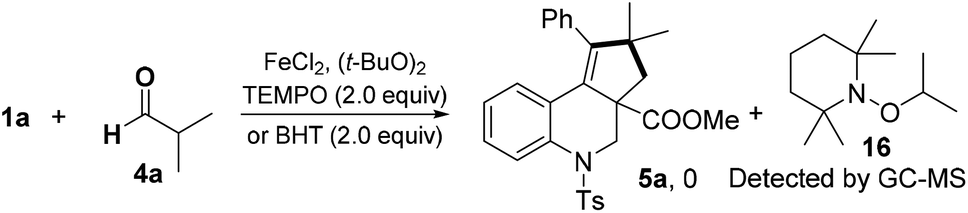 | ||
| Scheme 4 Radical trapping experiments. | ||
On the basis of the abovementioned results and previous reports,5–10 a possible mechanism for this decarbonylation-initiated selective C(sp3)–H cyclization is depicted in Scheme 5. Initially, the aldehyde 2 or 4 undergoes oxidative decarbonylation to produce alkyl radical A. Then, radical tandem annulation of A with enyne 1 leads to the key vinyl radical intermediate B, an H-acceptor. In the case of tertiary aldehydes 2, 1,6-H abstraction takes place to afford intermediate C. The subsequent intramolecular radical cyclization and irreversible oxidation give a stable benzyl cation E. We hypothesized that an iron catalyst greatly promotes this oxidation step. Finally, deprotonation of E delivers the six-membered product 3. In contrast, when secondary aldehydes are applied, 1,5-H abstraction occurs exclusively and produces the five-membered product 5 followed by the same process.
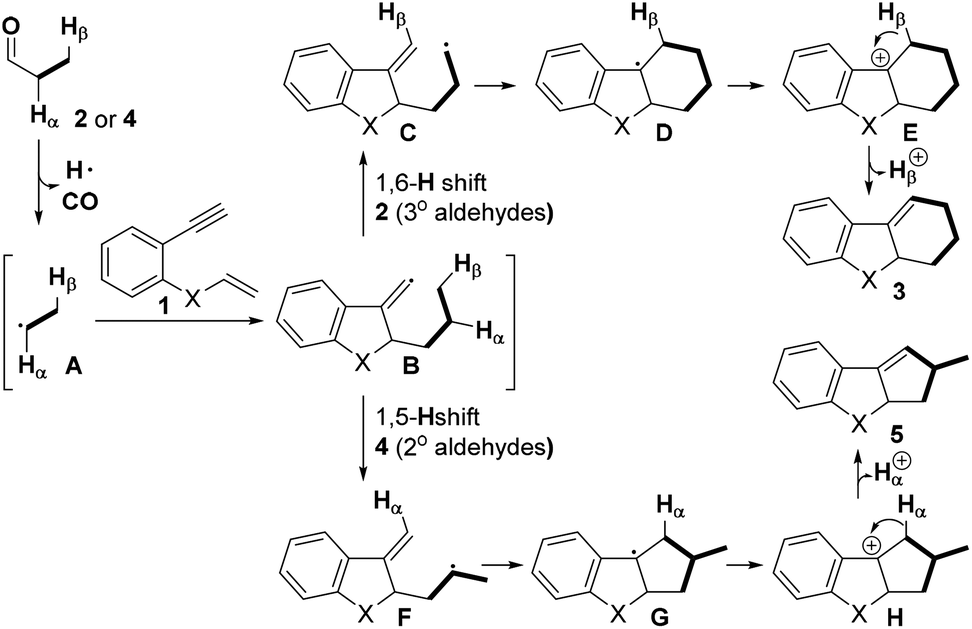 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In summary, we demonstrated a new iron-catalyzed decarbonylation initiated [2 + 2 + m] annulation of benzene-linked 1,n-enynes with aliphatic aldehydes. Tertiary aldehydes undergo decarbonylation/β-C(sp3)–H cleavage to afford [2 + 2 + 2] cyclization products, whereas secondary aldehydes undergo decarbonylation/α-C(sp3)–H cleavage to generate [2 + 2 + 1] cyclization products. The late stage functionalization of cholesterol metabolite was also reported. By the feasibilities of decarbonylation of aliphatic aldehydes and radical-initiated cyclization of enynes, the application of this protocol for inert alkyl C(sp3)–H bond functionalization is envisioned. Further studies on the scope and applications of this strategy are in progress.Experimental procedure
To a mixture of aliphatic aldehyde 2 or 4 (1.5 mmol), enyne 1 (0.3 mmol) and FeCl2 (1.0 mg, 2.5 mol%), chlorobenzene (1.0 mL) was added under a nitrogen atmosphere at room temperature. Then, pure di-tert-butyl peroxide (137 μL, 0.75 mmol) was dropped into the mixture. The resulting mixture was stirred at 120 °C for 2 hours. After the mixture was cooled to room temperature, the resulting solution was directly filtered through a pad of silica by EtOAc. The solvent was evaporated in vacuo to afford the crude products. NMR yields were determined by 1H NMR using dibromomethane as an internal standard. The residue was purified by flash column chromatography on silica gel (ethyl acetate/petroleum ether) to afford the pure product 3 or 5.Acknowledgements
Financial support from the National Science Foundation of China (21272267 and 21672259) and the Fundamental Research Funds for the Central Universities, the Research Funds of Renmin University of China (10XNL017) is greatly acknowledged.Notes and references
- For selected reviews, see: (a) C. Aubert, O. Buisine and M. Malacria, Chem. Rev., 2002, 102, 813 CrossRef CAS PubMed; (b) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268 CrossRef CAS PubMed; (c) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (d) A. Marinetti, H. Jullien and A. Voituriez, Chem. Soc. Rev., 2012, 41, 4884 RSC; (e) U. Wille, Chem. Rev., 2013, 113, 813 CrossRef CAS PubMed.
- For recent examples of 1,6-enynes cyclization, see: (a) S. Undeela, G. Ravikumar, J. B. Nanubolu, K. K. Singarapu and R. S. Menon, Chem. Commun., 2016, 52, 4824 RSC; (b) M. Hu, R.-J. Song and J.-H. Li, Angew. Chem., Int. Ed., 2015, 54, 608 CAS; (c) G.-B. Deng, Z.-Q. Wang, J.-D. Xia, P.-C. Qian, R.-J. Song, M. Hu, L.-B. Gong and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 1535 CrossRef CAS PubMed; (d) J.-Y. Luo, H.-L. Hua, Z.-S. Chen, Z.-Z. Zhou, Y.-F. Yang, P.-X. Zhou, Y.-T. He, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2014, 50, 1564 RSC.
- For recent examples of 1,7-enynes cyclization, see: (a) Y. An, Y. Kuang and J. Wu, Org. Chem. Front., 2016, 3, 994 RSC; (b) Y.-L. Zhu, B. Jiang, W.-J. Hao, A.-F. Wang, J.-K. Qiu, P. Wei, D.-C. Wang, G. Li and S.-J. Tu, Chem. Commun., 2016, 52, 1907 RSC; (c) A.-F. Wang, Y.-R. Zhu, S.-L. Wang, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, J. Org. Chem., 2016, 81, 1099 CrossRef CAS PubMed; (d) F. Gao, C. Yang, N. Ma, L.-G. Gao, D. Li and W. Xia, Org. Lett., 2016, 18, 600 CrossRef CAS PubMed; (e) X.-H. Ouyang, R.-J. Song, Y. Liu, M. Hu and J.-H. Li, Org. Lett., 2015, 17, 6038 CrossRef CAS PubMed; (f) Y.-L. Zhu, B. Jiang, W.-J. Hao, J.-K. Qiu, J. Sun, D.-C. Wang, P. Wei, A.-F. Wang, G. Li and S.-J. Tu, Org. Lett., 2015, 17, 6078 CrossRef CAS PubMed; (g) Y. Liu, J.-L. Zhang, R.-J. Song and J.-H. Li, Org. Lett., 2014, 16, 5838 CrossRef CAS PubMed; (h) Y. Liu, J.-L. Zhang, R.-J. Song, P.-C. Qian and J.-H. Li, Angew. Chem., Int. Ed., 2014, 53, 9017 CrossRef CAS PubMed.
- For selected reviews on iron catalysis, see: (a) L. Lv and Z. Li, Top. Curr. Chem., 2016, 374 DOI:10.1007/s41061-016-0038-y; (b) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (c) F. Jia and Z. Li, Org. Chem. Front., 2014, 1, 194 RSC; (d) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (e) A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC; (f) C. Bolm, J. Legros, J. L. Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed.
- J.-K. Qiu, B. Jiang, Y.-L. Zhu, W.-J. Hao, D.-C. Wang, J. Sun, P. Wei, S.-J. Tu and G. Li, J. Am. Chem. Soc., 2015, 137, 8928 CrossRef CAS PubMed.
- M. Hu, J.-H. Fan, Y. Liu, X.-H. Ouyang, R.-J. Song and J.-H. Li, Angew. Chem., Int. Ed., 2015, 54, 9577 CrossRef CAS PubMed.
- (a) M. Hu, R.-J. Song, X.-H. Ouyang, F.-L. Tan, W.-T. Wei and J.-H. Li, Chem. Commun., 2016, 52, 3328 RSC; (b) Y. Li, B. Liu, R.-J. Song, Q.-A. Wang and J.-H. Li, Adv. Synth. Catal., 2016, 358, 1219 CrossRef CAS.
- For reviews of 1,5-H shift, see: (a) L. Wang and J. Xiao, Top. Curr. Chem., 2016, 374 DOI:10.1007/s41061-016-0018-2; (b) M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010 CrossRef CAS PubMed; (c) B. Peng and N. Maulide, Chem. – Eur. J., 2013, 19, 13274 CrossRef CAS PubMed.
- L. Lv and Z. Li, Org. Lett., 2016, 18, 2264 CrossRef CAS PubMed.
- For oxidative decarbonylaiton of aliphatic aldehydes to generate alkyl radicals, see: (a) P. Biswas, S. Paul and J. Guin, Angew. Chem., Int. Ed., 2016, 55, 7756 CrossRef CAS PubMed; (b) X.-H. Ouyang, R.-J. Song, B. Liu and J.-H. Li, Adv. Synth. Catal., 2016, 358, 1903 CrossRef CAS; (c) L. Yang, W. Lu, W. Zhou and F. Zhang, Green Chem., 2016, 18, 2941 RSC; (d) Z. Zong, W. Wang, X. Bai, H. Xi and Z. Li, Asian J. Org. Chem., 2015, 4, 622 CrossRef CAS.
- During the preparation of this manuscript, Li and coworkers reported a metal-free oxidative decarbonylative [2 + 2 + 2] carbocyclization using tertiary and secondary alkyl aldehydes as a two carbon unit for assembling six-membered carbocycle-fused polycycles. Y. Li, G.-H. Pan, M. Hu, B. Liu, R. J. Song and J.-H. Li, Chem. Sci., 2016 10.1039/C6SC02451C.
- The structure of one major isomer of 3m was identified by X-ray diffraction. CCDC (1494076) contains the supplementary crystallographic data for this paper.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed.
- S. Wilsey, P. Dowd and K. N. Houk, J. Org. Chem., 1999, 64, 8801 CrossRef CAS PubMed.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR and 13C NMR Spectra for new compounds. CCDC 1494076. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00429f |
| This journal is © the Partner Organisations 2016 |