
Bisimino-functionalized dibenzo[a,c]acridines as highly conjugated pincer frameworks for palladium(II): synthesis, characterization and catalytic performance in Heck coupling†
Qaiser
Mahmood
ad,
Erlin
Yue
a,
Wenjuan
Zhang
ab,
Gregory A.
Solan
ac,
Tongling
Liang
a and
Wen-Hua
Sun
 *ad
*ad
aKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: whsun@iccas.ac.cn; Fax: +86-10-62618239; Tel: +86-10-62557955
bSchool of Materials Science and Engineering, Beijing Institute of Fashion Technology, Beijing 100029, China
cDepartment of Chemistry, University of Leicester, University Road, Leicester LE1 7RH, UK
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 27th September 2016
Abstract
A new pair of highly conjugated ligands, 10-[1-(arylimino)ethyl]-14-[(arylimino)methyl]dibenzo[a,c]acridine (aryl = 2,6-Me2Ph L1, 2,6-Et2Ph L2) incorporating both aldimine and ketimine units, have been prepared by a straightforward sequence of organic transformations from phenanthrene-9,10-dione. Cyclopalladation occurs readily at ambient temperature on treating L1 or L2 with PdCl2(NCCH3)2 in aprotic solvents to afford exclusively (NketimineNC)Pd(II) chloride pincer complexes Pd1 or Pd2, respectively. By contrast in methanol, Pd1 or Pd2 are isolated as the minor product with aldehyde-containing Pd3 or Pd4 as the major one, the result of hydrolysis of the pendant aldimine units in Pd1 and Pd2, respectively. All the ligands and palladium complexes have been characterized by FT-IR, 1H and 13C NMR spectroscopy, mass spectrometry and elemental analysis; the molecular structures for L1, Pd1, Pd2, Pd3 and Pd4 are also reported. Using low catalyst loadings (0.0005–0.002 mol%) and elevated temperatures (140–200 °C), Pd1–Pd4 are able to efficiently mediate the coupling of haloarenes with vinyl-containing substrates with turnover numbers as high as 174![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; the effects of steric/electronic variation within the substrate and NNC-pincer complex on catalyst performance are examined.
000; the effects of steric/electronic variation within the substrate and NNC-pincer complex on catalyst performance are examined.
Introduction
Palladium complexes bearing neutral and monoanionic pincer ligands have established themselves as a powerful tool for a wide range of organic transformations such as the aldol reaction, Michael addition, allylation of aldehydes and imines as well as a range of cross-coupling reactions.1–4 With regard to Heck-type coupling, numerous pincer catalysts have been developed incorporating two neutral donors and an anionic aryl-C donor within the terdentate ligand set. In particular those based on symmetrical EC−1E type (E = neutral donor) (A, Chart 1)5 framework have been at the forefront of research while those involving unsymmetrical ligand frames such as EC−1E′ (B, Chart 1)6 and EEC−1 (C, Chart 1)7 have also started to emerge as efficient catalysts for this transformation. The presence of a Pd–C σ-bond as the central or exterior donor no doubt imparts the complex with a high level of thermal stability and tolerance of many functional groups, hence inhibiting facile deactivation pathways and thus leading to highly productive catalysts.8 Indeed, pincer complexes have proved exceptional in determining this necessary balance between stability and reactivity.9 Consequently, it can be viewed that an ideal pincer catalyst would deliver high turnover numbers, while allowing low catalyst loading and exhibiting good thermal stability.10 Furthermore, it needs to be amenable to rational design, allowing for systematic tuning of the ligand to enable subtle control over the catalytic properties of the central metal atom.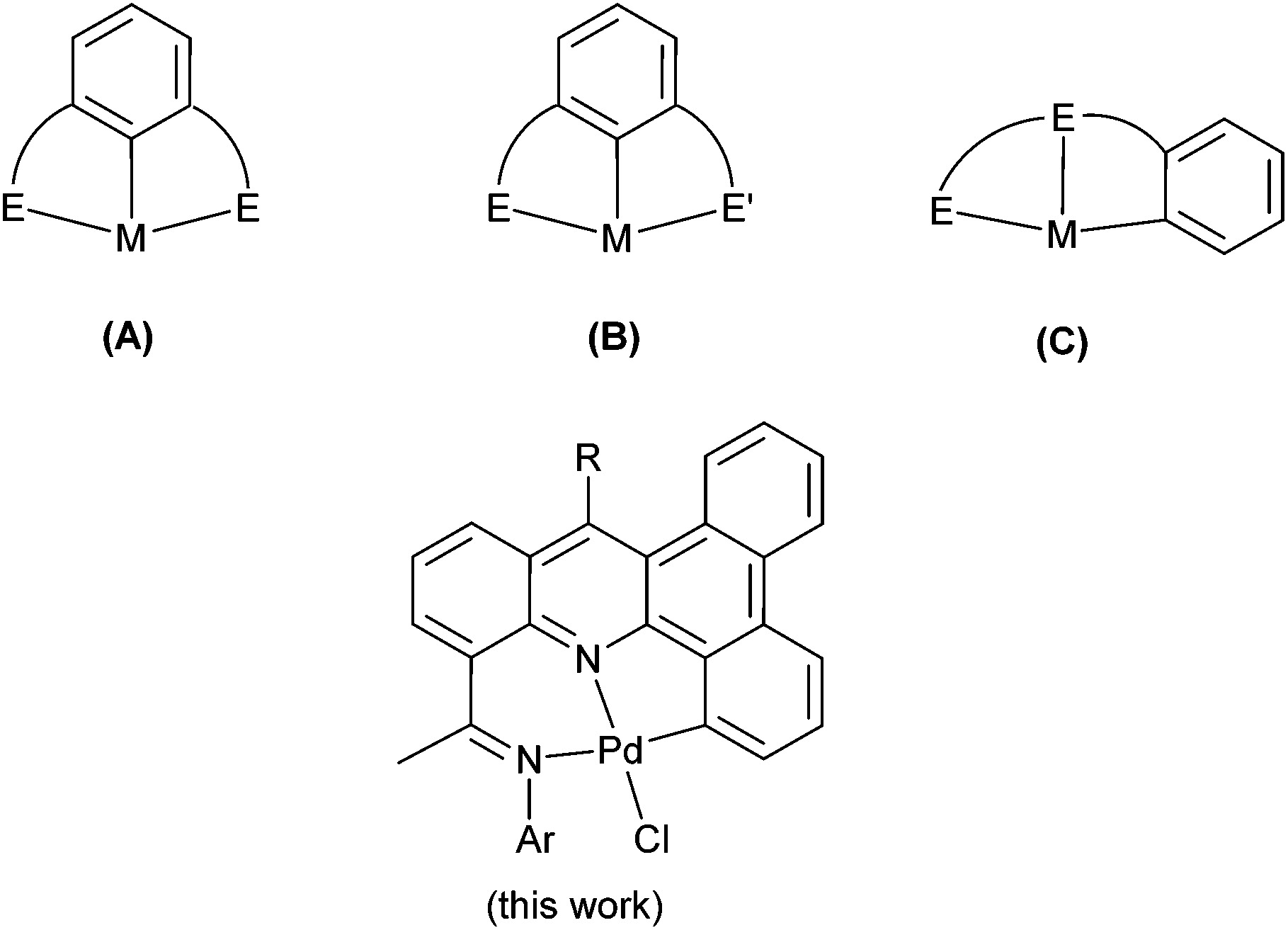 | ||
| Chart 1 Monoanionic pincer frameworks reported (A–C) along with our current work; E and E′ refer to neutral donors. | ||
With a view to enhancing the thermal stability further of an NNC-type palladium pincer complex, we have been interested in developing a means of increasing the conjugation of the ligand backbone through the introduction of suitably fused arene and heteroarene rings. In this report we devise an implement a synthetic approach to develop novel bisimino-functionalized benzo[a,c]acridines that are conducive to ready steric and electronic variation (see Chart 1). Owing to the planarity of the pentacyclic skeleton, cyclopalladation proceeds efficiently generating NNC complexes which have been used in the coupling of haloarenes to vinyl-containing substrates. Full synthetic details are reported as is an in depth discussion of the catalytic evaluation of the complexes and substrate scope.
Results and discussion
Synthesis of L1 and L2
Ligands L1 and L2 have been prepared using a six step procedure in reasonable overall yield from commercially available phenanthrene-9,10-dione (Scheme 1). The intermediate compound, 10-ethyl-14-methyldibenzo[a,c] acridine (2), has been previously reported using a rather cumbersome low yielding route.11 We have found that 2 can be more conveniently prepared by firstly treating phenanthrene-9,10-dione with a slight excess of 2,6-diethylaniline in the presence of para-toluenesulfonic acid to form spiro-[7-ethyl-3-methylindoline-2,10-phenanthren-9-one] (1) and then completing the heterocyclization by heating 1 at reflux in 1,2-dichlorobenzene. Mono-bromination at both the ethyl and methyl groups in 2 with two equivalents of N-bromosuccinimide (NBS) in carbon tetrachloride gave 3 in near quantitative yield.12 Substitution of the two bromide groups in 3 for OH groups to give 4 occurred readily by using a silver nitrate assisted hydrolysis in acetone/water (3:1 ratio),12 which when followed by a pyridinium chlorochromate (PCC) oxidation afforded the mixed aldehyde-ketone 5 in excellent yield.13 Finally, the target ligands, 10-[1-(arylimino)ethyl]-14-[(arylimino)methyl]dibenzo[a,c]acridine (aryl = 2,6-Me2Ph L1, 2,6-Et2Ph L2), could be obtained by the Schiff base condensation of 5 with either 2,6-dimethylaniline or 2,6-diethylaniline in toluene, respectively in reasonable yield.14,15 All organic compounds have been characterized by 1H, 13C NMR, IR spectroscopy, HR mass spectrometry or elemental analysis.
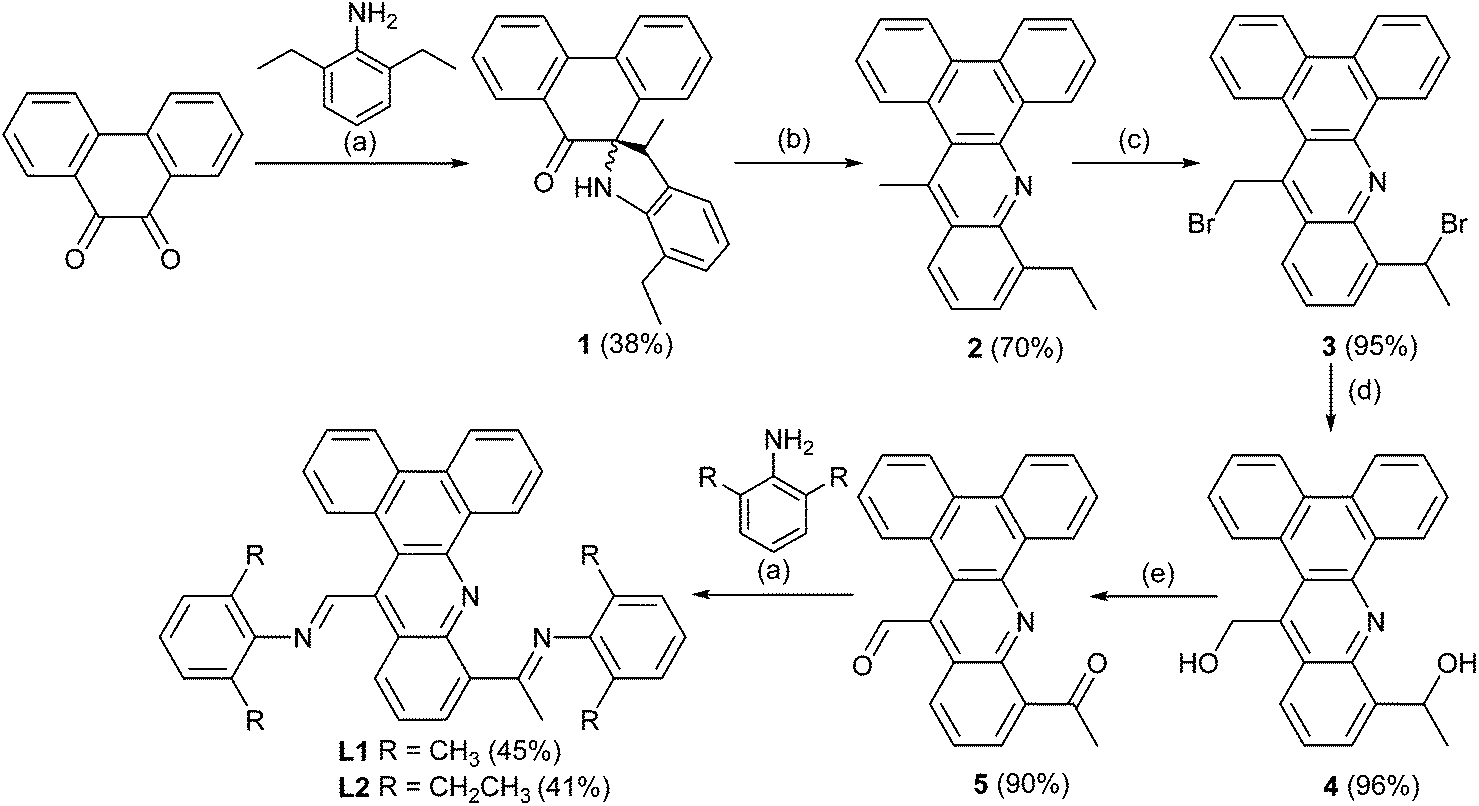 | ||
| Scheme 1 Reagents and conditions: (a) p-TsOH, toluene, reflux, 12 h; (b) p-TsOH, 1,2-dichlorobenzene, reflux, 12 h; (c) NBS, dibenzoyl peroxide, CCl4, reflux, 6 h; (d) AgNO3, acetone/water, 50 °C, 6 h; (e) PCC, CH2Cl2, molecular sieves, RT, 6 h. | ||
It is noteworthy that the formation of 1 was unexpected and, to the knowledge of the authors, has not been observed as a product (ca. 38%) in the condensation reaction of phenanthrene-9,10-dione with 2,6-substituted anilines; only small quantities of the expected mono- and bis-imine derivatives of phenanthrene-9,10-dione could be detected under these conditions.16,17 The thermally driven rearrangement and ring closing steps involved in the formation of 2 (70%) from 1 are likely mediated by a combination of the relief in ring strain in 1 and the thermodynamic stability of the highly conjugated aromatic 2. A proposed mechanism for the formation of 2 is depicted in Scheme 2 highlighting the role of acid and involving the elimination of water. Further confirmation of the structures of 1 and 2 was provided in the form of single crystal X-ray structures (see ESI†).
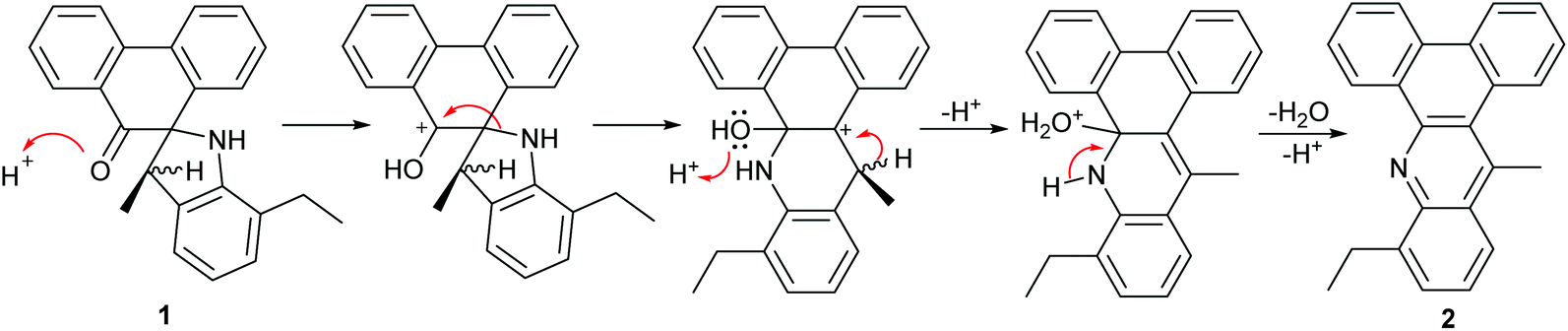 | ||
| Scheme 2 Proposed mechanism for the formation of 2 from 1. | ||
The 1H NMR spectra of L1 and L2 recorded in CDCl3 feature prominent singlets for their –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N protons at δ 8.84av. and –CMeN protons at δ 2.51av., while the 13C NMR spectra show –CN peaks at δ 171.5av. (ketimine) and δ 163.4av. (aldimine) for L1 and for L2. In their ToF mass spectra peaks corresponding to their protonated molecular ions are seen for each. In addition, the structure of L1 has been the subject of a single X-ray diffraction study. Suitable crystals for the study were grown by layering a dichloromethane solution of L1 with heptane. A view of L1 is shown in Fig. 1; selected bond distances and angles are collected in the figure caption.
N protons at δ 8.84av. and –CMeN protons at δ 2.51av., while the 13C NMR spectra show –CN peaks at δ 171.5av. (ketimine) and δ 163.4av. (aldimine) for L1 and for L2. In their ToF mass spectra peaks corresponding to their protonated molecular ions are seen for each. In addition, the structure of L1 has been the subject of a single X-ray diffraction study. Suitable crystals for the study were grown by layering a dichloromethane solution of L1 with heptane. A view of L1 is shown in Fig. 1; selected bond distances and angles are collected in the figure caption.
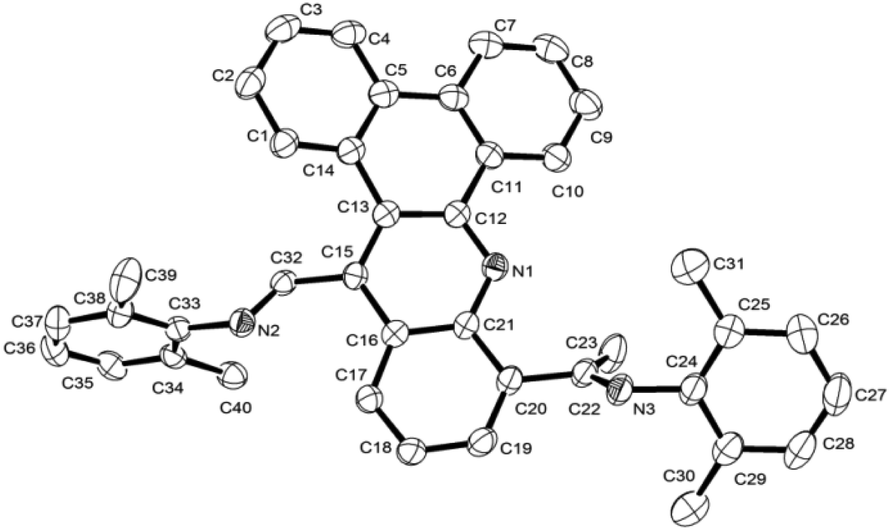 | ||
| Fig. 1 ORTEP diagram of L1 with the thermal ellipsoids set at 50% probability level; all hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): N(1)–C(12) 1.324(2), N(1)–C(21) 1.353(2), N(3)–C(22) 1.271(2), N(3)–C(24) 1.427(2), C(32)–N(2) 1.274(2), N(2)–C(33) 1.426(2), C(11)–C(10) 1.405(2), C(10)–C(9) 1.378(3), C(12)–N(1)–C(21) 119.07(14), C(32)–N(2)–C(33) 117.86(14), C(22)–N(3)–C(24) 121.21(15), N(3)–C(22)–C(20) 116.38(15), N(3)–C(22)–C(23) 125.38(16), C(9)–C(10)–C(11) 120.63(18). | ||
The structure consists of a slightly twisted dibenzo[a,c] acridine polycycle based on five fused aromatic rings. At the C15- and C20-carbons of the dibenzo[a,c]acridine are located the aldimine and ketimine units, respectively, with the ketimine fragment positioned on the same side as the acridine nitrogen. The two imino C–N bond distances are quite typical for CN double bond distances and are indeed similar to one another [C32–N2 1.274(2); C22–N3 1.271(2) Å]. The N-aryl ring of the aldimine unit is oriented towards orthogonality with respect to the heterocyclic ring, N1–C12–C13–C15–C16–C21, with a dihedral angle of ca. 77.53°, whilst the N-aryl ring of the ketimine adopts a shallower angle of ca. 32.85°.
Synthesis of NNC Pd pincer complexes
With a view to preparing organo-palladium complexes based on L1 or L2, we envisaged that, due to the presence of more than one nitrogen-based directing group, two sites for Caryl–H activation on the dibenzo[a,c]acridine could conceivably occur, namely at the C2-position or the C11-position (see Scheme 3 for numbering system). To explore these possibilities, L1 and L2 were treated with PdCl2(NCMe)2 under different reaction conditions (Table 1). Hence, the equimolar reaction of L1 or L2 with PdCl2(NCCH3)2 in aprotic solvents such as dichloromethane or toluene at room temperature gave Pd1 and Pd2, respectively, as the only isolable product in reasonable yield (Scheme 3). However, on reacting L1 or L2 in a mixture of methanol/toluene at 50 °C, two types of complexes were isolable for each namely Pd1/Pd3 or Pd2/Pd4, respectively.18 Indeed, Pd3 and Pd4 were formed as the major products while Pd1 and Pd2 the minor ones. Addition of sodium bicarbonate to the reaction of L1 with PdCl2(NCMe)2 in toluene gave some modest increase in the yield of Pd1. Complexes Pd1, Pd2, Pd3 and Pd4 were characterized by 1H NMR, 13C NMR, FT-IR spectroscopy, ESI mass spectrometry and by elemental analysis. In addition, all four complexes were the subject of single crystal X-ray diffraction studies.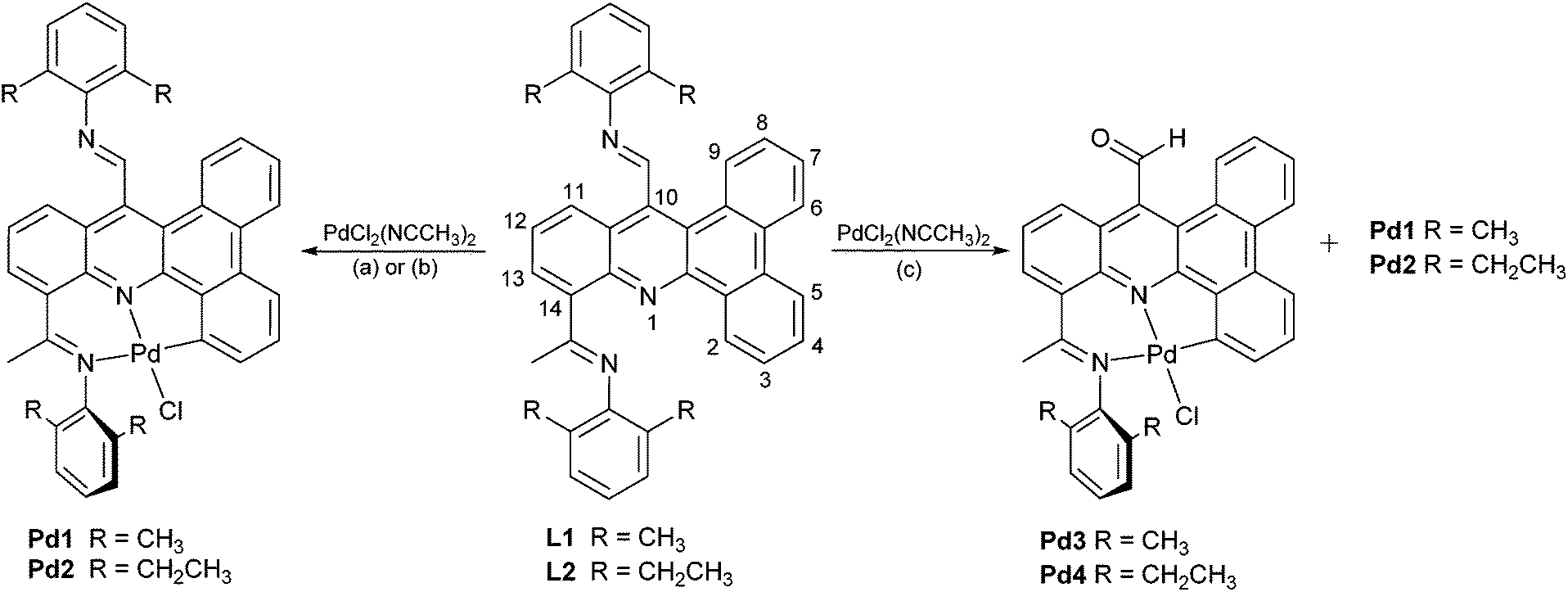 | ||
| Scheme 3 Synthesis of NNC-Pd pincer complexes: (a) dichloromethane or toluene, RT; (b) toluene, NaHCO3 (cat.), 60 °C, 6 h; (c) methanol/toluene, 50 °C, 6 h. | ||
| Entry | Ligand | Solvent | Base | Temp. (°C) | Product 1 (%) | Product 2 (%) |
|---|---|---|---|---|---|---|
| 1 | L1 | Dichloromethane | — | 25 | Pd1 (42) | — |
| 2 | L2 | Dichloromethane | — | 25 | Pd2 (38) | — |
| 3 | L1 | Toluene | — | 25 | Pd1 (36) | — |
| 4 | L2 | Toluene | — | 25 | Pd2 (30) | — |
| 5 | L1 | Toluene | NaHCO3 (cat.) | 60 | Pd1 (52) | — |
| 6 | L1 | Methanol/toluene | — | 50 | Pd1 (12) | Pd3 (46) |
| 7 | L2 | Methanol/toluene | — | 50 | Pd2 (15) | Pd4 (41) |
Single crystals of Pd1 and Pd2 were grown by layering chloroform solutions of the complex with heptane. The molecular structures of both are similar and will be discussed together. As a representative example a view of Pd1 is depicted in Fig. 2; selected bond distances and angles are listed in Table 2. Each structure reveals a mononuclear species in which the palladium(II) center occupies a tridentate cavity within the ligand frame with the ketimine nitrogen, the acridine nitrogen and an aryl carbon serving as the donor atoms. The geometry of the palladium can be best described as distorted square planar with a chloride ligand filling the fourth coordination site. Within the NNC-palladium unit both five- and six-membered chelate rings are present with the carbon donor incorporated into the smaller metallacycle. Due to the constraints of the NNC ligand the C1–Pd1–N2 angles are less than the desired 180°: 167.25(19)° for Pd1, 167.09(13)° for Pd2. The Pd(1)–C(1) bond distances in Pd1 [1.979(5) Å] and Pd2 [1.986(3) Å], are comparable with those found in related NNC pincer complexes19 and are the shortest of the three NNC contacts to palladium; this reflects the ionic contribution to the Pd–C bond. Little significant bond variation is seen between the bound CNketimine bond length and the CNaldimine bond lengths in either structure and indeed similar to that found in L1. Some puckering of the six-membered chelate ring is apparent with the bound CNketimine unit some way of co-planar with the pyridine plane (dihedral angle = 30.1° Pd1, 34.6° Pd2); the corresponding angle for the pendant aldimine is close to 45° (dihedral angle = 44.8° Pd1, 43.8° Pd2). For both complexes the N-aryl rings are inclined towards perpendicular (Pd1 88.28°, 85.59°; Pd2 81.63°, 89.44°) with respect to the C1–N1–N2–Cl1 coordination plane, presumably to minimize steric interactions.
 | ||
| Fig. 2 ORTEP diagram of Pd1 with thermal ellipsoids set at 50% probability level; all hydrogen atoms are omitted for clarity. | ||
| Pd1 | Pd2 | Pd3 | Pd4 | |
|---|---|---|---|---|
| Pd(1)–C(1) | 1.979(5) | 1.986(3) | 1.978(3) | 1.9834(19) |
| Pd(1)–N(1) | 2.028(4) | 2.027(3) | 2.027(2) | 2.0254(16) |
| Pd(1)–N(2) | 2.123(4) | 2.115(3) | 2.111(2) | 2.1276(16) |
| Pd(1)–Cl(1) | 2.3100(17) | 2.3009(9) | 2.3039(8) | 2.3030(8) |
| C(32)–N(3)aldimine | 1.264(7) | 1.275(4) | — | — |
| C(22)–N(2)ketimine | 1.294(7) | 1.281(4) | — | — |
| C(32)–O(1)aldehyde | — | — | 1.205(4) | 1.207(2) |
| C(1)–Pd(1)–N(1) | 82.38(19) | 82.98(13) | 82.94(10) | 82.96(7) |
| N(1)–Pd(1)–N(2) | 90.44(17) | 89.57(10) | 91.55(8) | 91.11(6) |
| C(1)–Pd(1)–N(2) | 167.25(19) | 167.09(13) | 170.18(10) | 166.88(7) |
| N(1)–Pd(1)–Cl(1) | 173.29(12) | 173.55(8) | 172.84(6) | 170.39(4) |
| C(1)–Pd(1)–Cl(1) | 93.17(16) | 92.48(11) | 92.05(8) | 92.80(6) |
| N(2)–Pd(1)–Cl(1) | 94.83(13) | 95.71(8) | 94.11(6) | 94.73(5) |
The 1H NMR spectra of Pd1 and Pd2 are supportive of their solid state structures being maintained in solution. The presence of only 16 protons in the aromatic region is consistent with cyclopalladation having occurred in L1 or L2 (cf. 17 Ar–H protons). The bound CMeN resonance is shifted ca. 0.3 ppm upfield on coordination for each complex while the chemical shift of the pendant CHN remains relatively unchanged. Surprisingly, no discernible difference in ν(CN)imine stretching frequency in the IR spectrum could be observed for the two types of imine. The ESI mass spectra for each complex recorded in acetonitrile show strong [M − Cl]+ peaks as their MeCN adducts.
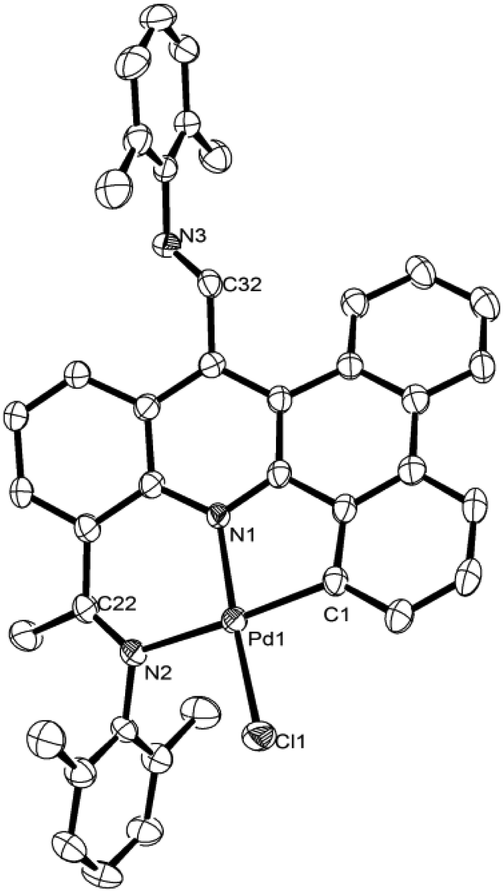
Crystals of Pd3 and Pd4 suitable for the X-ray determination were grown by layering dichloromethane solutions with heptane at room temperature. A perspective view of representative Pd4 is given in Fig. 3; selective bond lengths and bond angles for Pd3 and Pd4 are collected alongside Pd1 and Pd2 in Table 2. The structures closely resemble Pd1 and Pd2 with the main difference being that an aldehyde group is linked to the para-position of the pyridine unit rather than an aldimine group. The presence of the aldehyde on the back of the ligand frame has little effect on the NNC bond parameters with the Pd–Nimine distance again the longest [Pd3 2.111(2) Å, Pd4 2.1276(16) Å] and the Pd–C distance the shortest [Pd3 1.978(3) Å, Pd4 1.9834(19) Å]. As with the aldimine unit in Pd1 and Pd2, the aldehyde unit is tilted with regard to the pyridine plane (dihedral angle = 42.1° Pd3, 54.8° Pd4). The presence of the aldehyde groups is further confirmed in the 1H NMR spectra for Pd3 and Pd4 with downfield signals for the –CHO protons visible at δ 10.63 and δ 10.62 respectively, while in the 13C NMR spectra the –CHO carbons appear at δ 193.2 and 192.1 ppm. In the IR spectra bands corresponding to both ν(CO)aldehyde and ν(CN)imine at 1697 and 1614 cm−1, respectively are clearly observable for both. As with Pd1 and Pd2 the ESI mass spectrum for each complex recorded in acetonitrile show strong [M − Cl]+ peaks as their MeCN adducts.
 | ||
| Fig. 3 ORTEP diagram of Pd4 with thermal ellipsoids set at 50% probability level; all hydrogen atoms and are omitted for clarity. | ||
The preferential C–H activation of the C2–H proton (see Scheme 3 for numbering system) of the dibenzo[a,c]acridine unit over the C11–H position is likely due to the extra chelate ring stability afforded when Pd1 and Pd2 are formed. Attempts to react Pd1 or Pd2 with a further equivalent of PdCl2(NCMe)2, with a view to initiating a second C–H activation, were unsuccessful. It is assumed the C–H activation step to form Pd1 or Pd2 follows an AMLA type mechanism with a chloride ligand acting as the intramolecular base.20 Nevertheless the ready activation of this C–H bond at room temperature with palladium(II) chloride is worthy of note and is seems likely that this is made more facile by the close and fixed proximity of the aryl-H bond that is imposed by the conjugated ligand framework. With regard to the unexpected formation of Pd3 and Pd4, it would seem likely that the use of protic MeOH as solvent is initiating the hydrolysis of the corresponding aldimine groups.
Heck coupling reactions
To probe the capacity of NNC pincer complexes Pd1–Pd4 to mediate a range of Heck-type coupling reactions, we initially explored the use of Pd1 to establish the most suitable conditions for the coupling of bromobenzene with styrene. Using a 0.002 mol% catalyst loading we explored the effect of base, solvent, temperature and reaction duration on the conversion to stilbene; the results are collected in Table 3.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Temp. (°C) | Time (h) | Conv.b (%) | TONc |
| a Reaction conditions: 4 × 10−5 mmol Pd1, 2.0 mmol bromobenzene, 2.4 mmol styrene, 2.2 mmol base, 4.0 mL solution. b Determined by GC. c TON: mol stilbene/mol Pd. d 2.0 mmol iodobenzene used in place of bromobenzene. e 1 × 10−5 mmol Pd1 (0.0005 mol%), 2.0 mmol iodobenzene used in place of bromobenzene. | ||||||
| 1 | Na2CO3 | DMA | 60 | 8 | 0 | 0 |
| 2 | Na2CO3 | DMA | 100 | 8 | 0 | 0 |
| 3 | Na2CO3 | DMA | 120 | 8 | 0 | 0 |
| 4 | Na2CO3 | DMA | 140 | 4 | 21 | 10500 |
| 5 | Na2CO3 | DMA | 140 | 8 | 29 | 14500 |
| 6 | Na2CO3 | DMA | 140 | 12 | 41 | 20500 |
| 7 | K2CO3 | DMA | 140 | 8 | 6 | 3000 |
| 8 | NaHCO3 | DMA | 140 | 8 | 21 | 10500 |
| 9 | NaOH | DMA | 140 | 8 | 6 | 3000 |
| 10 | Na2CO3 | Toluene | 100 | 8 | 0 | 0 |
| 11 | Na2CO3 | DMF | 140 | 8 | 67 | 33500 |
| 12 | Na2CO3 | CH3CN | 60 | 8 | 0 | 0 |
| 13d | Na2CO3 | DMF | 140 | 8 | 90 | 45000 |
| 14d | Na2CO3 | DMF | 200 | 8 | 88 | 44000 |
| 15e | Na2CO3 | DMF | 140 | 8 | 87 | 174000 |

On inspection of the data, the most suitable reaction parameters were found when the coupling was performed in dimethylformamide (DMF) at 140 °C in the presence of Na2CO3 for eight hours resulting in a 67% conversion to stilbene (Table 3, entry 11). Some other solvents such as dimethylacetamide (DMA) (entries 1–6, Table 3), toluene (entry 10, Table 3) and acetonitrile (entry 12, Table 3) were also examined under similar conditions but these all proved less effective. Likewise, a variety of different bases such as K2CO3 (entry 7, Table 3), NaHCO3 (entry 8, Table 3) and NaOH (entry 9, Table 3) were screened but these again gave less promising results. Notably, the high temperature of 140 °C proved vital as performing the reactions at temperatures below 120 °C gave no conversion (entries 1–3, Table 3).
As expected, using more activated arenes led to higher conversions. For example with iodobenzene 90% conversion (entry 13, Table 3) was achieved, while with 4-iodotoluene close to quantitative conversion was noted (Table S1a†). Prolonging the reaction time gave only modest improvements in the conversion (Table S1b†). Most notably, Pd1 allowed 88% conversion to stilbene even at 200 °C (entry 14, Table 3) which highlights the exceptional balance between thermal stability and reactivity displayed by this catalyst.9,10 Reducing the catalyst loading from 0.002 to 0.0005 mol%, showed only a slight drop in conversion (87%, TON = 174000) (Table 3, entry 15). Compared with previous reports that show similarly high conversions in Heck reactions, Pd1 is at the lower end of the range in terms of catalyst loading; its high thermal robustness represents a notable advantage.6e,7b–d
To examine the role of electronic and steric effects on the catalytic efficiency, Pd2, Pd3 and Pd4 were additionally evaluated under the optimized reaction conditions established for Pd1; the results are compiled in Table 4. Several points emerge from examining the data. Firstly, both aldehyde-containing complexes Pd3 and Pd4 (entries 3 and 4 respectively) gave improved catalytic efficiency when compared with their imine counterparts Pd1 and Pd2, respectively. Secondly, the more sterically protected Pd2 gave a lower conversion to stilbene (entry 2, Table 4) compared to Pd1. Thirdly, the use of palladium(II) chloride alone gave only 20% conversion; albeit at higher catalyst loading. These observations highlight the role of the ligand in influencing catalytic performance. Both steric and electronic factors present within the ligand frame further affect turnover with a combination of good electron withdrawing groups and less sterically hindered aryl groups leading to optimal performance.
|
|
|||
|---|---|---|---|
| Entry | Complex | Conversionb (%) | TONc |
| a Reaction conditions: 4 × 10−5 mmol complex, 2.0 mmol bromobenzene, 2.4 mmol styrene, 2.2 mmol Na2CO3, 4.0 mL DMF, 140 °C for 8 h. b Determined by GC. c TON: mol stilbene/mol Pd. d 2 × 10−3 mmol Pd. | |||
| 1 | Pd1 | 67 | 33500 |
| 2 | Pd2 | 62 | 31000 |
| 3 | Pd3 | 70 | 35000 |
| 4 | Pd4 | 66 | 33000 |
| 5d | PdCl2 | 20 | 200 |
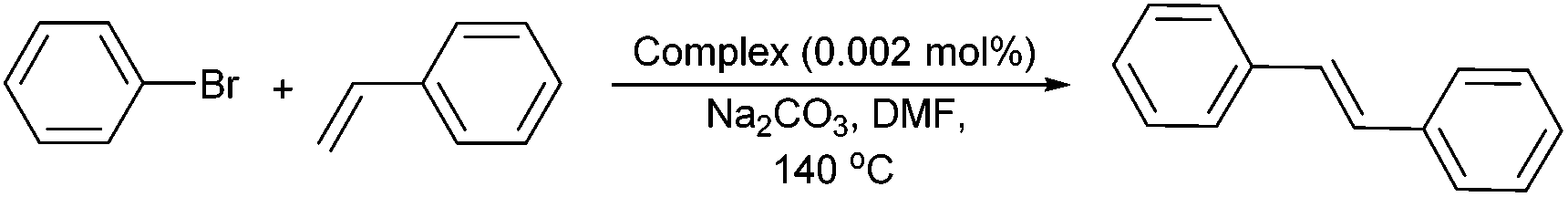
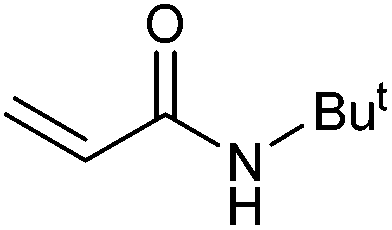
On the basis of the encouraging results obtained for the Heck coupling of bromobenzene with styrene, we decided to explore the potential of Pd3 for the coupling reaction of other haloarenes with styrene and N-tert-butylacrylamide; the results are documented in Table 5. Under similar conditions (viz., 0.002 mol% of catalyst, 8 h reaction at 140 °C, DMF as solvent and Na2CO3 as base), Pd3 showed a broad substrate scope with respect to the haloarenes employed (Table 5). In all the reactions involving styrene (entries 1–5 and 7–9), good to very good TON values were observed21 with the iodoarenes the best. However 2,6-dimethylbromobenzene appeared the least active substrate (entry 6) which can be attributed to the detrimental effect of the ortho-methyl substituents due to their steric properties, similar observations have been seen elsewhere.22,23 No negative effects on conversion were observed with either electron withdrawing (–CN) or donating (–NH2) groups positioned on the ortho-position of the bromoarenes, in fact some conversion rates were improved (entries 3 and 7). Meanwhile electron donating groups on the para-position enhanced the coupling reaction (entries 1, 4, 8 and 9). Using N-tert-butylacrylamide as the vinyl substrate with Pd3 also resulted in good to excellent conversions albeit with a narrower range of turnover numbers (entries 10–16). 4-Iodotoluene gave the highest activity (essentially quantitative conversion) of all the reactions screened (entry 16) and slightly higher than observed when styrene was employed as the vinyl-containing substrate (entry 8), an observation that has been noted previously.24
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar–X | Vinyl substrate | Conv.b (%) | Yieldc (%) | TONd |
| a Reaction conditions: 4 × 10−5 mmol Pd3, 2.0 mmol Ar–X, 2.4 mmol styrene or N-tert-butylacrylamide, 2.2 mmol Na2CO3, 4.0 mL DMF, 140 °C for 8 h. b Determined by GC. c Isolated yield, their 1H and 13C NMR spectra were compared with literature reports.25–27 d TON: mol product/mol Pd. | |||||
| 1 |
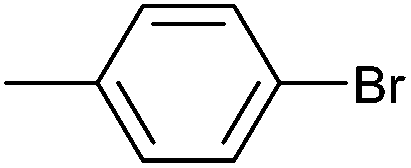
|
Styrene | 49 | 38 | 24500 |
| 2 |
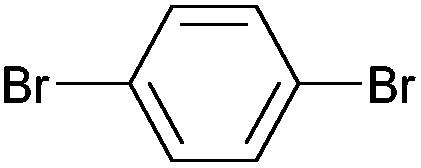
|
Styrene | 60 | 51 | 30000 |
| 3 |
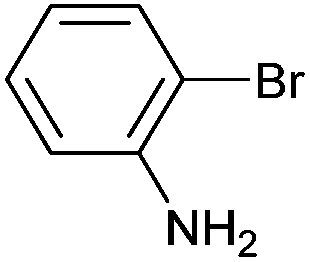
|
Styrene | 45 | 36 | 24500 |
| 4 |
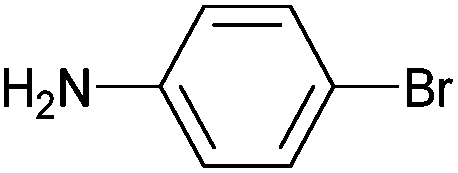
|
Styrene | 78 | 70 | 39000 |
| 5 |
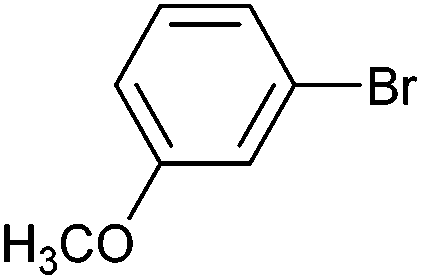
|
Styrene | 50 | 44 | 25000 |
| 6 |
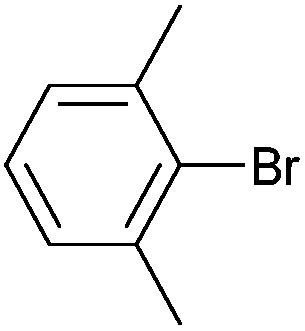
|
Styrene | 19 | 12 | 9500 |
| 7 |
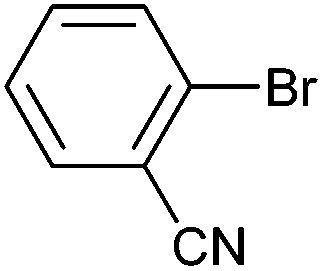
|
Styrene | 42 | 35 | 21000 |
| 8 |
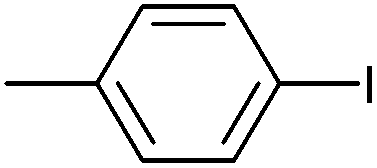
|
Styrene | 97 | 90 | 45000 |
| 9 |
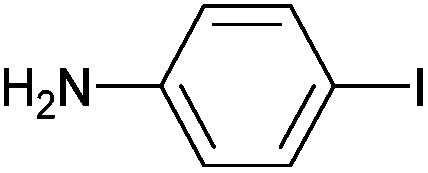
|
Styrene | 97 | 91 | 48500 |
| 10 |
|
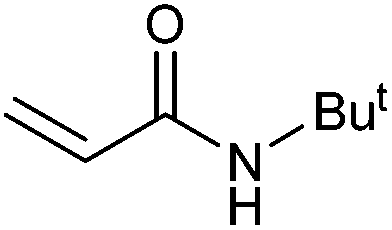
|
50 | 41 | 25000 |
| 11 |
|
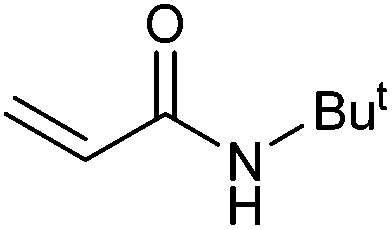
|
78 | 70 | 34000 |
| 12 |
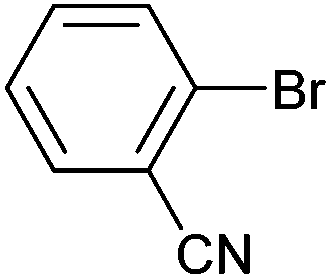
|
|
43 | 30 | 21500 |
| 13 |
|
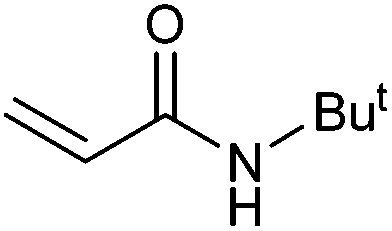
|
55 | 40 | 27500 |
| 14 |
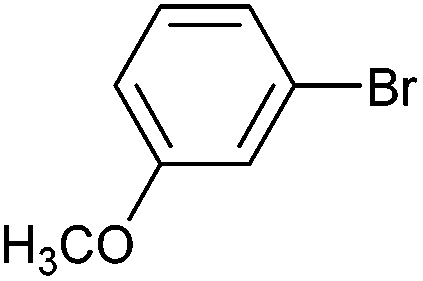
|
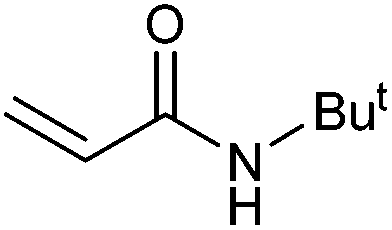
|
57 | 45 | 28500 |
| 15 |
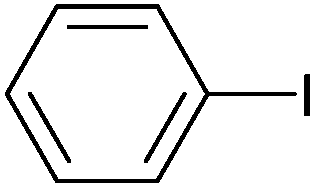
|
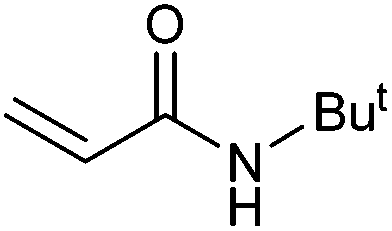
|
94 | 87 | 47000 |
| 16 |
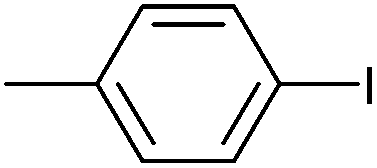
|
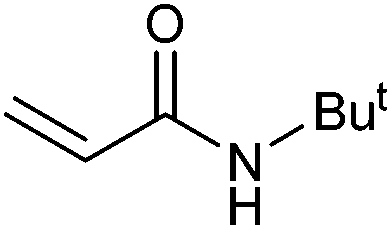
|
>99 | 93 | 49500 |
Conclusions
In summary, we have reported the synthesis of two highly conjugated mixed aldimine-ketimine ligands, L1 and L2, based on the dibenzo[a,c]acridine motif by a series of simple steps including heterocyclization of an unusual spirocycle. NketimineNC–palladium pincer complexes, Pd1 and Pd2, are readily accessible at room temperature on reaction of L1 or L2 with PdCl2(NCMe)2 in aprotic solvents while the introduction of methanol leads to hydrolysis of the non-coordinated aldimine unit in Pd1 and Pd2 to give aldehyde-containing Pd3 and Pd4. All four complexes display good to very good performance for the metal-mediated coupling of bromobenzene with styrene at elevated temperatures (ca. 140 °C) with Pd3 the standout performer. The effectiveness of Pd3 to catalyze the coupling of a series of electronically and sterically distinct bromobenzenes with styrene and N-tert-butylacrylamide has also shown the general applicability of this NNC pincer complex. Further studies are underway to determine the scope of these thermally robust ligand frames to support other metal-mediated catalytic processes.Experimental
General consideration
All manipulations of ligands were carried out under an open air atmosphere while those involving complexes were performed using standard Schlenk techniques under a nitrogen atmosphere. Solvents were distilled under nitrogen from appropriate drying agents immediately prior to use. 1H and 13C NMR spectra were recorded on a Bruker DMX 400 MHz spectrometer (400 MHz for 1H, 100 MHz for 13C) at ambient temperature using TMS as an internal standard; δ values were given in ppm and J values in Hz. Fourier Transform IR spectra were recorded on a Perkin-Elmer System 2000 FT-IR spectrometer. Elemental analysis was performed using an HPMOD 1106 microanalyzer. GC measurements were performed on a VARIAN CP-3800 GC containing a chrompack capillary column (cp-sil 5 CB). The electrospray (ESI) mass spectra for the complexes and the high resolution mass spectra (MALDI TOF) for all new organic compounds were measured using Bruker 9.4 T Solarix (FT-ICR-MS) instrument. Phenanthrene-9,10-dione, 2,6-diethylaniline, 2,6-dimethylaniline, the haloarenes, styrene and N-tert-butylacrylamide were purchased from Aldrich and Acros and used without further purification. PdCl2(NCCH3)2 was prepared using the literature method.28 Other general reagents were purchased from Aldrich, Acros or local suppliers.Synthesis of pincer Ligands
O) s), 1596 (s), 1478 (s), 1478 (s), 1393 (m), 1267 (w), 1064 (m), 987 (w), 898 (m), 733 (s). HRMS (MALDI) (m/z) calcd for C24H21NO [M + H]+ 340.17014; found 340.16953.
O) m), 1391 (s), 1344 (m), 797 (s), 760 (m), 739 (s). HRMS (MALDI) (m/z) calcd for C24H15NO2 [M + H]+ 350.11810; found 350.11755.
:1, v/v) as the eluent affording L1 as a deep yellow powder (0.60 g, 45%). Mp: 195–196 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 9.69 (d, J = 8.8 Hz, 1H, Ph–H), 9.33 (d, J = 7.6 Hz, 1H, Ph–H), 8.84 (s, 1H, CHN), 8.59 (d, J = 8.0 Hz, 1H, Ph–H), 8.52 (d, J = 8.0 Hz, 1H, Ph–H), 8.22 (d, J = 8.0 Hz, 1H, Ph–H), 8.17 (d, J = 8.0 Hz, 1H, Ph–H), 7.82–7.686 (m, 4H, Ph–H), 7.53 (t, J = 8.0 Hz, 1H, Ph–H), 7.18 (t, J = 6.4 Hz, 4H, Ph–H), 7.08–6.99 (m, 2H, Ph–H), 2.52 (s, 3H, CH3CN), 2.37 (s, 12H, 4 × CH3). 13C NMR (100 MHz, CDCl3, TMS): δ 171.5, 163.7, 151.0, 148.8, 147.4, 145.4, 141.6, 135.3, 132.1, 131.8, 131.2, 130.0, 128.9, 128.8, 128.6, 128.3, 128.2, 128.1, 128.0, 127.6, 127.0, 126.6, 126.3,126.0, 124.5, 124.4, 124.2, 123.9, 123.0, 122.7, 31.9, 28.9, 23.4, 22.7, 18.5, 18.1, 17.5, 14.0. FT-IR (KBr, cm−1): 2959 (w), 2918 (w), 2855 (m), 1646 (ν(HCN) s), 1628 (ν(CH3CN) s), 1375 (s), 1347 (s), 1292 (m), 1202 (m), 854 (s), 797 (s), 768 (s). Anal. Calcd for C40H33N3 (555): N, 7.56; C, 86.45; H, 5.99. Found: N, 7.61; C, 86.38; H, 6.09. HRMS (MALDI) (m/z) calcd for C44H33N3 [M + H]+ 556.27527; found 556.27472.
N), 8.59 (d, J = 8.0 Hz, 1H, Ph–H), 8.52 (d, J = 7.6 Hz, 1H, Ph–H), 8.19 (d, J = 8.0 Hz, 1H, Ph–H), 8.13 (d, J = 8.0 Hz, 1H, Ph–H), 7.82–7.76 (m, 2H, Ph–H), 7.73–7.68 (m, 2H, Ph–H), 7.51 (t, J = 8.0 Hz, 1H, Ph–H), 7.23–7.21 (m, 4H, Ph–H), 7.17–7.13 (m, 2H, Ph–H), 2.88–2.69 (m, 8H, 4 × CH2), 2.52 (s, 3H, CH3CN), 1.34 (t, J = 7.6 Hz, 6H, 2 × CH3), 1.26 (t, J = 7.6 Hz, 6H, 2 × CH3). 13C NMR (100 MHz, CDCl3, TMS): δ 171.4, 163.3, 149.9, 147.8, 147.5, 145.4, 141.8, 134.8, 133.3, 132.2, 132.0, 131.9, 131.8, 131.2, 130.0, 129.0, 128.8, 128.2, 128.1, 127.7, 127.1, 126.7, 126.6, 126.4, 125.9, 124.8, 124.3, 123.9, 123.3, 12.7, 53.4, 24.6, 23.6, 15.2, 14.0. FT-IR (KBr, cm−1): 2963 (w), 2925 (w), 2866 (m), 1652 (ν(HCN) s), 1620 (ν(CH3CN) s), 1456 (w), 1347 (s), 1290 (m), 1256 (m), 856 (s), 799 (s), 766 (s). Anal. Calcd for C44H41N3 (611): N, 6.87; C, 86.38; H, 6.75. Found: N, 6.76; C, 86.07; H, 6.86. HRMS (MALDI) (m/z) calcd for C44H41N3 [M + H]+ 612.33787; found 612.33732.
Synthesis of NNC pincer complexes
Pd1: Based on either of the procedures described in (a), (b) and (c), Pd1 was obtained as an orange solid in the corresponding yield given in Table 1. 1H NMR (400 MHz, CDCl3, TMS): δ 9.75 (d, J = 8.4 Hz, 1H, Ph–H), 8.83 (s, 1H, CHN), 8.48 (d, J = 8.0 Hz, 1H, Ph–H), 8.41 (d, J = 7.6 Hz, 1H, Ph–H), 8.19 (d, J = 7.2 Hz, 1H, Ph–H), 8.03–7.98 (m, 2H, Ph–H), 7.78 (t, J = 8.4 Hz, 1H, Ph–H), 7.71 (t, J = 7.2 Hz, 1H, Ph–H), 7.50–7.42 (m, 2H, Ph–H), 7.21–7.11 (m, 6H, Ph–H), 2.40 (s, 6H, 2 × CH3), 2.37 (s, 6H, 2 × CH3), 2.34 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3, TMS): δ 169.4, 162.3, 156.7, 150.3, 148.6, 141.5, 140.2, 137.2, 136.6, 133.4, 132.6, 132.5, 132.2, 131.5, 131.2, 130.0, 128.8, 128.1, 127.9, 126.9, 126.6, 126.5, 126.4, 125.7, 125.1, 124.3, 118.3, 53.3, 52.9, 23.0, 18.9, 18.5. FT-IR (KBr, cm−1): 2961 (w), 2914 (w), 2850 (w), 1618 (ν(HCN), (CH3CN) broad), 1588 (m), 1567 (m), 1368 (m), 1268 (m), 1235 (m), 1186 (w), 849 (w), 756 (m), 698 (s). ESI MS (MeCN): m/z 701 [M − Cl + MeCN]. Anal. Calcd for C40H32N3PdCl (695): N, 6.03; C, 68.97; H, 4.63. Found: N, 6.11; C, 68.91; H, 4.55.
Pd2: Based on either of the procedures outlined in (a) and (c), Pd2 was obtained as an orange solid in the corresponding yield given in Table 1. 1H NMR (400 MHz, CD2Cl2, TMS): δ 9.78 (d, J = 8.4 Hz, 1H, Ph–H), 8.81(s, 1H, CHN), 8.5 (d, J = 8.0 Hz, 1H, Ph–H), 8.43 (d, J = 8.0 Hz, 1H, Ph–H), 8.05–8.01(m, 3H, Ph–H), 7.79 (t, J = 8.4 Hz, 1H, Ph–H), 7.72 (t, J = 7.6 Hz, 1H, Ph–H), 7.49 (t, J = 7.6 Hz, 1H, Ph–H), 7.41 (t, J = 7.6 Hz, 1H, Ph–H), 7.23–7.14 (m, 6H, Ph–H), 2.97–2.88 (m, 2H, CH2), 2.7–2.6 (m, 6H, 3 × CH2), 2.3 (s, 3H, CH3), 1.29–1.21 (m, 12H, 4 × CH3). 13C NMR (100 MHz, CD2Cl2, TMS): δ 170.3, 162.1, 149.3, 147.8, 140.3, 137.6, 136.4, 133.4, 133.3, 132.6, 132.6, 131.4, 131.0, 130.1, 127.3, 126.7, 126.6, 125.7, 125.4, 125.3, 124.2, 118.3, 24.5, 24.5, 23.7, 15.2, 13.3. FT-IR (KBr, cm−1): 2961 (w), 2927 (w), 2869 (w), 1612 (ν(HCN), (CH3CN) broad), 1564 (ν(CH3CN) w), 1350 (w), 1269 (s), 1239 (s), 1177 (m), 857 (w), 759 (s), 709 (s). ESI MS (MeCN): m/z 757 [M − Cl + MeCN]. Anal. Calcd for C44H40N3PdCl (751): N, 5.36; C, 70.21; H, 5.58. Found: N, 5.28; C, 70.91; H, 5.55.
Pd3: Based on the procedure outlined in (c), Pd3 was obtained as an orange solid in the yield given in Table 1. 1H NMR (400 MHz, CD2Cl2, TMS): δ 10.63 (s, 1H, CHO), 8.88 (d, J = 8.0 Hz, 1H, Ph–H), 8.57 (d, J = 8.0 Hz, 1H, Ph–H), 8.44 (d, J = 7.6 Hz, 1H, Ph–H), 8.05 (t, J = 7.6 Hz, 2H, Ph–H), 7.88–7.78 (m, 3H, Ph–H), 7.69 (t, J = 7.6 Hz, 1H, Ph–H), 7.43 (t, J = 7.6 Hz, 1H, Ph–H), 7.17 (t, J = 7.6 Hz, 3H, Ph–H), 2.33 (s, 6H, 2 × CH3), 2.29 (s, 3H, CH3). 13C NMR (100 MHz, CD2Cl2, TMS): δ 193.1, 169.9, 156.8, 148.8, 138.3, 137.9, 133.1, 132.9, 132.5, 132.1, 130.8, 129.8, 128.7, 128.1, 127.9, 127.2, 126.7, 125.5, 125.3, 124.3, 123.6, 118.5, 29.3, 22.9, 18.5. FT-IR (KBr, cm−1): 2973 (m), 2916 (w), 2851 (w), 1696 (ν(CO) s), 1614 (ν(CN) s), 1567 (w), 1349 (w), 1265 (m), 1190 (m), 1050 (m), 853 (w), 754 (s), 724 (s). ESI MS (MeCN): m/z 598 [M − Cl + MeCN]. Anal. Calcd for C32H23N2OPdCl (593): N, 4.72; C, 64.77; H, 3.91 Found: N, 4.72; C, 64.91; H, 4.90.
Pd4: Based on the procedure outlined in (c), Pd4 was obtained as an orange solid in the yield given in Table 1. NMR (400 MHz, CD2Cl2, TMS): δ 10.62 (s, 1H, CHO), 8.87 (d, J = 8.0 Hz, 1H, Ph–H), 8.55 (d, J = 8.0 Hz, 1H, Ph–H), 8.43 (d, J = 7.6 Hz, 1H, Ph–H), 8.04 (d, J = 7.6 Hz, 2H, Ph–H), 7.86–7.77 (m, 3H, Ph–H), 7.68 (d, J = 7.6 Hz, 1H, Ph–H), 7.41(d, J = 7.6 Hz, 1H, Ph–H), 7.20 (s, 3H, Ph–H), 2.94–2.85 (m, 2H, CH2), 2.6–2.56 (m, 2H, CH2), 2.29 (s, 3H, CH3), 1.28–1.24 (m, 6H, 2 × CH3). 13C NMR (100 MHz, CD2Cl2, TMS): δ 193.2, 170.2, 156.9, 147.7, 141.2, 140.0, 138.3, 138.0, 136.4, 133.3, 132.8, 132.0, 131.4, 131.2, 131.2, 130.8, 127.5, 127.1, 126.6, 125.8, 124.3, 123.7, 118.5, 24.5, 23.6, 13.2. FT-IR (KBr, cm−1): 2962 (w), 2912 (w), 2839 (w), 1689 (ν(CO) s), 1611 (ν(CN) s), 1584 (m), 1346 (m), 1265 (w), 1178 (m), 1096 (s), 854 (m), 755 (s), 724 (m). ESI MS (MeCN): m/z 626 [M − Cl + MeCN]. Anal. Calcd for C34H27N2OPdCl (620): N, 4.51; C, 65.71; H, 4.38 Found: N, 4.52; C, 65.91; H, 4.38.
General procedure for Heck reaction
The Heck coupling reactions of haloarenes with styrene or N-tert-butylacrylamide were carried out in the presence of catalytic amounts of the palladium pincer complex. In a typical procedure (entry 11, Table 3) a mixture of bromobenzene (0.21 mL, 2.0 mmol), styrene (0.25 mL, 2.4 mmol), DMF (4 mL), anhydrous Na2CO3 (0.23 g, 2.2 mmol) was loaded into an oven dried Schlenk flask under a nitrogen atmosphere. Pd1 (100 μL, 4 × 10−5 mmol, 2 × 10−3 mol%), taken from a 4 × 10−4 M stock solution in DMF (10 mL), was added to the Schlenk flask via syringe and the reaction mixture stirred and heated at 140 °C. After the desired reaction time, the mixture was allowed to cool to room temperature, diluted with water and ethyl acetate. The organic layers were combined, washed with brine and then dried over anhydrous MgSO4. All volatiles were removed under reduced pressure and the residue purified by flash chromatography on silica gel using petroleum ether/ethyl acetate as the eluent affording stilbene (0.24 g, 67%).X-Ray structure determination
X-ray data were collected with a Rigaku MM007-HF Saturn 724 + CCD diffractometer using Mo Kα radiation (λ = 0.71073 Å), employing a confocal mirror monochromator as a radiation source for L1 whilst graphite-monochromator as a radiation source was employed for complexes Pd1, Pd2, Pd3 and Pd4. Cell parameters were obtained by global refinement of the positions of all collected reflections. Intensities were corrected for Lorentz and polarization effects and empirical absorption. The structures were determined by direct methods and refined by full-matrix least squares on F2. All the hydrogen atoms were placed in calculated positions. The molecular structure determination and their refinement were performed by the SHELXL-97 package.29 The disorder behavior of two ethyl groups in complex Pd2 was also processed by the SHELXL-97 software. During the structure refinement, free solvent (chloroform) was squeezed (Pd2) with PLATON software.30 Details of the crystal data and structure refinements for all five structures are shown in Table 6.| L1 | Pd1·2CHCl3 | Pd2 | Pd3·CH2Cl2 | Pd4 | |
|---|---|---|---|---|---|
| Empirical formula | C40 H33N3 | C42H34N3PdCl7 | C44H40N3PdCl | C33H25N2OPdCl3 | C34H27N2OPdCl |
| Formula weight | 555.69 | 935.29 | 751.63 | 678.30 | 621.43 |
| Temperature/K | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) |
| Wavelength/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic, | Triclinic | Trigonal | Monoclinic | Monoclinic |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
P21/c | P21/n |
| a/Å | 11.910(2) | 10.641(2) | 36.490(5) | 11.611(2) | 14.187(3) |
| b/Å | 32.793(7) | 10.978(2) | 36.490(5) | 19.188(4) | 12.380(3) |
| c/Å | 9.4655(19) | 17.744(3) | 21.305(4) | 13.466(3) | 14.674(3) |
| α/° | 90 | 100.04(3) | 90.00 | 90.00 | 90.00 |
| β/° | 113.37(3) | 106.43(3) | 90.00 | 113.15(3) | 92.59(3) |
| γ/° | 90 | 93.41(3) | 120.00 | 90.00 | 90.00 |
| Volume/Å3 | 3393.6(12) | 1944.7(6) | 24568(7) |
2758.5(10) | 2574.7(9) |
| Z | 4 | 2 | 18 | 25 | 4 |
| D calcd/(g cm−3) | 1.088 | 1.597 | 0.916 | 2.782 | 1.603 |
| μ/mm−1 | 0.064 | 0.994 | 0.412 | 4.624 | 0.857 |
| F(000) | 1176 | 944 | 6984 | 2125 | 1264 |
| Crystal size/mm | 0.23 × 0.14 × 0.10 | 0.54 × 0.38 × 0.12 | 0.33 × 0.33 × 0.30 | 0.32 × 0.21 × 0.12 | 0.20 × 0.19 × 0.11 |
| θ range (°) | 1.96 to 27.50 | 1.22 to 27.47 | 2.24 to 54.82 | 4.48 to 54.96 | 3.9 to 54.98 |
| Limiting indices | −15 ≤ h ≤ 15 | −13 ≤ h ≤ 13 | −47 ≤ h ≤ 47 | −14 ≤ h ≤ 15 | −18 ≤ h ≤ 18 |
| −42 ≤ k ≤ 42 | −14 ≤ k ≤ 1 | −47 ≤ k ≤ 47 | −24 ≤ k ≤ 24 | −16 ≤ k ≤ 16 | |
| −11 ≤ l ≤ 12 | −23 ≤ l ≤ 23 | −27 ≤ l ≤ 27 | −17 ≤ l ≤ 17 | −19 ≤ l ≤ 19 | |
| No. of rflns collected | 23288 |
24363 |
102155 |
19114 |
32464 |
| No. unique rflns | 7780 | 8870 | 12432 |
6237 | 5894 |
| R(int) | 0.0518 | 0.0370 | 0.0899 | 0.0262 | 0.0422 |
| No. of params | 393 | 483 | 469 | 364 | 355 |
| Completeness to θ | 99.6 | 99.5 | 100 | 98.8 | 99.7 |
| Goodness of fit on F2 | 1.047 | 1.024 | 1.072 | 1.107 | 1.084 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0633, wR2 = 0.1406 | R 1 = 0.0765, wR2 = 0.2212 | R 1 = 0.0652, wR2 = 0.2055 | R 1 = 0.0369, wR2 = 0.0834 | R 1 = 0.0281, wR2 = 0.0705 |
| R indices (all data) | R 1 = 0.0814, wR2 = 0.1510 | R 1 = 0.0811, wR2 = 0.2352 | R 1 = 0.0755, wR2 = 0.2162 | R 1 = 0.0392, wR2 = 0.0850 | R 1 = 0.0286, wR2 = 0.0708 |
| Largest diff. peak, and hole (e Å−3) | 0.219/−0.164 | 1.946/−2.206 | 0.72/−0.92 | 1.02/−0.84 | 0.64/−0.58 |
Acknowledgements
This work is supported by National Natural Science Foundation of China (No. 51273202 and U1362204). QM thanks CAS-TWAS president's fellowship for financial support. GAS is grateful to the Chinese Academy of Sciences for a Visiting Scientist Fellowship.References
- For reviews see: (a) J. T. Singleton, Tetrahedron, 2003, 59, 1837–1857 CrossRef CAS; (b) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2571 CrossRef CAS PubMed; (c) K. J. Szabó, Synlett, 2006, 811–824 CrossRef; (d) J. M. Serrano-Becerra and D. Morales-Morales, Curr. Org. Synth., 2009, 6, 169–192 CrossRef CAS; (e) X. Chen, K. M. Engle, D.-H. Wong and J.-Q. Tu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- (a) The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. M. Jensen, Elsevier, Amsterdam, 2007 Search PubMed; (b) Organometallic Pincer Chemistry, in Top. Organomet. Chem, ed. G. van Koten and D. Milstein, Springer-Verlag, Berlin, Heidelberg, 2013, vol. 40 Search PubMed; (c) Palladacycles, ed. J. Dupont and M. Pfeffer, Wiley VCH, Weinheim, 2008 Search PubMed.
- (a) New Trends in Cross-coupling: Theory and applications, ed. T. J. Colacot, RSC Catalysis series no. 21, 2015 Search PubMed; (b) Metal-catalyzed Cross-coupling reactions, ed. A. D. Mejeire and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- (a) N. Selander and and K. J. Szabó, Dalton Trans., 2009, 6267–6279 RSC; (b) N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS; (c) E.-I. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2017 CrossRef CAS PubMed; (d) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (e) H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed; (f) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC; (g) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed; (h) J. L. Bras and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef PubMed.
- (a) A. Kumar, G. K. Ra and A. K. Singh, RSC Adv., 2012, 2, 12552–12574 RSC; (b) H. Zhang and A. Lei, Dalton Trans., 2011, 40, 8745–8754 RSC; (c) V. Farina, Adv. Synth. Catal., 2004, 346, 1553–1582 CrossRef CAS; (d) I. G. Jung, S. U. Son, K. H. Park, K.-C. Chung, J. W. Lee and Y. K. Chung, Organometallics, 2003, 22, 715–4720 CrossRef; (e) Y. Zhang, G. Song, G. Ma, J. Zhao, C.-L. Pan and X. Li, Organometallics, 2009, 28, 3233–3238 CrossRef CAS; (f) E. Díez-Barra, J. Guerra, V. Hornillos, S. Merino and J. Tejeda, Organometallics, 2003, 22, 4610–4612 CrossRef; (g) M. S. Yoon, D. Ryu, J. Kim and K. H. Ahn, Organometallics, 2006, 25, 2409–2411 CrossRef CAS; (h) K. Takenaka, M. Minakawa and Y. J. Uozumi, J. Am. Chem. Soc., 2005, 127, 12273–12281 CrossRef CAS PubMed; (i) Q.-L. Luo, J.-P. Tan, Z.-F. Li, Y. Qin, L. Ma and D.-R. Xiao, Dalton Trans., 2011, 40, 3601–3609 RSC.
- (a) R. SanMartin, B. Inés, M. J. Moure, M. T. Herrero and E. Domínguez, Helv. Chim. Acta, 2012, 95, 955–962 CrossRef CAS; (b) R. B. Bedford, Chem. Commun., 2003, 1787–1796 RSC; (c) M. R. Eberhard, Org. Lett., 2004, 6, 2125–2128 CrossRef CAS PubMed; (d) M. P. Singh, F. Saleem, G. K. Rao, S. Kumar, H. Joshi and A. K. Singh, Dalton Trans., 2016, 45, 6718–6725 RSC; (e) N. Ghavale, S. T. Manjare, H. B. Singh and R. J. Butcher, Dalton Trans., 2015, 44, 11893–11900 RSC; (f) X.-Q. Hao, Y.-N. Wang, J.-R. Liu, K.-L. Wang, J.-F. Gong and M.-P. Song, J. Organomet. Chem., 2010, 695, 82–89 CrossRef CAS.
- (a) C.-T. Chen, Y.-S. Chan, Y.-R. Tzeng and M.-T. Chen, Dalton Trans., 2004, 2691–2696 RSC; (b) M. Bröring, C. Kleeberg and S. Köhler, Inorg. Chem., 2008, 47, 6404–6412 CrossRef PubMed; (c) J. Bravo, C. Cativiela, R. Navarro and E. P. Urriolabeitia, J. Organomet. Chem., 2002, 650, 157–172 CrossRef CAS; (d) S. Roy, S. Pramanik, T. Ghorui and K. Pramanik, RSC Adv., 2015, 5, 22544–22559 RSC.
- W. Baratta, M. Ballico, S. Baldino, G. Chelucci, E. Herdtweck, K. Siega, S. Magnolia and P. Rigo, Chem. – Eur. J., 2008, 14, 9148–9160 CrossRef CAS PubMed.
- H. A. Younus, N. Ahmad, W. Su and F. Verpoort, Coord. Chem. Rev., 2014, 276, 112–152 CrossRef CAS.
- N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed.
- D. N. Nicolaides and E. K. Litinas, J. Heterocycl. Chem., 1991, 28, 139–143 CrossRef CAS.
- T. Hayashi, K. Hayashizaki, T. Kiyoi and Y. Ito, J. Am. Chem. Soc., 1988, 110, 8153–8156 CrossRef CAS.
- Novartis Pharma GMBH, PatWO2007/65662, 2007, 75–76 Search PubMed.
- W.-H. Sun, Q. Xing, J. Yu, E. Novikova, W. Zhao, X. Tang, T. Liang and C. Redshaw, Organometallics, 2013, 32, 2309–2318 CrossRef CAS.
- F. Huang, Q. Xing, T. Liang, Z. Flisak, B. Ye, X. Hu, W. Yang and W.-H. Sun, Dalton Trans., 2014, 43, 16818–16829 RSC.
- B. Gao, X. Luo, W. Gao, L. Huang, S.-M. Gao, X. Liu and Q. Wu, Dalton Trans., 2012, 41, 2755–2763 RSC.
- B. Gao, D. Zhao, X. Li, Y. Cui, R. Duan and X. Pang, RSC Adv., 2015, 5, 440–447 RSC.
- G. Hamasaka, F. Sakurai and Y. Uozumi, Chem. Commun., 2015, 51, 3886–3888 RSC.
- (a) W. B. Cross, E. G. Hope, Y.-H. Lin, S. A. Macgregor, K. Singh, G. A. Solan and N. Yahya, Chem. Commun., 2013, 49, 1918–1920 RSC; (b) S.-W. Lai, T.-C. Cheung, M. C. Chan, K.-K. Cheung, S. M. Peng and C.-M. Che, Inorg. Chem., 2000, 39, 255–262 CrossRef CAS PubMed; (c) C. Bianchini, G. Lenoble, W. Oberhauser, S. Parisel and F. Zanobini, Eur. J. Inorg. Chem., 2005, 4794–4800 CrossRef CAS; (d) T. Wang, X.-Q. Hao, X.-X. Zhang, J.-F. Gong and M.-P. Song, Dalton Trans., 2011, 40, 8964–8976 RSC; (e) J. Singh, M. Deb and A. J. Elias, Organometallics, 2015, 34, 4946–4951 CrossRef CAS; (f) T. Wang, X.-Q. Hao, J.-J. Huang, K. Wang, J.-F. Gong and M.-P. Song, Organometallics, 2014, 33, 194–205 CrossRef CAS; (g) F. Neve, A. Crispini, C. D. Pietro and S. Campagna, Organometallics, 2002, 21, 3511–3518 CrossRef CAS.
- (a) A. D. Ryabov, I. K. Sakodinskaya and A. K. Yatsimirsky, J. Chem. Soc., Dalton Trans., 1985, 2629–2638 RSC; (b) P. Burger and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 10462–10463 CrossRef CAS; (c) A. J. Canty and G. van Koten, Acc. Chem. Res., 1995, 28, 406–413 CrossRef CAS; (d) M. Gómez, J. Granell and M. Martinez, Organometallics, 1997, 16, 2539–2546 CrossRef; (e) M. Gómez, J. Granell and M. Martinez, J. Chem. Soc., Dalton Trans., 1998, 37–44 RSC; (f) A. Vigalok, O. Uzan, L. J. W. Shimon, Y. Ben-David, J. M. L. Martin and D. Milstein, J. Am. Chem. Soc., 1998, 120, 12539–12544 CrossRef CAS; (g) C. E. Webster, Y. Fan, M. B. Hall, D. Kunz and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 858–859 CrossRef CAS PubMed; (h) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed; (i) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed; (j) D. García-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2007, 129, 6880–6886 CrossRef PubMed.
- D. Das, P. Singh, M. Singh and A. K. Singh, Dalton Trans., 2010, 39, 10876–10882 RSC.
- Q. Yao, E. P. Kinney and C. Zheng, Org. Lett., 2004, 6, 2997–2999 CrossRef CAS PubMed.
- B. Ye, L. Wang, X. Hu, C. Redshaw and W.-H. Sun, Inorg. Chim. Acta, 2013, 407, 281–288 CrossRef CAS.
- J. L. Bolliger, O. Blacque and C. M. Frech, Chem. – Eur. J., 2008, 14, 7969–7977 CrossRef CAS PubMed.
- Z. Wang, Q. Ding, X. He and J. Wu, Org. Biomol. Chem., 2009, 7, 863–865 CAS.
- D. Sawant, Y. Wagh, K. Bhatte, A. Panda and B. Bhanage, Tetrahedron Lett., 2011, 52, 2390–2393 CrossRef CAS.
- M. L. Kantam, P. V. Reddy, P. Srinivas and S. Bhargava, Tetrahedron Lett., 2011, 52, 4490–4493 CrossRef CAS.
- M. S. Inkpen, A. J. P. White, T. Albrecht and N. J. Long, Chem. Commun., 2013, 49, 5663–5665 RSC.
- G. M. Sheldrick, SHELXTL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data. CCDC 1496370 (1), 1496371 (2), 1496372 (Pd1), 1496373 (Pd2), 1496374 (Pd3), 1496375 (Pd4) and 1496529 (L1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00469e |
| This journal is © the Partner Organisations 2016 |