
Role of graphene oxide as a heterogeneous acid catalyst and benign oxidant for synthesis of benzimidazoles and benzothiazoles†
Kiran B. Dhoptea,
Rahul S. Zambarea,
Anand V. Patwardhana and
Parag R. Nemade *ab
*ab
aDepartment of Chemical Engineering, Institute of Chemical Technology, Nathalal Parekh Marg, Matunga, Mumbai, Maharashtra 400019, India. E-mail: pr.nemade@ictmumbai.edu.in; Fax: +91 22 3361 1020; Tel: +91 22 3361 2027
bDepartment of Oils, Oleochemicals and Surfactant Technology, Institute of Chemical Technology, Nathalal Parekh Marg, Mumbai, Maharashtra 400 019, India
First published on 11th January 2016
Abstract
We report the synthesis of benzothiazoles and benzimidazoles using graphene oxide as an effective catalyst with good yields and easy recyclability. Graphene oxide plays the dual role of a metal-free acid catalyst and an oxidizing agent. The mechanism of action of graphene oxide as a catalyst was confirmed using X-ray diffraction, Fourier transform infrared spectroscopy, Raman spectroscopy and transmission electron microscopy. Good yields were obtained at 60 °C as well as under ultrasonic irradiation at 35 °C with methanol as the solvent. Additionally, this is also the first report in which the regeneration of partially-reduced spent graphene oxide has been carried out to restore the oxygen containing functional groups and provide a path for the reuse of graphene oxide as the catalyst and oxidizing agent.
Introduction
Synthesis of fused five membered heterocyclic ring systems elicits interest due to their applicability in cell metabolism, active pharmaceutical ingredients, drug intermediates, etc.1–3 Benzothiazole and benzimidazole moieties are particularly relevant due to their potential activity and use as a key intermediate in the synthesis of antiviral, antibacterial and other drugs.4–7 The oxidative cyclization pathway is one of the facile routes for the synthesis of these fused heterocyclic rings. Numerous metal-based heterogeneous catalysts have been reported over the years for benzothiazole and benzimidazole synthesis using the oxidative cyclization pathway.8–12 Heavy metal toxicity and leaching concerns associated with metal oxidants, especially for the synthesis of drug intermediates have led researchers to search for metal-free pathways. Oxidizing agents such as hydrogezn peroxide or cyanuric chloride13 in conjunction with an acid catalyst such as ion exchange resins,14–16 or ultrasound mediated pathways,17,18 have been reported with good yield, selectivity and short reaction times. However, the homogeneous nature of most oxidants and their application in a stoichiometric amount is an area of concern.19–22Among metal free catalysts, graphene oxide (GO) also termed as “carbocatalyst”, has been reported to facilitate several organic transformations replacing hazardous chemical reagents.23–29 Graphene oxide presents large surface area, abundant functional sites, low toxicity, ease of synthesis and reuse potential. On account of presence of large number of oxygen containing functional groups such as carboxylic acid and hydroxyl groups, graphene oxide exhibits highly acidic character. GO has also been shown to act as benign oxidizing agent. The emphasis of current work is to develop a metal free cyclization pathway to reduce environmental hazards and overcome the shortcomings of reported pathways. We report use of graphene oxide as a metal free catalyst for synthesis of benzimidazoles and benzothiazoles. Our present methodology expands the role of graphene oxide as an acid catalyst utilizing the surface bound functional groups as well as an oxidizing agent for synthesis of benzothiazoles and benzimidazoles under ultrasonic irradiation and heating conditions. Studies on recyclability of graphene oxide and the effect on the GO morphology has also been carried out. Further, we report re-oxidization of spent graphene oxide that gets partially reduced to restore oxygenated functional groups and the activity of GO catalyst for the first time.
Experimental
Materials
Natural graphite powder (325 mesh, Alfa Aesar), sulphuric acid (98% assay with 99% purity, Merck), nitric acid (68–70% assay with 99.99% purity, Merck), hydrogen peroxide (30%, S D Fine Chem. Ltd.), o-phenylenediamine (99% Sigma Aldrich), o-aminothiophenol (99%, Sigma Aldrich), aromatic aldehydes (Sigma Aldrich) were procured and used without any further purification.General procedure for synthesis of graphene oxide
Graphene oxide (GO) was prepared from thermal exfoliation of natural graphite powder by modified Hummers method.26 Natural graphite particles were intercalated in a mixture of concentrated sulphuric acid and concentrated nitric acid followed by thermal exfoliation at 800 °C. Powdered exfoliated graphite was oxidized using KMnO4 in conc. H2SO4 followed by several cycles of washing with DI water and bleaching with 30% H2O2 giving crude GO, which was then purified for the removal of MnO2 impurities by sonication in HCl followed by DI water wash. GO suspension was dried under vacuum to give GO flakes that were used as catalyst. Functional sites present on graphene oxide surface were identified using FTIR spectra on Bruker-VERTEX 80V instrument aligned with Ultra-Scan interferometer with a resolution of 1 cm−1. The lamellar structure of graphene oxide was investigated using X-ray diffraction in a wide angle (2θ = 5° to 80°, Bruker X-ray Diffractometer, D8-Advance) having monochromatized Cu Kα radiation (λ = 1.5406 Å) with a data scanning rate of 0.016 s per step. The surface morphology of graphene oxide was determined using high resolution transmission microscopy (HR-TEM, JEOL), where images were collected with an operating voltage of 200 kV. Raman spectra (HR-800, Horiba Scientific) of the GO catalysts were analysed to identify any structural changes. Elemental composition of graphene oxide was determined using Thermo Finnigan, (Italy) FLASH EA 1112 analyser.General procedure for synthesis of benzothiazole and benzimidazole
A mixture of aromatic aldehyde (1 mmol), o-phenylenediamine (1 mmol)/o-aminothiophenol (1 mmol) and graphene oxide (20 mg) in methanol (3 cm3) was either heated to 60 °C, or placed in an ultrasonic irradiation chamber (33 kHz, 150 W output) at 35 °C under constant stirring for appropriate time. The progress of reaction was monitored using thin layer chromatography using a mixture hexane and ethyl acetate (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) as mobile phase. During work up, the catalyst was separated using filtration. 10 mL of water and 10 mL of dichloromethane was added. The organic layer was separated and solvent evaporated to afford crude product. Pure product was isolated by column chromatography using silica gel (mesh 60–120) and a mixture of hexane and ethyl acetate (7:3) as mobile phase. Structure of pure compound was confirmed using 1H-NMR, 13C-NMR, mass spectroscopy, FTIR and elemental analysis, melting point, etc.
:3) as mobile phase. During work up, the catalyst was separated using filtration. 10 mL of water and 10 mL of dichloromethane was added. The organic layer was separated and solvent evaporated to afford crude product. Pure product was isolated by column chromatography using silica gel (mesh 60–120) and a mixture of hexane and ethyl acetate (7:3) as mobile phase. Structure of pure compound was confirmed using 1H-NMR, 13C-NMR, mass spectroscopy, FTIR and elemental analysis, melting point, etc.
General procedure for GO regeneration
40 mg of recovered GO (recycle-5) catalyst was gradually added to 1 mL H2SO4 maintained at 0 °C under continuous stirring. 128 mg of KMnO4 was added to this mixture under continuous stirring with the temperature being maintained below 10 °C. The reaction mixture was allowed to warm to 30 °C and stirred for 3 h. The reaction mixture was then cooled below 10 °C and diluted by adding 7 mL deionized water and 6 mL H2O2 and stirred for 24 h. Subsequently, the reaction mixture was washed with deionized water, centrifuged and supernatant discarded. Resulting crude GO was washed with 1 M HCl, 6 times to remove any traces of metal impurities. This brown coloured mass (ESI Fig. S1†) was washed with DI water until neutral pH was obtained. This mass was dried at ambient temperature under reduced pressure to give regenerated reoxidized GO. Reoxidized GO was characterized by FTIR spectroscopy (Fig. 2) to confirm the changes in functional groups after reoxidation compared with GO recycled after 5th recycle.Results and discussion
Graphene oxide synthesized by oxidation of thermally exfoliated natural graphite powder, contains hydroxyl, epoxide as well as carboxyl functional groups on graphene sheets. Elemental analysis indicated 50.8% carbon, 3.8% hydrogen and 45.3% oxygen in graphene oxide, indicating large number of oxygen containing groups.25 Activity of GO for cyclization of benzaldehydes and o-phenylenediamine/o-aminothiophenols to give benzimidazoles and benzothiazoles was studied.Table 1 lists the catalytic activity of graphene oxide in comparison with catalysts reported earlier. No conversion was observed after 4 h at room temperature and in absence of catalyst, while low conversion (30%) was observed at 60 °C after 4 h. However in presence of GO, hetero-cyclization proceeds with 81% yield at 60 °C within 4 h. Higher activity was observed for reaction between benzaldehyde and o-aminothiophenol giving 82% yield after only 3 h. On the other hand, reduced graphene oxide, which does not contain as many carboxylic or hydroxyl groups led to only marginal increase in the yield, indicating role of acidic oxygen groups present in graphene oxide for catalysing cyclization. The activity of graphene oxide compares favourably with concentrated HCl, 70% sulphuric acid a homogeneous acid catalysts. Moreover, charring was observed during reaction containing concentrated sulphuric acid. Yield obtained with GO is higher than that obtained using much higher quantity of Indion-652 cation exchange resin with carboxylic acid functional groups. Yield reported for strongly acidic cation exchange resins Dowex 50W,14 Indion-190,15 and Amberlite-120 containing sulphonic acid functional groups were similar to the yield obtained with GO but at substantially higher catalyst loading.
|
||||
|---|---|---|---|---|
| Sr. No. | Catalyst | Time (h) | Catalyst loading | Yieldb (%) |
| a Reaction conditions: benzaldehyde (1 mmol), o-phenylenediamine (1 mmol), methanol (3 mL), temperature: 60 °C.b Isolated yield.c Reaction conditions: benzaldehyde (1 mmol), o-aminothiophenol (1 mmol), methanol (3 mL), temperature: 60 °C.d Temperature: 70 °C. | ||||
| 1 | No catalyst | 4 | — | 30 |
| 2 | Graphene oxide | 4 | 20 mg | 81 |
| 3 | Graphene oxide | 3 | 20 mg | 82c |
| 4 | Graphene oxide | 4 | 5 mg | 40 |
| 5 | Reduced graphene oxide | 4 | 20 mg | 45 |
| 6 | Indion-652 | 4 | 50 mg | 53 |
| 7 | Dowex-20 | 12 | 53 mg | 85d,15 |
| 8 | Indion-190 | 4 | 50 mg | 89 (ref. 16) |
| 9 | Amberlite IR-120 | 4 | 50 mg | 80 |
| 10 | Conc. HCl | 4 | 109.5 mg (3 mmol) | 30 |
| 11 | H2SO4 (70%) | 4 | 98 mg (1 mmol) | 52 |

High-intensity sonication has proven to be an important tool for various organic transformations, reducing reaction time without need for high temperatures or pressures. In order to cover scope of current methodology, cyclization reactions were carried out at 60 °C as well as under ultrasonic irradiation (Table 2). Graphene oxide shows excellent dispersion under ultrasonic irradiation within short time at room temperature. Higher yield was noted for all reactions carried out under ultrasonic irradiation at 35 °C in just 1 h compared to that obtained at 60 °C under stirring after 3 h. Therefore, ultrasonic irradiation leads to shorter reaction time with better yield compared to yield obtained under elevated temperature conditions. Heterocyclization proceeds well under solvent free conditions, however, the reaction mass becomes progressively viscous as conversion rises, leading to difficulty in keeping the catalyst and reaction mass well mixed. The reaction mass was diluted with solvents such as acetonitrile, dimethylformamide (DMF), ethanol, water, toluene, 1,4-dioxane etc. to overcome demixing due to higher viscosity and the effect on yield studied. The yield of reaction in non-polar solvents was poor as GO does not disperse well in non-polar solvents. Higher yield was obtained for reaction in polar solvents with the exception of water. Low yield is perhaps due to lower solubility of reactants in water. The yield of reaction in polar protic solvents was higher than in polar aprotic solvents.
|
|||||
|---|---|---|---|---|---|
| Sr. No. | Catalyst | Solvent | Heating | Ultrasonic irradiation | |
| Temp °C | Yieldb (%) | Yieldc (%) | |||
| a Reaction conditions: benzaldehyde (1 mmol), o-aminothiophenol (1 mmol), methanol: 3 cm3, graphene oxide: 20 mg.b Isolated yield, time: 3 h.c Isolated yield, ultrasonic irradiation output of 150 W at (33 kHz), temperature: 35 °C, time: 1 h.d Reaction conditions: benzaldehyde (1 mmol), o-phenelenediamine (1 mmol), graphene oxide: 20 mg. | |||||
| 1 | — | Methanol | 35 | — | 25 |
| 2 | GO | — | 60 | 45 | 70 |
| 3 | GO | — | 60 | 35 | 60d |
| 4 | GO | Methanol | 30 (RT) | 35 | — |
| 5 | GO | Methanol | 60 | 82 | 89 |
| 6 | GO | Ethanol | 60 | 81 | 85 |
| 7 | GO | Water | 60 | — | — |
| 8 | GO | Acetonitrile | 60 | 71 | 75 |
| 9 | GO | Dichloromethane | 39.6 (reflux) | 62 | 80 |
| 10 | GO | Toluene | 60 | 40 | 50 |
| 11 | GO | 1,4-Dioxane | 60 | 50 | 63 |
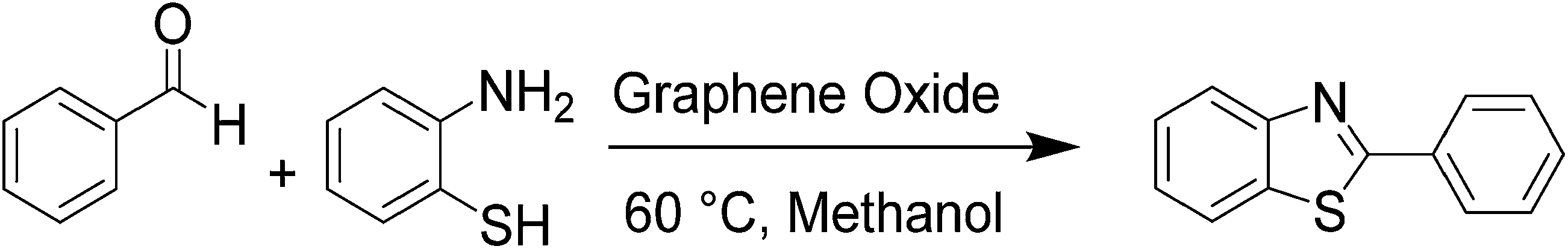
The effect of electron withdrawing and electron donating groups on the yield of reaction was studied and is shown in Table 3. Aldehydes bearing electron withdrawing groups (Table 3, entry 2–5, 17–19) as well as electron donating groups (Table 3, entry 7–11, 20) and amine with electron donating group (Table 3, entry 21) react cleanly with excellent yield under both heating and ultrasonic irradiation. The yield of the reaction does not appear to be substantially influenced by either electron withdrawing or electron donating groups on aldehyde. Unactivated aliphatic aldehyde shows poor reactivity (Table 3, entry 13). Cinnamaldehyde gives good yield under both refluxing conditions as well as under ultrasonic irradiation (Table 3, entry 14). However, aromatic aldehydes substituted with nitro group and hydroxyl group (entry 6, 12 and 16) exhibit low yield with prolonged reaction time possibly due to interaction or adsorption of the compounds with graphene oxide surface during reaction, however the exact reasons are unknown.
| Sr. No. | –R (aldehyde) | –X (amine) | Product | Timeb (h) | Yieldd (%) | Timec (min) | Yieldd (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: aldehyde (1 mmol), amine (1 mmol) methanol (3 cm3), graphene oxide (20 mg).b Reaction temperature: 60 °C.c Ultrasonic irradiation, reaction temperature: 35 °C.d Isolated yield. | |||||||
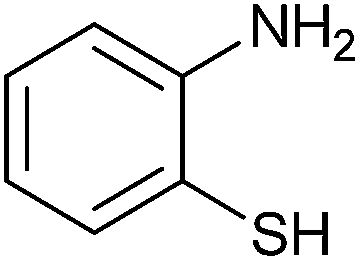
| 1 |  |
 |
 |
3 | 82 | 60 | 89 |
| 2 |  |
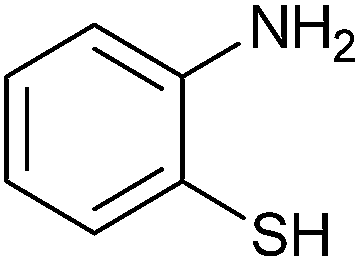 |
 |
3.5 | 81 | 60 | 88 |
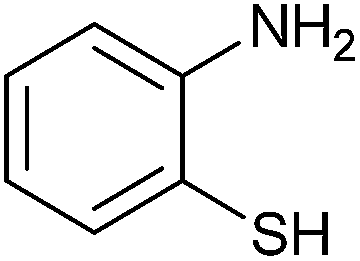
| 3 |  |
 |
 |
4 | 80 | 70 | 84 |
| 4 |  |
 |
 |
3.5 | 80 | 60 | 85 |
| 5 |  |
 |
 |
3 | 81 | 60 | 85 |
| 6 |  |
 |
 |
5.5 | 70 | 70 | 79 |
| 7 |  |
 |
 |
4.5 | 78 | 65 | 80 |
| 8 |  |
 |
 |
3.5 | 80 | 60 | 82 |
| 9 | 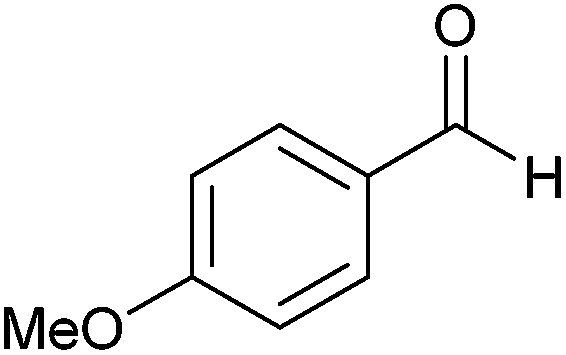 |
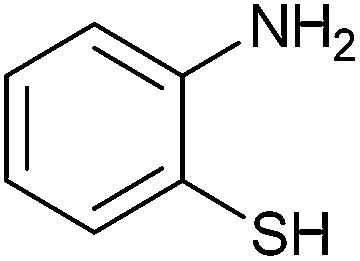 |
 |
3.5 | 81 | 60 | 84 |
| 10 | 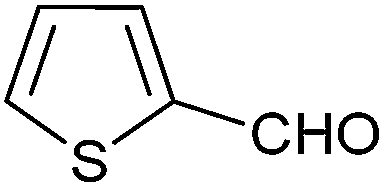 |
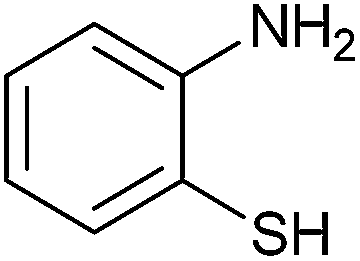 |
 |
4 | 78 | 75 | 81 |
| 11 |  |
 |
 |
4 | 76 | 70 | 81 |
| 12 |  |
 |
 |
5 | 70 | 80 | 75 |
| 13 |  |
 |
 |
4.5 | 78 | 90 | 80 |
| 14 |  |
 |
 |
6 | — | 120 | 42 |
| 15 |  |
 |
 |
4 | 81 | 70 | 86 |
| 16 |  |
 |
 |
4.5 | 80 | 70 | 83 |
| 17 |  |
 |
 |
4 | 78 | 75 | 84 |
| 18 |  |
 |
 |
4 | 81 | 70 | 84 |
| 19 |  |
 |
 |
4 | 80 | 70 | 83 |
| 20 |  |
 |
 |
5 | 70 | 80 | 77 |
| 21 |  |
 |
 |
4 | 81 | 80 | 83 |
Graphene oxide, being heterogeneous catalyst, can easily be separated from reaction mixture and reused. Catalytic activity of graphene oxide upon recycle was studied for 5 reaction cycles after initial reaction to ascertain reuse potential of GO for model reaction, synthesis of 2-(4-methoxyphenyl)benzothiazole from p-methoxy benzaldehyde and o-aminothiophenol in methanol under ultrasonic irradiation at 35 °C for 1 h. The catalyst was isolated after each recycle, washed with methanol and re-used. A marginal decrease in yield is observed with each recycle indicating slight loss of activity (Fig. 1).
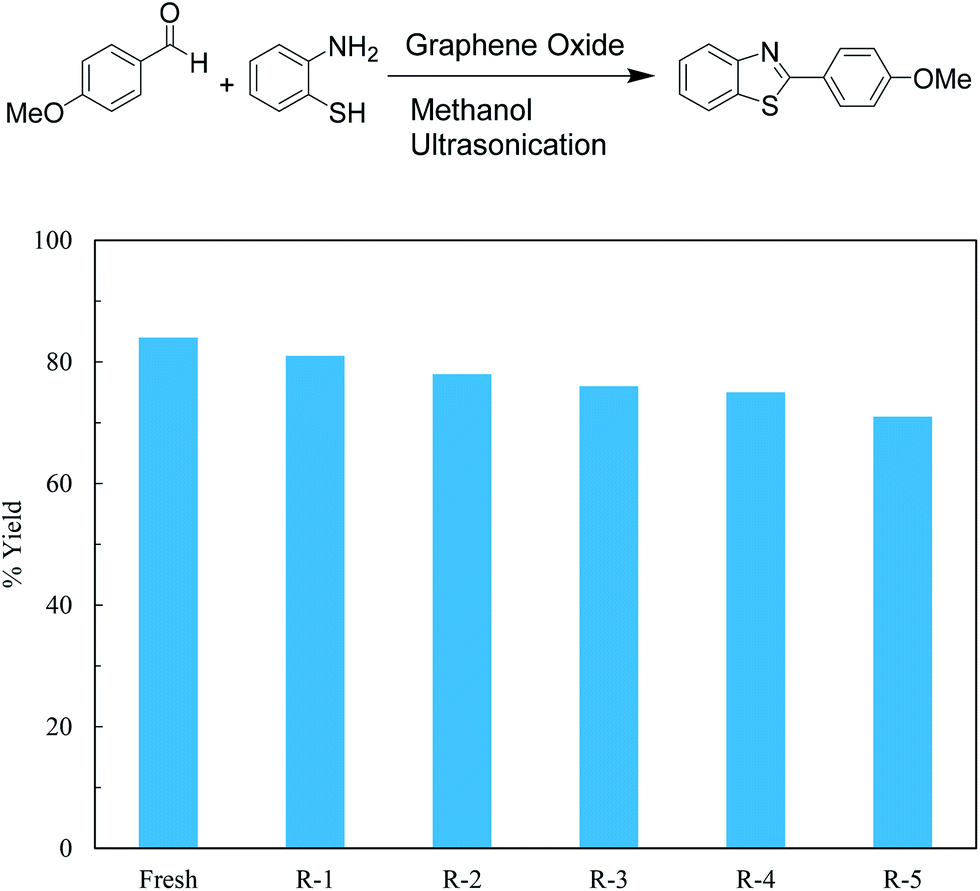 | ||
| Fig. 1 Recyclability study of GO for synthesis of 2-(4-methoxyphenyl)benzothiazole. | ||
Structural changes in GO arising due to the catalytic activity were analysed using Raman spectroscopy, XRD, FTIR, elemental analysis and HR-TEM. Raman spectroscopic analysis (Fig. 2) was carried out to determine changes in structural configuration of the catalyst. Graphene oxide shows a peak, characteristic of D band, at (1326 cm−1) from defects caused due to sp3 hybridized carbon atoms and an another peak, characteristic of G band, at (1587 cm−1) associated with in plane vibration of sp2 carbon atom.30,31 After 5th recycle slight shift is observed in G band from (1587 cm−1) to (1576 cm−1) strongly indicates partial reduction of graphene oxide after reuse. Moreover, ID/IG ratio of the catalyst increased from 1.31 for pristine GO to 1.63 for GO-R5 which confirms that partial reduction of graphene oxide occurs during reaction.32 Fig. 3 shows the XRD spectra of pristine GO, and GO catalyst after 5th recycle (GO-R5). XRD spectra of graphite is also added for reference. A comparison of pristine and recycled catalyst indicates a reduction in the intensity of first peak (2θ = 11, d spacing = 9.1), a characteristic peak of graphene oxide, and appearance of a new slightly broad peak at 2θ = 25 (d spacing = 3.4, crystallite size = 3.8 nm), which is a characteristic of reduced graphene oxide (rGO) upon reuse. These results show that reduction of functional groups on GO has occurred during reaction giving the catalyst rGO character.
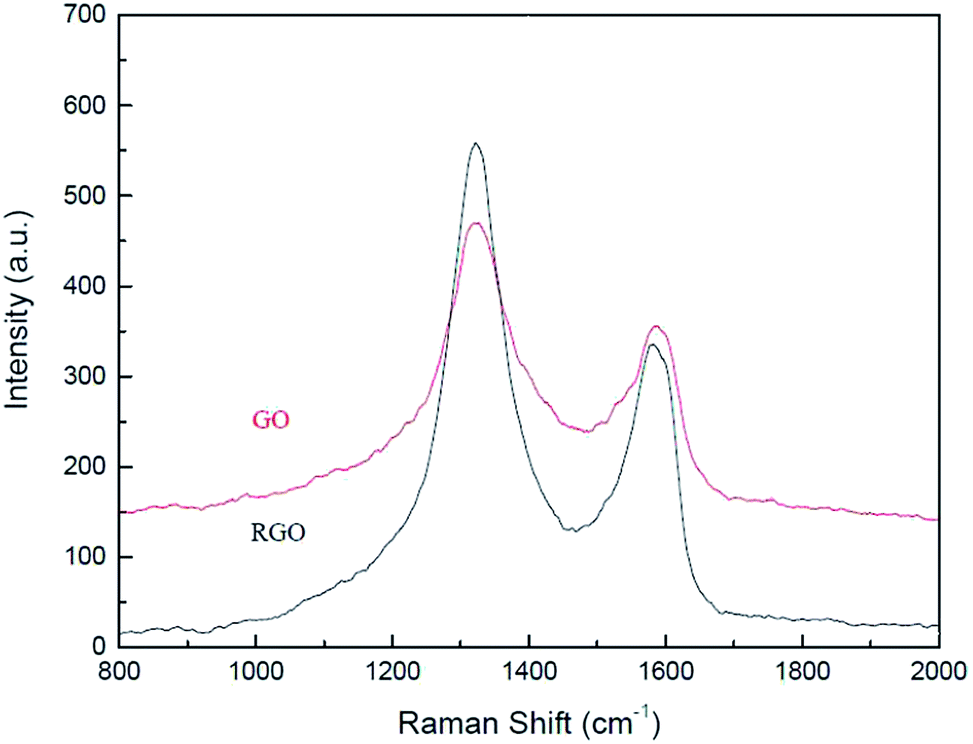 | ||
| Fig. 2 Raman spectra of GO and GO after 5th recycle. | ||
 | ||
| Fig. 3 XRD spectra of graphite, GO and GO after 5th recycle. | ||
Further investigation of catalyst using FTIR analysis revealed that peak at 1720 cm−1, corresponding to carbonyl groups, in GO had disappeared completely in GO-R5 (Fig. 4). Moreover, on recycle, overall peak intensity of broad peak at 3400 cm−1, corresponding to hydroxyl groups, decreased when compared to the intensity of peak at 1030 cm−1 corresponding to C–O groups. Ultrasonic irradiation33 and amino groups have tendency to reduce GO giving rGO and as GO is reduced, cyclization reaction is enhanced as observed by comparing entry 1 and entry 5 in Table 2. Contribution of oxygen containing functional groups during cyclization was confirmed by elemental analysis after subsequent reuse. The carbon content was increased from 50.8% (pure GO) to 74.9% (recovered after 5th recycle), whereas oxygen content decreased from 45.3% (pure GO) to 17.4% (recovered after 5th recycle). Thus O/C ratio decreased from 0.893 in GO to 0.232 in GO-R5. This decrease in oxygen content and detection of 5.6% nitrogen confirm the role of graphene oxide as an oxidizing agent during the cyclization reaction. Morphological study of GO and GO-R5 was carried out using HR-TEM microscopy to investigate disintegration of GO-sheets due to reactions (Fig. 5). After reuse, GO sheets appear to have disintegrated into smaller sheets along with slight aggregation. Therefore, as GO catalyses the reaction, its reduction to rGO as well as continuous exposure to ultrasonic irradiation leads to its disintegration into smaller sheets, possibly explaining the broad nature of rGO peak observed in GO-R5.
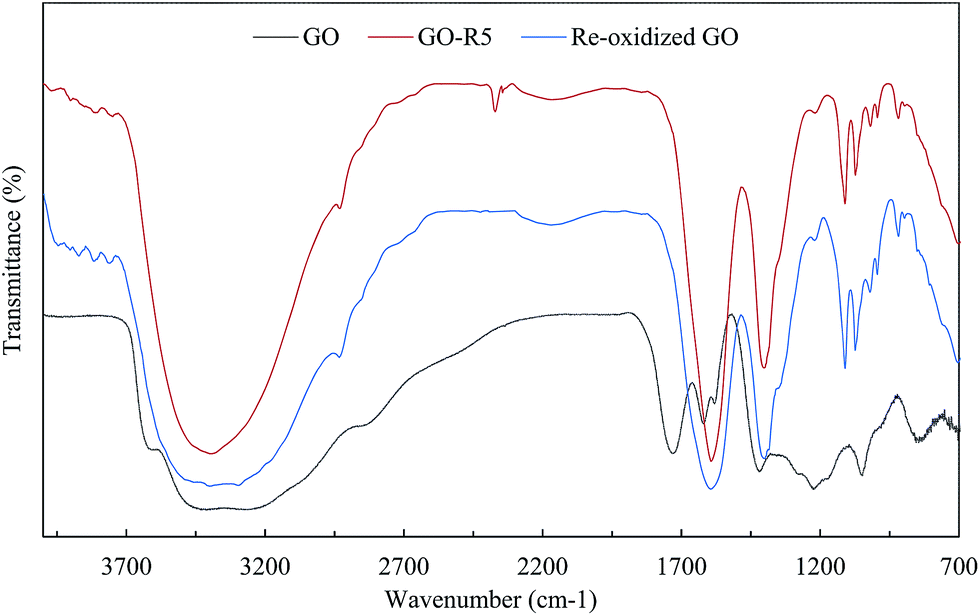 | ||
| Fig. 4 FTIR spectra of GO, GO after 5th recycle and re-oxidized GO. | ||
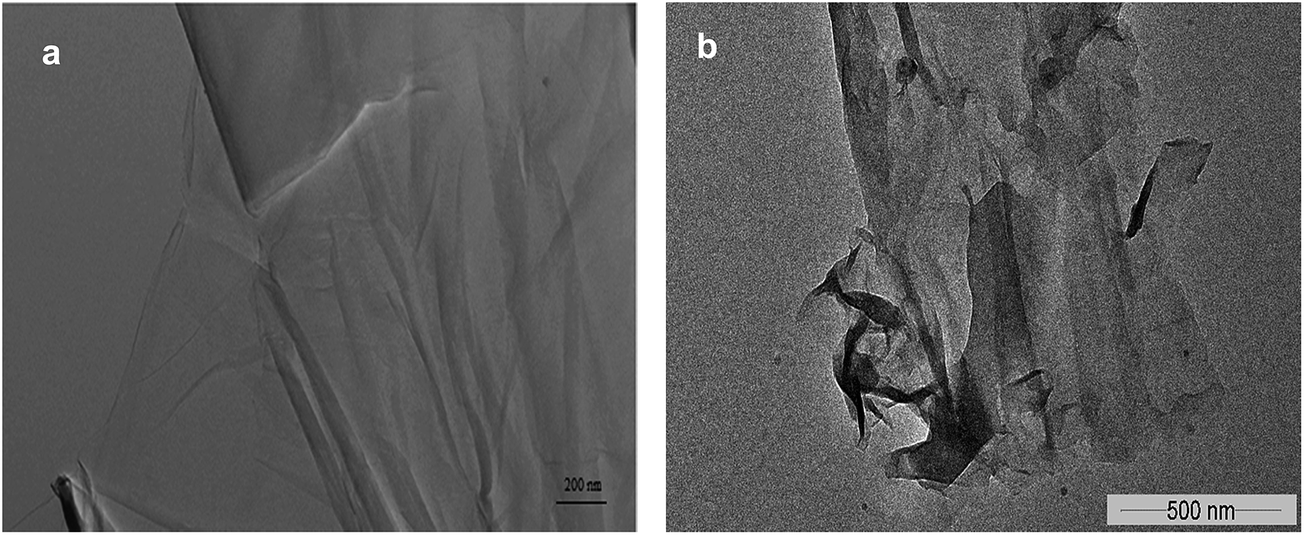 | ||
| Fig. 5 HR-TEM images of (a) GO and (b) GO after 5th recycle. | ||
Surface bound oxygen functional groups on GO play important role during benzimidazole and benzothiazole formation. The reaction proceeds through formation of imine intermediate by reaction between aldehyde and amine group under suitable experimental conditions (Fig. 6). Acid groups in GO have been reported to efficiently catalyse formation of imine from aromatic aldehyde and aromatic amine.25 In order to confirm our mechanistic approach, we monitored the reaction using gas chromatography. Under ultrasonic irradiation, almost all of aldehyde, the limiting reactant, was consumed within first 15 min coinciding with appearance of peak for imine intermediate in the gas chromatogram. The presence of imine intermediate confirms role of graphene oxide as an acid catalyst. Imine further undergoes oxidative cyclization mediated by graphene oxide, which itself is partially reduced to give reduced graphene oxide. Good yield of cyclization obtained under ultrasonic irradiation in total absence of oxygen and under N2 atmosphere confirms the oxidizing potential of GO. Thus functional groups present on graphene oxide play a key role in product formation even in absence of atmospheric oxygen. Oxygen containing functional groups are consumed as the reaction is catalysed. Therefore, GO activity goes on decreasing. For long term usage as a catalyst, facile regeneration must be possible. We performed regeneration of spent graphene oxide and the yield obtained with regenerated graphene oxide was almost similar to that obtained with pristine GO (Table 4). Therefore GO, which initially acts as proton donor catalysing imine formation, later on mediates cyclization of imine as an oxidizing agent to give benzothiazole and benzimidazole derivatives.
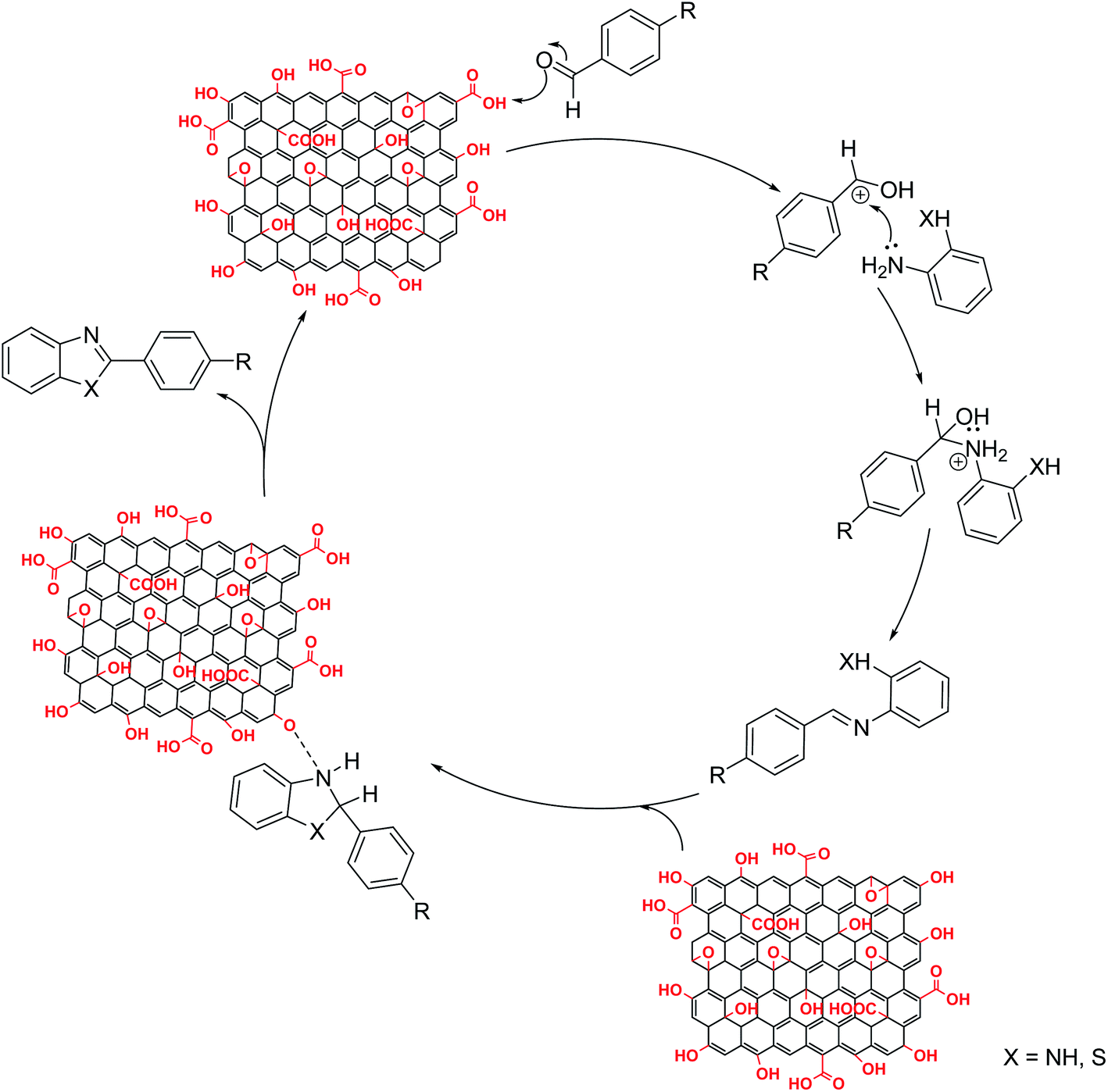 | ||
| Fig. 6 Proposed mechanism for graphene oxide catalysed synthesis of benzimidazole/benzothiazole. | ||
| Sr. No. | Catalyst | Time (h) | Catalyst loading | Yielda (%) |
|---|---|---|---|---|
| a Isolated yield.b Reaction conditions: benzaldehyde (1 mmol), o-phenylenediamine (1 mmol), methanol (3 mL), ultrasonic irradiation, reaction temperature: 35 °C.c Reaction conditions: benzaldehyde (0.5 mmol), o-phenylenediamine (0.5 mmol), methanol (2 mL), ultrasonic irradiation, reaction temperature: 35 °C, catalyst: graphene oxide regenerated after five recycles. | ||||
| 1 | Graphene oxide | 1 | 20 mg | 86b |
| 2 | Re-oxidized graphene oxide | 1 | 10 mg | 83c |
Conclusion
In conclusion, graphene oxide as metal free acid catalyst facilitates synthesis of benzothiazole and benzimidazole with good yield, short reaction time and easy recovery. Good yields are obtained under both heating and ultrasonic irradiation conditions. To the best of our knowledge, this is the first report establishing concurrent use of graphene oxide as acid catalyst and as oxidizing agent for the cyclization reaction. The reaction time was reduced drastically even at lower temperature with good yield of desired product under ultrasonic irradiation. Graphene oxide is readily recovered and was used effectively for 6 times with only marginal decrease in yield demonstrating reuse potential. We successfully carried out the re-oxidation of partially reduced GO, in order to further increase the reuse potential of catalyst, which gives nearly the same yield as fresh catalyst. Thus we have demonstrated carbocatalyst based metal-free heterogeneous catalytic pathway for cyclization for synthesis of benzimidazoles and benzothiazoles with potential for large scale application replacing metal catalysts.Acknowledgements
K. B. D. is thankful to the University Grants Commission of India for support under Special Assistance Program. Authors would also like to thank the Department of Science and Technology (DST), India for financial support under FIST program. Authors would like to acknowledge the Sophisticated Analytical Instrumentation Facility at Indian Institute of Technology Bombay for assistance with sample analysis.References
- M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef CAS PubMed.
- A. Geronikaki, E. Babaev, J. Dearden, W. Dehaen, D. Filimonov, I. Galaeva, V. Krajneva, A. Lagunin, F. Macaev, G. Molodavkin, V. Poroikov, S. Pogrebnoi, V. Saloutin, A. Stepanchikova, E. Stingaci, N. Tkach, L. Vlad and T. Voronina, Bioorg. Med. Chem., 2004, 12, 6559–6568 CrossRef CAS PubMed.
- C. Liu, J. Lin, S. Pitt, R. F. Zhang, J. S. Sack, S. E. Kiefer, K. Kish, A. M. Doweyko, H. Zhang, P. H. Marathe, J. Trzaskos, M. Mckinnon, J. H. Dodd, J. C. Barrish, G. L. Schieven and K. Leftheris, Bioorg. Med. Chem. Lett., 2008, 18, 1874–1879 CrossRef CAS PubMed.
- T. Hisano, M. Ichikawa, K. Tsumoto and M. Tasaki, Annu. Rep. Fac. Pharm. Pharm. Sci., Fukuyama Univ., 1982, 1, 17–25 Search PubMed.
- R. Paramashivappa, P. Phani Kumar, P. V. Subba Rao and A. Srinivasa Rao, Bioorg. Med. Chem. Lett., 2003, 13, 657–660 CrossRef CAS PubMed.
- D. Seenaiah, P. R. Reddy, G. M. Reddy, A. Padmaja, V. Padmavathi and N. Siva Krishna, Eur. J. Med. Chem., 2014, 77, 1–7 CrossRef CAS PubMed.
- P. Xiang, T. Zhou, L. Wang, C.-Y. Sun, J. Hu, Y.-L. Zhao and L. Yang, Molecules, 2012, 17, 873–883 CrossRef CAS PubMed.
- A. Dandia, V. Parewa and K. S. Rathore, Catal. Commun., 2012, 28, 90–94 CrossRef CAS.
- R. Fazaeli and H. Aliyan, Appl. Catal., A, 2009, 353, 74–79 CrossRef CAS.
- A. Pramanik, R. Roy, S. Khan, A. Ghatak and S. Bhar, Tetrahedron Lett., 2014, 55, 1771–1777 CrossRef CAS.
- S. Sadjadi and H. Sepehrian, Ultrason. Sonochem., 2011, 18, 480–483 CrossRef CAS PubMed.
- R. Shelkar, S. Sarode and J. Nagarkar, Tetrahedron Lett., 2013, 54, 6986–6990 CrossRef CAS.
- B. Maleki, D. Azarifar, S. F. Hojati, H. Veisi, M. Gholizadeh, H. Salehabadi and M. K. Moghadam, J. Heterocycl. Chem., 2011, 48, 449–453 CrossRef CAS.
- C. Mukhopadhyay and A. Datta, J. Heterocycl. Chem., 2009, 46, 91–95 CrossRef CAS.
- V. S. Padalkar, V. D. Gupta, K. R. Phatangare, V. S. Patil, P. G. Umape and N. Sekar, Green Chem. Lett. Rev., 2012, 5, 139–145 CrossRef CAS.
- S. D. Sharma and D. Konwar, Synth. Commun., 2009, 39, 980–991 CrossRef.
- G.-F. Chen, H.-M. Jia, L.-Y. Zhang, B.-H. Chen and J.-T. Li, Ultrason. Sonochem., 2013, 20, 627–632 CrossRef CAS PubMed.
- Y.-Q. Yuan and S.-R. Guo, Synth. Commun., 2011, 41, 2169–2177 CrossRef CAS.
- K. Bahrami, M. M. Khodaei and F. Naali, J. Org. Chem., 2008, 73, 6835–6837 CrossRef CAS PubMed.
- Y.-X. Chen, L.-F. Qian, W. Zhang and B. Han, Angew. Chem., 2008, 120, 9470–9473 CrossRef.
- Y. Nagasawa, Y. Matsusaki, T. Hotta, T. Nobuta, N. Tada, T. Miura and A. Itoh, Tetrahedron Lett., 2014, 55, 6543–6546 CrossRef CAS.
- C. Praveen, K. H. Kumar, D. Muralidharan and P. T. Perumal, Tetrahedron, 2008, 64, 2369–2374 CrossRef CAS.
- C. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275–2285 CrossRef CAS PubMed.
- D. R. Dreyer, H.-P. Jia and C. W. Bielawski, Angew. Chem., 2010, 122, 6965–6968 CrossRef.
- T. Bhattacharya, B. Majumdar, D. Dey and T. K. Sarma, RSC Adv., 2014, 4, 45831–45837 RSC.
- K. B. Dhopte, D. S. Raut, A. V. Patwardhan and P. R. Nemade, Synth. Commun., 2015, 45, 778–788 CrossRef CAS.
- M. Mirza-Aghayan, M. Alizadeh, M. Molaee Tavana and R. Boukherroub, Tetrahedron Lett., 2014, 55, 6694–6697 CrossRef CAS.
- D. R. Dreyer, H.-P. Jia, A. D. Todd, J. Geng and C. W. Bielawski, Org. Biomol. Chem., 2011, 9, 7292 CAS.
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, Tetrahedron, 2011, 67, 4431–4434 CrossRef CAS.
- K. Ai, Y. Liu, L. Lu, X. Cheng and L. Huo, J. Mater. Chem., 2011, 21, 3365–3370 RSC.
- E.-Y. Choi, T. H. Han, J. Hong, J. E. Kim, S. H. Lee, H. W. Kim and S. O. Kim, J. Mater. Chem., 2010, 20, 1907–1912 RSC.
- Z. Bo, X. Shuai, S. Mao, H. Yang, J. Qian, J. Chen, J. Yan and K. Cen, Sci. Rep., 2014, 4, 4684 Search PubMed.
- A. Bonanni, A. Ambrosi, C. K. Chua and M. Pumera, ACS Nano, 2014, 8, 4197–4204 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19066e |
| This journal is © The Royal Society of Chemistry 2016 |