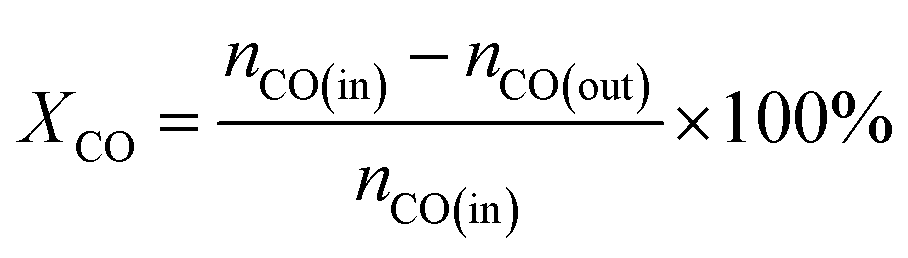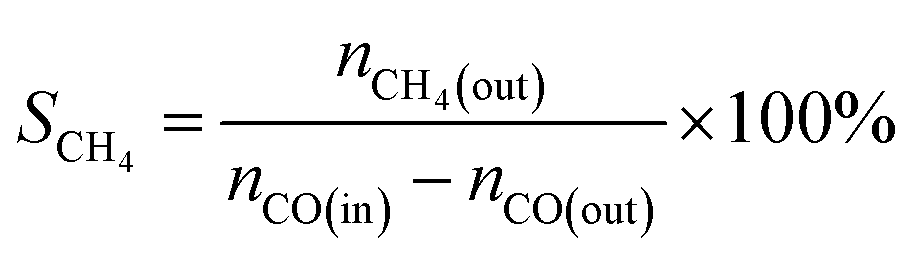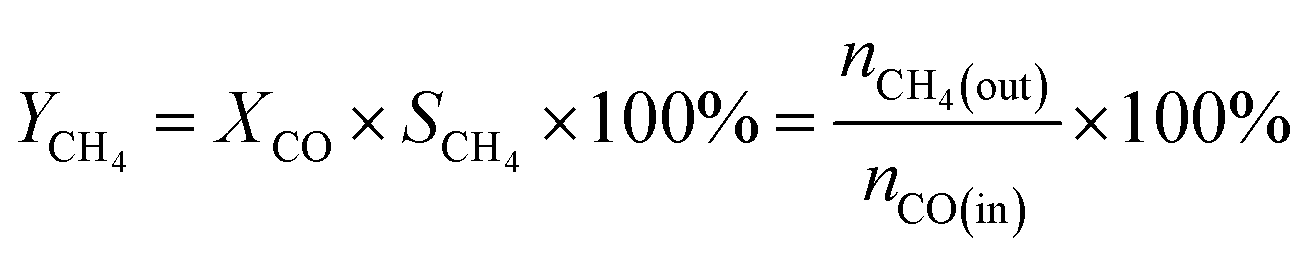DOI:
10.1039/C5RA19748A
(Paper)
RSC Adv., 2016,
6, 677-686
Ni-based catalyst derived from Ni/Al hydrotalcite-like compounds by the urea hydrolysis method for CO methanation
Received
24th September 2015
, Accepted 1st December 2015
First published on 7th December 2015
Abstract
The catalytic methanation of CO was investigated at atmospheric pressure over Ni-based catalysts derived from Ni/Al hydrotalcite-like compounds. The catalysts were prepared by the urea hydrolysis method and characterized by using XRD, TGA, N2 adsorption–desorption, H2-TPR, H2-TPD and TEM. The H2-TPD analysis revealed an increase in the Ni surface area, from 14.9 to 21.8 m2 g−1, as the Ni/Al molar ratio was increased from 0.5 to 2, and a nearly constant Ni surface area, of about 22.3 m2 g−1, at Ni/Al molar ratios > 2. A similar dependence on Ni/Al molar ratio was also found for CO conversion. The NA1 (Ni/Al molar ratio to 1) catalyst exhibited excellent stability at 600 °C for 1800 min on the stream. The NA1 catalyst prepared from Ni/Al hydrotalcite-like compounds exhibited higher catalytic stability due to higher Ni dispersion and stronger resistance to coke deposition than did the impregnated NA1-I catalyst.
1. Introduction
Natural gas, as a clean energy carrier, has attracted increasing attention worldwide due to its emissions of sulfur, nitrogen and dust being lower than these emissions from coal.1,2 In the last decade, much attention has been paid to the production of synthetic natural gas (SNG) from coal, which provides a clean way for utilizing coal in regions rich in coal and lacking natural gas, such as Korea and China.3–5 This process includes coal gasification to syngas, the water–gas shift reaction and methanation of syngas, in which methanation is the most critical step as shown in eqn (1) and (2).6 Even though the CO methanation reaction has been widely applied for gas purification processes in ammonia plants as well as for removing traces of CO in H2-rich gas in the fuel cell industry,7–10 there are still many challenges for its application in the conversion of coal to natural gas.| | |
3H2 + CO → CH4 + H2O ΔH = −206.28 kJ mol−1
| (1) |
| | |
4H2 + CO2 → CH4 + 2H2O ΔH = −164.94 kJ mol−1
| (2) |
From a thermodynamic point of view, carbon monoxide methanation is feasible.11,12 However, methanation is a highly exothermic reaction and carbon deposition may result in deactivation of the catalyst. As reported, the catalysts used for methanation include noble metals such as Ru and non-noble metals such as Ni. It is well known that Ru-based catalysts exhibit excellent catalytic activity and resistance to coke formation during CO methanation. Unfortunately, Ru-based catalysts are expensive, and the use of these catalysts is not economically feasible in large-scale commercial projects. Compared with Ru-based catalysts, Ni-based catalysts have nearly comparable activity but with extra advantages such as low cost and high methane selectivity.5,13 However, Ni-based catalysts are vulnerable to being deactivated by coke formation. Therefore, enhancing the resistance of Ni catalysts to coke formation is a prerequisite for their successful commercial application in the methanation of CO.
Hydrotalcite-like compounds (HTlc), also known as layered double hydroxides (LDH) with the general formula [M(II)1−xM(III)x(OH)2]x+[(An−)x/n·yH2O]x−, constitute a class of two-dimensional nanostructured anionic clays. The substitution of M2+ (M = Mg, Fe, Co, Cu, Ni, or Zn) by M3+ (M = Al, Cr, Ga, Mn or Fe) results in a net positive charge, attracting charge-balancing anions to the interlayer region.14–18 Complex oxides prepared from HTlc have been known to exhibit large metal surface areas, high thermal stability and uniform distributions of the active species.19–22 As reported, the formation of small metallic nickel crystallites (6 nm) dispersed over NiO–alumina derived from HTlc can be achieved, which has been shown to preserve the high activity of carbon dioxide methanation in 500 h lifetime tests even at high space velocity (268![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 mL g−1 h−1), with only a little increase of Ni crystallite size (8.6 nm) and 11% coke deposit.23 Feng et al.24 pointed out that for the material prepared by calcination of NiAl–HTlc, NiO was reduced at higher temperatures due to the stronger interaction between NiO and Al2O3. And higher nickel dispersion was achieved than that achieved using the Ni/Al2O3 catalyst prepared by the impregnation method.
800 mL g−1 h−1), with only a little increase of Ni crystallite size (8.6 nm) and 11% coke deposit.23 Feng et al.24 pointed out that for the material prepared by calcination of NiAl–HTlc, NiO was reduced at higher temperatures due to the stronger interaction between NiO and Al2O3. And higher nickel dispersion was achieved than that achieved using the Ni/Al2O3 catalyst prepared by the impregnation method.
There have been few published reports, however, about the use of the Ni–Al2O3 catalyst derived from HTlc for carbon monoxide methanation. In this study, Ni–Al2O3 catalysts prepared from HTlc with a series of Ni/Al molar ratios were investigated for CO methanation. The samples were characterized by X-ray diffraction (XRD), thermogravimetric analysis (TGA), N2 adsorption–desorption, transmission electron microscopy (TEM), H2 temperature-programmed reduction (H2-TPR) and H2 temperature-programmed desorption (H2-TPD). The catalytic activity and stability were investigated to derive the relationship between structure and activity for this set of catalysts.
2. Experimental
2.1 Catalyst preparation
The nickel aluminum composite oxides derived from HTlc used in this study were prepared by the urea hydrolysis method (UHM). Specified amounts of Ni(NO3)2·6H2O (98.0%, Tianjin Kermel Chemical Co., Ltd.), Al(NO3)3·9H2O (99.0%, Tianjin Kermel Chemical Co., Ltd.) with specific Ni/Al molar ratios, and urea as the precipitant (99.0%, Tianjin Kermel Chemical Co., Ltd.) were dissolved in deionized water, by stirring for 30 min. Then the solution was transferred into an autoclave for hydrothermal treatment at 110 °C for 10 h. The solid obtained was separated by filtration, washed with deionized water until the filtrate solution became neutral (pH = 7), and dried at 110 °C for 24 h. Finally, the catalyst precursor was calcined at 650 °C for 6 h in air to obtain the nickel–aluminum composite oxide. The Ni–Al composite oxide is denoted as NAx, in which x represents the Ni/Al molar ratio and was investigated at values of 0.5, 1, 2, 3 and 4. A reference Ni/Al2O3 catalyst with a Ni/Al molar ratio of 1 was prepared by the conventional impregnation method (IPM), and was denoted as NA1-I. The Al2O3 support (Yixing Qianye Non-metallic Materials Co., Ltd.) was pretreated at 500 °C for 4 h. Specified amounts of Ni(NO3)2·9H2O were dissolved in deionized water, and an Al2O3 support was added to this solution. The resulting slurry was stirred at room temperature for 12 h, and then dried at 120 °C for 12 h in an oven. The solid sample was calcined at 650 °C for 6 h in air.
2.2 Catalyst characterization
XRD was conducted using a Rigaku C/max-2500 diffractometer and graphite filtered Cu Kalpha radiation (k = 1.5406 Å) at 40 kV and 100 mA. The scanning rate was 5° min−1 and the scanning range was from 2θ = 5–90°. The average size of the Ni particle was calculated using the Scherrer equation. The TGA measurement of the sample was performed using an STA449F3 (NETZSCH Corp.). The sample was heated under a 50 mL min−1 flow of air from room temperature to 800 °C at 10 °C min−1. The reduction behavior of the oxides was studied by H2-TPR using a Micromeritics AutoChem 2910 system equipped with a thermal conductivity detector (TCD). The catalyst (100 mg) was loaded into a U-shaped quartz tube and heated at 10 °C min−1 to 1000 °C in 10% H2/Ar at a flow rate of 30 mL min−1. H2-TPD was performed using a Micromeritics AutoChem2910 system. The catalyst (100 mg) was reduced at 650 °C for 1 h under hydrogen flow (flow rate = 30 mL min−1) and then cooled to 100 °C under hydrogen flow. Then the flow of hydrogen was switched to argon at the same temperature and kept for 30 min to remove the weakly adsorbed hydrogen. The temperature was then raised to 650 °C at a heating rate of 10 °C min−1 under Ar flow (flow rate = 30 mL min−1). The Ni metal surface area and dispersion were calculated by H2-TPD.25 N2 adsorption–desorption measurements were taken at −196 °C using a Micromeritics Tristar 3000 system. Before analysis, samples were outgassed at 300 °C for 4 h in a degas port of the analyzer. The surface area was calculated by the standard Brunauer–Emmet–Teller (BET) method, and the pore size distribution was determined from the Barrett–Joyner–Halenda (BJH) analysis based on the Kelvin equation.26 Morphologies of the catalyst were characterized by TEM (Philips TECNAI G2F20). Samples for analysis were prepared by ultrasonic dispersion in ethanol and deposited on a micro-mesh copper grid.
2.3 Catalytic activity measurement
The methanation of CO was carried out at atmospheric pressure in a fixed bed quartz tube (i.d. = 8 mm, length = 550 mm) reactor. The catalyst bed was filled with 50 mg (40–60 mesh) catalyst diluted by 500 mg of quartz sand (40–60 mesh). Prior to the reaction, the catalyst was reduced at 650 °C in situ for 1 h in a pure H2 stream (45 mL min−1) and then cooled down to the starting reaction temperature in N2 (25 mL min−1). Then the N2/H2/CO gas mixture with a fixed molar ratio of 1/6.75/2.25 (total flow rate = 250 mL min−1) was fed through the catalyst bed at the GHSV of 300000 mL g−1 h−1. The effluent product gases were cooled in a condenser followed by a dryer, and these gases were then quantitatively analyzed for the presence of N2, CO, CH4 and CO2 by using online gas chromatography with a TDX-01 packed column.
The CO methanation performance of the catalyst was evaluated in terms of CO conversion (XCO), CH4 selectivity (SCH4), and CH4 yield (YCH4) by the formulas
| |
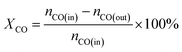 | (3) |
| |
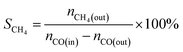 | (4) |
| |
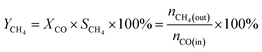 | (5) |
where
ni is the molar flow rate of component
i and subscripts in and out refer to the gas streams at the inlet and outlet.
3. Results and discussion
3.1 Characterization of catalyst precursors
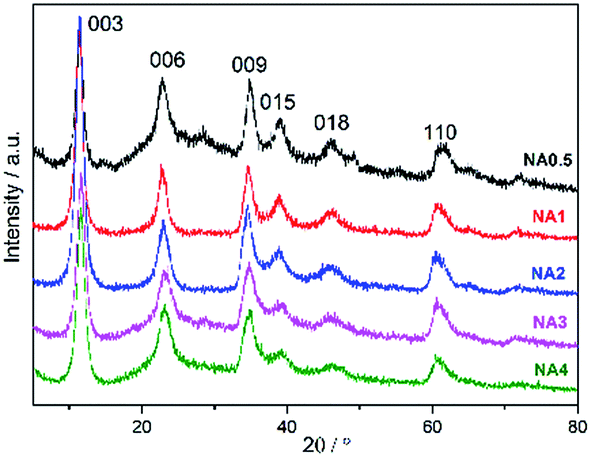
All NAx precursors yielded XRD patterns (Fig. 1) with reflections at 2θ = 11.4°, 22.9°, 34.8°, 39.0°, 46.2°, and 61.3°, which correspond to the (003), (006), (009), (015), (018), and (110) HTlc planes, respectively.27,28 The peaks were observed to be complete and sharp, indicative of a well-ordered crystallite HTlc structure. However, the weak signals at 2θ = 14.4°, 28.1°, and 48.9° resulting from AlO(OH) (JCPDS no. 49-0133) were found only for NA0.5, suggesting that another phase appeared for the catalyst at this low Ni/Al molar ratio. The corresponding crystal structure parameters (d spacing and lattice size) are listed in Table 1. The layer spacings d003, d006 and d009 of the catalyst precursors were calculated using Bragg's law, and were found to be similar to the values reported by Feng et al.24 The values of the lattice parameters a (= 2d110) and c (= d003 + 2d006 + 3d009) of the catalyst precursors were found to be close to those of the pristine HTlc. The XRD analysis demonstrates that the NAx catalyst precursors have the typical HTlc structure.
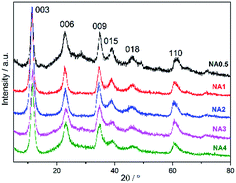 |
| | Fig. 1 XRD patterns of the NAx precursors. | |
Table 1 Analysis of XRD patterns for the NAx precursors
| Catalyst precursors |
d003 (nm) |
d006 (nm) |
d009 (nm) |
d110 (nm) |
Lattice parameters |
| a (nm) |
c (nm) |
| NiAl–HTlc24 |
0.76 |
0.38 |
0.24 |
0.15 |
0.30 |
2.28 |
| NA0.5 |
0.78 |
0.39 |
0.26 |
0.15 |
0.30 |
2.35 |
| NA1 |
0.78 |
0.39 |
0.26 |
0.15 |
0.30 |
2.34 |
| NA2 |
0.78 |
0.39 |
0.26 |
0.15 |
0.30 |
2.34 |
| NA3 |
0.78 |
0.39 |
0.26 |
0.15 |
0.30 |
2.34 |
| NA4 |
0.77 |
0.39 |
0.26 |
0.15 |
0.30 |
2.33 |
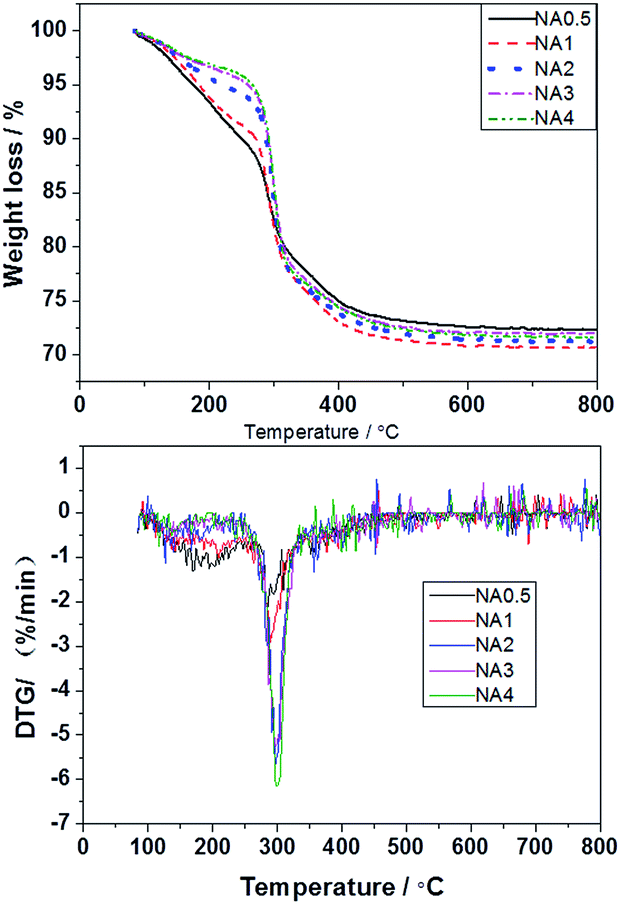
The thermal decomposition of the NAx precursors was evaluated by TGA/DTG and the results are presented in Fig. 2. The NAx precursors were previously shown to decompose in two stages: at approximately 160–270 °C and 270–500 °C.29,30 The decomposition at the lower temperature range was principally due to loss of attached waters, hydroxyls, carbon species such as CO2, etc., while that at the higher temperature range resulted from the removal of species located in the interlaminar region. This decomposition of the interlayer was accompanied by the loss of hydroxyls in the form of H2O and the loss of carbonate and nitrate anions in the form of CO2 and NOx.31 The TGA/DTG curves became horizontal above 500 °C, showing that the precursors had at that stage totally decomposed. These results indicate that the layered structure of the precursors was destroyed at temperatures over 500 °C. At these high temperatures, composite NiAl oxides were obtained. In this paper, we chose 650 °C as the calcination temperature for the catalyst preparation.
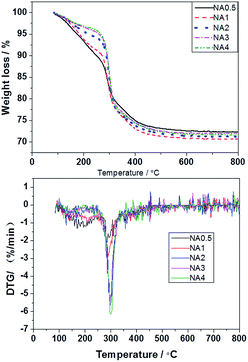 |
| | Fig. 2 The TGA/DTG curves of the NAx precursors. | |
3.2 Characterization of catalysts after calcination
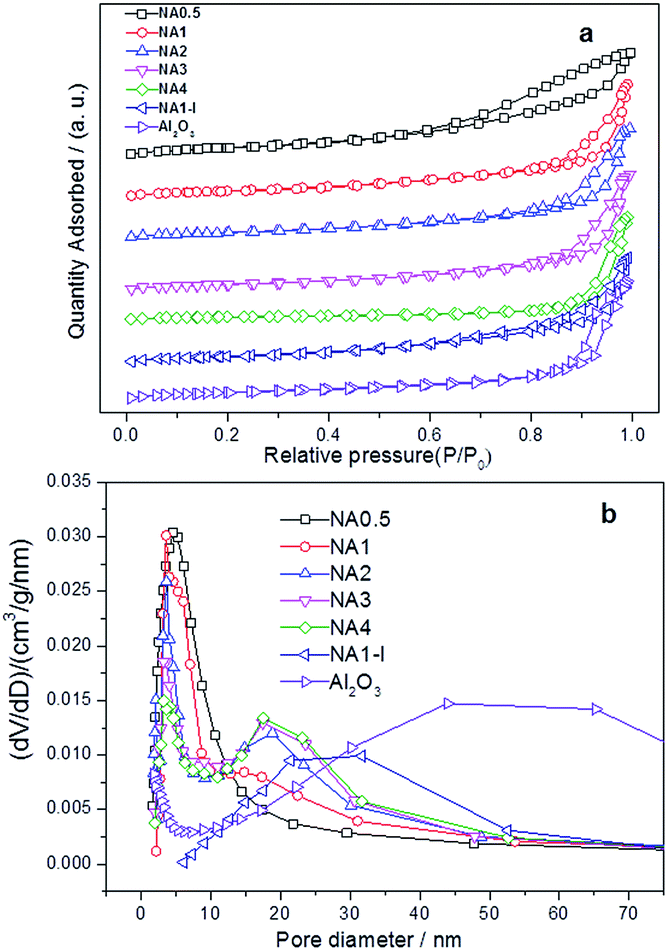
N2 adsorption–desorption isotherms and pore size distribution profiles of the calcined samples are provided in Fig. 3. Fig. 3(a) shows the hysteresis loops of the NAx catalysts to be typical characteristic of mesoporous material (type IV). The hysteresis loops exhibited characteristics of both Hl type and H3 type isotherms, demonstrating formation of both tubular and parallel slit-shaped capillary pores due to the generated gas (CO2, NOx and vapor) diffusion in the calcination process and the stacking of alumina microcrystallites.24 NA0.5 and NA1-I catalysts showed monomodal pore size distribution profiles while NAx catalysts (x = 1–4) showed bimodal pore size distributions in Fig. 3(b). The textural properties including specific surface areas, pore volumes and sizes of the Ni-based catalysts after calcination are summarized in Table 2. As the Ni/Al molar ratio of the NAx catalyst was increased, the average pore size increased while the surface area decreased. The BET surface area, pore volume and average pore size of NA1-I were smaller than those of Al2O3. The NiO particles attached on the walls and/or in the mouth of the pores of Al2O3 may account for decrease of the BET area, inducing decreases of pore volume and average pore size.
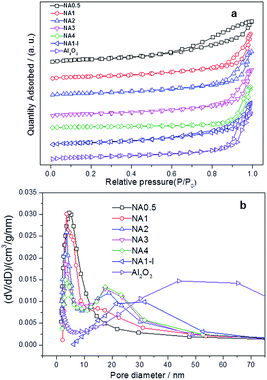 |
| | Fig. 3 Textural properties of the catalysts after calcination (a) N2 adsorption–desorption isotherm, (b) the pore size distribution. | |
Table 2 The physisorption data of catalysts after calcination
| Samples |
BET surface area (m2 g−1) |
BJH pore volume (cm3 g−1) |
Average pore size (nm) |
| NA0.5 |
161.8 |
0.46 |
8.1 |
| NA1 |
131.0 |
0.47 |
11.7 |
| NA2 |
123.3 |
0.50 |
13.1 |
| NA3 |
116.0 |
0.48 |
13.8 |
| NA4 |
111.7 |
0.51 |
15.3 |
| NA1-I |
103.1 |
0.44 |
29.2 |
| Al2O3 |
136.8 |
1.36 |
33.5 |
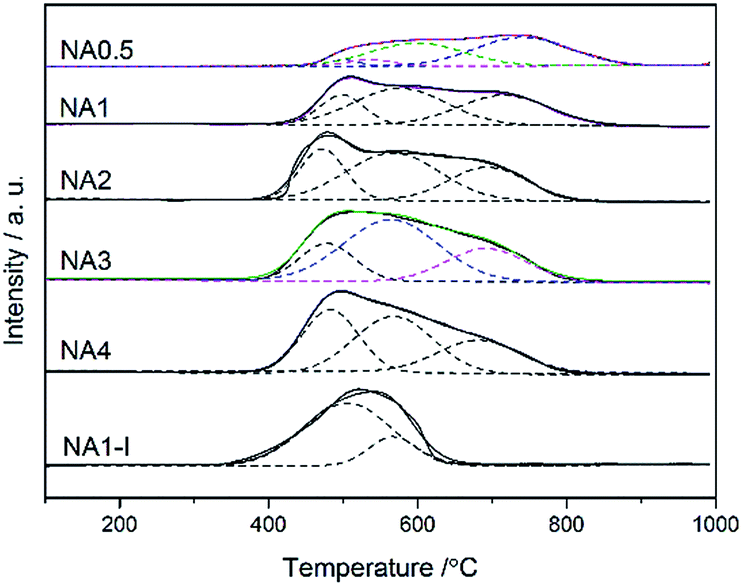
H2-TPR curves for all the Ni-based catalysts are shown in Fig. 4. One broad hydrogen consumption peak was found for all the catalysts corresponding to reduction of nickel oxide to metallic nickel. The H2-TPR measurements can be used to investigate the metal-support interaction and the reducibility of catalysts. Based on the metal-support interaction analysis, the reducible NiO species in the catalysts can be classified to several types: α-type, β-type and γ-type.32 The low-temperature peak from 340 °C to 510 °C arose from the reduction of α-type NiO species that has a weak interaction with the support.33,34 The middle peaks were assigned to the reduction of β-type NiO species that has a stronger interaction with support than does the α-type NiO species.35,36 The β-type NiO can be further classified into β1-type (530–580 °C) and β2-type (580–730 °C). The β1-type NiO species with its Ni-rich mixed oxide phase is more reducible than the β2-type NiO species with its Al-rich mixed oxide phases.32,34,35 The high-temperature peak (730–880 °C) was assigned to the γ-type NiO species, and was attributed to the stable nickel aluminate with a spinel structure.33–35 The H2 consumption peaks were fitted by a Gaussian-type function in order to determine the proportion of different NiO species and the results are listed in Table 3. The impregnated NA1-I catalyst showed a reduction peak in the 340–640 °C range belonging to the α-type and β1-type without β2-type and γ-type NiO species found, which displayed a weaker interaction between the metal and support than did the NA1 catalyst derived from HTlc. The NA0.5 catalyst yielded a broad peak (430–880 °C), which by applying a Gaussian-type function can be resolved into four peaks: α-type, β1-type, β2-type and γ-type NiO (3.6%, 5.5%, 40.3% and 50.6% in Table 3, respectively), implying the relatively strong interaction of NiO with Al2O3, which could lead to the difficulty in reduction37,38 compared to that exhibited by the NAx catalysts (x = 1–4). The NAx (x = 1–4) catalysts exhibited broad peaks in the range of 380–850 °C including the α-type, β1-type and β2-type NiO species without γ-type NiO species in accordance with the XRD results (Fig. 5). Cui et al. found that a moderate level of metal-support interaction is necessary to ensure the high activity and stability of the Ni/Al2O3 catalyst for syngas methanation at high temperatures.39 NAx (x = 1–4) catalysts had much more β-type reducible NiO species, in contrast to NA0.5 and NA1-I, which should show the higher catalytic activity.
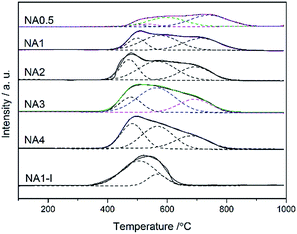 |
| | Fig. 4 H2-TPR profiles of the NAx catalysts and NA1-I catalyst after calcination. | |
Table 3 H2-TPR quantitative data of the catalysts
| Catalysts |
Temperature (°C) |
Integrated area |
Fraction of total area (%) |
Reducibilitya (%) |
| α |
β1 |
β2 |
γ |
Aα |
Aβ1 |
Aβ2 |
Aγ |
α |
β1 |
β2 |
γ |
| Calculated by: (Aα + Aβ1)/(Aα + Aβ1)NA4 × 100%, especially reducibility of NA0.5 calculated by: (Aα + Aβ1 + Aβ2)/(Aα + Aβ1)NA4 × 100%. |
| NA0.5 |
500 |
535 |
598 |
740 |
0.53 |
0.82 |
5.98 |
7.51 |
3.6 |
5.5 |
40.3 |
50.6 |
33.5 |
| NA1 |
498 |
576 |
716 |
— |
3.41 |
7.12 |
8.87 |
— |
17.6 |
36.7 |
45.7 |
— |
48.1 |
| NA2 |
471 |
566 |
694 |
— |
6.34 |
12.21 |
7.11 |
— |
24.7 |
47.6 |
27.7 |
— |
84.7 |
| NA3 |
478 |
564 |
692 |
— |
5.56 |
15.69 |
7.27 |
— |
19.5 |
55 |
25.5 |
— |
97.1 |
| NA4 |
482 |
567 |
682 |
— |
9.58 |
12.31 |
7.14 |
— |
33 |
42.4 |
24.6 |
— |
100.0 |
| NA1-I |
505 |
567 |
— |
— |
14.83 |
3.8 |
— |
— |
79.6 |
20.4 |
— |
— |
85.1 |
 |
| | Fig. 5 XRD patterns of the catalysts after calcination at 650 °C. | |
The reduction degree of the catalyst can be correlated to the integrated area of the reduction peaks below 650 °C. To better understand the reduction degree of the catalysts, the reducibility of NA4 calculated from the total integrated area of the first two reduction peaks (below 650 °C) was defined as 100%. The relative reducibility of the catalyst was the ratio of the sum of its reduction peak areas below 650 °C to the sum of the reduction peak areas below 650 °C of the NA4 catalyst. As listed in Table 3, the reduction degree of the NAx catalyst increased with an increase of the Ni/Al molar ratio due to the weakened metal-support interaction. For a Ni/Al molar ratio of 1, the reducibility of NA1-I was higher than that of NA1, on account of the larger Ni particle size of NA1-I and the weaker metal-support interaction.40
H2-TPD was used to investigate the nickel dispersion and the nickel surface area for the Ni-based catalysts. The dispersion of the Ni species was observed to decrease in the order: NA0.5 > NA1 > NA2 > NA3 > NA4 (Table 4). This decrease may be attributed to the decreasing specific surface area with the increase of Ni/Al molar ratio in the NAx catalysts. So, the dispersion of the Ni atom declined despite the higher Ni/Al molar ratio.41,42 The nickel surface area, which contributes active sites for the reaction, is responsible for the catalytic performance. From Table 4, a significant increase of nickel surface area was found as the Ni/Al molar ratio was increased from 0.5 to 2, and then the surface area was observed to stay nearly constant with a further increase of the Ni/Al molar ratio from 2 to 4. It is noteworthy that both the nickel dispersion and nickel surface area of the NA1 catalyst derived from HTlc were much higher than those of the NA1-I catalyst.
Table 4 Physical and chemical properties of the catalysts
| Samples |
Da (%) |
Nickel surface areaa (m2 gCat−1) |
Particle size (nm) |
Activity per Ni surface area at 300 °C (mmol (CO) (m2 gCat−1)−1 min−1) |
| dNiOb |
dNic |
dNid |
dNie |
dNif |
| Determined by H2-TPD. Determined according to the NiO (200) facet of the calcinated catalysts. Determined according to the Ni (111) facet of the reduced catalysts. Determined according to the Ni (111) facet of the used catalysts at 600 °C for 1800 min. Mean size of the Ni particle based on TEM analysis of the reduced catalysts. Mean size of the Ni particle based on TEM analysis of the used catalysts at 600 °C for 1800 min. |
| NA0.5 |
5.9 |
14.9 |
8.2 |
13.8 |
— |
— |
— |
3.79 |
| NA1 |
5.4 |
18.6 |
8.7 |
14.8 |
19.5 |
15.4 |
20.4 |
6.66 |
| NA2 |
4.7 |
21.8 |
9.0 |
15.2 |
— |
15.8 |
— |
6.47 |
| NA3 |
4.4 |
22.3 |
9.2 |
15.3 |
— |
16.6 |
— |
6.17 |
| NA4 |
4.0 |
22.4 |
9.5 |
16.1 |
— |
— |
— |
6.27 |
| NA1-I |
3.6 |
12.7 |
21.4 |
22.5 |
27 |
29.2 |
34.1 |
2.29 |
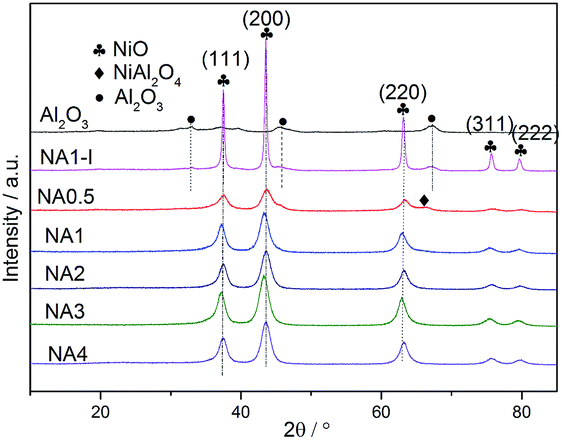
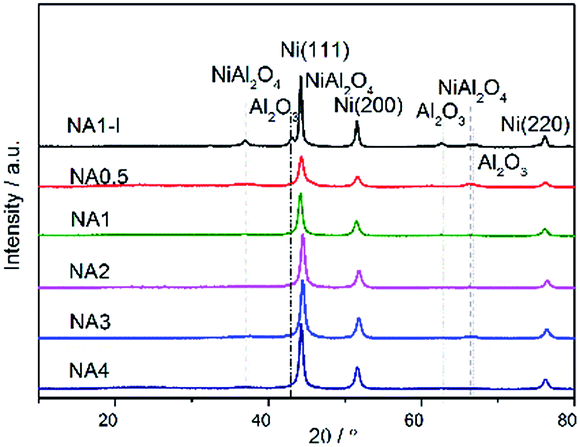
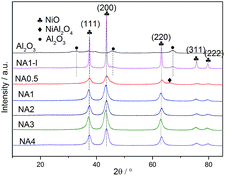
The XRD patterns of the Ni-based catalysts after calcination are shown in Fig. 5. Diffraction peaks at 2θ = 37.4°, 43.3°, 63.3°, 75.4° and 79.3° were detected for all the catalysts, corresponding to NiO (111), (200), (220), (311) and (222) crystal planes, respectively.31 The results demonstrated that the HTlc structure completely collapsed after calcination at 650 °C and the NiO crystalline phases formed. The catalyst NA0.5, i.e., that with a Ni/Al molar ratio of 0.5, could theoretically form stoichiometric NiAl2O4. Among the NAx catalysts with different Ni/Al molar ratios, the peak corresponding to the spinel phase NiAl2O4 was only observed for the NA0.5 catalyst according to the H2-TPR results. In contrast, the other catalysts with Ni/Al molar ratios from 1 to 4 only showed diffraction peaks corresponding to NiO crystals without peaks indicative of Al2O3 or NiAl2O4 crystals; these catalysts have excess Ni and have difficulty forming stoichiometric NiAl2O4. Seo et al.43 reported that the diffraction peaks corresponding to Al2O3 or NiAl2O4 were absent for the catalysts with Ni/Al molar ratios of 0.59 → 2.03 after calcination at 650 °C for 6 h. However, the NiAl2O4 diffraction peak appeared when the calcination temperature was raised to 800 °C, and the associated crystallinity increased significantly with the increased calcination temperature, suggesting a higher formation energy for the NiAl2O4 phase than for the NiO phase for Ni–Al2O3 catalysts derived from HTlc. In contrast to the NA1 catalyst, diffraction peaks at 32.9°, 45.9° and 67.3°, corresponding to Al2O3, were found for NA1-I. The NiO particles on NA1-I were determined, according to the sharp NiO peak on NA1-I, to be much larger than those on the NA1 catalyst. All of these results proved that the UMP method is more favorable for improving NiO dispersion on the catalyst than is the IPM method, which is in accordance with the characterizations indicated by H2-TPD.
3.3 Characterization of the reduced catalysts
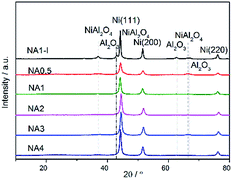
Fig. 6 shows the XRD patterns of the Ni-based catalysts after being reduced at 650 °C for 1 h in the H2 stream. Diffraction peaks were detected at 44.2°, 51.6° and 76.1°, corresponding to the Ni (111), Ni (200) and Ni (220) crystal planes. Ni particle sizes were calculated from the Scherrer formula and the results are listed in Table 4. Clearly, the Ni particle size on the NAx catalyst increased with increasing Ni/Al ratio. In addition, a smaller Ni particle size was obtained on the NA1 catalyst derived from HTlc than that on NA1-I (listed in Table 4).
 |
| | Fig. 6 XRD patterns of the catalysts after reduction. | |
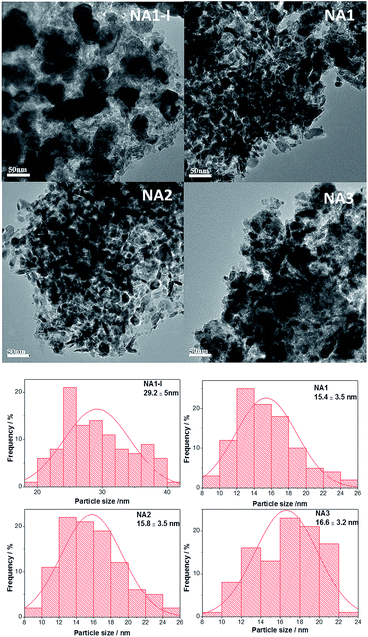
The morphologies of the Ni particles were examined by TEM analysis, and TEM images with particle size distribution histograms are presented in Fig. 7. The reduced NAx catalysts displayed nearly spherical Ni particles and the average Ni particle diameters were 15.4, 15.8, and 16.6 nm respectively for NAx (x = 1–3) catalysts, while the Ni particle size of NA1-I was 29.2 nm. The TEM results further demonstrate that NA1 from HTlc can yield Ni-based catalyst with smaller Ni particles than those from the NA1-I impregnated catalyst.
 |
| | Fig. 7 TEM images of the fresh catalysts after reduction. | |
3.4 Catalyst evaluation
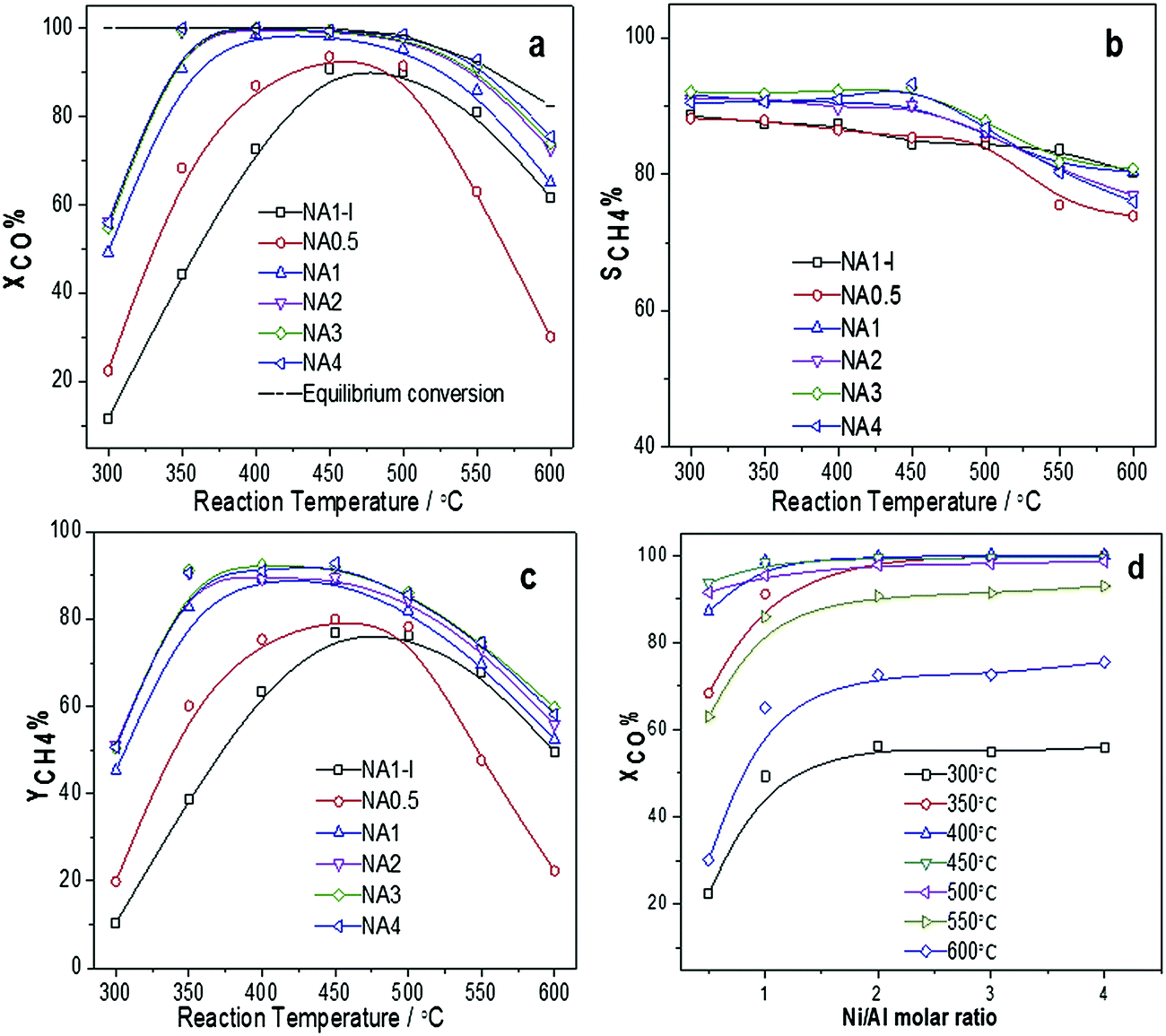
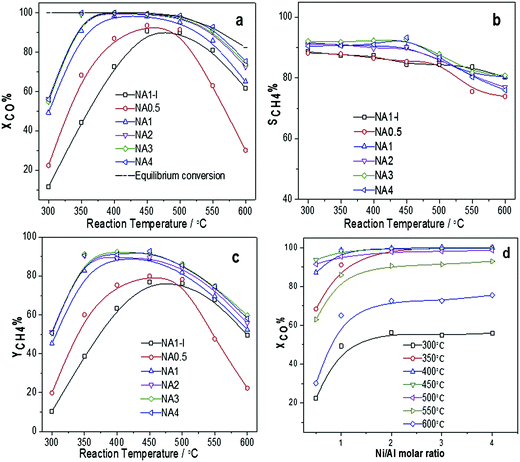
3.4.1 Catalytic CO methanation activity on different catalysts. The catalytic activities of the Ni-based catalysts at 0.1 MPa and different temperatures (300–600 °C) are shown in Fig. 8. Fig. 8(a) shows that a maximum CO conversion was observed at reaction temperatures between 400 °C and 500 °C for the NAx catalysts and nearly 100% CO conversion was achieved when using the NAx (x = 1–4) catalysts. At lower temperatures, CO conversion increased with temperature due to kinetics factors. However, at higher temperatures, CO conversion decreased due to the thermodynamic equilibrium limitation. Among NAx (x = 0.5–4) catalysts, the NA0.5, i.e., that with the lowest Ni/Al molar ratio, exhibited the lowest activity. The CH4 yield curves for all the catalysts are shown in Fig. 8(c). CH4 yield first increased and then declined as the reaction temperature was increased. CO conversion as a function of Ni/Al molar ratio is shown in Fig. 8(d). It is clear that CO conversion increased as the Ni/Al molar ratio increased from 0.5 to 2, and then remained nearly unchanged with a further increase of the Ni/Al molar ratio from 2 to 4. From the consistency between the dependence of CO conversion on the Ni/Al molar ratio and that of the Ni surface area listed in Table 4, we can deduce that the Ni surface area is a key parameter affecting the catalytic activity43 in CO methanation, with greater Ni surface areas giving rise to higher catalytic activities. In addition, the NA1 catalyst displayed better catalytic performance than did the NA1-I catalyst between 300 °C to 600 °C. This observation was also attributed to the more extensive Ni surface area, the higher Ni dispersion and the smaller Ni particle size of the NA1 catalyst than those of the NA1-I catalyst as listed in Table 4. In addition, the activity level per Ni surface area of the catalysts toward CO methanation at 300 °C were obtained, and followed the order: NA1 > NA2 > NA4 > NA3 > NA0.5 > NA1-I. All of these results illustrated that NAx catalysts derived from HTlc displayed better methanation performances than did the impregnated catalyst.
 |
| | Fig. 8 Catalytic performances of Ni catalysts at 0.1 MPa and different reaction temperatures: (a) CO conversion, (b) CH4 selectivity, (c) CH4 yield; (d) CO conversion as a function of the Ni/Al molar ratio. | |
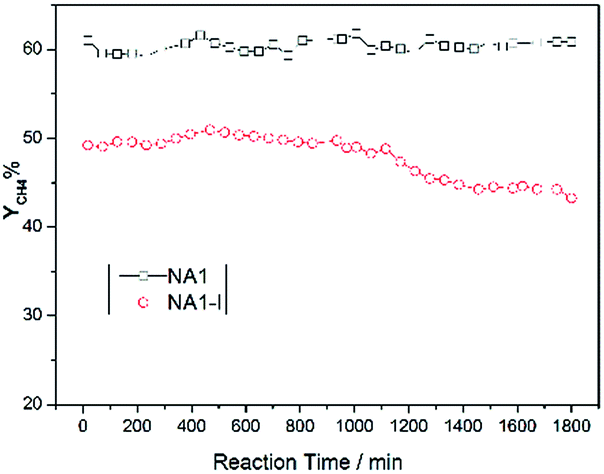
3.4.2 Stability test of CO methanation. The catalysts were expected to show not only high activity but also good stability. In particular, Ni-based catalysts usually undergo carbon deposition and/or Ni aggregation at high reaction temperatures, which may deteriorate the catalytic stability.44 Therefore, such stability is a critical parameter for the CO methanation performance of Ni-based catalysts. For these reasons, methane yields as a function of time over NA1 and NA1-I catalysts were tested (at 600 °C and 0.1 MPa) and the results are shown in Fig. 9. The NA1 catalyst was found to be more stable than the NA1-I catalyst. Over a time period of 1800 min, the methane yield resulting from the NA1 catalyst was nearly unchanged while the methane yield from the NA1-I catalyst decreased from 50% to 44%. These results demonstrated that the NA1 catalyst derived from HTlc exhibited greater high-temperature stability than did the impregnated NA1-I catalyst.
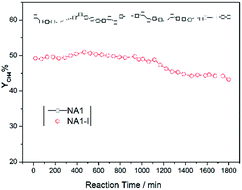 |
| | Fig. 9 Stability of CO methanation using NA1 and NA1-I catalysts at 600 °C and 0.1 MPa. | |
3.5 Characterization of the used catalyst
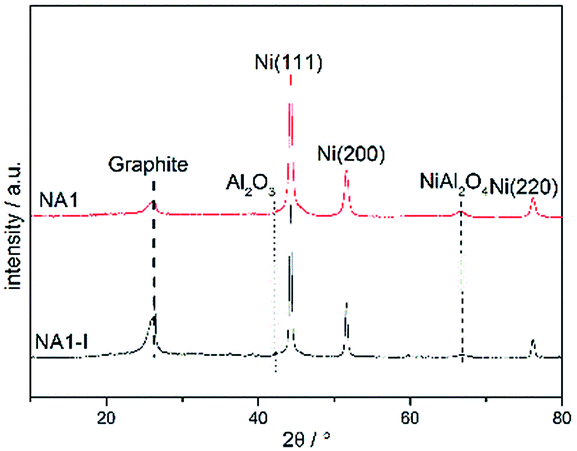
The catalysts that were subjected to the above 1800 min stability test at 600 °C and 0.1 MPa were then characterized using XRD, TEM and TGA. The XRD patterns of these used catalysts are shown in Fig. 10. The used catalysts yielded the signals corresponding to Ni (111), Ni (200) and Ni (220), which were also detected in the reduced catalyst. The used catalysts, however, also yielded a diffraction peak at 2θ = 26.4°. This peaks is characteristic of graphite, implying that carbon deposition occurred during the CO methanation process. The graphite peak was more pronounced for the spent NA1-I catalyst than that for the spent NA1 catalyst, suggesting that carbon deposition was more severe on the spent NA1-I catalyst. Also, the used catalysts yielded more intense and sharper Ni diffraction peaks than did the reduced catalysts shown in Fig. 6, demonstrating that aggregation of Ni particles occurred to some extent during the CO methanation process. As listed in Table 4, the Ni particle size increased from 14.8 nm and 22.5 nm to 19.5 nm and 27 nm, respectively, for the used NA1 and NA1-I catalysts.
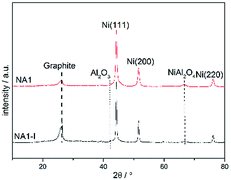 |
| | Fig. 10 XRD patterns of the used catalysts. | |
The morphologies of the used catalysts are shown in Fig. 11. Compared with the corresponding reduced NA1 and NA1-I catalysts as shown in Fig. 7, the used catalysts (NA1 and NA1-I) exhibited increased Ni particle size, in accordance with the results of XRD. It is clear that the used NA1-I catalyst was encapsulated by deposited carbon. Carbon deposition was not distinctly observed on this NA1 catalyst surface. The Ni particles on the used NA1 catalyst remained mainly exposed. These observations are consistent with the maintenance of the original, catalytic performance of NA1 at 600 °C after 1800 min. Combining the results of XRD and TEM, it could be seen that the deactivation of NA1-I was mainly due to the deposition of large amounts of carbon species on the catalyst.
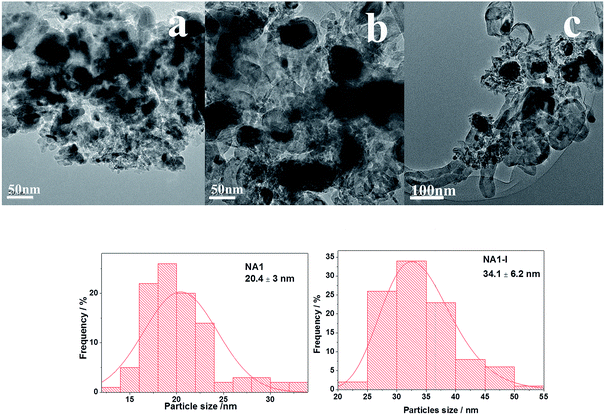 |
| | Fig. 11 TEM images of the used catalysts: (a): NA1, (b) and (c): NA1-I. | |
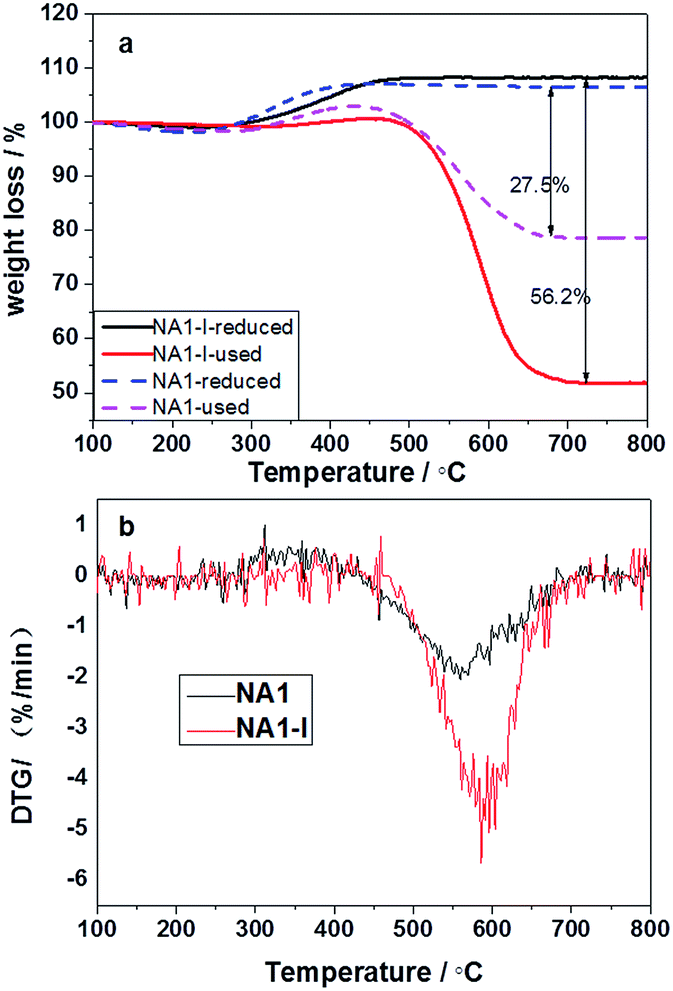
TG profiles of the reduced and used catalysts, and the DTG profiles of the used catalysts are shown in Fig. 12. As shown in Fig. 12(a), the reduced catalysts gained a little weight over the temperature range 300–500 °C owing to oxidation of the reduced Ni particles.45 As shown in Fig. 12, the used catalysts displayed two mass loss peaks due to coke combustion. It could be deduced that two kinds of carbon formed on the used catalysts: easily oxidized carbon species at temperatures below 300 °C and relatively inactive carbon species oxidized above 500 °C.46 The carbon deposited on the catalyst could have originated from CO disproportionation and CH4 decomposition.47 The deposited carbon species corresponding to the weight loss below 300 °C was relatively active and oxidizable, and originated from CO disproportionation; the deposited carbon species corresponding to the weight loss above 500 °C was inert, could not be removed except at high temperatures, and derived from CH4 decomposition.44 The carbon deposited on the used catalysts thus mainly derived from CH4 decomposition. The carbon deposition amount (56.2%) on the used NA1-I catalyst was found to be higher than that (27.5%) on the used NA1 catalyst. Therefore, NA1 appears to be more resistant than NA1-I to carbon deposition. The weight loss temperature of NA1 (in Fig. 12(b)) was found to be 50 °C lower than that of NA1-I, indicating that carbon deposited on NA1 could be more easily removed.
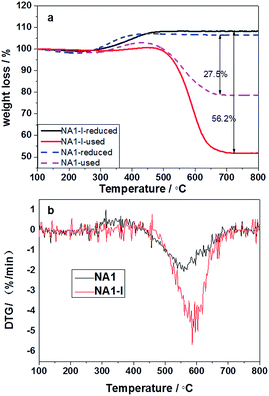 |
| | Fig. 12 TG profiles of the reduced and used catalysts (a) and DTG profiles of the used catalysts (b). | |
The Ni particle size was a key factor in carbon deposition. Larger nickel particles were reported to be more favorable for coke formation.48–51 Wang et al. found that coke formation took place mainly on large Ni particles and not on small ones during methane dry reforming.52 Liu et al. reported that the small-particle Ni catalyst was more active and stable than the large-particle Ni catalyst during CO methanation.4 The driving force for the dissociation of carbon and its diffusion should be relatively low for small Ni particles, and it was apparently more difficult for the carbon to lift these Ni particles from the support because of their stronger metal-support interactions.49,53 Compared with the NA1 catalyst, the NA1-I catalyst had larger Ni particles and weaker interactions between the Ni and the Al2O3 support. Carbon deposited on the Ni particles of NA1-I was easily dissociated and formed a lot of graphite, which nucleated quickly to form encapsulating carbon. It is therefore not surprising that deposition of coke on the NA1-I catalyst is a serious issue. Meanwhile, strong metal-support interactions may also lead to more efficient electron transfer between the metal and support to achieve a stable catalytic performance.54,55 Rossetti et al.56 reported that a strong metal-support interaction could account for high glycerol conversion and sufficient stability of the catalyst they used, whereas a weak interaction could lead to a decrease in the catalytic activity. The NA1-I catalyst had larger Ni particles, a weaker interaction between the metal and support, and more severe carbon deposition than did the NA1 catalyst. Consequently, the NA1 catalyst can generate strong metal-support interactions with good Ni dispersion, and show strong resistance to formation of coke, which contributes to its good stability.
4. Conclusion
A Ni catalyst derived from HTlc precursors was developed for methanation of carbon monoxide. XRD analysis demonstrated the precursor of the NAx catalyst to have the HTlc structure. N2 adsorption–desorption and H2-TPD results revealed both the specific surface area and Ni dispersion of the NAx catalyst to decrease with increasing Ni/Al molar ratio. But the Ni surface area only increased as the Ni/Al ratio was increased from 0.5 to 2, and then remained nearly unchanged as the Ni/Al ratio was increased further. A nearly 100% conversion of CO by Ni-based catalysts with different Ni/Al molar ratios (1–4) was observed at reaction temperatures between 400 °C and 500 °C. The NA1 catalyst derived from HTlc exhibited better catalytic stability than the NA1-I catalyst, due to higher Ni dispersion, smaller Ni particle size and the stronger metal-support interaction leading to stronger resistance to coke deposition.
References
- Y. Zhang, G. Zhang, L. Wang, Y. Xu and Y. Sun, J. Ind. Eng. Chem., 2012, 18, 1590–1597 CrossRef CAS.
- S. Hwang, J. Lee, U. G. Hong, J. C. Jung, D. J. Koh, H. Lim, C. Byun and I. K. Song, J. Ind. Eng. Chem., 2012, 18, 243–248 CrossRef CAS.
- V. M. Lebarbier, R. A. Dagle, L. Kovarik, K. O. Albrecht, X. Li, L. Li, C. E. Taylor, X. Bao and Y. Wang, Appl. Catal., B, 2014, 144, 223–232 CrossRef CAS.
- Q. Liu, J. Gao, F. Gu, X. Lu, Y. Liu, H. Li, Z. Zhong, B. Liu, G. Xu and F. Su, J. Catal., 2015, 326, 127–138 CrossRef CAS.
- Q. Liu, J. Gao, M. Zhang, H. Li, F. Gu, G. Xu, Z. Zhong and F. Su, RSC Adv., 2014, 4, 16094–16103 RSC.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS.
- P. Panagiotopoulou, D. I. Kondarides and X. E. Verykios, Appl. Catal., A, 2008, 344, 45–54 CrossRef CAS.
- F. B. Derekaya and G. Yaşar, Catal. Commun., 2011, 13, 73–77 CrossRef CAS.
- E. Kok, J. Scott, N. Cant and D. Trimm, Catal. Today, 2011, 164, 297–301 CrossRef CAS.
- H. Habazaki, M. Yamasaki, B. P. Zhang, A. Kawashima, S. Kohno, T. Takai and K. Hashimoto, Appl. Catal., A, 1998, 172, 131–140 CrossRef CAS.
- R. B. Anderson, C. B. Lee and J. C. Machiels, Can. J. Chem. Eng., 1976, 54, 590–594 CrossRef CAS.
- R. B. Anderson, J. Phys. Chem., 1986, 90, 4806–4810 CrossRef CAS.
- J. Gao, Q. Liu, F. Gu, B. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 22759–22776 RSC.
- K. Li, G. Wang, D. Li, Y. Lin and X. Duan, Chin. J. Chem. Eng., 2013, 21, 453–462 CrossRef CAS.
- F. R. Costa, M. Saphiannikova, U. Wagenknecht, G. Heinrich and B. Springer-Verlag, in Wax Crystal Control: Nanocomposites, Stimuli-Responsive Polymers, 2008, vol. 210, pp. 101–168 Search PubMed.
- F. Leroux and C. Taviot-Gueho, J. Mater. Chem., 2005, 15, 3628–3642 RSC.
- C. Taviot-Gueho and F. Leroux, in Layered Double Hydroxides, ed. X. Duan and D. G. Evans, 2006, vol. 119, pp. 121–159 Search PubMed.
- F. Leroux and J. P. Besse, Chem. Mater., 2001, 13, 3507–3515 CrossRef CAS.
- F. Zhang, X. Xiang, F. Li and X. Duan, Catal. Surv. Asia, 2008, 12, 253–265 CrossRef CAS.
- K. Takehira and T. Shishido, Catal. Surv. Asia, 2007, 11, 1–30 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- S. Liu, D. Chen, K. Zhang, J. Li and N. Zhao, Int.
J. Hydrogen Energy, 2008, 33, 3736–3747 CrossRef CAS.
- S. Abelló, C. Berrueco and D. Montané, Fuel, 2013, 113, 598–609 CrossRef.
- J.-T. Feng, Y.-J. Lin, D. G. Evans, X. Duan and D.-Q. Li, J. Catal., 2009, 266, 351–358 CrossRef CAS.
- S. Velu and S. K. Gangwal, Solid State Ionics, 2006, 177, 803–811 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183–190 CrossRef CAS.
- M. Li, X. Wang, S. Li, S. Wang and X. Ma, Int. J. Hydrogen Energy, 2010, 35, 6699–6708 CrossRef CAS.
- Z. P. Xu and H. C. Zeng, J. Phys. Chem. B, 2000, 104, 10206–10214 CrossRef CAS.
- L. Chmielarz, P. Kuśtrowski, A. Rafalska-Łasocha and R. Dziembaj, Thermochim. Acta, 2002, 395, 225–236 CrossRef.
- L. Zhao, X. Li, Z. Qu, Q. Zhao, S. Liu and X. Hu, Sep. Purif. Technol., 2011, 80, 345–350 CrossRef CAS.
- J. Zhang, H. Xu, X. Jin, Q. Ge and W. Li, Appl. Catal., A, 2005, 290, 87–96 CrossRef CAS.
- Y. Xu, H. Long, Q. Wei, X. Zhang, S. Shang, X. Dai and Y. Yin, Catal. Today, 2013, 211, 114–119 CrossRef CAS.
- D. Hu, J. Gao, Y. Ping, L. Jia, P. Gunawan, Z. Zhong, G. Xu, F. Gu and F. Su, Ind. Eng. Chem. Res., 2012, 51, 4875–4886 CrossRef CAS.
- A. Zhao, W. Ying, H. Zhang, H. Ma and D. Fang, Catal. Commun., 2012, 17, 34–38 CrossRef CAS.
- K. Y. Koo, H.-S. Roh, Y. T. Seo, D. J. Seo, W. L. Yoon and S. Bin Park, Int. J. Hydrogen Energy, 2008, 33, 2036–2043 CrossRef CAS.
- J. Gao, C. Jia, J. Li, F. Gu, G. Xu, Z. Zhong and F. Su, Ind. Eng. Chem. Res., 2012, 51, 10345–10353 CrossRef CAS.
- J. Zhang, Z. Xin, X. Meng, Y. Lv and M. Tao, Ind. Eng. Chem. Res., 2013, 52, 14533–14544 CrossRef CAS.
- D. Cui, J. Liu, J. Yu, J. Yue, F. Su and G. Xu, RSC Adv., 2015, 5, 10187–10196 RSC.
- A. Y. Khodakov, A. Griboval-Constant, R. Bechara and F. Villain, J. Phys. Chem. B, 2001, 105, 9805–9811 CrossRef CAS.
- Y.-S. Jung, W.-L. Yoon, Y.-S. Seo and Y.-W. Rhee, Catal. Commun., 2012, 26, 103–111 CrossRef CAS PubMed.
- Y.-S. Jung, W.-L. Yoon, T.-W. Lee, Y.-W. Rhee and Y.-S. Seo, Int. J. Hydrogen Energy, 2010, 35, 11237–11244 CrossRef CAS.
- Y.-S. Seo, Y.-S. Jung, W.-L. Yoon, I.-G. Jang and T.-W. Lee, Int. J. Hydrogen Energy, 2011, 36, 94–102 CrossRef CAS.
- C. Guo, Y. Wu, H. Qin and J. Zhang, Fuel Process. Technol., 2014, 124, 61–69 CrossRef CAS.
- G. Wu, S. Li, C. Zhang, T. Wang and J. Gong, Appl. Catal., B, 2014, 144, 277–285 CrossRef CAS.
- D. Liu, X. Y. Quek, W. N. E. Cheo, R. Lau, A. Borgna and Y. Yang, J. Catal., 2009, 266, 380–390 CrossRef CAS.
- M. K. Nikoo and N. A. S. Amin, Fuel Process. Technol., 2011, 92, 678–691 CrossRef CAS.
- V. M. Gonzalez-DelaCruz, J. P. Holgado, R. Pereñíguez and A. Caballero, J. Catal., 2008, 257, 307–314 CrossRef CAS.
- D. Chen, K. O. Christensen, E. Ochoa-Fernández, Z. Yu, B. Tøtdal, N. Latorre, A. Monzón and A. Holmen, J. Catal., 2005, 229, 82–96 CrossRef CAS.
- K. O. Christensen, D. Chen, R. Lødeng and A. Holmen, Appl. Catal., A, 2006, 314, 9–22 CrossRef CAS.
- A. J. Vizcaíno, A. Carrero and J. A. Calles, Catal. Today, 2009, 146, 63–70 CrossRef.
- N. Wang, W. Chu, T. Zhang and X. S. Zhao, Int. J. Hydrogen Energy, 2012, 37, 19–30 CrossRef CAS.
- N. Wang, K. Shen, X. Yu, W. Qian and W. Chu, Catal. Sci. Technol., 2013, 3, 2278–2287 CAS.
- V. Nichele, M. Signoretto, F. Menegazzo, A. Gallo, V. Dal Santo, G. Cruciani and G. Cerrato, Appl. Catal., B, 2012, 111–112, 225–232 CrossRef CAS.
- A. Iriondo, V. L. Barrio, J. F. Cambra, P. L. Arias, M. B. Guemez, M. C. Sanchez-Sanchez, R. M. Navarro and J. L. G. Fierro, Int. J. Hydrogen Energy, 2010, 35, 11622–11633 CrossRef CAS.
- I. Rossetti, A. Gallo, V. Dal Santo, C. L. Bianchi, V. Nichele, M. Signoretto, E. Finocchio, G. Ramis and A. Di Michele, ChemCatChem, 2013, 5, 294–306 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.