
A new avenue to the synthesis of highly substituted pyrroles: synthesis from N-propargylamines
Esmail Vessally
Department of Chemistry, Payame Noor University, Tehran, Iran. E-mail: vessally@yahoo.com
First published on 5th January 2016
Abstract
Pyrroles have attracted much attention due to their potential biological activities. Developing more efficient methods for the generation of pyrrole cores with unusual substitution patterns e.g. 2,4-disubstituted pyrroles is particularly interesting. This review gives an overview of new developments in the synthesis of highly substituted pyrroles from N-propargylamines in recent years.
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from Tabriz University, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his Ph.D. degree in 2009 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as Associate Professor. His research interests include Theoretical Organic Chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
The pyrrole compound is not only prevalent in a wide variety of important classes of natural products1 and synthetic pharmaceuticals2 but also used as a building block in organic synthesis.3 Consequently, many efforts have been devoted to the design of expedient and efficient synthetic routes to this heterocycle. Some of the most popular methods for their preparation include the Knorr,4 Pall-Knorr,5 Hantzsch,6 aza-Wittig,7 metal-catalyzed cross-coupling,8 and especially multicomponent reactions.9 Widespread use of these methods is limited by the requirement of expensive metal catalysts, by the production of harmful waste streams, or both.The N-propargylamine motifs are privileged scaffolds in chemistry due to their presence in a large number of natural and unnatural compounds with important properties, both in pharmacology and materials science.10 Overall, they are highly useful building blocks in organic synthesis and have been abundantly used as precursors in the synthesis of heterocyclic compounds11 and complex natural products.12 In this regard, the synthesis of pyrrole cores from N-propargylamines have undergone an explosive growth in recent years. Synthesis of pyrroles from N-propargylamines provides a novel avenue to titled compounds that in the most cases, have many advantages over more conventional methodologies, which can be summarized as follows:
(1) Nontoxic by-products.
(2) High atom economy.
(3) Ease of handling.
(4) Environmentally friendly processes.
(5) High yielding, wide in scope.
(6) Mild synthetic route for compounds with unusual substitution patterns (e.g. 2,4-disubstituted pyrroles).
To the best of our awareness, a comprehensive review has not appeared on synthesis of pyrroles from N-propargylamines in literature so far. In this review, we have classified these reactions based on the type (e.g. intra- and intermolecular reactions) and the starting materials (e.g. cyclization of N-vinylpropargylamines, N-allylpropargylamines, and N-propargylpropargylamine). The main methods for synthesis of titled compounds from N-propargylamines are summarized in Fig. 1.
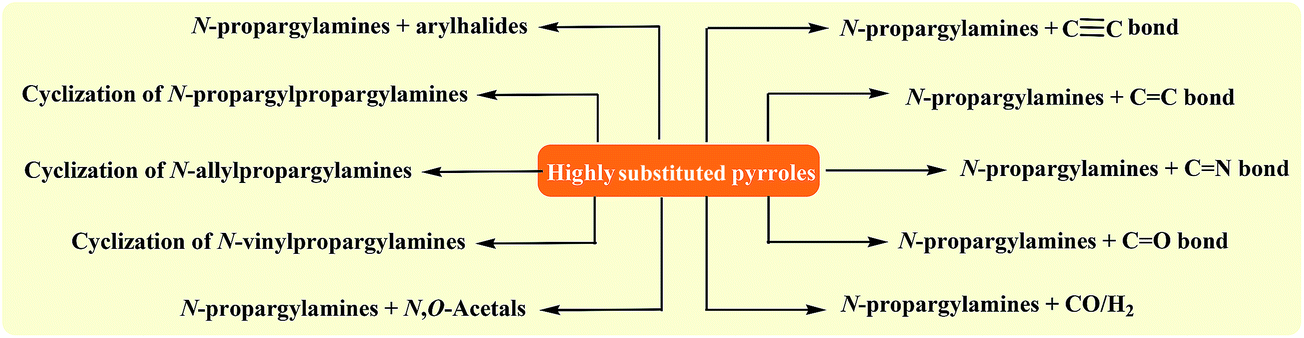 | ||
| Fig. 1 The main approaches for synthesis of highly substituted pyrroles from N-propargylamines. | ||
2. Synthesis of highly substituted pyrroles from N-propargylamines via intermolecular reactions
2.1. From N-propargylamines and carbonyl compounds
In 1988, Tsuda, and co-workers reported an example of cycloaddition of N-propargylamines with aldehydes. They showed that N-(pent-2-ynyl)-N-propylpent-2-yn-1-amine 1 underwent a cyclization reaction with benzaldehyde in the presence of Ni(COD)2 as catalyst, PPh3 as ligand in THF at 120 °C. The desired product 3 was obtained in excellent yield of 97%. They probed the mechanism of the reaction and found that the reaction proceeded by generation of a 1,2 bis(alkylidene)cycloalkane intermediate B, followed by cycloisomerization of the resulting dihydropyrrole C from rearrangement of B to corresponding pyrrole 3 (Fig. 2).13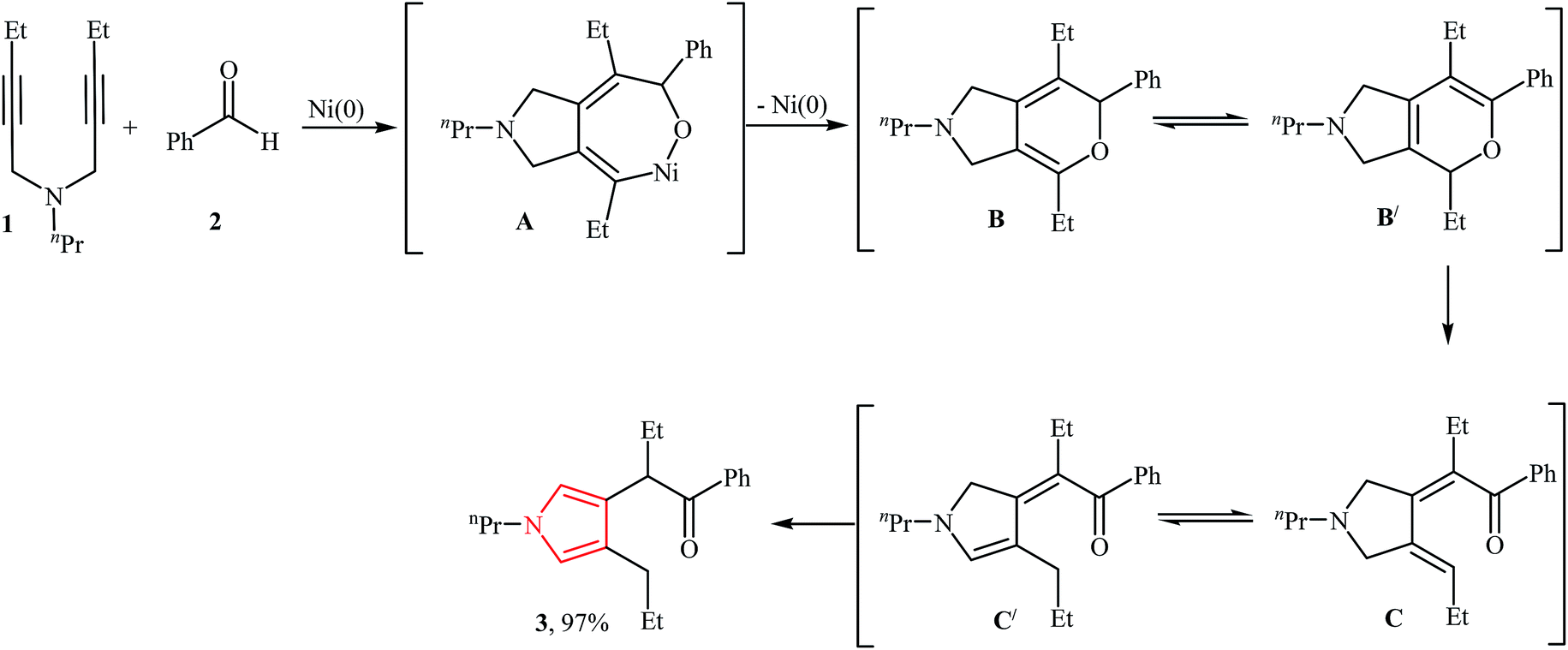 | ||
| Fig. 2 Proposed mechanism for synthesis of pyrrole 3 via cycloaddition of N-propargylamine 1 with aldehyde 2. | ||
Comprehensive synthesis of a diverse collection of highly substituted pyrroles 6 from treatment of N-propargylamines 4 with aldehydes 5 was reported by Bremner and Organ. The reaction was undertaken at 200 °C under microwave irradiation in presence of 4 Å molecular sieves in DMF. The reaction scope appears to be quite broad as alkyl, aryl, and hetaryl groups were tolerated at various substitution sites of both reaction components and gave corresponding pyrroles 6 in good yields (Scheme 1a). According to the proposed mechanism, the reaction involves: (1) the condensation of 4 with 5 which results the intermediate enynamine A; (2) [3,3]-pericyclic rearrangement of A to form the imino-allene intermediate B; and (3) cyclization of B to afford pyrrole 6 (Scheme 1b).14
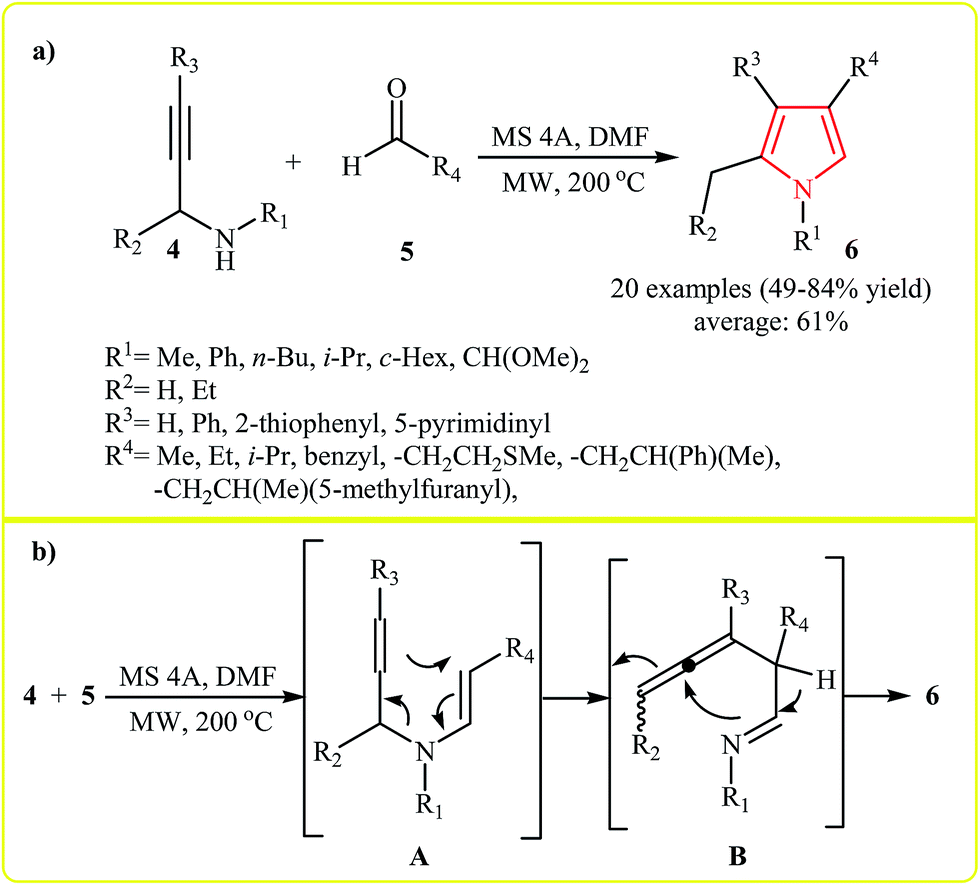 | ||
| Scheme 1 (a) Microwave-assisted synthesis of pyrroles 6 from treatment of N-propargylamines 4 with aldehydes 5. (b) Proposed mechanism for formation of 6. | ||
Müller's group described the synthesis of 2-substituted N-Boc-4-iodopyrroles 9 via an efficient Pd/Cu-catalyzed one-pot three component reaction of acid chlorides 7, N-Boc-protected propargylamine 8, and NaI. The reaction starts with the formation of an alkynone intermediate A via Sonogashira cross-coupling reaction of 7 with 8, and then the addition-cyclocondensation of A with NaI furnishes corresponding pyrroles in good yields (Table 1). The authors extended the applicability of this protocol for the synthesis of 4-alkynyl-N-Boc-pyrroles 11 by addition of another terminal alkyne 10 to the reaction mixture (Scheme 2).15
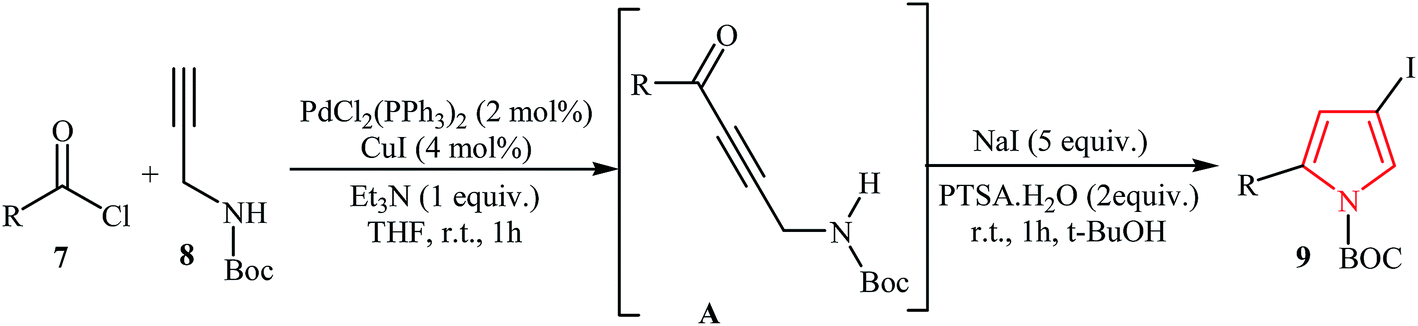
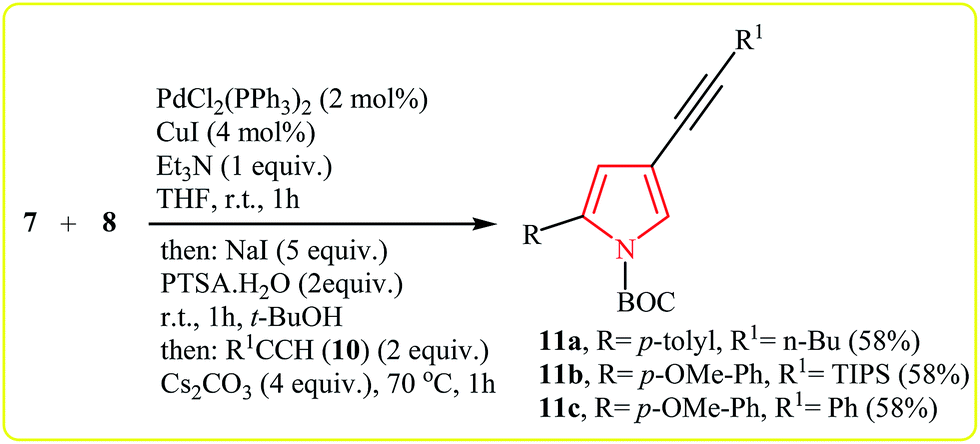 | ||
| Scheme 2 Coupling–addition–cyclocondensation–coupling sequence to 4-alkynyl-N-Boc-pyrroles 11. | ||
2.2. From N-propargylamines and C–C double or triple bond
An interesting approach toward the synthesis of highly substituted pyrroles via cycloaddition of N-propargylamines and C–C double bonds was developed by Zhao et al. (Scheme 3a and b). Thus, a variety of trisubstituted pyrroles 14 were synthesized via the base catalyzed [2 + 3]-cycloaddition of propargylamines 12 and α-acylketene dithioacetals 13 in DMF (Scheme 3a). According to the proposed mechanism, the reaction starts with intramolecular Michael addition of 12 to 13 to form the intermediates A and B. Subsequently the 5-exo-dig cyclization of B-isomer give intermediate C that undergoes a sequential deacetylation and aromatization to afford expected pyrroles 14 (Scheme 3b).16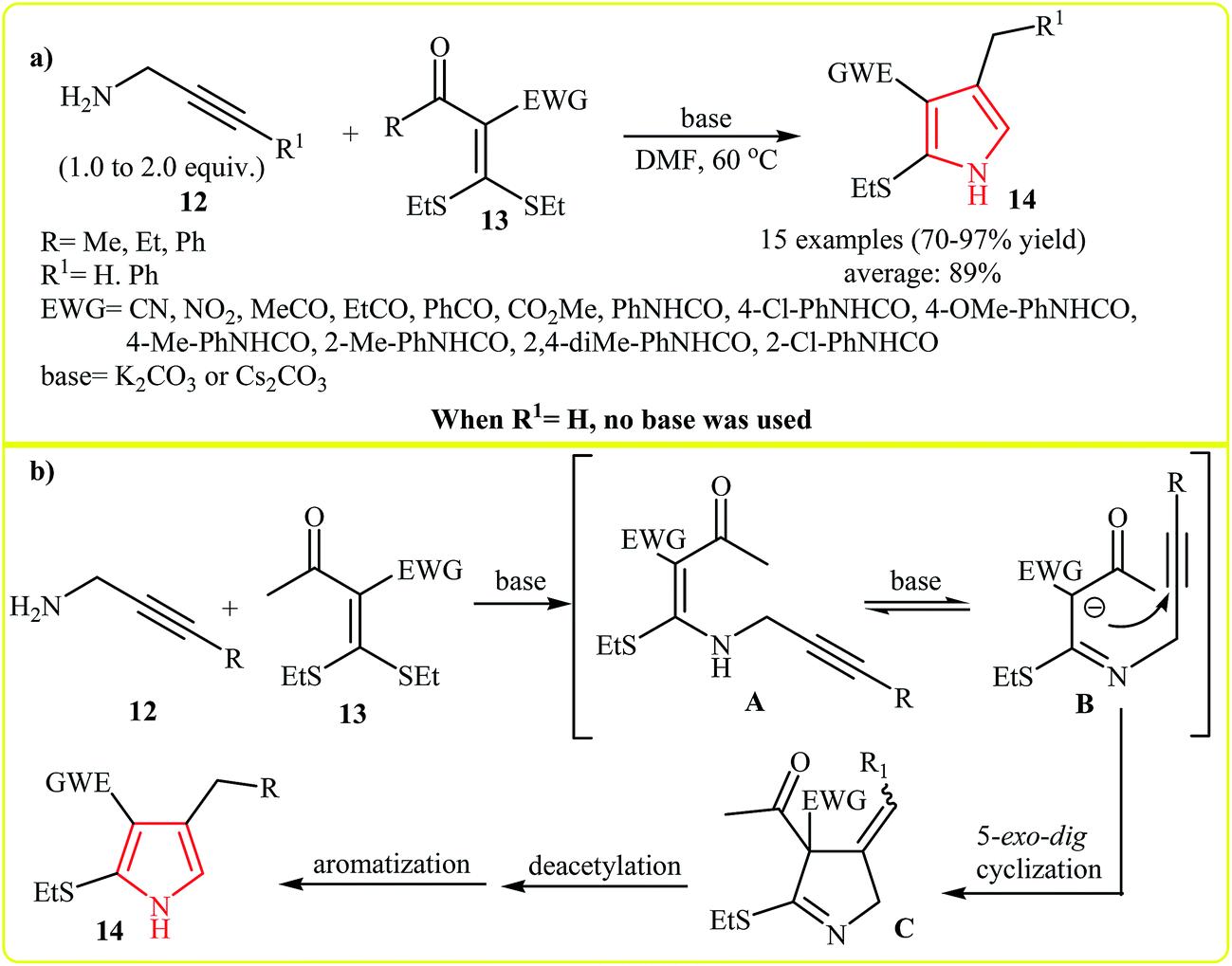 | ||
| Scheme 3 (a) [3 + 2] cycloaddition of propargylamines 12 and α-acylketene dithioacetals 13 to pyrroles 14. (b) Proposed mechanism for formation of 14. | ||
Shortly after, the same group expanded this methodology to the synthesis of 1,2,3,4-tetrasubstituted pyrroles 17, 18 by cycloaddition of acetyl ketene dithioacetals 15 and secondary propargylamine (N-methylprop-2-yn-1-amine) 16 in water. Interestingly, this protocol showed different reaction behaviors depending on the addition or absence of an external base. In the presence and absence of an external base, the reaction gave the 1,2,3,4-tetrasubstituted pyrroles bearing a acetyl group and ethylthio group at the C2 of the pyrrole core, respectively (Scheme 4).17 A plausible mechanisms for formation of 17 and 18 is depicted in Scheme 5.
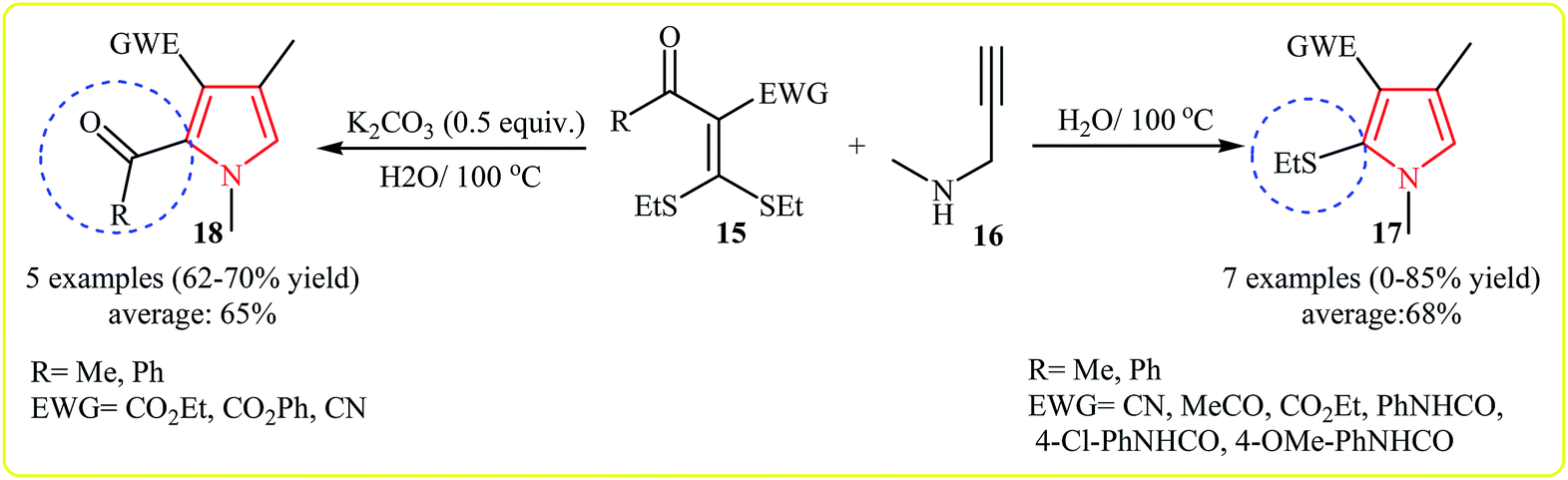 | ||
| Scheme 4 Synthesis of 1,2,3,4-tetrasubstituted pyrroles by cycloaddition of N-methylprop-2-yn-1-amine and acetyl ketene dithioacetals. | ||
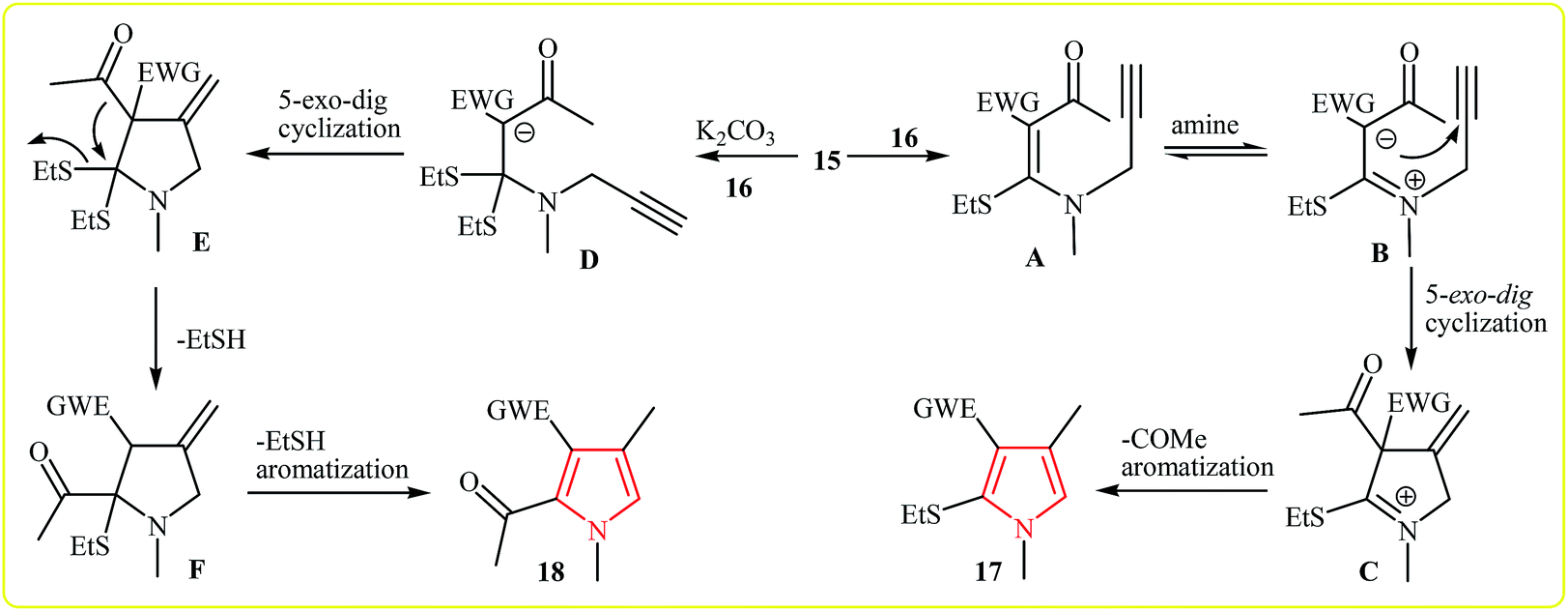 | ||
| Scheme 5 Proposed mechanisms for formation of 3 and 4. | ||
Very recently, Castagnolo and co-workers reported an interesting method for synthesis of 1,2,3-substituted pyrroles 20 via enyne cross metathesis of propargylamines 19 with ethyl-vinyl ether in the presence of Grubbs' catalyst under microwave irradiation. It is noted that the presence of CuSO4 as promoter is vital for this reaction. Under optimized condition, the reaction worked well with both alkyl and aryl substituted amines. However, the scope of the reaction is limited to electron-poor amines and sterically hindered N-propargylamine failed to react under aforementioned conditions (Scheme 6).18
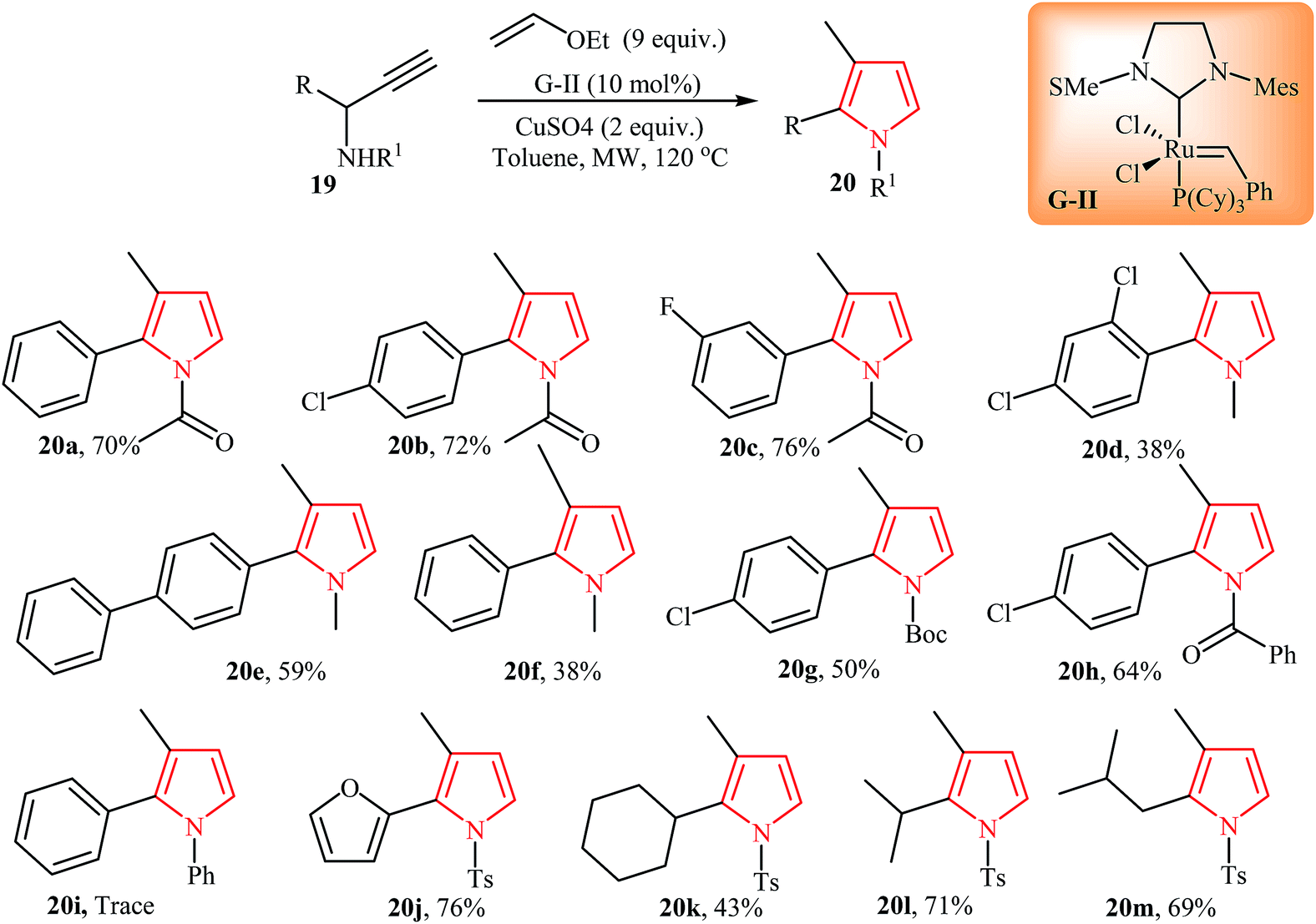 | ||
| Scheme 6 Synthesis of 1,2,3-substituted pyrroles 20 from propargylamines 19. | ||
In 2011, Trost et al. developed the synthesis of pyrroles 23 by Pd(II)-catalyzed cascade reaction of N-propargylamines and alkynes. Thus, the reaction of tert-butyl 3-(methoxycarbonyl)prop-2-ynylcarbamate 21 and alkynes 22 furnishes 2,4-disubstituted pyrroles 23 in good to excellent yields (Scheme 7).19 The reaction proceeds via addition of alkyne to 21 followed by a 5-endo-dig-cyclization and tautomerization of the ynenoate intermediate into pyrrole 23. It is interesting to note that the electronic character of the substituents in the alkynes had little effect on the facility of reaction. Generally, all of electron-rich, electron-poor and branched alkynes can efficiently react under optimized condition. Furthermore, the reaction is tolerant toward a wide variety of functional groups such as amino, hydroxyl, carbonyl, alkoxide and halogens that can undergo in further reaction to produce unique pyrrole scaffolds.
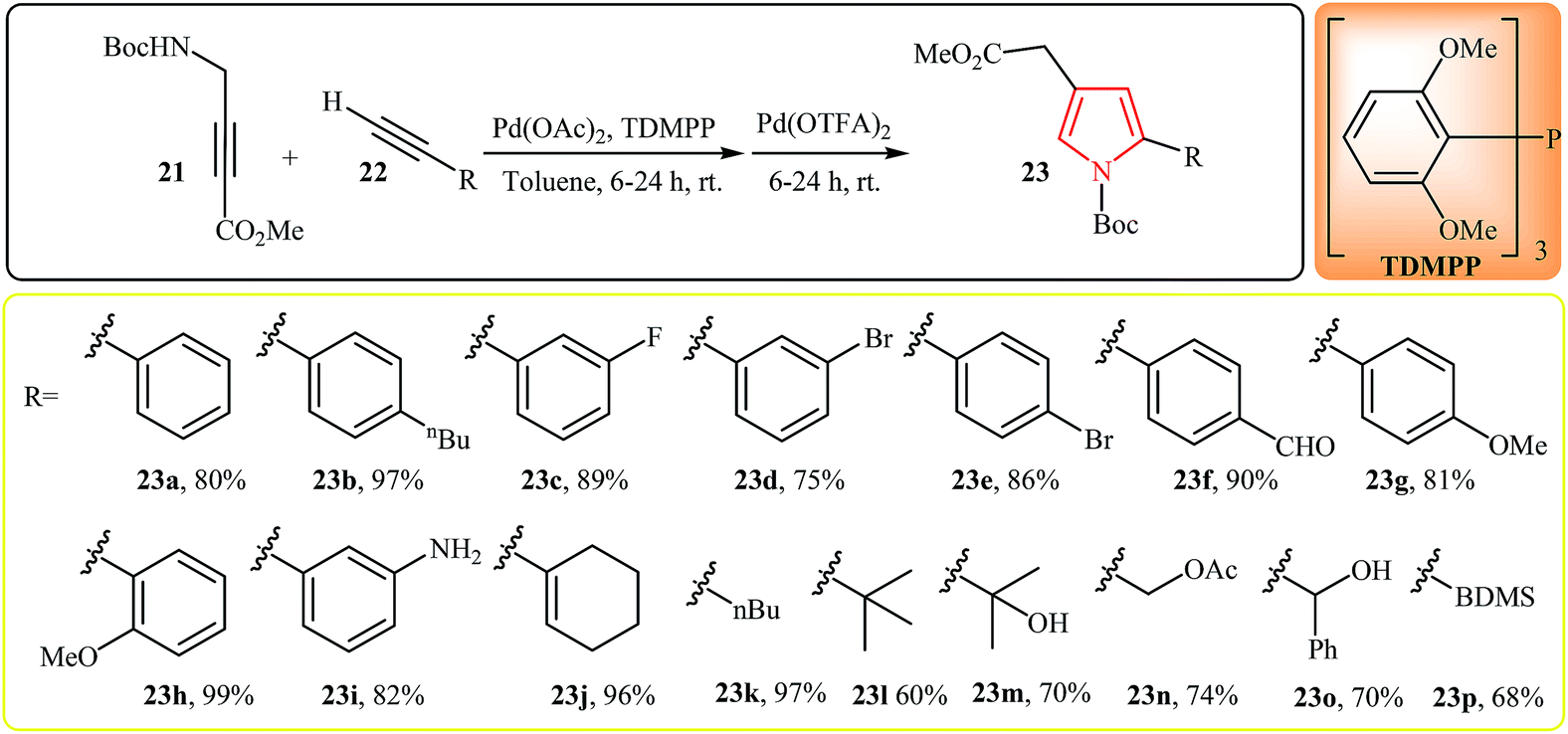 | ||
| Scheme 7 One-pot synthesis of pyrroles 23 via Pd(II)-catalyzed cascade reaction of N-propargylamine 21 and alkynes 22. | ||
Very recently, an efficient transition-metal-free reaction between activated alkynes 24 with primary and secondary N-propargylamines 25 that leads to polysubstituted (tri-, tetra-, and penta-substituted) pyrroles 26 using K3PO4 as catalyst in DMSO was reported by Jin et al. (Scheme 8). Interestingly, when the base was changed to CsF the reaction of 24 (with R1 = Me) with N-propargylamines 25′ via a Michael addition/aza-Claisen rearrangement/cyclization sequential process afforded pyrroles 27 as desired product with a different substituent pattern (Table 2).20
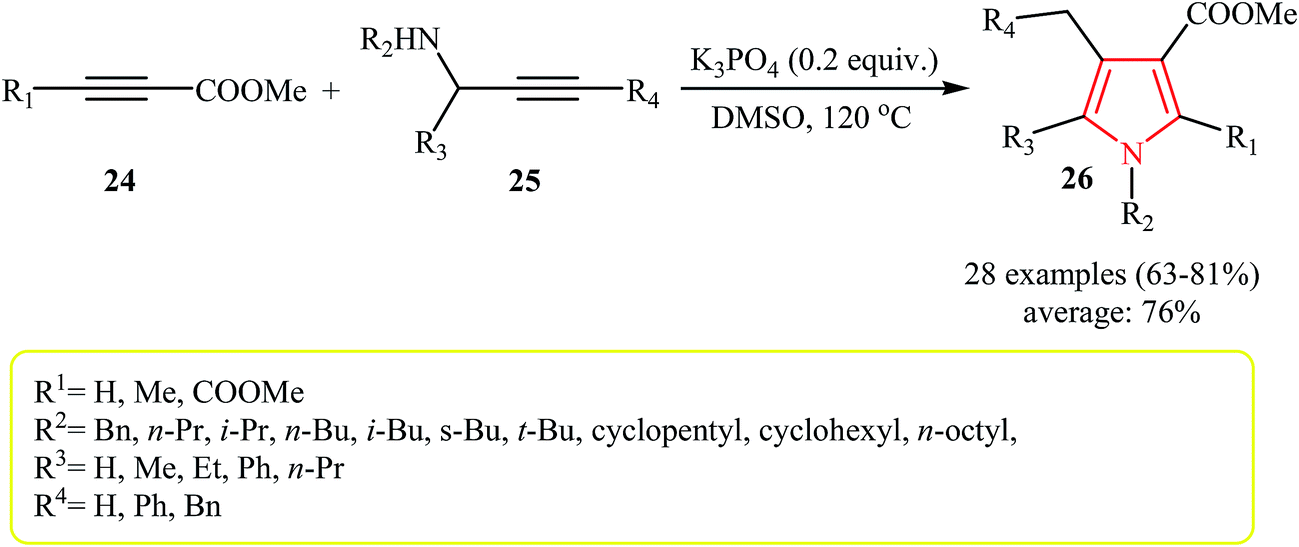 | ||
| Scheme 8 Cascade synthesis of polysubstituted pyrroles 26. | ||
|
||||
|---|---|---|---|---|
| Entry | R2 | R3 | Yield (%) | |
| 26 | 27 | |||
| 1 | Bn | H | 13 | 52 |
| 2 | n-Bu | H | 20 | 40 |
| 3 | i-Bu | H | 21 | 42 |
| 4 | n-Octyl | H | 20 | 40 |
| 5 | Bn | Me | 12 | 48 |
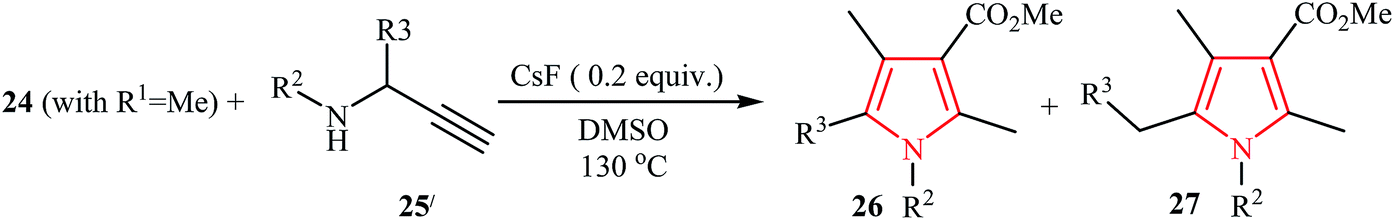
2.3. From hydroformylation of N-propargylamines
The hydroformylation reaction is the simultaneous addition of one mole each of hydrogen and carbon monoxide to a carbon–carbon double or triple bond by transition-metal catalyst, to produce two new C–C and C–H bonds.21 Tons of chemicals are produced every year via this transformation and the production capacities is growing day by day.22 In 1991, Campi and co-workers reported a different application for this reaction, when the propargylamines 28 underwent a hydroformylation and then cyclization reaction with CO/H2 in the presence of [Rh(OAC)2]2/PPh3 as catalytic system to form β-arylpyrroles 29 in good to high yields (Scheme 9). However, the reaction does not work well with alkylpropargylamines, due to the formation of significant amounts of furan-2-ones as side products (18–23%). Mechanistically, this transformation involves carbonyl reduction and removal of the amine function by hydrogenolysis.23 To the best of our awareness this is the only example of N-propargylamine hydroformylation reported so far.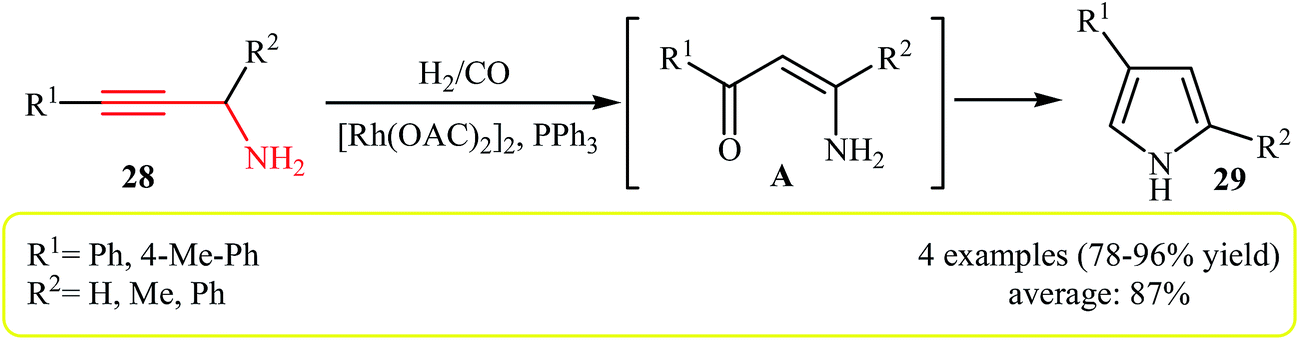 | ||
| Scheme 9 Synthesis of 2,4-disubstituent pyrroles 29 via hydroformylation of N-propargylamines 28. | ||
2.4. Miscellaneous
In 2010, Meng, Hu and Wang developed the synthesis of 1,4,5-trisubstituted pyrroles 32 via Pd(II)-catalyzed coupling/cycloisomerization of N-allyl-4-methyl-N-(3-phenylprop-2-ynyl)benzenesulfonamide 30 and bromobenzenes 31, using PPh3 as ligand, (n-Bu)3N as base and DMF as solvent at 140 °C. The electronic character of the aryl halides had remarkably strong effect on the reaction. The reaction tolerates electron-donating substituents at meta and para positions of aryl moiety and gave corresponding coupling products in good to high yields, but extension of the reaction to electron-withdrawing aryl rings was failed (Scheme 10). Interestingly, when substituent (Ph) at the terminus of the alkynes was changed to methyl, instead of pyrroles, the reaction afforded nonaromatic heterocycle derivatives 34 in moderate to good yields (8 examples with average yield of 65%). The authors proposed the below mechanism for this reactions (Scheme 11).24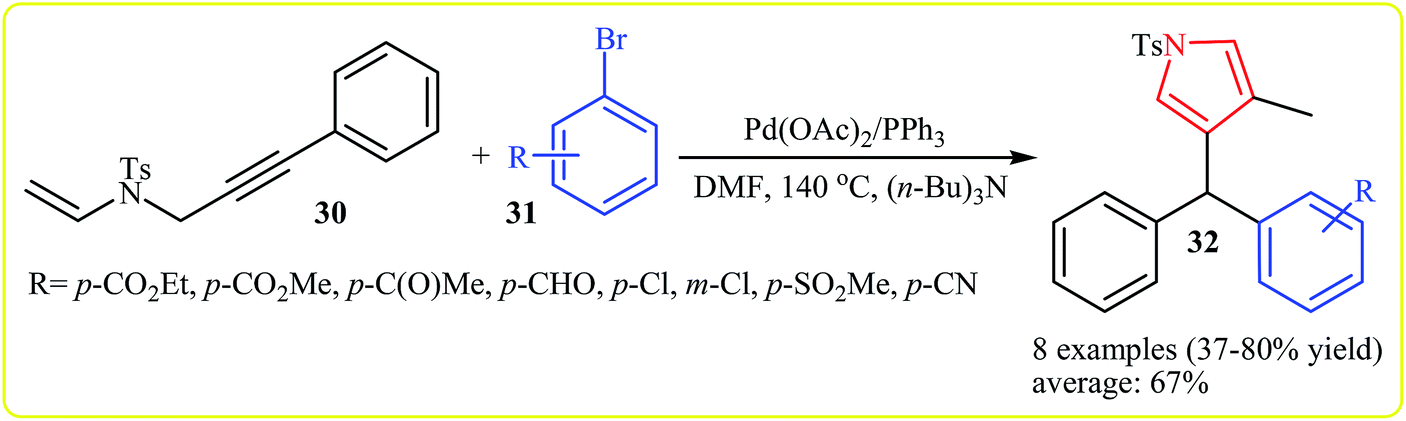 | ||
| Scheme 10 Synthesis of trisubstituted pyrroles 32 from 30 and aryl halides 31. | ||
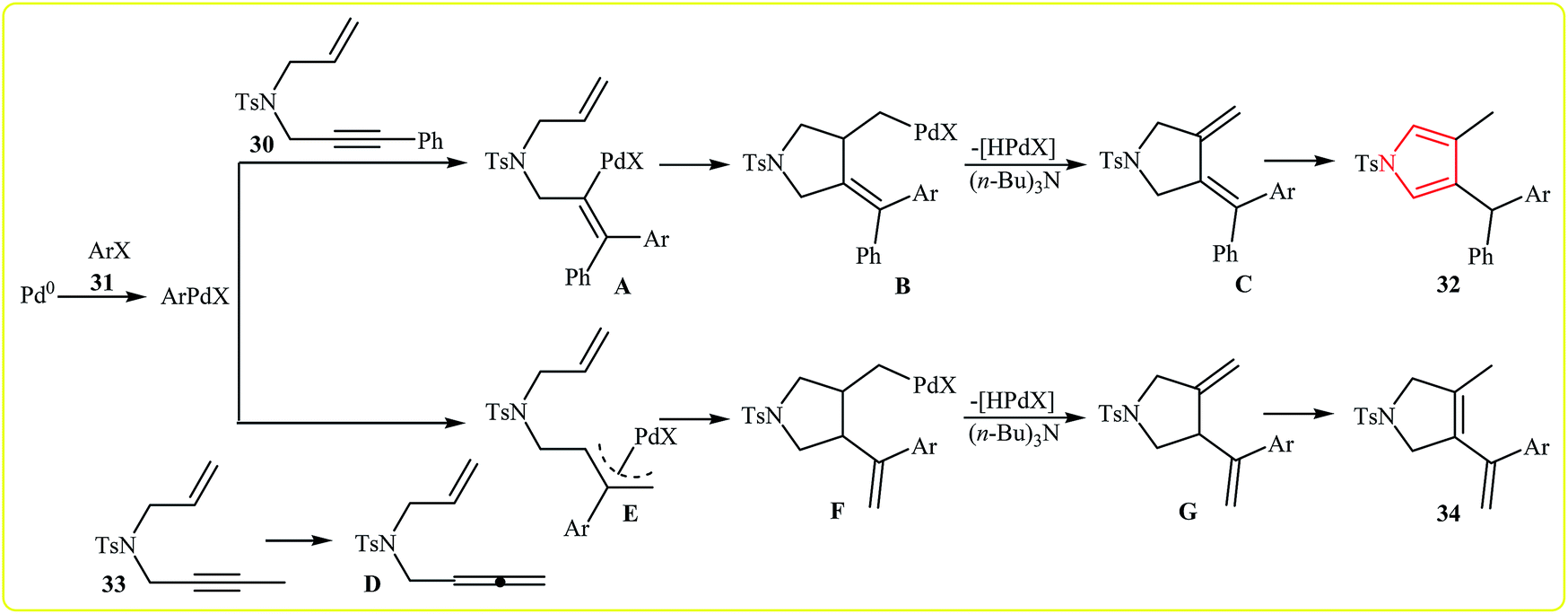 | ||
| Scheme 11 Proposed mechanistic pathways for the formation of different heterocyclic compounds 32 and 34. | ||
An efficient synthesis of derivatives of the 1,2,3,5-tetraaryl pyrrole scaffold has been developed by Wan et al. The treatment of imines 35 with N-propargylamines 36 in presence of bis(trimethylsilyl)amide (LiHMDS) as base and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA) as the additive in THF, was found to afford pyrroles 37. A broad scope of substituted imines, regardless of the electronic effects and the position of the substituents, and a variety of N-propargylamines, such as aryl and heteroarylated propargylamines could efficiently be employed in this reaction (Scheme 12).25
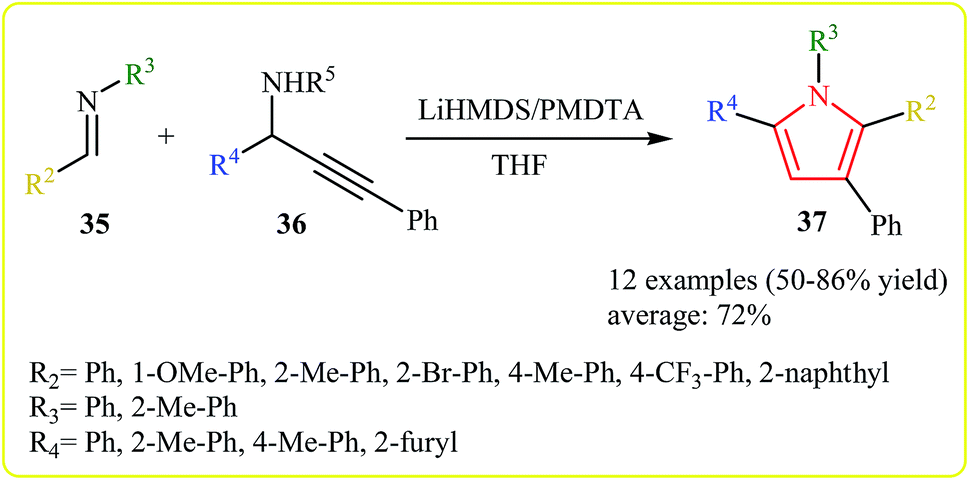 | ||
| Scheme 12 Synthesis of pyrrole 37 via treatment of imines 35 with N-propargylamines 36. | ||
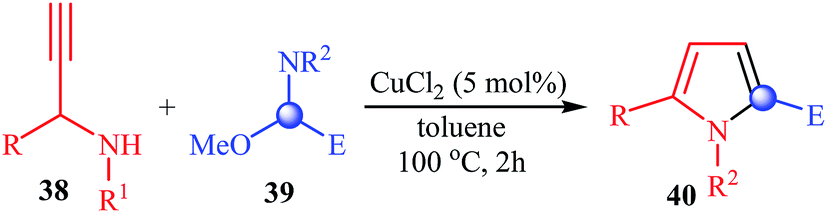
More recently, Sakai and co-workers reported a very beautiful example of a copper-catalyzed synthesis of substituted pyrroles 40 from N-propargylamines 38 and N,O-acetals 39. Conceptually, the reaction is based on the [4 + 1] annulation that N,O-acetals function as a C1 unit. The reaction tolerates both primary and secondary propargylamines and a variety of N,O-acetals that have an enolizable substituent adjacent to the central sp3-carbon (Table 3).26
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | R1 | R2 | E | Yield (%) | Entry | R | R1 | R2 | E | Yield (%) |
| a CM: complex mixture.b ND: not determined. | |||||||||||
| 1 | H | 2-Me–Ph | –(CH2)5– | CO2Me | 86 | 13 | H | H | –(CH2)5– | CO2Me | NDb |
| 2 | H | 3-Me–Ph | –(CH2)5– | CO2Me | 80 | 14 | H | Ph | Et | CO2Me | 85 |
| 3 | H | 4-Me–Ph | –(CH2)5– | CO2Me | 80 | 15 | H | Ph | –(CH2)5– | COPh | 43 |
| 4 | H | 4-NMe2–Ph | –(CH2)5– | CO2Me | 81 | 16 | H | Ph | –(CH2)5– | CONR2 | 58 |
| 5 | H | 4-OMe–Ph | –(CH2)5– | CO2Me | 90 | 17 | H | Ph | –(CH2)5– | Ph | NDb |
| 6 | H | 2-OH–Ph | –(CH2)5– | CO2Me | CMa | 18 | H | Ph | –(CH2)5– | H | NDb |
| 7 | H | 4-F–Ph | –(CH2)5– | CO2Me | 81 | 19 | (CH2)2Ph | Ph | –(CH2)5– | CO2Me | 70 |
| 8 | H | 4-Cl–Ph | –(CH2)5– | CO2Me | 87 | 20 | c-Hexyl | Ph | –(CH2)5– | CO2Me | 60 |
| 9 | H | 4-Br–Ph | –(CH2)5– | CO2Me | 83 | 21 | Ph | Ph | –(CH2)5– | CO2Me | 90 |
| 10 | H | 4-CF3–Ph | –(CH2)5– | CO2Me | 84 | 22 | 4-Me–Ph | Ph | –(CH2)5– | CO2Me | 91 |
| 11 | H | 4-Ac–Ph | –(CH2)5– | CO2Me | 77 | 23 | 4-Cl–Ph | Ph | –(CH2)5– | CO2Me | 77 |
| 12 | H | 4-CN–Ph | –(CH2)5– | CO2Me | 70 | 24 | H | t-Bu | –(CH2)5– | CO2Me | 31 |
3. Synthesis of highly substituted pyrroles from N-propargylamines via intramolecular reactions
3.1. From N-vinylpropargylamines
The thermal rearrangement of N-vinylpropargylamines into pyrroles was first introduced by Cossy and co-workers in 1996. It was demonstrated that various annulated[b]pyrroles 42 could be prepared in moderate to good yields via a tandem aza-Claisen rearrangement–cyclization reaction of N-vinylpropargylamines 41 (Scheme 13).27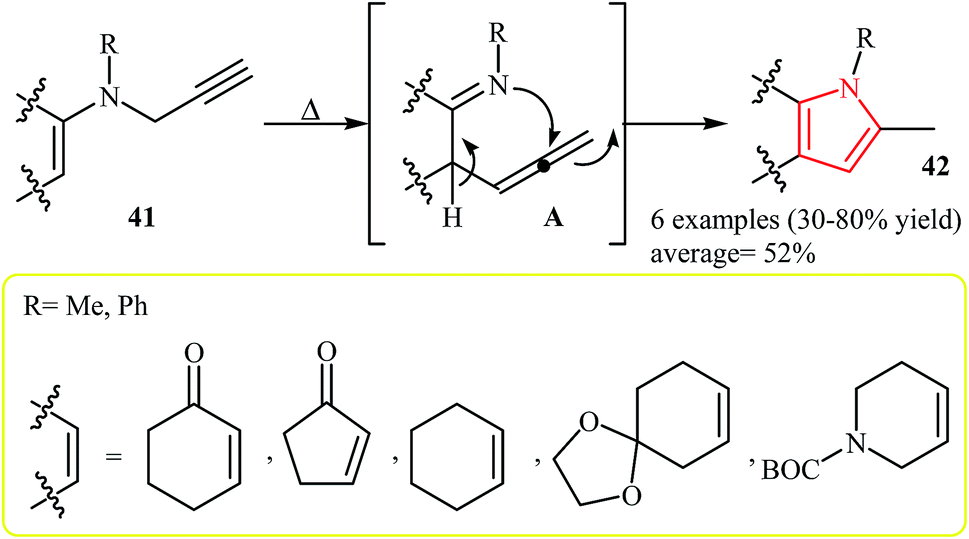 | ||
| Scheme 13 The thermal rearrangement of N-vinylpropargylamines 41 into pyrroles 42. | ||
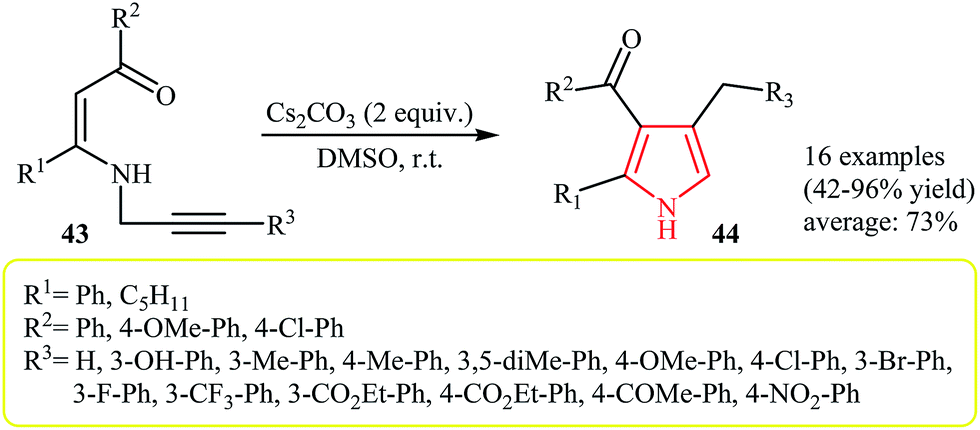
Later, in 2008, Cacchi and co-workers extended this chemistry to an intramolecular cyclization–protonation–isomerization cascade of N-vinylpropargylamines 43 to N–H free 2,3,4-trisubstituted pyrroles 44 using Cs2CO3 as catalyst in anhydrous DMSO at room temperature (Scheme 14).28 The scope of the Au-catalyzed version of this reaction was investigated by Saito et al. Thus, it was shown that fully substituted pyrroles possessing an ester functional group at C-3 position could efficiently be synthesized from N-vinylpropargylamines using the [(IP)Au(MeCN)]BF4/HFIP system at room temperature.29
 | ||
| Scheme 14 Synthesis of NH free polysubstituted pyrroles 44 from 43. | ||
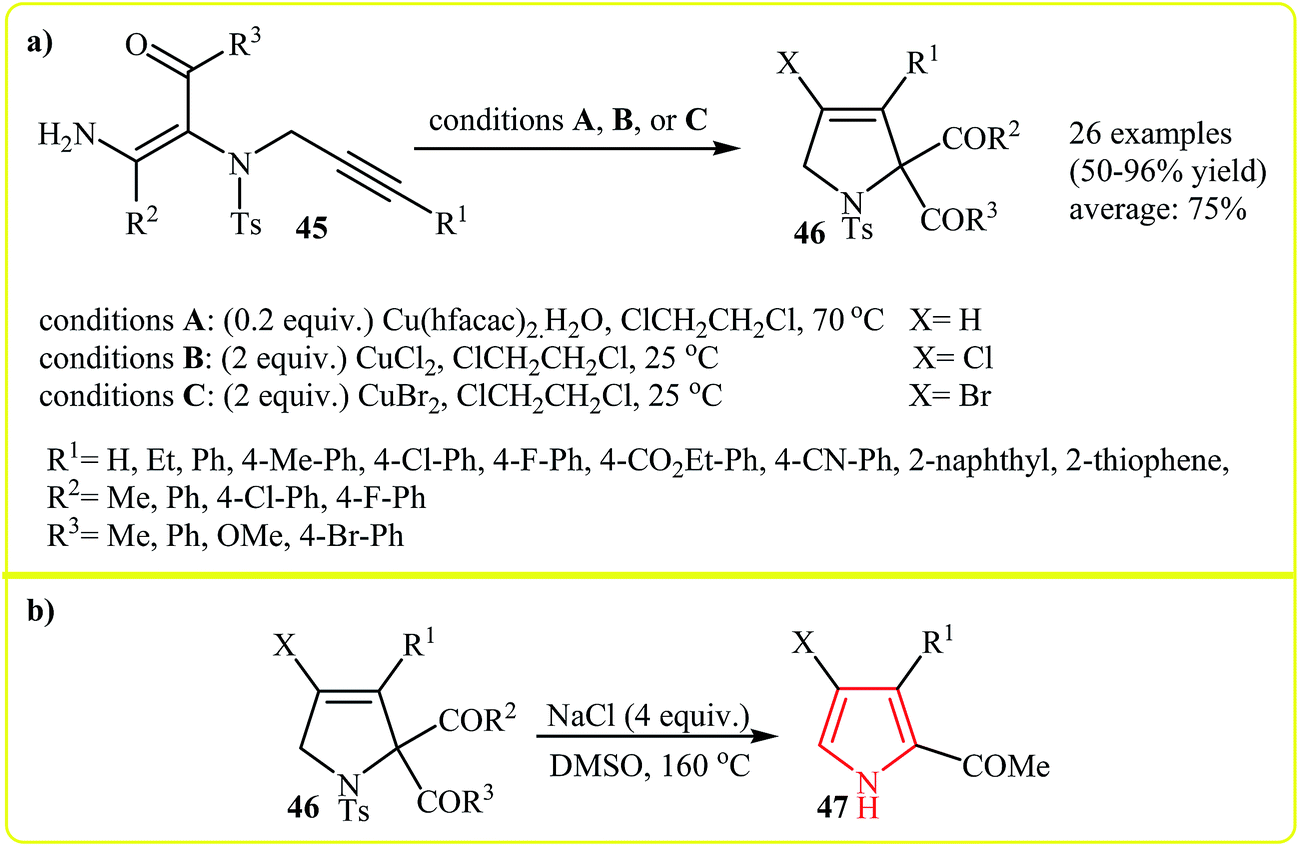
Along this line, very recently Wang and co-workers reported the copper(II)-mediated electrophilic cyclization transformation of N-protected N-vinylpropargylamines 45 into highly substituted 3-pyrrolines 46 (Scheme 15a)30 which can be easily converted to trisubstituted pyrroles 47 by treatment with sodium chloride (Scheme 15b).31
 | ||
| Scheme 15 (a) Construction of highly substituted 3-pyrrolines 46 from N-protected N-vinylpropargylamines 45, (b) convert of 46 into trisubstituted pyrroles 47. | ||
3.2. From N-allylpropargylamines
In 2005, Yamamoto and co-workers developed the synthesis of polycyclic pyrrole-2-carboxylates 54 from acetylenes 48, ethyl glyoxylate 49, benzylallylamine 50, and activated alkenes 53 via a semi one-pot Mannich reaction/isomerization/Diels–Alder reaction/dehydrogenative aromatization sequence. Thus, at the first step, the Cu-catalyzed three-component (acetylenes 48, ethyl glyoxylate 49, benzylallylamine 50) Mannich reaction gives N-allylpropargylamine 51, which undergoes an Ir-catalyzed cycloisomerization into diene 52. The formed diene 52 is converted to pyrrole 54 upon a subsequent Diels–Alder reaction with dienophile 53, followed by a dehydrogenative aromatization (Scheme 16).32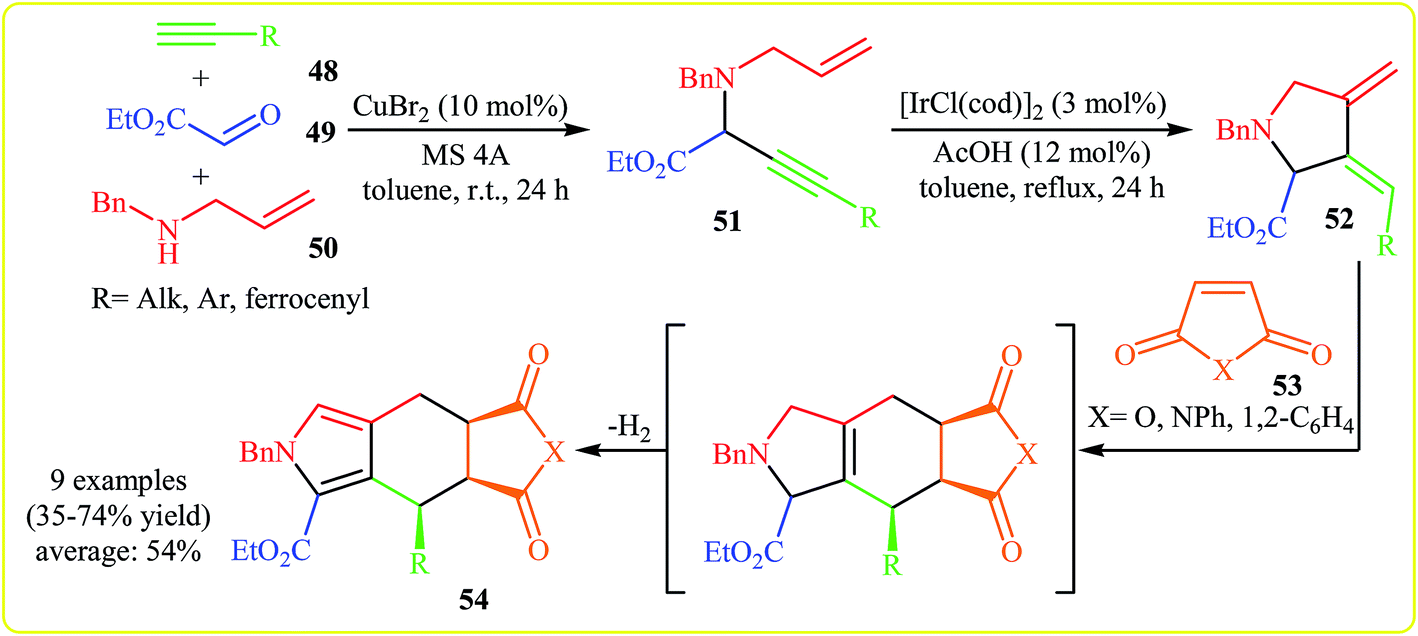 | ||
| Scheme 16 Synthesis of polycyclic pyrrole-2-carboxylates 54 via a transition metal-catalyzed four-component coupling approach. | ||
Follow this work, the Stevens group suggested the RCM/oxidation protocol for synthesis of 2-phosphono pyrroles 57 from the corresponding N-allylpropargylamine 55 using second-generation Grubbs catalyst 56 and tetrachloro-1,4-benzoquinone (TCQ) as an oxidant in benzene under reflux. However, it is limited to substrates bearing small substituents on C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (Scheme 17).33
C bond (Scheme 17).33
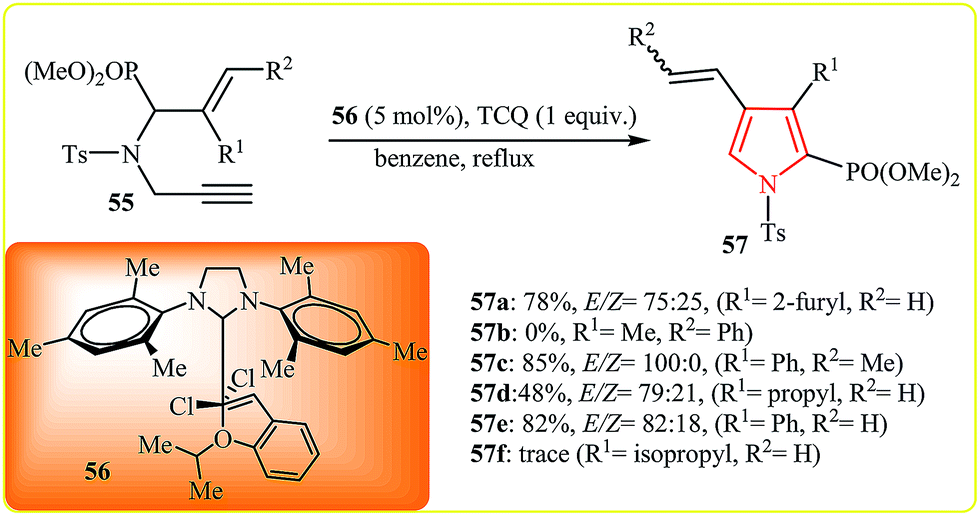 | ||
| Scheme 17 Metathesis–oxidation sequence for the synthesis of 2-phosphono pyrroles 57 from N-allylpropargylamine 55. | ||
3.3. From N-propargylpropargylamines
Gleiter and Ritter developed an efficient Pd-catalyzed synthesis of N,N′-dialkyl-3,3′-bispyrroles 59 from N-propargylamines 58 in methanol at 140 °C (Scheme 18). According to the proposed mechanism, the reaction based on two allylic rearrangements and two dehydrogenation steps. It should be noted that the aforementioned temperature is vital for this reaction, because at lower temperatures than 140 °C the yield of pyrroles 59 is decreased in favor of the dihydro-60 and the tetrahydro-61 derivatives.34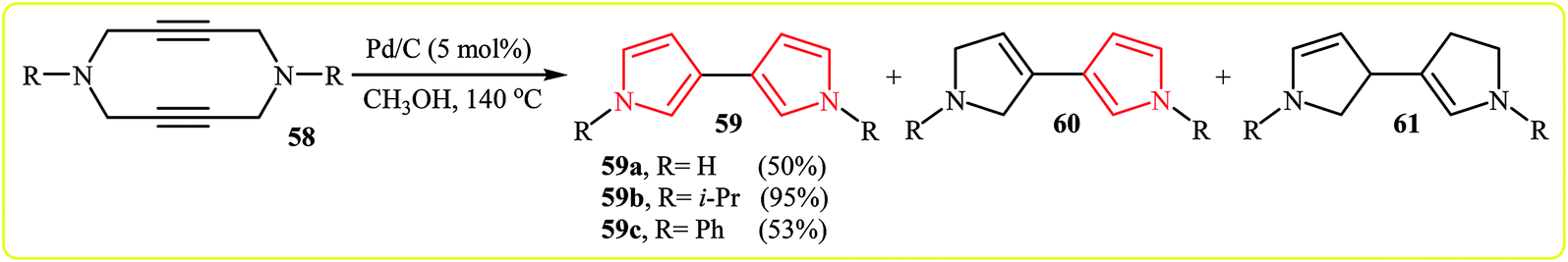 | ||
| Scheme 18 The Pd-catalyzed rearrangement of N-propargylamines 58. | ||
After this work, in 2007, Tanaka et al. reported that vinylpyrroles 64 could be prepared via the Rh-catalyzed cycloisomerization of the corresponding 1,6-diynes 62. The authors proposed the Rh-catalyzed mechanism shown in Fig. 3. First, Rh(I) complex A is formed by reaction between the cationic Rh(I)/Segphos complex and 1,2-cyclohexanedione 63. This intermediate reacts with the diyne 62 leading to hodacyclopentadiene intermediate B. Finally, the β-hydride elimination and double-bond isomerization of B affords the observed product.35
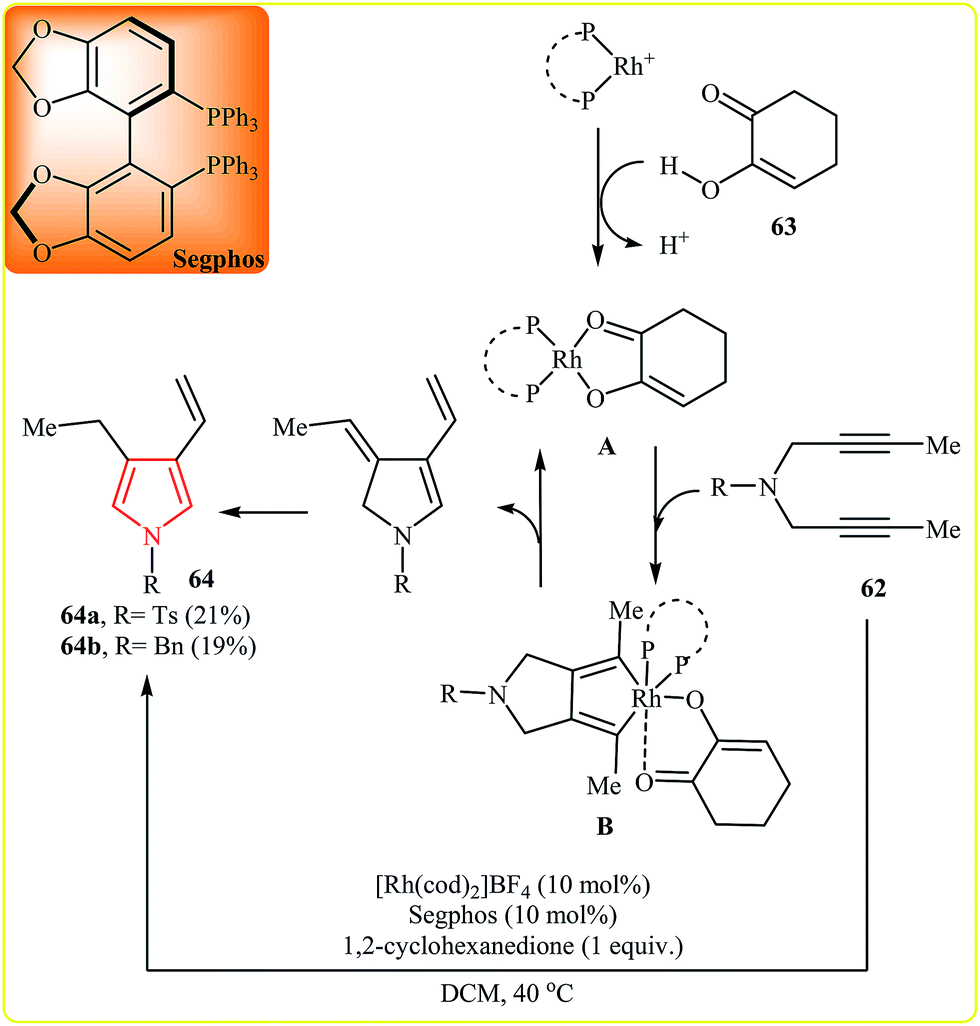 | ||
| Fig. 3 Rh-catalyzed synthesis of vinylpyrroles 64 from 1,6-diynes 62. | ||
3.4. Miscellaneous
An interesting and rare example for synthesis of bicyclic pyrroles was reported by Wuonola and Smallheer in 1993. Thus, in refluxing 1,3,5-triisopropylbenzene, an intramolecular Diels–Alder reaction of imidazolecarboxamide 65, between N-propargylamine motif and imidazole, afforded pyrrole 67 in yield of 70%. The mechanism proposed by the authors to explain this reaction is based on the formation of the isoquinoline system 66, followed by expulsion of a molecule of HCN (Scheme 19).36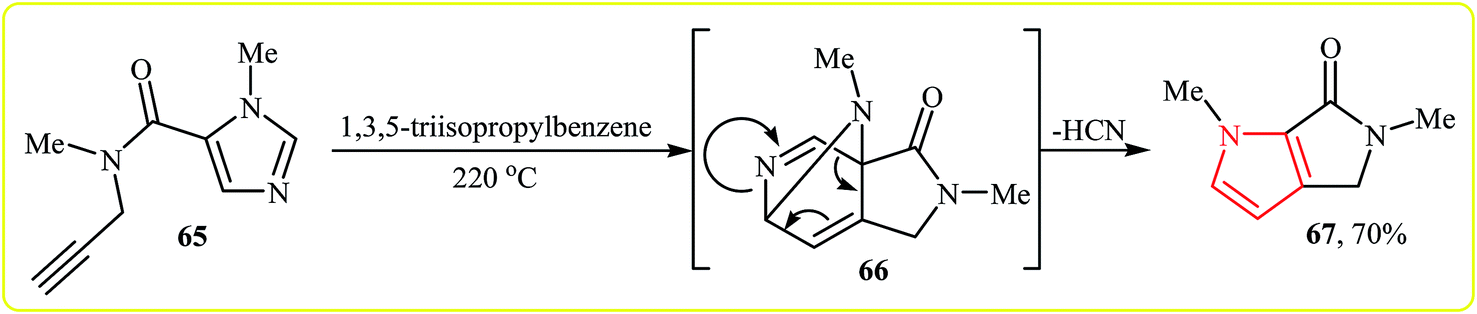 | ||
| Scheme 19 Synthesis of bicyclic pyrrole 67 by intramolecular Diels–Alder reaction to the imidazole nucleus 65. | ||
A process for the synthesis of N–H free trisubstituted pyrroles 74 involves the addition of arganocuprates 69 to a silylated propargylamine 68 to led vinyl cuprate 70 followed by addition of acid chlorides 71 to give intermediate 72. Finally, a cyclization–elimination sequence of 72 which resulted in the formation of the expected pyrroles 74 (Scheme 20). The (Me3)2SiN group play three successive roles in this one pot reaction: (1) it is a protected primary amino group allowing the cuprate reaction. (2) It directs the stereochemistry of the cuprate addition by stabilizing the trans-adduct via chelation. (3) It is reactive enough to cause cyclization upon nucleophilic attack at the cis-orientated carbonyl group.37
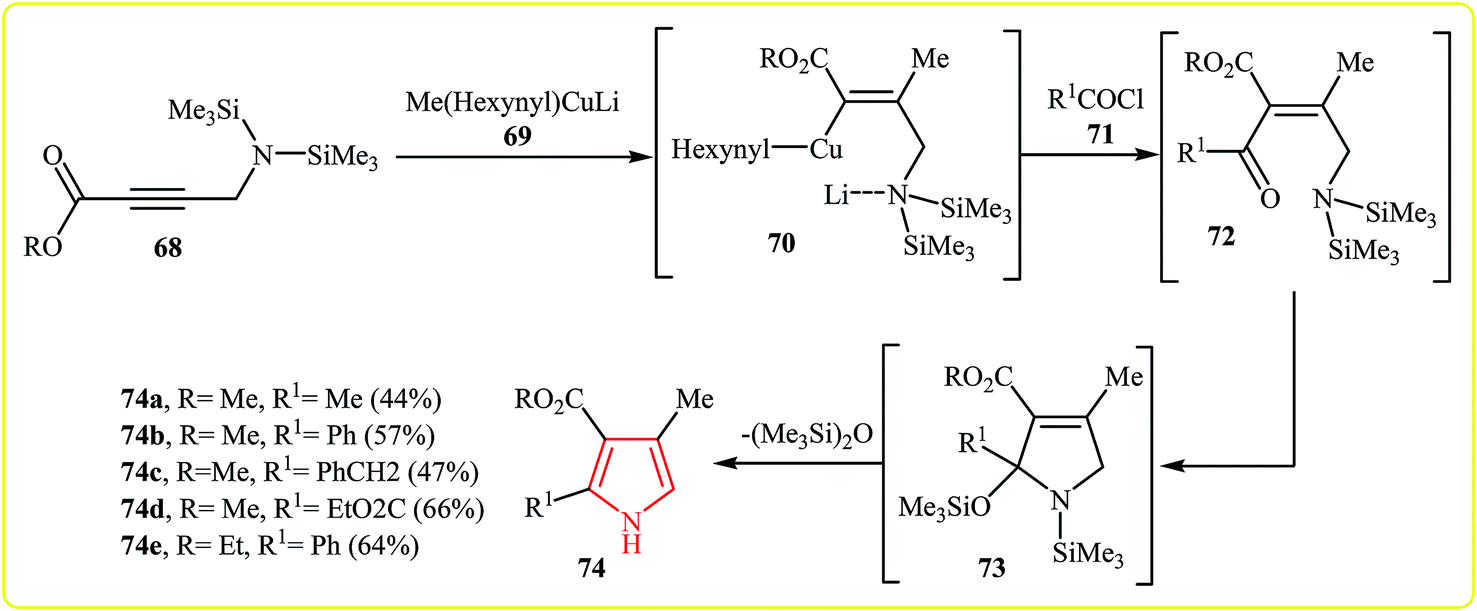 | ||
| Scheme 20 Synthesis of pyrroles 74 from silylated propargylamines 68. | ||
Lee and co-workers showed that Boc-protected furfuryl propargylamine 75 underwent a spontaneous intramolecular Diels–Alder reaction and then ring opening that afforded the bicyclic pyrrole 77 in yield of 63% in the presence of a base in t-BuOH (Scheme 21).38
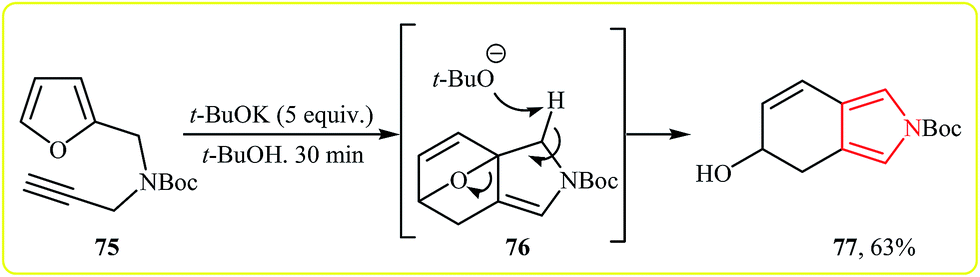 | ||
| Scheme 21 One-pot synthesis of bicyclic pyrrole 77 from furfuryl propargylamine 75. | ||
In 2009, Zhao and co-workers reported the synthesis of trisubstituted pyrroles 79 via the Au(III)-catalyzed hydroamination (5-endo-dig cyclization) of the corresponding amino-functionalized enynes 78. The reaction provides N-alkyl-, N-arylsulfonyl-, and carbamoyl and benzoyl-protected pyrroles 79 bearing a C2-aminomethyl group, in good yields (Scheme 22).39
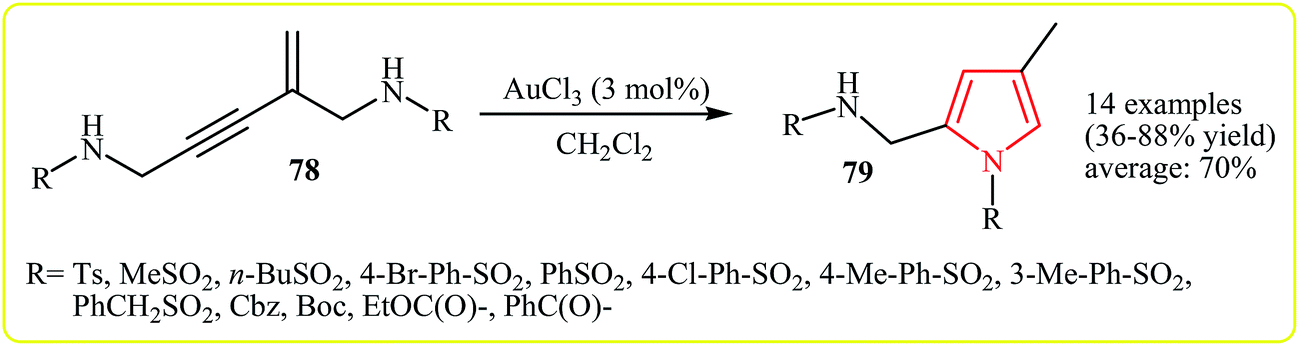 | ||
| Scheme 22 Synthesis of N-protected pyrroles 79 via the Au(III)-catalyzed hydroamination of amino-functionalized enynes 78. | ||
Recently, Yeh et al. introduced a sequential reaction for the synthesis of 2,3-disubstituted pyrroles 83 from 3,5,5-trimethyl-2,3-epoxycyclohexan-1-ones incorporating a (3-arylpropargyltosylamino)methyl tether at the C-2 position 80, beginning with activation of the oxirane by TMSOTf occurred to give the semipinacol rearrangement product 81, which upon intramolecular [2 + 2] cycloaddition and [2 + 2] cycloreversion (alkyne–ketone metathesis) steps furnished N-tosyldihydropyrrole derivative 82. A subsequent oxidation followed by basic treatment occurred to generate pyrrole 83 (Fig. 4).40 It is noted that the reaction tolerates electron-neutral and -rich substituents at aryl moiety and gave corresponding pyrroles in good yields, but it could not be extended to electron-poor rings.
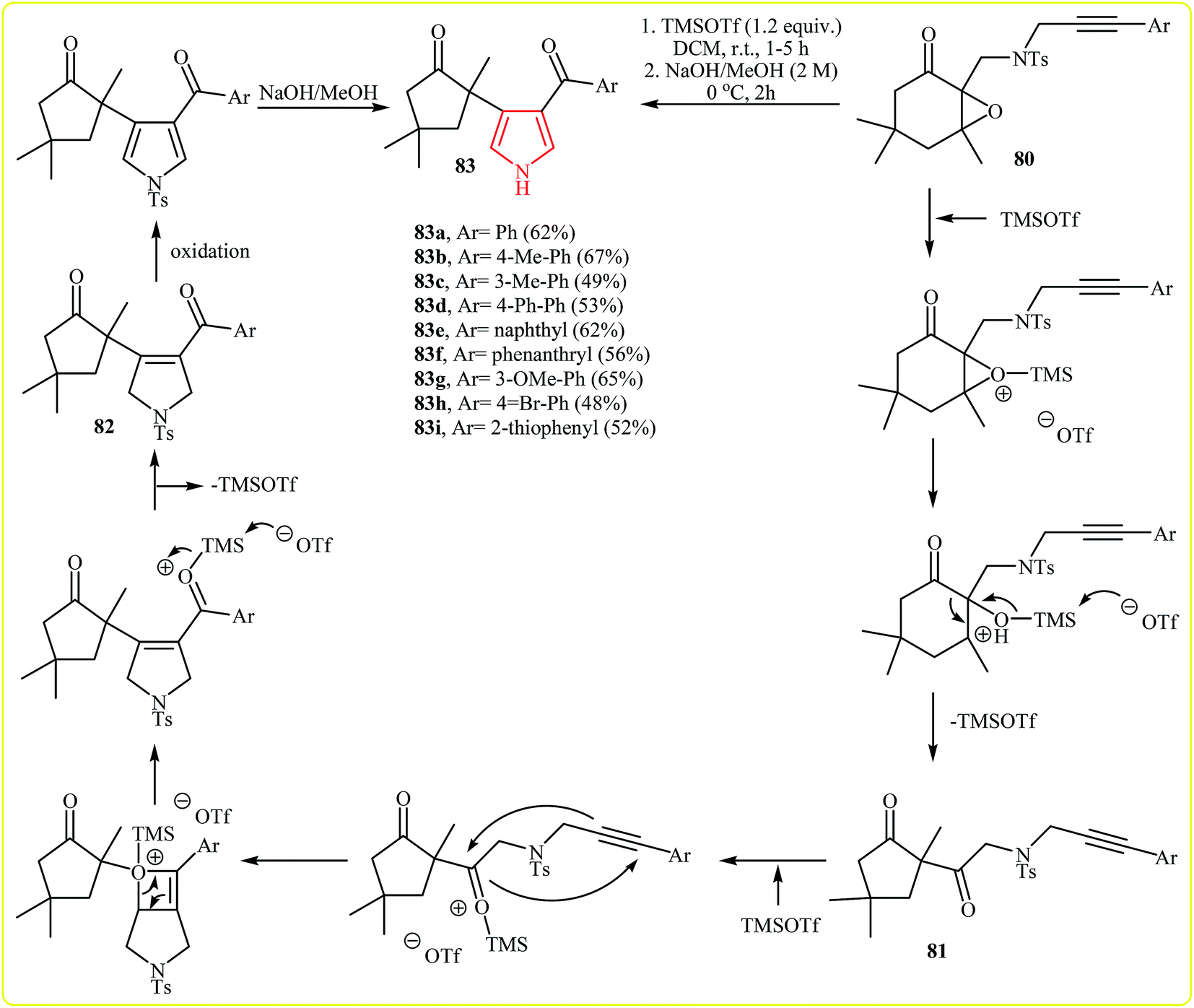 | ||
| Fig. 4 The synthesis of 2,3-disubstituted pyrroles 83 via TMSOTf-assisted cyclization reaction of 80. | ||
More recently, Zhao and co-workers reported an example of base-catalyzed intramolecular cyclization reaction of N-cyanopropargylamines. They showed that N-cyanopropargylamine 84 underwent cyclization–decyanation–aromatization in the presence of NaH as catalyst in DMF at 130 °C. The corresponding pyrrole 85 was obtained in yield of 86%. Interestingly, when the reaction was performed at 110 °C, pyrrole 86 was produced in 70% yield along with 10% of 85 (Scheme 23). It should be mentioned that the scope of the reaction is limited to internal alkynes only, because the substrates with terminal alkynes 87 gave dihydro pyrroles 88 instead of pyrroles (Scheme 24).41
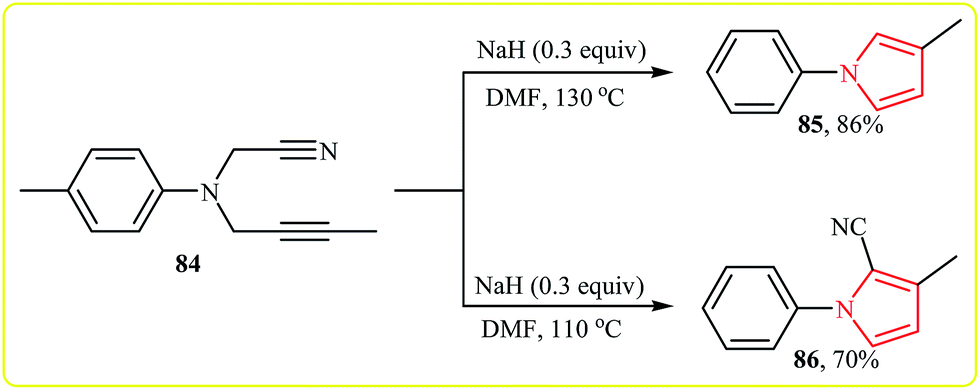 | ||
| Scheme 23 NaH-catalyzed intramolecular cyclization reaction of 85. | ||
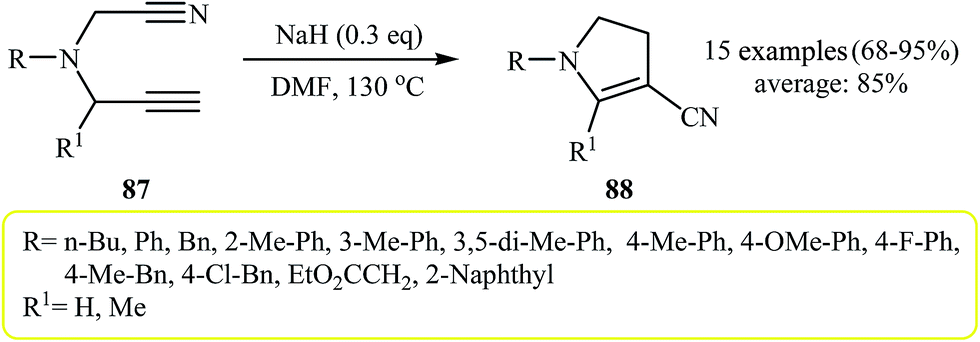 | ||
| Scheme 24 Cycloisomerization of 5-cyano-pentyne derivatives 87 to 3-cyano-4,5-dihydro-1H-pyrroles 88. | ||
4. Summary and outlook
In conclusion, this review provides concise overview on the synthesis of highly substituted pyrroles from N-propargylamines via intra- or intermolecular cyclization reaction. In many cases, the use of this avenue for synthesis of pyrrole core provides milder conditions and simpler procedures than previously reported examples. This research area has still further possibilities for growth (for instance, by expanding of the substrates scope to N-propargylsulfamates or N-propargylsulfonamides for synthesis of special pyrroles) and we believed that the highly versatile and extremely effective procedures for the synthesis of highly substituted pyrroles from N-propargylamines will be attainable in the near future.Acknowledgements
This work was supported by the “Sandoogh Hemayate as Pajuoheshgharane Keshvare” Iran.References
- (a) A. Grube, E. Lichte and M. Köck, J. Nat. Prod., 2006, 69, 125–127 CrossRef CAS PubMed; (b) B. M. Trost and G. Dong, Org. Lett., 2007, 9, 2357–2359 CrossRef CAS PubMed; (c) S. T. Handy and Y. Zhang, Org. Prep. Proced. Int., 2005, 37, 411–445 CrossRef CAS; (d) A. Fürstner, Synlett, 1999, 10, 1523–1533 CrossRef; (e) L. Naumovski, J. Ramos, M. Sirisawad, J. Chen, P. Thiemann, P. Lecane, D. Magda, Z. Wang, C. Cortez and G. Boswell, Mol. Cancer Ther., 2005, 4, 968–976 CrossRef CAS PubMed; (f) T. Bando and H. Sugiyama, Acc. Chem. Res., 2006, 39, 935–944 CrossRef CAS PubMed; (g) Y. Arikawa, H. Nishida, O. Kurasawa, A. Hasuoka, K. Hirase, N. Inatomi, Y. Hori, J. Matsukawa, A. Imanishi and M. Kondo, J. Med. Chem., 2012, 55, 4446–4456 CrossRef CAS PubMed.
- V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC.
- (a) F. Bellina and R. Rossi, Tetrahedron, 2006, 62, 7213–7256 CrossRef CAS; (b) V. Blangy, C. Heiss, V. Khlebnikov, C. Letondor, H. Stoeckli-Evans and R. Neier, Angew. Chem., Int. Ed., 2009, 48, 1688–1691 CrossRef CAS PubMed; (c) M. Takase, N. Yoshida, T. Narita, T. Fujio, T. Nishinaga and M. Iyoda, RSC Adv., 2012, 2, 3221–3224 RSC; (d) P. Liu, X. Wang and Y. Wang, ACS Sustainable Chem. Eng., 2014, 2, 1795–1801 CrossRef CAS.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 1635–1642 CrossRef.
- C. Paal, Ber. Dtsch. Chem. Ges., 1885, 18, 367–371 CrossRef.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1890, 23, 1474–1476 CrossRef.
- (a) A. R. Katritzky, J. Jiang and P. J. Steel, J. Org. Chem., 1994, 59, 4551–4555 CrossRef CAS; (b) I. Freifeld, H. Shojaei and P. Langer, J. Org. Chem., 2006, 71, 4965–4968 CrossRef CAS PubMed; (c) P. Langer, Synthesis, 2009, 2009, 227–242 CrossRef; (d) A. A. Fesenko and A. D. Shutalev, J. Org. Chem., 2013, 78, 1190–1207 CrossRef CAS PubMed.
- (a) M. N. Zhao, Z. H. Ren, Y. Y. Wang and Z. H. Guan, Chem.–Eur. J., 2014, 20, 1839–1842 CrossRef CAS PubMed; (b) M.-N. Zhao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2014, 16, 608–611 CrossRef CAS PubMed.
- (a) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2010, 39, 4402–4421 RSC; (b) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2014, 43, 4633–4657 RSC.
- (a) M. Namicinski, L. Pulaski, A. Kochman, J. Skolimowski, G. Bartosz and D. Metodiewa, In Vivo, 2004, 18, 171–180 Search PubMed; (b) S. Dragoni, V. Porcari, M. Valoti, M. Travagli and D. Castagnolo, J. Pharm. Pharmacol., 2006, 58, 561–565 CrossRef CAS PubMed; (c) Y. Kleiner, O. Bar-Am, T. Amit, A. Berdichevski, E. Liani, G. Maor, I. Reiter, M. B. Youdim and O. Binah, J. Cardiovasc. Pharmacol., 2008, 52, 268–277 CrossRef CAS PubMed; (d) F. T. Zindo, J. Joubert and S. F. Malan, Future Med. Chem., 2015, 7, 609–629 CrossRef CAS PubMed; (e) R. A. Hauser, Int. J. Neurosci., 2011, 121, 53–62 CrossRef CAS PubMed; (f) I. Bolea, A. Gella and M. Unzeta, J. Neural Transm., 2013, 120, 893–902 CrossRef CAS PubMed; (g) M. Yogev-Falach, O. Bar-Am, T. Amit, O. Weinreb and M. B. Youdim, FASEB J., 2006, 20, 2177–2179 CrossRef CAS PubMed; (h) P. L. McCormack, CNS Drugs, 2014, 28, 1083–1097 CrossRef CAS PubMed; (i) M. Xue, H. Huang, Y. Ke, C. Chu and X. Liang, J. Chromatogr. A, 2009, 1216, 8623–8629 CrossRef CAS PubMed; (j) H. Huang, Y. Jin, M. Xue, L. Yu, Q. Fu, Y. Ke, C. Chu and X. Liang, Chem. Commun., 2009, 6973–6975 RSC; (k) Y. S. Gal, B. Jung, W. C. Lee and S. K. Choi, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2657–2662 CrossRef CAS.
- (a) F. J. Fananás, T. Arto, A. Mendoza and F. Rodriguez, Org. Lett., 2011, 13, 4184–4187 CrossRef PubMed; (b) T. P. Lebold, A. B. Leduc and M. A. Kerr, Org. Lett., 2009, 11, 3770–3772 CrossRef CAS PubMed; (c) D. F. Harvey and D. M. Sigano, J. Org. Chem., 1996, 61, 2268–2272 CrossRef CAS; (d) B. Nilsson and U. Hacksell, J. Heterocycl. Chem., 1989, 26, 269–275 CrossRef CAS; (e) A. Arcadi, S. Cacchi, L. Cascia, G. Fabrizi and F. Marinelli, Org. Lett., 2001, 3, 2501–2504 CrossRef CAS PubMed; (f) M. Zhu, W. Fu, G. Zou, C. Xun, D. Deng and B. Ji, J. Fluorine Chem., 2012, 135, 195–199 CrossRef CAS; (g) Z. Jiang, P. Lu and Y. Wang, Org. Lett., 2012, 14, 6266–6269 CrossRef CAS PubMed.
- (a) B. Jiang and M. Xu, Angew. Chem., Int. Ed., 2004, 43, 2543–2546 CrossRef CAS PubMed; (b) J. J. Fleming and J. Du Bois, J. Am. Chem. Soc., 2006, 128, 3926–3927 CrossRef CAS PubMed.
- T. Tsuda, T. Kiyoi, T. Miyane and T. Saegusa, J. Am. Chem. Soc., 1988, 110, 8570–8572 CrossRef CAS.
- W. S. Bremner and M. G. Organ, J. Comb. Chem., 2007, 10, 142–147 CrossRef PubMed.
- E. Merkul, C. Boersch, W. Frank and T. J. Müller, Org. Lett., 2009, 11, 2269–2272 CrossRef CAS PubMed.
- Y. L. Zhao, C. H. Di, S. D. Liu, J. Meng and Q. Liu, Adv. Synth. Catal., 2012, 354, 3545–3550 CrossRef CAS.
- C.-Q. Ren, C.-H. Di, Y.-L. Zhao and J.-P. Zhang, Tetrahedron Lett., 2013, 54, 1478–1481 CrossRef CAS.
- H. Chachignon, N. Scalacci, E. Petricci and D. Castagnolo, J. Org. Chem., 2015, 80, 5287–5295 CrossRef CAS PubMed.
- B. M. Trost, J.-P. Lumb and J. M. Azzarelli, J. Am. Chem. Soc., 2011, 133, 740–743 CrossRef CAS PubMed.
- J. Weng, Y. Chen, B. Yue, M. Xu and H. Jin, Eur. J. Org. Chem., 2015, 2015, 3164–3170 CrossRef CAS.
- (a) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (b) M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17–85 CrossRef CAS; (c) D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS; (d) R. S. Dickson, T. de Simone, E. M. Campi and W. R. Jackson, Inorg. Chim. Acta, 1994, 220, 187–192 CrossRef CAS.
- (a) H.-W. Bohnen and B. Cornils, Adv. Catal., 2002, 47, 1–64 CAS; (b) S. N. Bizzarri, S. Fenelon and M. Ishikawa-Yamaki, Chemical Economics Handbook, SRI International, Menlo Park, 1999, pp. 682–7000A Search PubMed; (c) S. Naqvi, Oxo Alcohols. Process Economics Program Report 21E, SRI Consulting, Menlo Park, CA, 2010 Search PubMed.
- (a) E. M. Campi, W. R. Jackson and Y. Nilsson, Tetrahedron Lett., 1991, 32, 1093–1094 CrossRef CAS; (b) E. Campi, G. Fallon, W. Jackson and Y. Nilsson, Aust. J. Chem., 1992, 45, 1167–1178 CrossRef CAS.
- T.-J. Meng, Y. Hu and S. Wang, J. Org. Chem., 2009, 75, 582–588 CrossRef PubMed.
- Y. Hu, C. Wang, D. Wang, F. Wu and B. Wan, Org. Lett., 2013, 15, 3146–3149 CrossRef CAS PubMed.
- N. Sakai, H. Hori and Y. Ogiwara, Eur. J. Org. Chem., 2015, 1905–1909 CrossRef CAS.
- J. Cossy, C. Poitevin, L. Sallé and D. G. Pardo, Tetrahedron Lett., 1996, 37, 6709–6710 CrossRef CAS.
- S. Cacchi, G. Fabrizi and E. Filisti, Org. Lett., 2008, 10, 2629–2632 CrossRef CAS PubMed.
- A. Saito, T. Konishi and Y. Hanzawa, Org. Lett., 2009, 12, 372–374 CrossRef PubMed.
- C. Zheng, Y. Wang and R. Fan, Org. Lett., 2015, 17, 916–919 CrossRef CAS PubMed.
- A. P. Krapcho and J. F. Weimaster, J. Org. Chem., 1980, 45, 4105–4111 CrossRef CAS.
- Y. Yamamoto, H. Hayashi, T. Saigoku and H. Nishiyama, J. Am. Chem. Soc., 2005, 127, 10804–10805 CrossRef CAS PubMed.
- N. Dieltiens, K. Moonen and C. V. Stevens, Chem.–Eur. J., 2007, 13, 203–214 CrossRef CAS PubMed.
- R. Gleiter and J. Ritter, Tetrahedron, 1996, 52, 10383–10388 CrossRef CAS.
- K. Tanaka, Y. Otake and M. Hirano, Org. Lett., 2007, 9, 3953–3956 CrossRef CAS PubMed.
- M. A. Wuonola and J. M. Smallheer, Tetrahedron Lett., 1992, 33, 5697–5698 CrossRef CAS.
- R. J. Corriu, G. Bolin, J. Iqbal, J. J. Moreau and C. Vernhet, Tetrahedron, 1993, 49, 4603–4618 CrossRef CAS.
- M. Lee, H. Moritomo and K. Kanematsu, J. Chem. Soc., Chem. Commun., 1994, 1535 RSC.
- H. M. Peng, J. Zhao and X. Li, Adv. Synth. Catal., 2009, 351, 1371–1377 CrossRef CAS.
- M.-C. P. Yeh, M.-N. Lin, C.-H. Hsu and C.-J. Liang, J. Org. Chem., 2013, 78, 12381–12396 CrossRef CAS PubMed.
- J. Meng, Y.-J. Li, Y.-L. Zhao, X.-B. Bu and Q. Liu, Chem. Commun., 2014, 50, 12490–12492 RSC.
| This journal is © The Royal Society of Chemistry 2016 |