
Synthesis and characterization of chiral PEDOT enantiomers bearing chiral moieties in side chains: chiral recognition and its mechanism using electrochemical sensing technology†
Liqi Dong,
Long Zhang,
Xuemin Duan*,
Daize Mo,
Jingkun Xu* and
Xiaofei Zhu
School of Pharmacy, Jiangxi Science & Technology Normal University, Nanchang, 330013, China. E-mail: duanxuemin@126.com; xujingkun@tsinghua.org.cn; Fax: +86-791-83823320; Tel: +86-791-88537967
First published on 18th January 2016
Abstract
In this work, we present a pair of chiral PEDOT derivatives named poly((R)-2-(chloromethyl)-2,3-dihydrothieno[3,4-b][1,4]dioxine) ((R)-PEDTC) and poly((S)-2-(chloromethyl)-2,3-dihydrothieno[3,4-b][1,4]dioxine) ((S)-PEDTC), which were employed as excellent chiral recognition materials for fabricating chiral sensors and then discriminating between 3,4-dihydroxyphenylalanine (DOPA) enantiomers. Importantly, the mechanism of the stereospecificity of the interaction between the DOPA enantiomers and chiral polymers was discussed specifically. A series of performances of corresponding polymers were characterized in some detail using different strategies including CV, CD, FT-IR, UV-vis, SEM, and TG. The CD spectrum indicated that (R)-PEDTC and (S)-PEDTC are mirror symmetric. CV shows that the polymers had superior redox reversibility in CH3CN–Bu4NPF6. Finally, different electrochemical methods including CV, square wave voltammetry (SWV) and differential pulse voltammetry (DPV) were introduced for the discrimination of DOPA enantiomers. Satisfactory measurement results demonstrated that (R)-PEDTC/GCE and (S)-PEDTC/GCE exhibited excellent enantioselectivity of DOPA enantiomers and the tendency was for anisotropic interaction between (R)-PEDTC and L-DOPA, and (S)-PEDTC and D-DOPA. This implied that the obtained polymer films could be promising candidates as enantioselective materials in the electrochemical sensor field.
1. Introduction
Chiral conducting polymers refer to a class of conducting polymers (CPs) that possess chirality and present some unique opportunities when used as chiral substrates or as chiral electrode materials.1 They could, for example, find applications in electrochemical chiral sensing or electrochemical asymmetric synthesis. Research on them began in 1985 via the electropolymerization of pyrrole monomers bearing chiral substituents (R*) covalently attached to pyrrole N centres by R. H. Baughman et al.2 Soon afterwards, chiral polythiophenes were synthesized by M. Lemaire et al. under an asymmetric reaction field consisting of chiral nematic (N*) liquid crystals.3 From then on, a great number of studies on chiral conducting polymers including polypyrrole (Ppy)4 polyanilines (PANI),5–7 polyacetylene,8 polyfluorenes (PFs),9,10 polythiophenes (PTs),11–14 and thienylene–phenylene copolymers15–17 have emerged rapidly. It is amazing that chiral CPs have undergone tremendous advancement, with such a short history, in developing various applications using their particular properties, such as chiral discrimination,18–20 chromatographic separation,21,22 asymmetric catalysis,23–25 optoelectronic devices and batteries,26 photoisomerization switching27 and controlled release drug delivery.28,29Among the chiral CPs, chiral poly(3,4-ethylenedioxythiophene) (PEDOT), emerging as a “superstar” material, has attracted tremendous attention due to its unique structure and electronic properties.30,31 Compared with racemic PEDOT, introduction of a chiral moiety into a side chain could reduce the rigidity of polymer chains and have a positive impact on the solubility, thermal stability, and orientation of liquid crystal materials.1,32,33 Despite the fact that chiral PEDOTs were synthesized in 2004 by K. Akagi et al.,31 immense challenges have been encountered for their applications, and only a few studies have been dedicated to forming nematic liquid crystals.34,35 However, some important applications, such as molecular recognition, enantioselective separation, microstructure control and asymmetric catalysis are still lacking basic exploration. Therefore, exploring the potential applications of chiral PEDOT will provide inspiration for the further design of novel chiral PEDOT derivatives, which will lead to innovative changes in the development of chiral PEDOT.
Some recent studies by our group showed that chiral PEDOT bearing chiral moieties in the side chains might be candidates to enable the recognition of enantiomers.36,37 With relative ease we can study and control the behavior of the polymers which make them very valuable in chiral recognition. On one hand, to the best of our knowledge, the chiroptical activity properties of PEDOTs due to incorporation chirality have already been reported.36 However, the field of using these properties for enantiomer recognition is still in its early days. The homochiral selective electrode for discriminating between enantiomers and its mechanism still remain unexplored and the gap between enantiomers also has a chance to get even larger. Hence, understanding the mechanism of chiral recognition, and then pursuing the larger gap between two enantiomers and more efficient chiral electrode materials to develop the application of chiral PEDOT is valuable and fascinating. On the other hand, to date, the dominant approaches for chiral recognition are chromatographic methods (including high performance liquid chromatography (HPLC),38–40 gas chromatography (GC),41–43 chiral ligand exchange chromatography (CLEC),44 and supercritical fluid chromatography (SFC)45,46), spectroscopic characterizations (including fluorescence detection (FL)47,48 and vibrational circular dichroism (VCD)49), capillary electrophoresis (CE)50,51 and molecular imprinting polymer techniques (MIP).52,53 Even if these techniques are fairly efficient, they are usually time consuming, require costly equipment and need sophisticated and extensive analysis procedures. However, electrochemical strategies offer some advantages over the other techniques such as low cost, simple equipment, on-line measurement and high sensitivity.54–58 Thus, the combination of the electrochemical molecular recognition approach and chiral PEDOT materials can be potentially utilized to fabricate a chiral electrochemical sensor.
In this article, we synthesized a pair of PEDOT enantiomers i.e. (R)-PEDTC and (S)-PEDTC and systematically investigated the electropolymerization behavior, electrochemical properties, structural characterization, circular dichroism, morphology and thermal stability. Finally, (R)-/(S)-PEDTC as chiral material modified glassy carbon electrodes (GCEs) were successfully employed to recognize 3,4-dihydroxyphenylalanine (DOPA) enantiomers. Satisfactory results implied that the obtained polymer films could be a promising candidate in the chiral recognition field.
2. Experimental section
2.1 Chemicals and materials
All materials were reagent grade and were used directly without further purification unless otherwise noted. 3,4-Dimethoxythiophene (98%, AR) was purchased from Vita Chemical Reagent Co. Ltd (Shanghai China). (R)-3-Chloro-1,2-propanediol (99%, 98% e.e.) and (S)-3-chloro-1,2-propanediol (99%, 98% e.e.) were purchased from Daicel Chiral Technologies Co., Ltd (Shanghai China). p-Toluenesulfonic acid monohydrate (p-TSA, 99%, AR), L-/D-3,4-dihydroxyphenylalanine (L-/D-DOPA, 99%, GR), ammonia solution (NH3·H2O, 25–28%, AR), potassium bromide (KBr, 99%, IR) and sulfuric acid (H2SO4, 98%, AR) were purchased from Aladdin Industrial Inc (Shanghai China). Acetonitrile (CH3CN, AR) was purchased from Sinopharm Chemical Reagent Co. Ltd. Dimethyl sulfoxide (DMSO, 99%, AR) was purchased from Lingfeng Chemical Reagent Co. Ltd. Toluene (AR; Xilong Chemical Co., Ltd.) was purified using sulfuric acid and dried with anhydrous calcium chloride. Indium tin oxide (ITO) coated glass was purchased from Zhuhai Kaivo Optoelectronic Technology Co., Ltd. Tetrabutylammonium hexafluorphosphate (Bu4NPF6, 99%, Energy Chemical Reagent Co., Ltd.) was dried under vacuum at 60 °C for 24 h before use. Double-distilled deionized water was used directly without further purification.2.2 Electrosynthesis and electrochemical tests
Electrochemical polymerization was carried out using 0.01 M (R)-/(S)-2-(chloromethyl)-2,3-dihydrothieno[3,4-b][1,4]dioxine ((R)-/(S)-EDTC) in CH3CN–Bu4NPF6 (0.1 M) systems. Prior to polymerization, all the solutions were deaerated with a dry nitrogen stream for 15 min and maintained under a slight overpressure during the electrochemical experiments to avoid the effects of oxygen. Film thickness was controlled by the total charge passed through the cell, which was read directly from the current–time (I–t) curves by a computer. After polymerization, the polymer films were washed repeatedly with acetonitrile to remove the electrolyte and monomer/oligomers.All of the electrochemical tests were conducted in monomer-free solvent-electrolytes in a one-compartment cell with the use of a CHI 660D electrochemistry workstation (Shanghai Chenhua Instruments Co., China) under computer control. Two Pt wires with a diameter of 1 mm were used as the working electrode and counter electrode. An Ag/AgCl electrode dipped in the solution served as the reference electrode, and it revealed sufficient stability during the experiments. The electrodes mentioned above were carefully polished with abrasive paper (1500 mesh) and cleaned with deionized water and acetone successively before each examination.
2.3 Apparatus of the chiral sensor
Cyclic voltammetry (CV), square wave voltammetry (SWV) and differential pulse voltammetry (DPV) measurements were achieved with a CHI 660D electrochemistry workstation. A conventional three-electrode system was employed, including a bare or modified glassy carbon electrode (GCE) as a working electrode, and a platinum wire and a saturated calomel electrode (SCE) as a counter electrode and reference electrode, respectively. (R)-PEDTC/GCE and (S)-PEDTC/GCE were obtained individually using one-step electropolymerization of 10 mM monomers on GCE at a constant potential of 1.1 V vs. SCE in CH3CN–Bu4NPF6 system, and the deposition time was 40 s. Then, the films were washed repeatedly with double-distilled deionized water to remove the electrolyte monomers and dried in air. All measurements were conducted at room temperature (25 ± 2 °C).2.4 Characterization
1H NMR spectra were recorded on a Bruker AV 400 NMR spectrometer with chloroform-d as the solvent and tetramethylsilane (TMS) as an internal standard. Infrared spectra (FT-IR) were recorded using a Bruker Vertex 70 Fourier spectrometer with samples in KBr pellets. The resolution ratio is 2 cm−1. Scanning electron microscopy (SEM) measurements were taken using a JSM-6701F cold field emission scanning electron microscope with the polymer deposited on the ITO-coated glass. Circular dichroism (CD) spectroscopy (JASCO J-720) was used to characterize the polymer films. Fluorescence spectra of the monomer and polymer were measured with an F-4500 fluorescence spectrophotometer (Hitachi). Thermogravimetric analysis (TGA) was performed with a Pyris Diamond TG/DTA thermal analyzer (Perkin-Elmer) under a nitrogen stream from 25 to 1100 °C at a heating rate of 10 °C min−1.To obtain a sufficient amount of polymer for characterization, Pt and stainless-steel sheets with a surface area of 4 and 6 cm2 were employed as the working and counter electrodes, respectively. Ag/AgCl electrodes directly immersed in the solutions served as the reference electrodes. For spectral analyses, the polymer films were de-doped with 25% hydrazine hydrate for 3 days and then washed repeatedly with pure water. Finally, the polymer film was dried at 60 °C under vacuum for 24 h.
2.5 Syntheses of (R)-/(S)-EDTC
To a well-stirred solution of 3,4-dimethoxythiophene (16.300 g, 113.05 mmol) in 280 mL of dry toluene, (R)-3-chloro-1,2-propanediol or (S)-3-chloro-1,2-propanediol (25.620 g, 231.77 mmol) and p-toluenesulfonic acid monohydrate (1.600 g, 9.29 mmol) were added under a nitrogen atmosphere. The solution was heated at 90 °C for 72 h. Then, another lot of (R)- or (S)-diol (25.620 g, 231.77 mmol) was added, and the solution was heated at 90 °C for another 3 h and was allowed to cool to room temperature. After removal of the solvent, the remaining crude product was isolated by column chromatography (silica gel, petroleum ether/acetic ether, 10/1, v/v) to give 12.525 g of a white solid (yield 58%) for (R)-EDTC or to give 10.666 g of a white solid (yield 49%) for (S)-EDTC.(R)-EDTC: [α]25D = −2.300 (c = 1 g/100 mL, in CHCl3) 1H NMR (400 MHz, CDCl3, ppm) δ: 6.27 (d, J = 4 Hz, 2H), 4.30–4.24 (m, 1H), 4.20–4.16 (m, 1H), 4.08–4.05 (m, 1H), and 3.65–3.54 (m, 2H). The 1H NMR spectrum is shown in ESI Fig. S1.†
(S)-EDTC: [α]25D = +2.300 (c = 1 g per 100 mL, in CHCl3) 1H NMR (400 MHz, CDCl3, ppm) δ: 6.27 (d, J = 4 Hz, 2H), 4.30–4.24 (m, 1H), 4.20–4.16 (m, 1H), 4.08–4.03 (m, 1H), and 3.65–3.54 (m, 2H). The 1H NMR spectrum is shown in ESI Fig. S2.†
3. Results and discussion
3.1 Syntheses of monomers
The racemic mixture 2-chloromethyl-2,3-dihydrothieno[3,4-b][1,4]dioxine (EDTC) has been reported.59–61 Based on the works of scientists previously, the EDTC was developed by reacting 3,4-dimethoxythiophene and 3-chloro-1,2-propanediol enantiomers in an acid-catalyzed trans-etherification reaction (Scheme 1). The chirality of EDTC was introduced by bringing in a chiral source i.e. (R)-/(S)-3-chloro-1,2-propanediol. Notably, the 3-chloro-1,2-propanediol enantiomers as a chiral source have already been confirmed, which means the chirality of (R)- or (S)-EDTC will not be changed in a synthetic process. Thus, two single chiral EDTC monomers were obtained. | ||
| Scheme 1 The synthesis route of (R)-/(S)-EDTC enantiomers and its electropolymerization. | ||
3.2 Electrochemical polymerization
The successive CVs of (R)-EDTC (A) and (S)-EDTC (B) in CH3CN–Bu4NPF6 systems at a potential scanning rate of 50 mV s−1 are illustrated in Fig. 1. In the first cycle of CVs, the current densities on the reverse scan were higher than those on the forward scan (in the region of −0.8 to 1.5 V for (R)-EDTC and −0.8 to 1.45 V for (S)-EDTC). The formation of this loop could be explained as a characteristic of the nucleation process.62 The redox peaks of (R)-EDTC and (S)-EDTC appeared near +0.25 V and +0.14 V, and −0.16 V and −0.33 V. These peaks were assigned to the p-doping/dedoping processes of the (R)-EDOTC and (S)-EDTC film formed in previous scans. Upon subsequent potential scanning, a progressive increase of the peak currents densities and a decrease of the oxidation onset potentials could be seen in subsequent scans, indicating the gradually growing amount of the polymer film with an increased number of monomeric units deposited onto the electrode surface (light-blue to blue-back as the deposit thickened).63 In fact, the formation of a compact and homogeneous film on the electrode surface was observed. Apparently, both the electrochemical polymerization of (R)-EDTC and (S)-EDTC and the onset oxidation potential (Eox) of (R)-EDTC (1.26 V, Fig. 1A inset) and (S)-EDTC (1.27 V, Fig. 1B inset) were extraordinarily analogous. In addition, the Eox of EDOT and other analogues such as EDTM and C4-EDOT–COOH were 0.92 V, 0.88 V, and 0.99 V,64,65 which were lower than (R)-/(S)-EDTC. The results indicated that the oxidation of (R)-/(S)-EDTC was harder than for EDOT and its analogues. | ||
| Fig. 1 Cyclic voltammograms (CVs) of (R)-EDTC (A) and (S)-EDTC (B) electropolymerized in CH3CN containing 0.1 M Bu4NPF6 and 0.01 M monomers at a potential scan rate of 50 mV s−1. Inset: the corresponding anodic polarization curve of (R)-/(S)-EDOTC enantiomers. | ||
 | ||
| Fig. 2 FT-IR spectra of (R)-PEDTC (A) and (S)-PEDTC (B): monomers (a) and polymers (b). | ||
3.3 Structural characterization
FT-IR spectra of the monomers and their corresponding polymers were recorded to elucidate their structure and interpret the polymerization mechanism (Fig. 2). The detailed peak assignment and comparison for (R)-EDTC, (R)-PEDTC, (S)-EDTC and (S)-PEDTC were summarized in ESI Table S1.† The results were similar to a previous report of a racemic mixture of EDTC.60 In the functional group region, the peak at 3111 cm−1 in the monomer spectrum of (R)-EDTC and (S)-EDTC is attributed to the![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching vibration which disappeared/weakened in the spectrum of (R)-PEDTC and (S)-PEDTC. This phenomenon is a direct proof of the electrochemical polymerization of (R)-EDTC and (S)-EDTC occurring at the α-positions of the thiophene ring. In the fingerprint region, the emergence of characteristic peaks located at 1021 cm−1 and 919 cm−1 for (R)-EDTC, 1016 cm−1 and 920 cm−1 for (S)-EDTC should be assigned to in-plane deformation vibration and out-of-plane deformation vibration. However, all of these peaks disappeared in the FT-IR spectrum of the polymers, which further confirmed that (R)-PEDTC and (S)-PEDTC were dominantly electropolymerized through the α,α′-coupling of EDOT units. Typically, for all the monomers and polymers, the peaks at approximately 1186–1102 cm−1 and 836–875 cm−1 could be attributed to asymmetric and symmetric C–O stretching vibrations, respectively. Furthermore, the vibration at nearly 1482 cm−1 is ascribed to the stretching mode of the CC bond, while the peaks at 1375, 1275, and 1055 cm−1 result from the stretching of a single C–C bond and the C–O–C vibration, respectively. For the chlorine atom, the absorption peaks originating from the C–Cl stretching and deformation vibrations could be observed at 770 and 543 cm−1, respectively.
C–H stretching vibration which disappeared/weakened in the spectrum of (R)-PEDTC and (S)-PEDTC. This phenomenon is a direct proof of the electrochemical polymerization of (R)-EDTC and (S)-EDTC occurring at the α-positions of the thiophene ring. In the fingerprint region, the emergence of characteristic peaks located at 1021 cm−1 and 919 cm−1 for (R)-EDTC, 1016 cm−1 and 920 cm−1 for (S)-EDTC should be assigned to in-plane deformation vibration and out-of-plane deformation vibration. However, all of these peaks disappeared in the FT-IR spectrum of the polymers, which further confirmed that (R)-PEDTC and (S)-PEDTC were dominantly electropolymerized through the α,α′-coupling of EDOT units. Typically, for all the monomers and polymers, the peaks at approximately 1186–1102 cm−1 and 836–875 cm−1 could be attributed to asymmetric and symmetric C–O stretching vibrations, respectively. Furthermore, the vibration at nearly 1482 cm−1 is ascribed to the stretching mode of the CC bond, while the peaks at 1375, 1275, and 1055 cm−1 result from the stretching of a single C–C bond and the C–O–C vibration, respectively. For the chlorine atom, the absorption peaks originating from the C–Cl stretching and deformation vibrations could be observed at 770 and 543 cm−1, respectively.
In addition, both the (R)- and (S)-PEDTC exhibit essentially identical (as expected) characteristic absorption bands, with peaks similar to those of racemic PEDTC prepared by electrochemical polymerization, indicating that the two films consist of repeating units of EDTC. Besides that, the absorption bands in the spectra of the doped polymers were obviously broadened in comparison with those monomers, similar to those of other conducting polymers.63,64,66 This phenomenon was probably due to the fact that the resulting product was composed of oligomers/polymers with wide chain dispersity.
3.4 Optical properties
The as-prepared PEDTC films in the doped state were dark blue in color. When the films were dedoped by 25% ammonia for 3 days, their colour changed to brownish yellow. However, the resulting PEDTC films were found to be incompletely soluble in DMSO or THF, and they also exhibited poor solubility in other solvents such as acetonitrile and acetone.UV-vis spectra of the monomers and polymers dissolved in DMSO were examined in Fig. 3. The monomers of (R)-EDTC and (S)-EDTC showed characteristic absorption peaks at about 265 and 271 nm, respectively (Fig. 3 inset). For (R)-PEDTC and (S)-PEDTC, the trend of the absorbance curves and the position of the peaks were similar to PEDOT.67 In the neutral state of polymers, the peak at about 590 nm was attributed to π–π* transitions. Meanwhile, in the oxidized state, the polymer films showed not only the absorption at 590 nm but also a broad peak at wavelengths longer than 940 nm. This wide peak is associated with the bipolaron band in the spectrum. This result confirmed the occurrence of electrochemical polymerization among the monomers and the formation of a conjugated polymer with broad molar mass distribution. Also, the absorption band at 590 nm in the neutral state and the broad band located from 900 to 1000 nm in the oxidized state are responsible for the light-blue and black-blue colours in the dedoped and doped films, respectively. In other words, the reversible electrochromic change occurs through electrochemical doping and dedoping. In addition, it should be noted that the spectra of doped and dedoped polymers were consistent, which is mainly because of the automatic dedoping process of the polymer.68
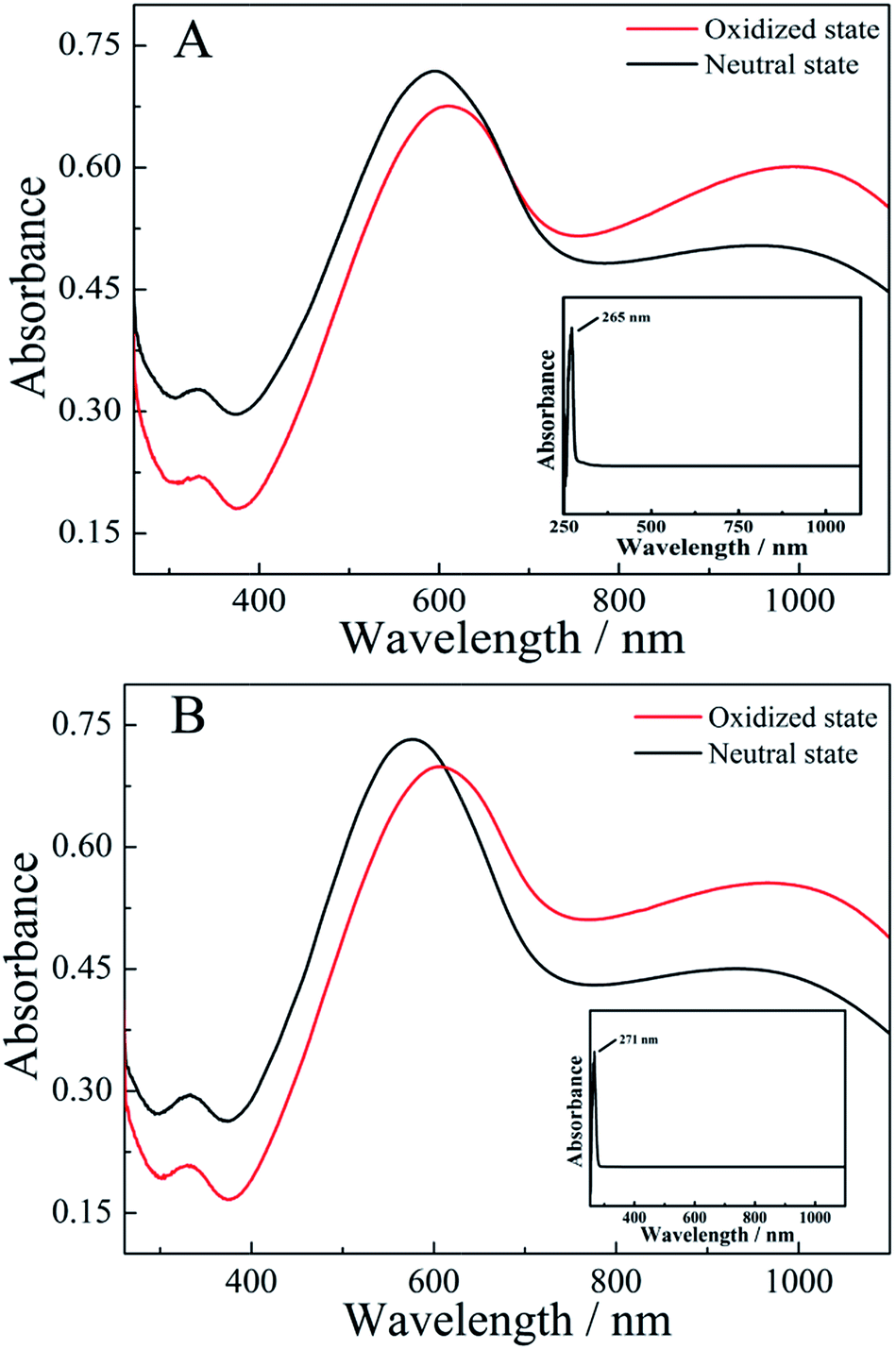 | ||
| Fig. 3 UV-vis spectra of (R)-PEDTC (A) and (S)-PEDTC (B) in neutral and oxidized states. Solvent: DMSO. | ||
Circular dichroism (CD) spectra of the polymers are shown in Fig. 4. As shown in Fig. 4, (R)- and (S)-PEDTC exhibit a strong Cotton effect in the neutral state, but no Cotton effect was observed in the oxidized state. In the neutral state, at a wavelength of about 300 nm (R)-PEDTC exhibits a negative Cotton effect while (S)-PEDTC displays a positive effect in CD measurements. That is to say, the CD spectra of (R)-PEDTC and (S)-PEDTC are mirror symmetric but otherwise essentially identical. In the oxidized state, the CD spectra of the polymers showed a decrease in intensity and an inversion of the sign of the Cotton effect. It is suggested that the changes of the CD bands in the neutral and oxidized states might be related to a chirality change in the polymers. Therefore, the CD bands that could be observed in the neutral polymers were ascribed to two factors: (i) the neutral polymers have a relatively high degree of freedom in terms of the internal rotation of the main chain, and (ii) conversely, when the polymer is oxidized through doping, the planarity of the main chain is enhanced due to the dopants (PF61−) being intercalated into the main chain.11,31,69
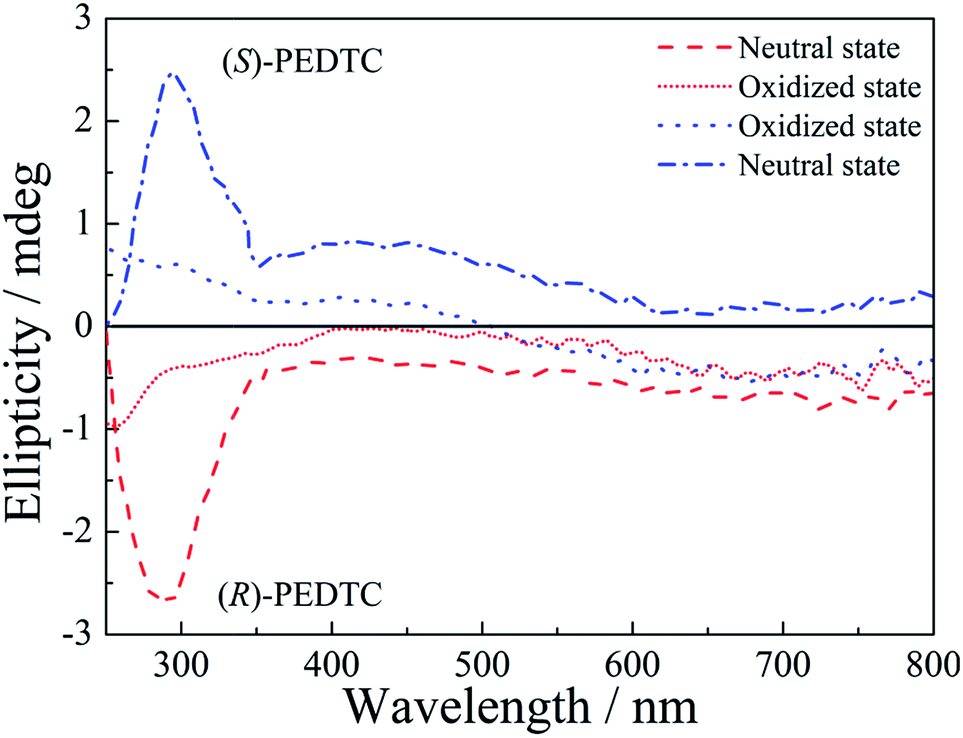 | ||
| Fig. 4 CD spectra of neutral and oxidized (R)-PEDTC and (S)-PEDTC. Solvent: DMSO. | ||
As can be seen in Fig. 4, the intensity of the CD spectra was weaker than other chiral polymers with similar structures reported previously.8,14,31 That was mainly attributed to the small chiral group (Cl atom) and regioirregular polymers. Theoretically, the regioregularity of polymers could form head-to-head (HH) or head-to-tail (HT), and it is assumed that such structural changes affect the optical properties of the polymer. However, control of the regioregularity of the polymer is difficult to achieve by this electrochemical polymerization method, which tends to form polymers with random HH and HT regularities.17
3.5 Electrochemistry properties
For a comparative study, the electrochemical behaviors of both (R)-PEDTC and (S)-PEDTC films were carried out in monomer-free CH3CN–Bu4NPF6, as shown in Fig. 5. Similar to other EDOT-based polymers, both (R)-PEDTC and (S)-PEDTC films exhibited broad anodic and cathodic peaks in the CH3CN–Bu4NPF6 system.62–64,68 Also, the electrochemical performance of (R)-PEDTC and (S)-PEDTC were extremely similar to each other. All peak current densities were well proportional to the potential scanning rate (Fig. 5C and D), which indicated that the redox process was non-diffusional and electroactive materials were anchored to the surface of the working electrode. Furthermore, the calculated jp,a/jp,c values (jp,a or jp,c is calculated as the ratio between anodic or cathodic peak current density and the potential scan rate) of (R)-PEDTC and (S)-PEDTC are both very close to 1.0, demonstrating that the polymers had superior redox reversibility in CH3CN–Bu4NPF6. It was also found that the discrepancies in the redox peak potentials of (R)-PEDTC and (S)-PEDTC were obvious. The main reasons that account for this phenomenon are usually as follows: slow heterogeneous electron transfer, local rearrangement of polymer chains, slow mutual transformation of various electronic species, and electronic charging of interfacial exchange corresponding to the metal/polymer and polymer/solution interfaces.70 It is worth noting that (R)-PEDTC and (S)-PEDTC display very similar electrochemical behavior, probably because the chirality of the polymers does not affect the redox behavior.17 | ||
| Fig. 5 CVs of (R)-PEDTC (A) and (S)-PEDTC (B)-coated Pt electrodes in monomer-free CH3CN–Bu4NPF6 (0.1 M) systems at potential scan rates from 300 to 25 mV s−1 with potential decrements of 25 mV s−1. (C, D) Plots of anodic (jp,a) and cathodic (jp,c) peak currents densities vs. potential scan rates | ||
As we all know, good stability of conducting polymers is a key property for their application in advanced technological applications.71 Therefore, the long-term stability of these polymers was investigated in a monomer-free electrolyte solution by applying potential pulses at a potential scan rate of 150 mV s−1, as shown in Fig. 6. On the basis of charge exchange, the electrochemical activities of (R)-PEDTC and (S)-PEDTC were preserved up to 84% and 78% after 100 cycles, respectively, whereas, the electrochemical activities of these polymers retained only about 37% of the charge exchange after 300 cycles. Overall, both (R)-PEDTC and (S)-PEDTC experienced an apparent degradation just after 100 cycles. (R)-PEDTC displayed better stability, compared to (S)-PEDTC. All the results demonstrate a relatively high electrochemical stability of (R)-PEDTC and (S)-PEDTC, which provides an excellent basis for using the polymers in chiral recognition experiments.54,56,57,64
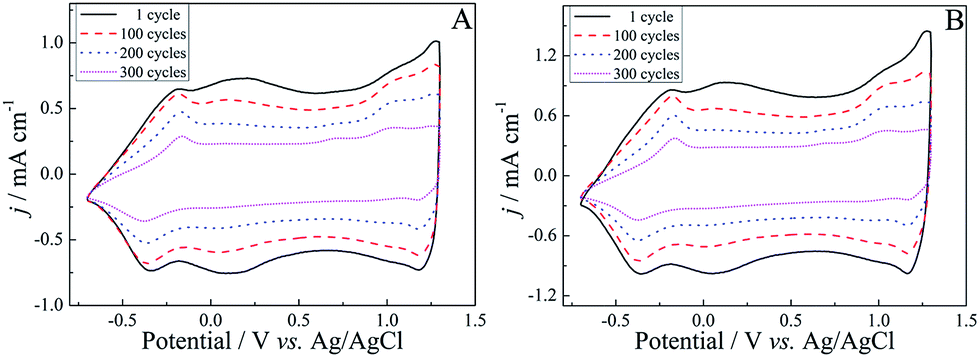 | ||
| Fig. 6 Long-term CVs of (R)-PEDTC (A) and (S)-PEDTC (B) in monomer-free CH3CN–Bu4NPF6 (0.1 M) upon repeated cycling at a scan rate of 150 mV s−1. | ||
3.6 Thermal properties and surface morphology
Thermogravimetric analytical experiments were performed under a nitrogen stream in the temperature range of 298–1260 K with a heating rate of 10 K min−1. According to Fig. 7A and B, it can be clearly observed that there were typically three-step weight losses for both (R)-PEDTC and (S)-PEDTC: (i) a 3.55% weight loss of the polymer occurred before 448 K, (ii) a rapid weight loss occurred in the range of temperature 449–654 K, and (iii) the total weight loss at 1260 K was calculated to be 75% based on the initial weight. The above results are comparable to those for the reported residue of PEDOT (20%).72 Compared with parent PEDOT prepared in aqueous micellar solution73 or CH2Cl2–Bu4NBF4,72, (R)-PEDTC and (S)-PEDTC films showed a reasonably good thermal stability. The detailed thermogravimetric parameters of (R)-/(S)-PEDTC were summarized in ESI Table S2.† | ||
Fig. 7 A, B: TG and DTG curves of (R)-PEDTC (A) and (S)-PEDTC (B) under a nitrogen atmosphere. C, D: SEM micrographs of (R)-PEDTC (C) and (S)-PEDTC (D) films deposited electrochemically on the ITO/glass electrode in CH3CN–Bu4NPF6 (0.1 M) systems containing 0.01 M monomers. Deposition time: 30 s; ITO/glass dimension: 0.5 × 0.5 × 0.1 cm3 (magnification: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×, inset: 50000×). 000×, inset: 50000×). | ||
For analyzing the surface morphology, constituents, and texture of (R)-PEDTC and (S)-PEDTC, scanning electron microscopy (SEM) was performed (Fig. 7). Macroscopically, as-formed (R)- and (S)-PEDTC films were compact, helical and blue in color. Microscopically, the (R)-PEDTC film had a rather compact, porous network structure, meanwhile the (S)-PEDTC film displayed a well-entangled and interconnected, loose, and porous network morphology. The morphologies of the (R)- and (S)-PEDTC films were parallel. However, at high magnifications (50000×), the (R)-PEDTC film appeared to be a little swollen and more compact (inset of Fig. 7C and D). In addition, these morphologies of the polymer films were extremely beneficial to the immobilization of biologically active species due to the large surface area and high absorption ability of the network morphology.74
3.7 Chiral recognition
According to other publications,54,56,75 0.25 M H2SO4 was selected as an electrolyte and employed in the recognition of DOPA enantiomers. It can be clearly seen in Fig. 8A and B (dotted line) that there was no redox electrochemical response at the modified electrode in 0.25 M H2SO4, which could eliminate the impact of the electrolyte on the experiment, whereas a pair of apparent redox peaks appeared at around 0.55 V when D-DOPA and L-DOPA were added in 0.25 M H2SO4, ascribed to two-electron-two-proton oxidation and reduction of the dopa/dopaquinone couple.76 That means that both (R)-PEDTC/GCE and (S)-PEDTC/GCE had a faradaic response to DOPA enantiomers and the redox peaks corresponded to the electrochemical behavior of DOPA.76 As illustrated in Fig. 8A and B, similar CV curves and little difference in the peak currents for D-DOPA and L-DOPA were observed. For (R)-PEDTC/GCE, the anodic peak current value of D-DOPA or L-DOPA was 11.56 or 12.47 μA, and the cathodic one was −9.76 or −10.42 μA. For (S)-PEDTC/GCE, the anodic peak current value of D-DOPA or L-DOPA was 12.74 or 10.13 μA, and the cathodic one was −10.34 or −8.17 μA. Although DOPA enantiomers can be recognized by CV technology, the gap between the peak currents was unremarkable. Thus, further analyses using SWV technology revealed striking evidence of chiral discrimination and larger electrochemical response signals were obtained for (R)-/(S)-PEDTC to the DOPA enantiomers (Fig. 8C and D). For (R)-PEDTC, the anodic peak current value of D-DOPA or L-DOPA was 24.09 or 27.37 μA in the forward scan, and the cathodic peak current value was −22.40 or −24.51 μA in the reverse scan. For (S)-PEDTC/GCE, the anodic peak current value of D-DOPA or L-DOPA was 28.31 or 19.16 μA (forward scan), and the cathodic one was −26.11 or −18.47 μA (reverse scan). These results clearly indicated that a clear chiral discrepancy was observed: (R)-PEDTC/GCE showed a higher peak current response towards L-DOPA; while the contrary phenomenon occurred on (S)-PEDTC/GCE. That is to say that the heterochiral interaction between (R)-/(S)-PEDTC and L-/D-DOPA was considerably more favorable than the homochiral interaction. Such differences in the current change make this sensor have a practical application potential for the enantioselective recognition of the DOPA enantiomers. | ||
| Fig. 8 CVs and SWVs of (R)-PEDTC (A and C) and (S)-PEDTC (B and D) modified electrodes in 0.25 M H2SO4 containing 0.25 mM L-DOPA (dashed line) or D-DOPA (solid line). CV: scan rate, 50 mV s−1; SWV: step height, 0.004 V; frequency, 5 Hz. | ||
In order to gain further evidence for (R)-PEDTC and (S)-PEDTC having enantioselectivity to DOPA enantiomers, DPV technology was chosen for further study. Also, distinctive differences in peak current values could be seen in Fig. 9: the peak current value of D-DOPA or L-DOPA on (R)-PEDTC was 80.09 or 108.24 μA, while the peak current values of D-DOPA or L-DOPA on (S)-PEDTC were 124.16 or 94.22 μA. The experimental results of SWVs and DPVs were essentially identical, confirming that (R)-PEDTC/GCE and (S)-PEDTC/GCE exhibited excellent enantioselectivity to DOPA enantiomers and the tendency was for anisotropic interactions between (R)-PEDTC and D-DOPA, and (S)-PEDTC, and L-DOPA.
 | ||
| Fig. 9 DPVs of (R)-PEDTC (A) and (S)-PEDTC (B) modified electrodes in 0.25 M H2SO4 containing 0.25 mM D-DOPA (solid line) or L-DOPA (dashed line). DPV: increasing potential, 0.004 V; amplitude, 5 Hz; pulse width, 0.2 s; pulse period, 0.5 s. | ||
4. Discussion
The elucidation of the chiral recognition mechanism is the key question for chiral recognition. Herein, we consider the mechanism of chiral recognition underlying DOPA enantiomers based on the present experimental results as shown in Chart 1. The electrochemical sensing process usually consists of two steps: analyte recognition and signal generation.19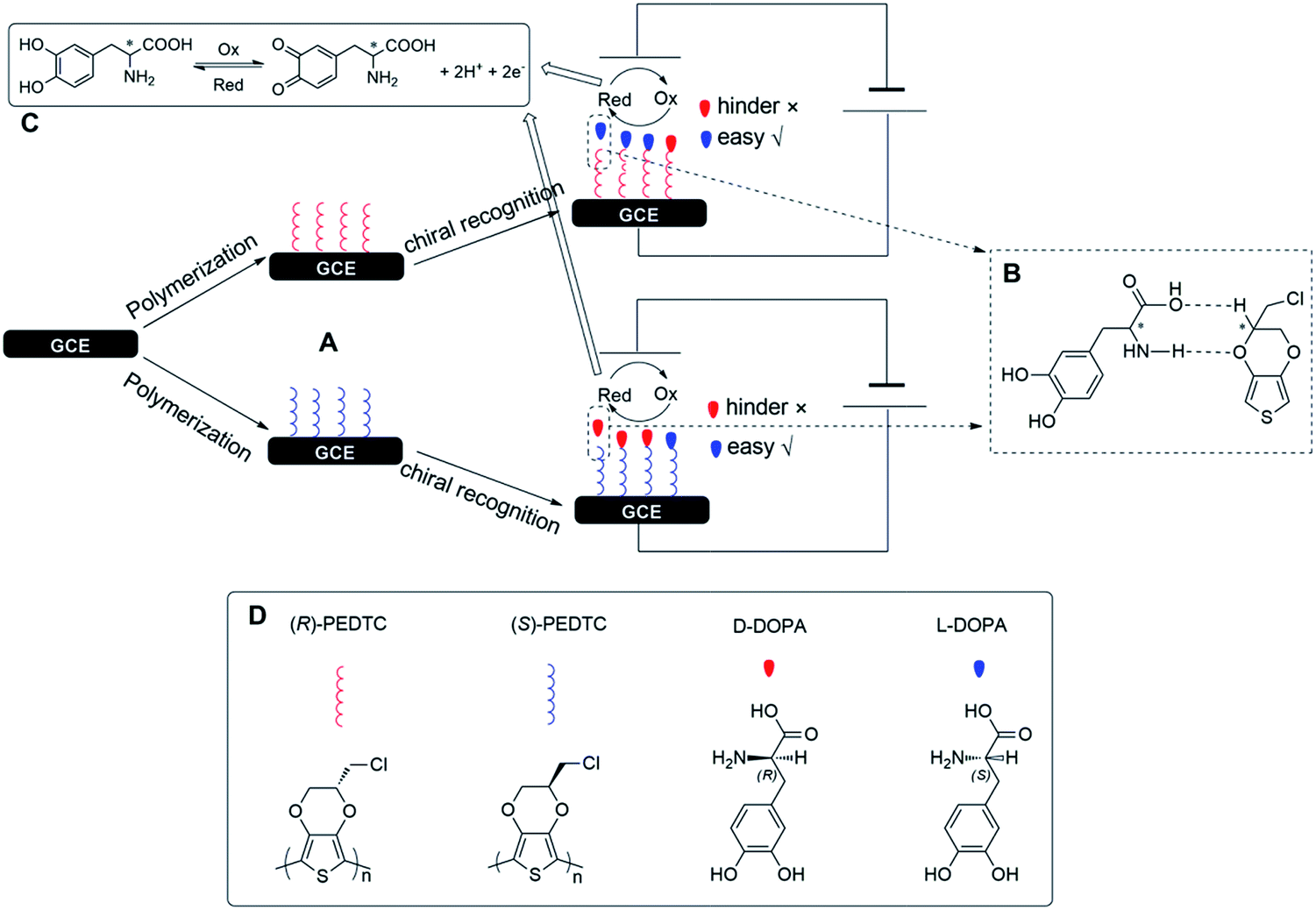 | ||
| Chart 1 (A) Fabrication process of the (R)-/(S)-PEDTC modified electrode. (B) The schematic illustration of the interaction between (R)-/(S)-PEDTC and DOPA enantiomers. (C) The redox reaction mechanism of DOPA. (D) Chemical structures of (R)-, (S)-PEDTC and D-, L-DOPA. | ||
At the stage of analyte recognition, the interaction between the polymer modified electrode and DOPA enantiomers, and the redox reaction of DOPA are two parts of this step. Marzieh Eslami et al.76 demonstrated that the amine and carboxyl groups of DOPA at low pHs will be protonated, and the molecules will be in acidic form. Therefore, the nucleophilic property of the amine group is removed through protonation. These efforts can be rationalized as a consequence of the formation of hydrogen bonds that link to the polymer surface (Chart 1B).77 Then, when DOPA enantiomers are attached to the surface of the modified electrode, the left-handed DOPA (L-DOPA) is easier to connect to the (R)-PEDTC/GCE (right-handed) while the right-handed DOPA (D-DOPA) is hindered.78 Conversely, (S)-PEDTC has the opposite pattern against the D-DOPA and L-DOPA. Therefore, the selective response may be attributed to the fact that the conformation of L-DOPA is more inclined to interact with the (R) form polymer, whereas the D-DOPA is more inclined to interact with (S)- form polymer. In other words, the heterochiral interaction between the polymer modified electrodes and DOPA enantiomers is considerably more favourable than the homochiral interaction. Since most of the DOPA molecules in strong acidic pH (0.25 M H2SO4) are in acidic form, they have a repulsion force with each other, and the probability of the dimerization chemical reaction is insignificant. The mechanism of the hydrolysis chemical reaction is shown in Chart 1C.54,76
During the signal generation of SWV and DPV, the staircase scan waveform could effectively extend the interaction time between the electrode and DOPA. After many cycles, the enantioselectivity is finally amplified and thus the DOPA enantiomers are discriminated.79 This is highly consistent with the fact that the chiral selectivity is attributed to the preferential interaction between the chiral selector and one of the enantiomers.54,56,75,78
5. Conclusions
In summary, a pair of chiral enantiomers of EDOT derivatives were synthesized. The corresponding polymers (R)-PEDTC and (S)-PEDTC were prepared for the first time by electrochemical polymerization, and the polymers showed good redox activity and stability in monomer-free electrolytes. The enantiomers of (R)-PEDTC and (S)-PEDTC were almost the same as each other in terms of electrochemical behavior, structural characterization, morphology and thermal stability. However, CD spectra showed a mirror image of (R)-PEDTC and (S)-PEDTC films in the neutral state. We have shown that the polymer-modified electrodes are distinctly different for the D and L enantiomers of DOPA through the electrochemical sensing technologies of SWV and DPV. Thereby, we established the mechanism of chiral recognition for the first examples of chiral PEDOT electrode materials. However, more intensive studies of this nature are required before a new model of the action of the chiral PEDOT derivatives in promoting enantioselectivity may be proposed.Acknowledgements
The authors would like to acknowledge the financial support of this work by the National Natural Science Foundation of China (51263010, 51303073, 51272096), the Natural Science Foundation of Jiangxi Province (20142BAB206028) and the Science and Technology Landing Plan of Universities in Jiangxi Province (KJLD14069) and Jiangxi Science and Technology Normal University (YC2014-X27).Notes and references
- L. A. Kane-Maguire and G. G. Wallace, Chem. Soc. Rev., 2010, 39, 2545–2576 RSC.
- R. L. Elsenbaumer, H. Eckhardt, Z. Iqbal, J. Toth and R. H. Baughman, Mol. Cryst. Liq. Cryst., 1985, 118, 111–116 CrossRef CAS.
- M. Lemaire, D. Delabouglise, R. Garreau, A. Guy and J. Roncali, J. Chem. Soc., Chem. Commun., 1985, 658–661 Search PubMed.
- M. Salmón and G. Bidan, J. Electrochem. Soc., 1985, 132, 1897–1899 CrossRef.
- M. R. Majidi, L. A. P. Kane-maguire and G. G. Wallace, Polymer, 1994, 35, 3113–3115 CrossRef CAS.
- E. E. Havinga, M. M. Bouman, E. W. Meijer, A. Pomp and M. M. J. Simenon, Synth. Met., 1994, 66, 93–97 CrossRef CAS.
- M. R. Majidi, L. A. P. Kane-Maguire and G. G. Wallace, Aust. J. Chem., 1998, 51, 23–30 CrossRef CAS.
- K. Akagi, G. Piao, S. Kaneko, K. Sakamaki, H. Shirakawa and M. Kyotani, Science, 1998, 282, 1683–1686 CrossRef CAS PubMed.
- M. Oda, H.-G. Nothofer, G. Lieser, U. Scherf, S. C. J. Meskers and D. Neher, Adv. Mater., 2000, 12, 362–365 CrossRef CAS.
- M. Oda, H. G. Nothofer, U. Scherf, V. Šunjić, D. Richter, W. Regenstein and D. Neher, Macromolecules, 2002, 35, 6792–6798 CrossRef CAS.
- H. Goto and K. Akagi, Macromolecules, 2005, 38, 1091–1098 CrossRef CAS.
- G. Bidan, S. Guillerez and V. Sorokin, Adv. Mater., 1996, 8, 157–160 CrossRef CAS.
- H. Goto, Y. Okamoto and E. Yashima, Macromolecules, 2002, 35, 4590–4601 CrossRef CAS.
- B. M. W. Langeveld-Voss, R. A. J. Janssen, M. P. T. Christiaans, S. C. J. Meskers, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1996, 118, 4908–4909 CrossRef CAS.
- S. Yorozuya, I. Osaka, A. Nakamura, Y. Inoue and K. Akagi, Synth. Met., 2003, 135–136, 93–94 CrossRef CAS.
- K. Watanabe, I. Osaka, S. Yorozuya and K. Akagi, Chem. Mater., 2012, 24, 1011–1024 CrossRef CAS.
- H. Goto, Y. S. Jeong and K. Akagi, Macromol. Rapid Commun., 2005, 26, 164–167 CrossRef CAS.
- Y. Sulaiman and R. Kataky, Analyst, 2012, 137, 2386–2393 RSC.
- P. R. Teasdale and G. G. Walfacet, Analyst, 1993, 118, 329–334 RSC.
- E. M. Sheridan and C. B. Breslin, Electroanalysis, 2005, 17, 532–537 CrossRef CAS.
- K. Shimomura, T. Ikai, S. Kanoh, E. Yashima and K. Maeda, Nat. Chem., 2014, 6, 429–434 CrossRef CAS PubMed.
- C. Zhang, H. Wang, Q. Geng, T. Yang, L. Liu, R. Sakai, T. Satoh, T. Kakuchi and Y. Okamoto, Macromolecules, 2013, 46, 8406–8415 CrossRef CAS.
- R. Liu, M. Shiotsuki, T. Masuda and F. Sanda, Macromolecules, 2009, 42, 6115–6122 CrossRef CAS.
- K. Akagi, S. Guo, T. Mori, M. Goh, G. Piao and M. Kyotani, J. Am. Chem. Soc., 2005, 127, 14647–14654 CrossRef CAS PubMed.
- S. Y. Oh, K. Akagi and H. Shirakawa, Macromolecules, 1993, 26, 6203–6206 CrossRef CAS.
- P. Novak, K. Muller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–281 CrossRef CAS PubMed.
- H. Hayasaka, T. Miyashita, K. Tamura and K. Akagi, Adv. Funct. Mater., 2010, 20, 1243–1250 CrossRef CAS.
- C. Song, C. Zhang, F. Wang, W. Yang and J. Deng, Polym. Chem., 2013, 4, 645–652 RSC.
- M. H. El-Newehy, A. S. Elsherbiny and H. Mori, J. Appl. Polym. Sci., 2013, 127, 4918–4926 CrossRef CAS.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- H. Goto and K. Akagi, Macromol. Rapid Commun., 2004, 25, 1482–1486 CrossRef CAS.
- M. Ding, Prog. Polym. Sci., 2007, 32, 623–668 CrossRef CAS.
- Y. S. Jeong and K. Akagi, J. Mater. Chem., 2011, 21, 10472–10481 RSC.
- H. Goto, J. Mater. Chem., 2009, 19, 4914–4921 RSC.
- T. Hatano, A. H. Bae, M. Takeuchi, N. Fujita, K. Kaneko, H. Ihara, M. Takafuji and S. Shinkai, Angew. Chem., Int. Ed., 2004, 43, 465–469 CrossRef CAS PubMed.
- L. Dong, B. Lu, X. Duan, J. Xu, D. Hu, K. Zhang, X. Zhu, H. Sun, S. Ming, Z. Wang and S. Zhen, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2238–2251 CrossRef CAS.
- D. Hu, L. Zhang, K. Zhang, X. Duan, J. Xu, L. Dong, H. Sun, X. Zhu and S. Zhen, J. Appl. Polym. Sci., 2015, 132, 41559 Search PubMed.
- D. W. Armstrong, W. Demond and B. P. Czech, Anal. Chem., 1985, 57, 481–484 CrossRef CAS.
- L. Zhang, M. Song, Q. Tian and S. Min, Sep. Purif. Technol., 2007, 55, 388–391 CrossRef CAS.
- Q. Fu, H. Sanbe, C. Kagawa, K.-K. Kunimoto and J. Haginaka, Anal. Chem., 2003, 75, 191–198 CrossRef CAS PubMed.
- K. Bodenhofer, A. Hierlemann, J. Seemann, G. Gauglitz, B. Koppenhoefer and W. Gopel, Nature, 1997, 387, 577–580 CrossRef CAS PubMed.
- V. Schurig and W. Buerkle, J. Am. Chem. Soc., 1982, 104, 7573–7580 CrossRef CAS.
- V. Schurig, J. Chromatogr. A, 2001, 906, 275–299 CrossRef CAS PubMed.
- Z. Chen, T. Nishiyama, K. Uchiyama and T. Hobo, Anal. Chim. Acta, 2004, 501, 17–23 CrossRef CAS.
- Y. Liu, A. Berthod, C. R. Mitchell, T. L. Xiao, B. Zhang and D. W. Armstrong, J. Chromatogr. A, 2002, 978, 185–204 CrossRef CAS PubMed.
- Y. Zhao, W. A. Pritts and S. Zhang, J. Chromatogr. A, 2008, 1189, 245–253 CrossRef CAS PubMed.
- A. Balamurugan, V. Kumar and M. Jayakannan, Chem. Commun., 2014, 50, 842–845 RSC.
- H. Ito and S. Shinoda, Chem. Commun., 2015, 51, 3808–3811 RSC.
- X. Liu and I. P. Hamilton, J. Am. Chem. Soc., 2014, 136, 17757–17761 CrossRef CAS PubMed.
- R. Kuhn, F. Erni, T. Bereuter and J. Haeusler, Anal. Chem., 1992, 64, 2815–2820 CrossRef CAS.
- L. Sánchez-Hernández, M. Guijarro-Diez, M. L. Marina and A. L. Crego, Electrophoresis, 2014, 35, 12–27 CrossRef PubMed.
- N. M. Maier and W. Lindner, Anal. Bioanal. Chem., 2007, 389, 377–397 CrossRef CAS PubMed.
- K. C. Ho, W. M. Yeh, T. S. Tung and J. Y. Liao, Anal. Chim. Acta, 2005, 542, 90–96 CrossRef CAS.
- Y. Huang, Q. Han, Q. Zhang, L. Guo, D. Guo and Y. Fu, Electrochim. Acta, 2013, 113, 564–569 CrossRef CAS.
- E. Zor, H. Bingol, A. Ramanaviciene, A. Ramanavicius and M. Ersoz, Analyst, 2015, 140, 313–321 RSC.
- Y. J. Kang, J. W. Oh, Y. R. Kim, J. S. Kim and H. Kim, Chem. Commun., 2010, 46, 5665–5667 RSC.
- Y. Huang, D. Guo, Q. Zhang, L. Guo, Y. Chen and Y. Fu, RSC Adv., 2014, 4, 33457–33461 RSC.
- S. Pleus and M. Schwientek, Synth. Met., 1998, 95, 233–238 CrossRef CAS.
- Y. Yao, L. Zhang, Z. Wang, J. Xu and Y. Wen, Chin. Chem. Lett., 2014, 25, 505–510 CrossRef CAS.
- L. Zhang, Y. Wen, Y. Yao, X. Duan, J. Xu and X. Wang, J. Appl. Polym. Sci., 2013, 130, 2660–2670 CrossRef CAS.
- L. Zhang, Y. Wen, Y. Yao, Z. Wang, X. Duan and J. Xu, Chin. Chem. Lett., 2014, 25, 517–522 CrossRef CAS.
- B. Lu, S. Zhang, L. Qin, S. Chen, S. Zhen and J. Xu, Electrochim. Acta, 2013, 106, 201–208 CrossRef CAS.
- Z. Wang, J. Xu, B. Lu, S. Zhang, L. Qin, D. Mo and S. Zhen, Langmuir, 2014, 30, 15581–15589 CrossRef CAS PubMed.
- L. Zhang, Y. Wen, Y. Yao, J. Xu, X. Duan and G. Zhang, Electrochim. Acta, 2014, 116, 343–354 CrossRef CAS.
- Y. Yao, Y. Wen, L. Zhang, Z. Wang, H. Zhang and J. Xu, Anal. Chim. Acta, 2014, 831, 38–49 CrossRef CAS PubMed.
- S. Ming, S. Zhen, K. Lin, L. Zhao, J. Xu and B. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 11089–11098 CAS.
- Q. Peil, G. Zuccarello, M. Ahlskog and O. Inganäs, Polymer, 1994, 35, 1347–1351 CrossRef.
- B. Lu, J. Yan, J. Xu, S. Zhou and X. Hu, Macromolecules, 2010, 43, 4599–4608 CrossRef CAS.
- Y. S. Jeong and K. Akagi, Macromolecules, 2011, 44, 2418–2426 CrossRef CAS.
- G. Inzelt, M. Pineri, J. W. Schultze and M. A. Vorotyntsev, Electrochim. Acta, 2000, 45, 2403–24221 CrossRef CAS.
- F. Uckert, Y. H. Tak, K. Müllen and H. Bässler, Adv. Mater., 2000, 12, 905–908 CrossRef CAS.
- R. Yue, B. Lu, J. Xu, S. Chen and C. Liu, Polym. J., 2011, 43, 531–539 CrossRef CAS.
- S. Zhang, J. Hou, R. Zhang, J. Xu, G. Nie and S. Pu, Eur. Polym. J., 2006, 42, 149–160 CrossRef CAS.
- Z. Wang, J. Xu, Y. Yao, L. Zhang, Y. Wen, H. Song and D. Zhua, Sens. Actuators, B, 2014, 196, 357–369 CrossRef CAS.
- Q. Han, Q. Chen, Y. Wang, J. Zhou and Y. Fu, Electroanalysis, 2012, 24, 332–337 CrossRef CAS.
- M. Eslami, M. Namazian and H. R. Zare, Electrochim. Acta, 2013, 88, 543–551 CrossRef CAS.
- R. B. Bazaco, R. Gomez, C. Seoane, P. Bauerle and J. L. Segura, Tetrahedron Lett., 2009, 50, 4154–4157 CrossRef CAS.
- L. Chen, F. Chang, L. Meng, M. Li and Z. Zhu, Analyst, 2014, 139, 2243–2248 RSC.
- K. Chun, T. H. Kim, O. S. Lee, K. Hirose, T. D. Chung, D. S. Chung and H. Kim, Anal. Chem., 2006, 78, 7597–7600 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of (R)-and (S)-EDTC, table of assignment for FT-IR spectra, and table of thermogravimetric parameters for (R)- and (S)-PEDTC. See DOI: 10.1039/c5ra20871h |
| This journal is © The Royal Society of Chemistry 2016 |