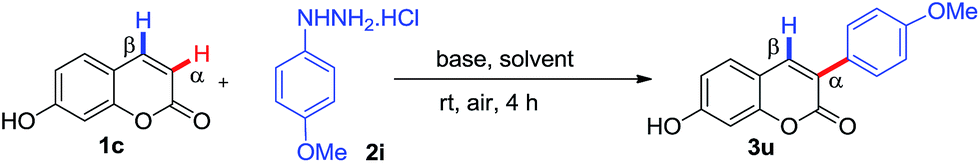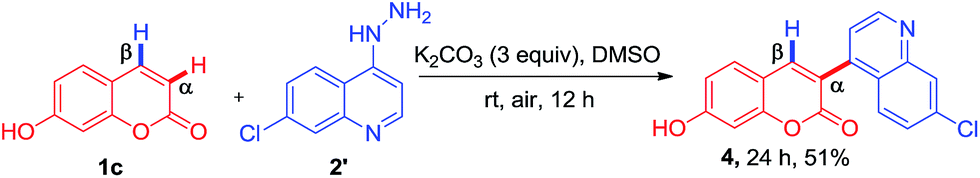DOI:
10.1039/C5RA20954D
(Paper)
RSC Adv., 2016,
6, 109-118
Regioselective α-arylation of coumarins and 2-pyridones with phenylhydrazines under transition-metal-free conditions†
Received
9th October 2015
, Accepted 10th December 2015
First published on 15th December 2015
Abstract
A facile regioselective metal-free direct α-arylation of coumarins and 2-pyridones is achieved by the reaction of coumarins and 2-pyridones with phenylhydrazine in good yields. The reaction proceeds at room temperature under mild conditions using inexpensive reagents and without the need for step intensive activating groups. The methodology is operationally simple, practically viable and also allows the coupling of similar nitrogen heterocycle aza-coumarins without prerequisite N-protection.
Metal-free direct C–H arylation processes have started to emerge as straightforward, atom-economical and environment-friendly research.1 These methods are an alternative to metal-catalyzed processes, which are associated with limitations such as step and cost intensive procedures along with the use of toxic, sensitive and environmentally hazardous metals and ligands.2 In the recent past, radical C–H functionalization of innately reactive heterocycles has re-emerged as an avenue for selective, early or late stage functionalization of pharmaceutically important precursors and products.3 Radical-mediated metal-free C–H arylations have been achieved substantially utilizing a variety of aryl coupling partners4 and among the notable radical-mediated aryl coupling partners, base-mediated arylhydrazine derived aryl radicals have emerged as powerful synthetic intermediate towards the direct C–H arylation under oxidative conditions with limited substrate scope.5 However, the utility of these intermediates with robust control of regioselectivity in oxidative C–H arylation reactions of biologically prominent and synthetically useful motifs is highly challenging and valuable.
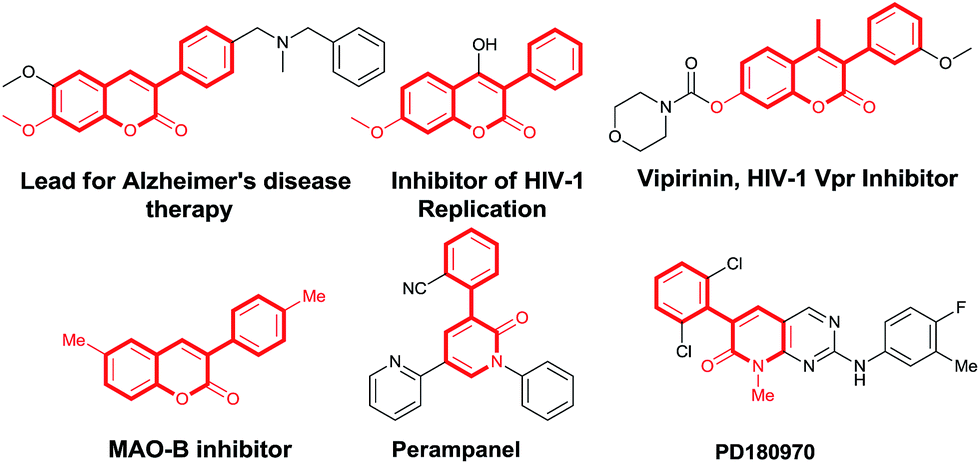
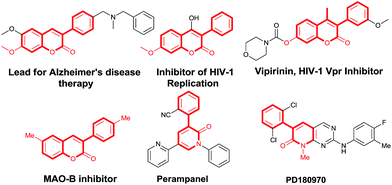
3-Arylcoumarins and 3-aryl-2-pyridones are important scaffolds present in many natural products and bioactive compounds.6 In particular, α-arylated coumarin derivatives have been shown to be potent, selective monoamine oxidase B inhibitors,7 active in neurological disorders8 and inhibitors of HIV-1 replication (Fig. 1).9 Furthermore, 3-arylcoumarins are also important class of fluorophores and found their application as fluorescent labels in complex biological systems.10 On the other hand, the 3-aryl-2-pyridones, which is key pharmacophore of pharmaceutically important molecules such as perampanel, and PD180970 (Fig. 1).11 Despite the importance of 3-aryl coumarins and 3-aryl-2-pyridones as biologically active scaffolds, the α-arylation of coumarins and 2-pyridones are difficult due to regioselectivity bias of β-arylation of coumarins and γ-arylation of 2-pyridones, which is expected in Heck-type reaction.12 Radical mediated C–H arylation methods proved as a convenient method because of the inherent reactivity of heterocycles towards aryl radical species. The α-regioselectivity in case of coumarins/aza-coumarins and 2-pyridones towards aryl radical is due to the formation of resonance stabilized benzylic radical13 and allylic radical14 intermediates respectively. Herein, we report a K2CO3/air promoted regioselective method for direct α-arylation of biologically prominent heterocycles such as coumarins, 2-pyridones and aza-coumarins without prerequisite N-protection under metal and ligand-free condition.
 |
| | Fig. 1 3-Arylcoumarin and 3-aryl-2-pyridone based bioactive molecules. | |
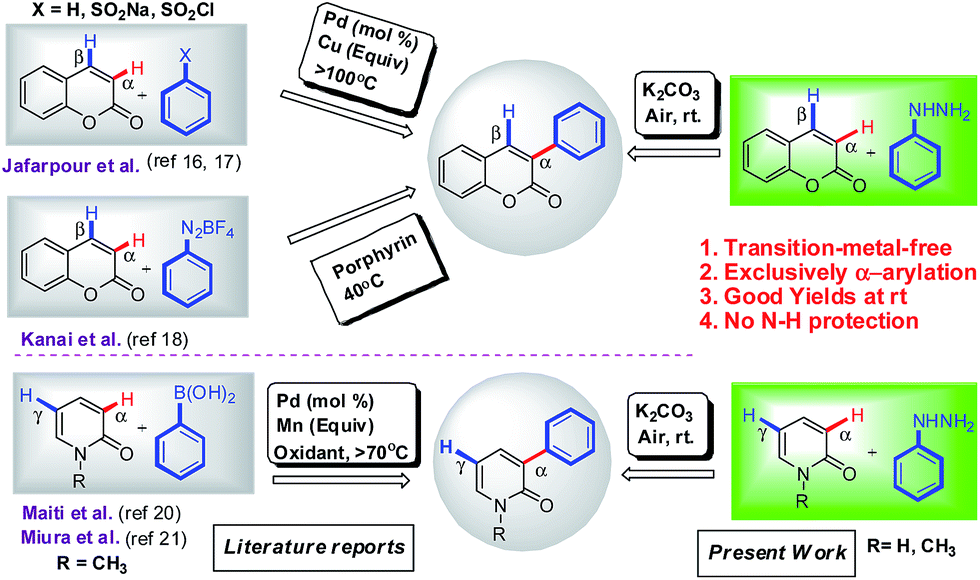
Mostly the α-arylation of coumarins was achieved either from suitable phenyl substituted precursors15 or require prefunctionalization at α-position such as in palladium-catalyzed Suzuki coupling of 3-halo coumarin with arylboronic acid16 and palladium-catalyzed decarboxylative coupling of coumarin-3-carboxylic acid with haloarenes.17 The most notable methodologies for regioselective α-arylation were desulfitative coupling of coumarins with arenesulfonyl chloride,18 dehydrogenative cross-coupling of coumarins with arenes via two-fold C–H activation under palladium catalyst19 and recently Kanai and co-workers reported porphyrin-mediated Meerwein arylation of coumarins which suffer from some disadvantages such as synthesis and solubility of organic electron mediators and use of unstable and explosive aryl coupling partners (arenediazonium salts).13 In recent years, only a few reports are available for metal-catalyzed regioselective α-arylation of N-protected 2-pyridones (i) Suzuki-coupling of 5-halouracil with arylboronic acid by Wnuk group,20 (ii) iron-catalyzed regioselective direct α-arylation of N-alkyl-2-pyridones with aryl boronic acid using K2S2O8 as an oxidant and K2CO3 as base at 70 °C by Maiti group,21 and manganese-mediated α-selective direct arylation of N-alkyl-2-pyridone with arylboronic acid under N2 condition at 90 °C by Miura group.14 Although all the above methodologies have been utilized effectively for α-arylation of coumarins and 2-pyridones, but are limited by the prefunctionalization, use of transition-metals (Cu or Ag was used in equivalents), use of ligands, high temperatures and low yields (Scheme 1). Thus, the development of an efficient transition-metal-free methodology towards regioselective α-arylation of coumarins and 2-pyridones is highly desirable.
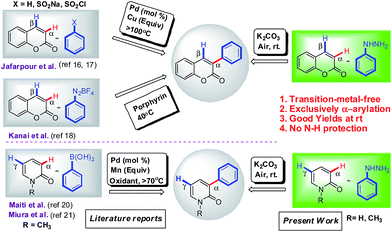 |
| | Scheme 1 Strategic approaches for the synthesis of 3-arylcoumarins and 3-aryl-2-pyridones. | |
As part of our continuous efforts for the development of efficient C–H arylation and coupling processes for biologically important motifs,5c,22 we pursued the synthetic challenge to identify a practically viable metal-free regioselective α-arylation of coumarin and 2-pyridone with phenylhydrazines. We initially investigated the α-arylation of 7-hydroxy coumarin 1c with 4-methoxy phenylhydrazine 2i in the presence of K2CO3 in DMSO (dimethyl sulfoxide) at room temperature under air for 4 hours. To our delight, the reaction yielded only α-arylated product 3u with 85% yield (Table 1, entry 1) and no C-4 arylated product was observed.
Table 1 Optimization of the reaction conditionsa
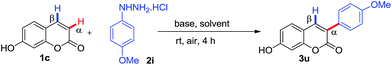
|
| Entry |
Base (3 equiv.) |
Solvent |
Yieldb (%) |
| Reaction condition: 1c (1.0 mmol), 2h (1.2 mmol), K2CO3 (3.0 mmol), DMSO (15 mL) at rt. Isolated yield. 70% aq. TBHP was used. Under nitrogen atmosphere. Under O2 atmosphere. |
| 1 |
K2CO3 |
DMSO |
85 |
| 2 |
Na2CO3 |
DMSO |
30 |
| 3 |
Ag2CO3 |
DMSO |
35 |
| 4 |
Cs2CO3 |
DMSO |
60 |
| 5 |
NaOEt |
DMSO |
55 |
| 6 |
KtOBt |
DMSO |
30 |
| 7 |
PIDA |
DMSO |
35 |
| 8 |
TBHP |
DMSO |
20c |
| 9 |
K2S2O8 |
DMSO |
30 |
| 10 |
DTBP |
DMSO |
— |
| 11 |
I2 |
DMSO |
— |
| 12 |
— |
DMSO |
— |
| 13 |
K2CO3 |
DMF |
35 |
| 14 |
K2CO3 |
DMA |
30 |
| 15 |
K2CO3 |
NMP |
20 |
| 16 |
K2CO3 |
MeOH |
43 |
| 17 |
K2CO3 |
DMSO |
24d |
| 18 |
K2CO3 |
DMSO |
84e |
Encouraged by above promising results in hand, further a set of experiments was performed to optimize the reaction conditions with various bases and oxidants (Table 1). We screened different bases such as Na2CO3, Ag2CO3, Cs2CO3, NaOEt and KtOBt (Table 1, entries 2–6) and none of them were found to be better than K2CO3. The efficiency of other oxidants such as (diacetoxyiodo)benzene (PIDA), tert-butyl hydroperoxide (TBHP), K2S2O8, di-tert-butyl peroxide (DTBP) and molecular iodine were checked, however, none of them were found to give better yield than K2CO3/air system (Table 1, entries 7–11 versus entry 1). In the absence of K2CO3 no product was observed (Table 1, entry 12), which clearly indicated that the base is essential for this reaction. Different solvents other than DMSO were also screened such as DMF (dimethylformamide), DMA (dimethylacetamide), NMP (N-methylpyrrolidone) and MeOH (methanol) but all of them produced lower yield of product 3u (Table 1, entries 1 and 13–16). To check the essentiality of air as an oxidant in this transformation, we performed the reaction with degassed DMSO under N2 atmosphere. The yield of product 3u decreased to 24%, which clearly indicated that oxygen is crucial for the reaction (Table 1, entry 17). In the presence of O2 (ballon) similar yield of product 3u was observed (Table 1, entry 18).
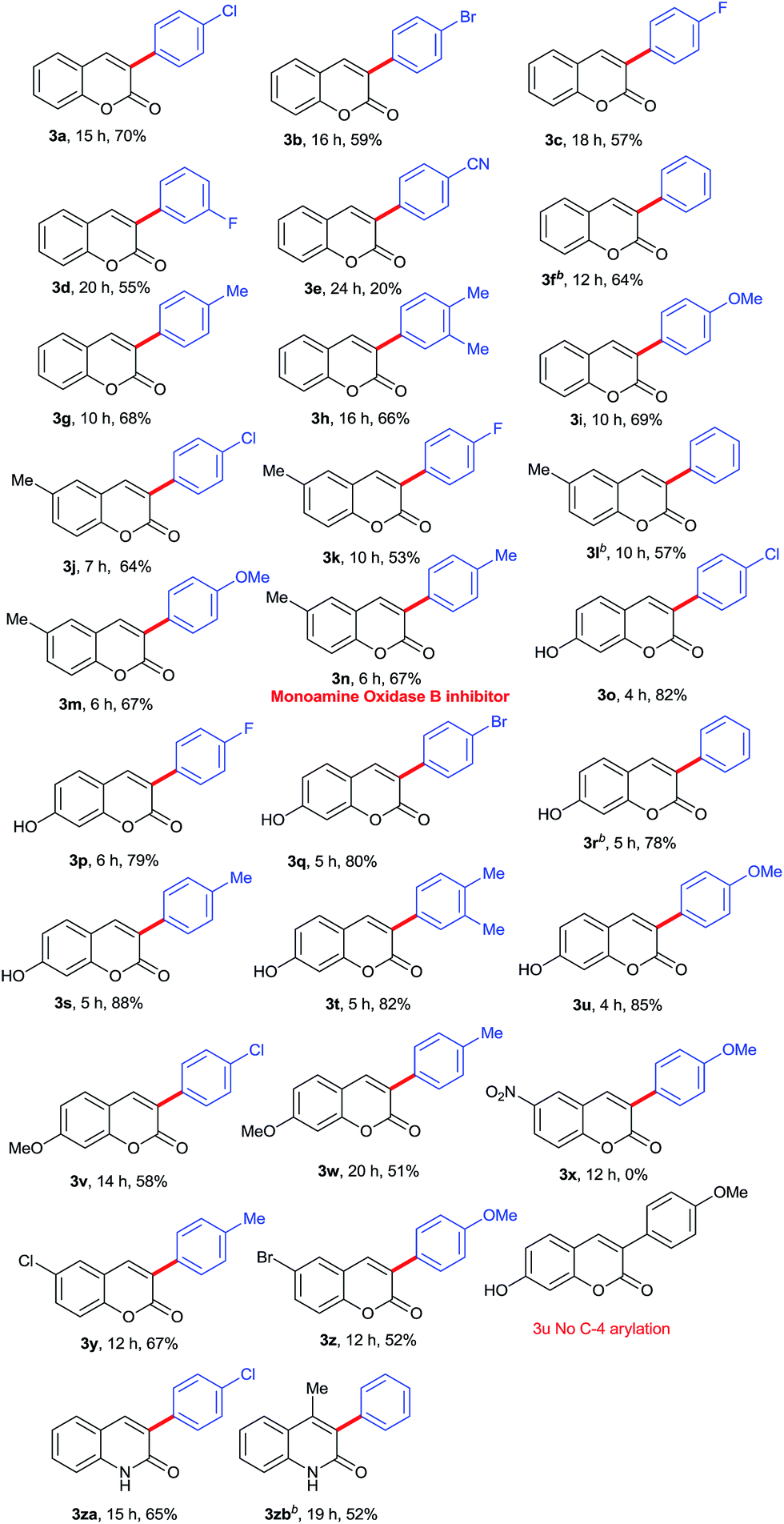
After optimization results in hand, we turned our attention towards the scope of the substrate in metal-free regioselective α-arylation of coumarins with various phenylhydrazines. The phenylhydrazines having electron-withdrawing groups such as 4-F, 4-Cl, 4-Br, 3F (Table 2, product 3a–3d) and electron-donating groups such as 4-Me, 3,4-diMe, 4-OMe (Table 2, product 3g–3i) respectively on the aromatic ring, afforded the α-arylated products in good to excellent yields. However, 4-cyano substituted phenylhydrazine produced the α-arylated product in 20% yield (Table 2, product 3e). Electron donating groups (such as OMe, Me) at C-4 of phenylhydrazine reduce the reaction time (Table 2, products 3g & 3i), however 4-CN function in the phenylhydrazine afforded only 20% yield in 24 h (Table 2, product 3e) and further no product was observed in the case of 4-nitrophenylhydrazine even after prolonged reaction time.
Table 2 Scope of regioselective α-arylation of coumarin and aza-coumarina
|
| Reaction condition: 1 (1.0 mmol), 2 (1.2 mmol), K2CO3 (3.0 mmol), DMSO (15 mL) at rt. Isolated yields are given. Phenylhydrazine was used as free base. |
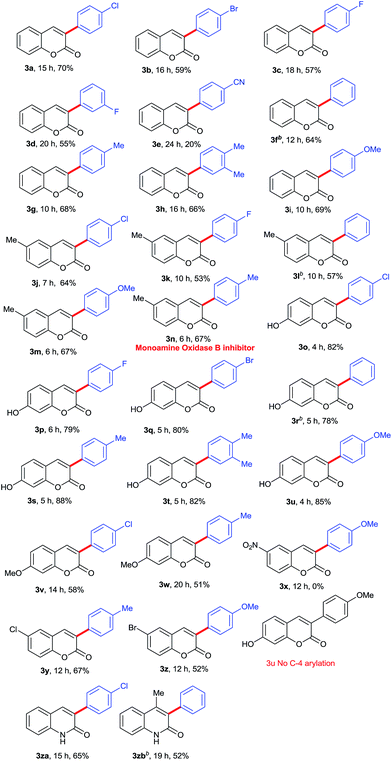 |
Further, the scope of the methodology was investigated with respect to coumarins. The coumarin moiety bearing electron-donating groups such as methyl, methoxy and hydroxyl at the C-6 or C-7 position afforded their corresponding α-arylated products in moderate to good yields (Table 2, products 3j–3w), however the reaction time in case of hydroxyl substituted coumarins at C-7 was considerably less (4–6 h) (Table 2, products 3o–3u) as compared to C-7 methoxy (14–20 h) (Table 2, products 3v & 3w) and C-6 methyl (6–10 h) (Table 2, products 3j–3n) substituted coumarins. Notably, the nitro group at 6-position of coumarin did not furnish the α-arylated product (Table 2, product 3x). The C-6 halogen (Cl and Br) containing coumarins with C-4 electron-donating (methyl and methoxy) phenylhydrazines afforded α-arylated product 67% and 52% respectively (Table 2, product 3y and 3z), however the substrates possessing electron withdrawing groups (Cl and Br) both on C-6 of coumarins and C-4 of arylhydrazines, drastically affect the reaction outcome where no product was observed. To check the feasibility of regioselective arylation on aza-coumarins, we tested the reaction of unprotected 2-hydroxyqyinolines with phenylhydrazines under optimized condition. Pleasingly, the reaction proceeded smoothly without N-protection and afforded regioselectively α-arylated products in moderate yields (Table 2, products 3za and 3zb).
Subsequently, the compatibility of present methodology was further tested with heteroaromatic hydrazine, since the heteroaromatic substitutions on bioactive molecules impart desirable pharmacological and pharmacokinetic properties. To achieve the unique substitution at α-position, we attempted the reaction of 4-hydrazinoquinoline with 7-hydroxycoumarin under the optimized condition. To our delight, the reaction afforded the compound 4 α-(7-chloroquinolin-4-yl)-7-hydroxy-2H-chromen-2-one, a unique hybrid of quinoline and coumarin in 51% yield (Scheme 2).
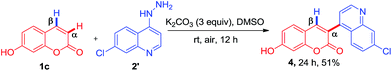 |
| | Scheme 2 Synthesis of 3-(7-chloroquinolin-4-yl)-7-hydroxy-2H-chromen-2-one. | |
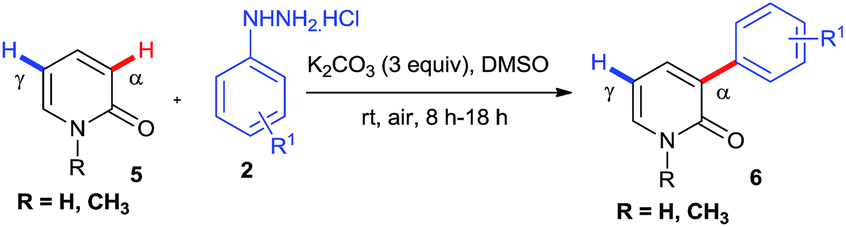
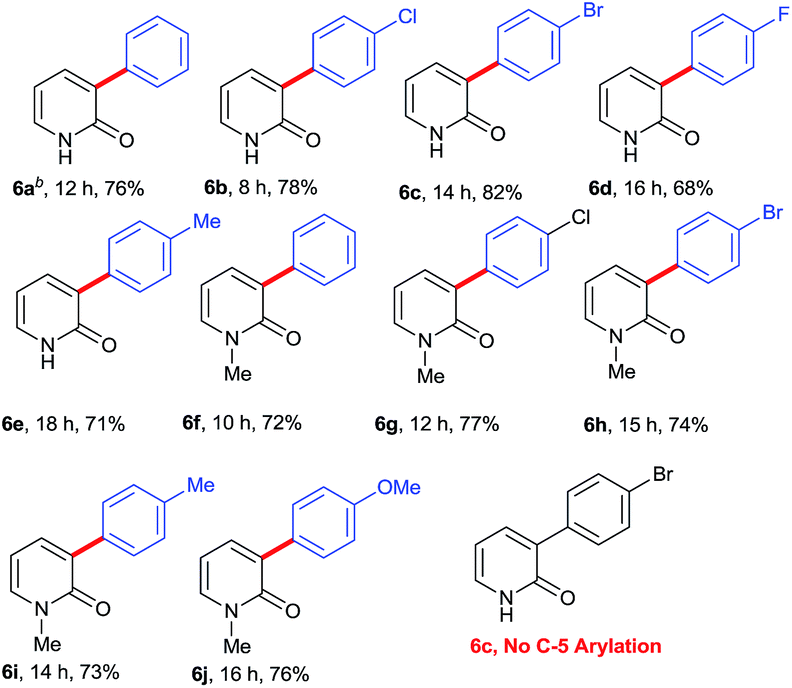
Successful installation of α-arylation in aza-coumarins inspired us to expand the applicability of the method towards metal-free regioselective arylation of the 2-pyridone moiety. To the best of our knowledge, this is the first metal-free report for α-C–H arylation of 2-pyridone. Gratifyingly the reaction of 2-pyridones with phenylhydrazines afforded exclusively α-arylated products in good to excellent yields (Table 3, product 6a–6j). It was noteworthy that the reaction proceeded with or without N–H protection of 2-pyridones at room temperature and also compatible with both electron withdrawing and electron donating substitutions on phenylhydrazine.
Table 3 Scope of regioselective α-arylation of 2-pyridonea
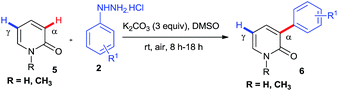
|
| Reaction condition: 5 (1.0 mmol), 2 (1.2 mmol), K2CO3 (3.0 mmol), DMSO (15 mL) at rt. Isolated yields are given. Phenylhydrazine was used as free base. |
 |
To gain insight into the mechanism, we performed control experiment for radical trapping. The reaction of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) with 4-cyano phenylhydrazine 2e afforded adduct 4-((2,2,6,6-tetramethylpiperidin-1-yl)oxy)benzonitrile 7 in 64% yield (Scheme 3).
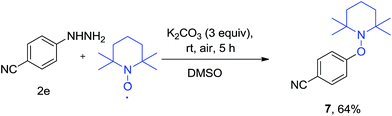 |
| | Scheme 3 Radical trapping experiment with TEMPO. | |
Based on our experiment, previous literature reports14,20,23–27 and our previous work,5c a possible mechanism has been proposed in Scheme 3. Initially, the treatment of phenylhydrazine hydrochloride with base afforded phenylhydrazine [A], which further undergoes oxidation in the presence of base/O2 to form aryl radical [B]24,27 by removal of N2 and then aryl radical addition to coumarin/aza-coumarin (1) and 2-pyridone (4) afforded benzylic radical13 intermediates [C] and allylic radical14 intermediate [D] respectively, which were stabilized by resonance. Upon further oxidation,26 intermediates [C] and [D] leads to α-arylated product of coumarin, aza-coumarin (3) and pyridone (5) (Scheme 4).
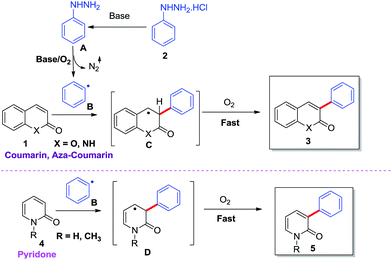 |
| | Scheme 4 Plausible mechanism. | |
Conclusions
In conclusion, we have achieved a transition-metal-free regioselective α-arylation of coumarins, 2-pyridones and aza-coumarin via base/air-promoted denitrogenative coupling of phenylhydrazines in moderate to good yields. High functional group tolerability with excellent regioselectivity and room temperature reaction condition are salient features of the present strategy.
Experimental
General information
All glass apparatus were oven dried prior to use. 1H NMR and 13C NMR spectra were recorded on Bruker Avance-400 using CDCl3 and DMSO-d6 as solvent and tetramethylsilane as internal reference. Splitting patterns are described as singlet (s), doublet (d), triplet (t), multiplet (m) and broad (br). IR spectra were recorded on a FT-IR spectrophotometer and melting points were determined on melting point apparatus and are uncorrected. Electrospray ionization mass spectrometry (ESI-MS) was obtained on Thermo LCQ Advantage Max Spectrometer and HRMS was recorded on Agilent 6520 Q-TOF. Reactions were monitored on silica gel TLC plates (coated with TLC grade silica gel, obtained from Merck). Detecting agents used (for TLC) were: iodine vapors and/or spraying with an aq. solution of vanillin in 10% sulfuric acid followed by heating at 100 °C. Column chromatography was performed over silica gel (230–400 mesh) by using Smart flash EPCLC AI-700X YAMAZEN with minimal amount of solvent. All chemicals and reagents were obtained from Aldrich, Alfa Aesar and were used without further purification.
General experimental procedures
Representative procedure for the synthesis of product 3a. A solution of coumarin 1a (146 mg, 1.0 mmol), phenylhydrazine 2a (215 mg, 1.2 mmol) and potassium carbonate (408 mg, 3.0 mmol) in dimethyl sulphoxide (15 mL) was stirred at room temperature for 15 hours. Next, the reaction was diluted with water (80 mL) and the aqueous layer was extracted with ethyl acetate (3 × 20 mL). The organic layer was further dried over sodium sulphate (anhydrous) and concentrated under reduced pressure to give crude product, which upon purification by column chromatography over silica gel (gradient elution with hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate, 9:1 to 7:3) afforded the desired product 179 mg (70%) of 3a as white solid. The remaining reactions were carried out following this procedure for products 3b–3zb.
:ethyl acetate, 9:1 to 7:3) afforded the desired product 179 mg (70%) of 3a as white solid. The remaining reactions were carried out following this procedure for products 3b–3zb.
Representative procedure for the synthesis of product 6b. A solution of 2-pyridone 5a (95 mg, 1.0 mmol), phenylhydrazine 2b (215 mg, 1.2 mmol) and potassium carbonate (408 mg, 3.0 mmol) in dimethyl sulphoxide (15 mL) was stirred at room temperature for 8 hours. Next, the reaction was diluted with water (80 mL) and the aqueous layer was extracted with ethyl acetate (3 × 20 mL). The organic layer was further dried over sodium sulphate (anhydrous) and concentrated under reduced pressure to give crude product, which was further purified by column chromatography over silica gel to afford the desired product 160 mg (78%) of 6b as white solid. The remaining reactions were carried out following this procedure for products 6a and 6c–6j.
Representative for the synthesis of product 7. A solution of phenylhydrazine 2e (169 mg, 1.0 mmol), 4-((2,2,6,6-tetramethylpiperidin-1-yl)oxy)benzonitrile (234 mg, 1.5 mmol) and potassium carbonate (408 mg, 3.0 mmol) in dimethyl sulphoxide (20 mL) was stirred at room temperature for 5 hours. Next, the reaction was diluted with water (100 mL) and the aqueous layer was extracted with ethyl acetate (3 × 20 mL). The organic layer was further dried over sodium sulphate (anhydrous) and concentrated under reduced pressure to give crude product, which was further purified by column chromatography over silica gel to afford the desired product 165 mg (64%) of 7 as white solid.
Compound characterization data
3-(4-Chlorophenyl)-2H-chromen-2-one (3a)28. Yield 70% as white solid: mp 180–182 °C; FT-IR (KBr, cm−1) 3019, 1618, 1210, 1076; 1H NMR (400 MHz, CDCl3) δ 7.29–7.33 (m, 1H), 7.36–7.39 (m, 1H), 7.41–7.44 (m, 2H), 7.53–7.57 (m, 2H), 7.66–7.68 (m, 2H), 7.82 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 116.54 (CH), 119.51 (C), 124.63 (CH), 127.20 (C), 127.98 (CH), 128.71 (2 × CH), 129.85 (2 × CH), 131.68 (CH), 133.10 (C), 134.96 (C), 139.91 (CH), 153.58 (C), 160.34 (C); ESI-MS (m/z) 257 (M + H)+; HRMS (ESI) calculated for C15H10ClO2 (M + H)+ 257.0369, found 257.0367.
3-(4-Bromophenyl)-2H-chromen-2-one (3b)28. Yield 59% as light yellow solid: mp 190–192 °C; FT-IR (KBr, cm−1) 3019, 1610, 1215, 1069; 1H NMR (400 MHz, CDCl3) δ 7.29–7.33 (m, 1H), 7.36–7.39 (m, 1H), 7.53–7.62 (m, 6H), 7.82 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 116.55 (CH), 119.50 (C), 123.21 (C), 124.65 (CH), 127.23 (C), 127.99 (CH), 130.12 (2 × CH), 131.67 (2 × CH), 131.72 (CH), 133.57 (C), 139.93 (CH), 153.58 (C), 160.27 (C); ESI-MS (m/z) 301 (M + H)+; HRMS (ESI) calculated for C15H10BrO2 (M + H)+ 300.9864, found 300.9862.
3-(4-Fluorophenyl)-2H-chromen-2-one (3c)28. Yield 57% as white solid: mp 158–160 °C; FT-IR (KBr, cm−1) 3021, 1609, 1215, 1113; 1H NMR (400 MHz, CDCl3) δ 7.11–7.17 (m, 2H), 7.29–7.33 (m, 1H), 7.37–7.39 (m, 1H), 7.52–7.56 (m, 2H), 7.69–7.72 (m, 2H), 7.79 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 115.60 (d, J = 21.47 Hz, 2 × CH), 116.49 (CH), 119.58 (C), 124.59 (CH), 127.32 (C), 127.91 (CH), 130.47 (d, J = 8.15 Hz, 2 × CH), 130.71 (C), 131.52 (CH), 139.69 (CH), 153.51 (C), 160.55 (C), 164.33 (d, J = 247.39 Hz, C–F); ESI-MS (m/z) 241 (M + H)+; HRMS (ESI) calculated for C15H10FO2 (M + H)+ 241.0665, found 241.0668.
3-(3-Fluorophenyl)-2H-chromen-2-one (3d)29. Yield 55% as white solid: mp 152–154 °C; FT-IR (KBr, cm−1) 3019, 1610, 1215, 1069; 1H NMR (400 MHz, CDCl3) δ 7.08–7.13 (m, 1H), 7.29–7.33 (m, 1H), 7.37–7.44 (m, 2H), 7.45–7.51 (m, 2H), 7.53–7.57 (m, 2H), 7.84 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 115.81 (d, J = 22.85 Hz, CH), 115.89 (d, J = 21.04 Hz, CH), 116.54 (CH), 119.42 (C), 124.19 (d, J = 2.86 Hz, CH), 124.65 (CH), 127.06 (C), 128.08 (CH), 130.03 (d, J = 8.37 Hz, CH), 131.82 (CH), 136.74 (d, J = 8.28 Hz, C), 140.40 (CH), 153.62 (C), 160.20 (C), 163.90 (d, J = 244.27 Hz, C–F); ESI-MS (m/z) 241 (M + H)+; HRMS (ESI) calculated for C15H10FO2 (M + H)+ 241.0665, found 241.0659.
4-(2-Oxo-2H-chromen-3-yl)benzonitrile (3e)30. Yield 20% as white solid: mp 152–154 °C; FT-IR (KBr, cm−1) 3019, 2230, 1614, 1215, 1068; 1H NMR (300 MHz, CDCl3) δ 7.31–7.41 (m, 2H), 7.57–7.62 (m, 2H), 7.72–7.76 (m, 2H), 7.83–7.87 (m, 2H), 7.89 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 112.46 (C), 116.68 (CH), 118.51 (C), 119.19 (C), 124.85 (CH), 126.47 (C), 128.29 (CH), 129.20 (2 × CH), 132.22 (2 × CH), 132.41 (CH), 139.18 (C), 141.31 (CH), 153.82 (C), 159.87 (C); ESI-MS (m/z) 248 (M + H)+; HRMS (ESI) calculated for C16H10NO2 (M + H)+ 248.0712, found 248.0687.
3-Phenyl-2H-chromen-2-one (3f)28. Yield 64% as white solid: mp 135–137 °C; FT-IR (KBr, cm−1) 3019, 1613, 1215, 1069; 1H NMR (400 MHz, CDCl3) δ 7.28–7.32 (m, 1H), 7.37–7.48 (m, 4H), 7.51–7.56 (m, 2H), 7.69–7.72 (m, 2H), 7.82 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 116.49 (CH), 119.71 (C), 124.50 (CH), 127.91 (CH), 128.42 (C), 128.49 (2 × CH), 128.55 (2 × CH), 128.88 (CH), 131.41 (CH), 134.73 (C), 139.86 (CH), 153.56 (C), 160.60 (C); ESI-MS (m/z) 223 (M + H)+; HRMS (ESI) calculated for C15H11O2 (M + H)+ 223.0759, found 223.0763.
3-(p-Tolyl)-2H-chromen-2-one (3g)28. Yield 68% as white solid: mp 150–152 °C; FT-IR (KBr, cm−1) 3019, 1618, 1210, 1076; 1H NMR (400 MHz, CDCl3) δ 2.41 (s, 3H), 7.27–7.32 (m, 3H), 7.37 (d, J = 8.2 Hz, 1H), 7.51–7.55 (m, 2H), 7.62 (d, J = 7.76 Hz, 2H), 7.79 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.30 (CH3), 116.44 (CH), 119.80 (C), 124.44 (CH), 127.80 (CH), 128.41 (C & 2 × CH), 129.19 (2 × CH), 131.19 (CH), 131.83 (C), 138.94 (C), 139.19 (CH), 153.46 (C), 160.69 (C); ESI-MS (m/z) 237 (M + H)+; HRMS (ESI) calculated for C16H13O2 (M + H)+ 237.0916, found 237.0897.
3-(3,4-Dimethylphenyl)-2H-chromen-2-one (3h). Yield 66% as light yellow solid: mp 110–112 °C; FT-IR (KBr, cm−1) 3019, 1609, 1215, 1069; 1H NMR (400 MHz, CDCl3) δ 2.31 (s, 3H), 2.33 (s, 3H), 7.21 (d, J = 7.80 Hz, 1H), 7.27–7.31 (m, 1H), 7.35–7.37 (m, 1H), 7.43–7.46 (m, 1H), 7.48–7.54 (m, 3H), 7.78 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 19.61 (CH3), 19.89 (CH3), 116.42 (CH), 119.83 (C), 124.40 (CH), 125.99 (CH), 127.77 (CH), 128.50 (C), 129.61 (CH), 129.76 (CH), 131.11 (CH), 132.26 (C), 136.69 (C), 137.65 (C), 139.12 (CH), 153.44 (C), 160.72 (C); ESI-MS (m/z) 251 (M + H)+; HRMS (ESI) calculated for C17H15O2 (M + H)+ 251.1072, found 251.1069.
3-(4-Methoxyphenyl)-2H-chromen-2-one (3i)28. Yield 69% as white solid: mp 140–142 °C; FT-IR (KBr, cm−1) 3019, 1610, 1215, 1026; 1H NMR (400 MHz, CDCl3) δ 3.86 (s, 3H), 6.97–6.99 (m, 2H), 7.27–7.31 (m, 1H), 7.35–7.37 (m, 1H), 7.49–7.54 (m, 2H), 7.66–7.69 (m, 2H), 7.76 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 55.38 (CH3), 113.94 (2 × CH), 116.40 (CH), 119.87 (C), 124.43 (CH), 127.10 (C), 127.70 (CH), 127.91 (C), 129.85 (2 × CH), 131.01 (CH), 138.48 (CH), 153.33 (C), 160.19 (C), 160.80 (C); ESI-MS (m/z) 253 (M + H)+; HRMS (ESI) calculated for C16H13O3 (M + H)+ 253.0865, found 253.0855.
3-(4-Chlorophenyl)-6-methyl-2H-chromen-2-one (3j). Yield 64% as white solid: mp 158–160 °C; FT-IR (KBr, cm−1) 3021, 1612, 1215, 1026; 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H), 7.25–7.27 (m, 1H), 7.33–7.36 (m, 2H), 7.39–7.43 (m, 2H), 7.64–7.67 (m, 2H), 7.75 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 20.80 (CH3), 116.25 (CH), 119.25 (C), 127.03 (C), 127.75 (CH), 128.68 (2 × CH), 129.85 (2 × CH), 132.75 (CH), 133.27 (C), 134.33 (C), 134.85 (C), 139.95 (CH), 151.73 (C), 160.57 (C); ESI-MS (m/z) 271 (M + H)+; HRMS (ESI) calculated for C16H12ClO2 (M + H)+ 271.0526, found 271.0518.
3-(4-Fluorophenyl)-6-methyl-2H-chromen-2-one (3k). Yield 53% as white solid: mp 158–160 °C; FT-IR (KBr, cm−1) 3019, 1603, 1215, 1084; 1H NMR (400 MHz, CDCl3) δ 2.42 (s, 3H), 7.11–7.15 (m, 2H), 7.25–7.27 (m, 1H), 7.32–7.35 (m, 2H), 7.67–7.71 (m, 2H), 7.73 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 20.80 (CH3), 115.57 (d, J = 21.49 Hz, 2 × CH), 116.22 (CH), 119.32 (C), 127.19 (C), 127.67 (CH), 130.45 (d, J = 8.17 Hz, 2 × CH), 130.87 (C), 132.57 (CH), 134.28 (C), 139.70 (CH), 151.67 (C), 160.76 (C), 164.29 (d, J = 247.4 Hz, C–F); ESI-MS (m/z) 255 (M + H)+; HRMS (ESI) calculated for C16H12FO2 (M + H)+ 255.0821, found 255.0823.
6-Methyl-3-phenyl-2H-chromen-2-one (3l)28. Yield 57% as light brown solid: mp 140–142 °C; FT-IR (KBr, cm−1) 3019, 1613, 1215, 1070; 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H), 7.28 (s, 1H), 7.33–7.35 (m, 2H), 7.38–7.47 (m, 3H), 7.68–7.71 (m, 2H), 7.76 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 20.80 (CH3), 116.20 (CH), 119.45 (C), 127.69 (CH), 128.26 (C), 128.47 (2 × CH), 128.55 (2 × CH), 128.78 (CH), 132.46 (CH), 134.17 (C), 134.90 (C), 139.89 (CH), 151.71 (C), 160.82 (C); ESI-MS (m/z) 237 (M + H)+; HRMS (ESI) calculated for C16H13O2 (M + H)+ 237.0916, found 237.0900.
3-(4-Methoxyphenyl)-6-methyl-2H-chromen-2-one (3m)19. Yield 67% as white solid: mp 134–136 °C; FT-IR (KBr, cm−1) 3019, 1611, 1215, 1069; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H), 3.82 (s, 3H), 6.92–6.96 (m, 2H), 7.20–7.23 (m, 1H), 7.27–7.28 (m, 2H), 7.62–7.65 (m, 2H), 7.67 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 20.80 (CH3), 55.38 (CH3), 113.92 (2 × CH), 116.11 (CH), 119.60 (C), 127.26 (C), 127.50 (CH), 127.76 (C), 129.83 (2 × CH), 132.07 (CH), 134.08 (C), 138.53 (CH), 151.48 (C), 160.12 (C), 161.03 (C); ESI-MS (m/z) 267 (M + H)+; HRMS (ESI) calculated for C17H15O3 (M + H)+ 267.1021, found 267.1012.
6-Methyl-3-(p-tolyl)-2H-chromen-2-one (3n)7b. Yield 67% as light yellow solid: mp 138–140 °C; FT-IR (KBr, cm−1) 3019, 1621, 1215, 1112; 1H NMR (400 MHz, CDCl3) δ 2.39 (s, 3H), 2.41 (s, 3H), 7.23–7.25 (m, 3H), 7.30–7.32 (m, 2H), 7.59 (d, J = 8.16 Hz, 2H), 7.72 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 20.79 (CH3), 21.29 (CH3), 116.14 (CH), 119.53 (C), 127.60 (CH), 128.19 (C), 128.41 (2 × CH), 129.16 (2 × CH), 131.99 (C), 132.24 (CH), 134.09 (C), 138.82 (C), 139.22 (CH), 151.61 (C), 160.90 (C); ESI-MS (m/z) 251 (M + H)+; HRMS (ESI) calculated for C17H15O2 (M + H)+ 251.1072, found 251.1073.
3-(4-Chlorophenyl)-7-hydroxy-2H-chromen-2-one (3o)31. Yield 82% as light yellow solid: mp 252–254 °C; FT-IR (KBr, cm−1) 3401, 3019, 1602, 1215, 1069; 1H NMR (400 MHz, DMSO-d6) δ 6.75 (d, J = 2.16 Hz, 1H), 6.82–6.84 (dd, J = 8.48, 2.24 Hz, 1H), 7.49–7.51 (m, 2H), 7.60 (d, J = 8.56 Hz, 1H), 7.72–7.75 (m, 2H), 8.19 (s, 1H), 10.65 (s, 1H); 13C NMR (DMSO-d6) δ 102.21 (CH), 112.35 (C), 113.96 (CH), 121.29 (C), 128.63 (2 × CH), 130.46 (2 × CH), 130.58 (CH), 133.13 (C), 134.39 (C), 141.85 (CH), 155.47 (C), 160.38 (C), 161.93 (C); ESI-MS (m/z) 273 (M + H)+; HRMS (ESI) calculated for C15H10ClO3 (M + H)+ 273.0318, found 273.0291.
3-(4-Fluorophenyl)-7-hydroxy-2H-chromen-2-one (3p)32. Yield 79% as light yellow solid: mp 245–247 °C; FT-IR (KBr, cm−1) 3394, 3019, 1608, 1215, 1024; 1H NMR (400 MHz, DMSO-d6) δ 6.75 (d, J = 2.24 Hz, 1H), 6.81–6.84 (dd, J = 8.52, 2.28 Hz, 1H), 7.25–7.29 (m, 2H), 7.59 (d, J = 8.56 Hz, 1H), 7.72–7.76 (m, 2H), 8.15 (s, 1H), 10.60 (s, 1H); 13C NMR (DMSO-d6) δ 102.19 (CH), 112.39 (C), 113.90 (CH), 115.60 (d, J = 21.31 Hz, 2 × CH), 121.63 (C), 130.45 (CH), 130.89 (d, J = 8.1 Hz, 2 × CH), 131.92 (C), 141.54 (CH), 155.38 (C), 160.56 (C), 161.75 (C), 163.55 (d, J = 243.92 Hz, C–F); ESI-MS (m/z) 257 (M + H)+; HRMS (ESI) calculated for C15H10FO3 (M + H)+ 257.0614, found 257.0593.
3-(4-Bromophenyl)-7-hydroxy-2H-chromen-2-one (3q)33. Yield 80% as light yellow solid: mp 228–230 °C; FT-IR (KBr, cm−1) 3410, 3019, 1602, 1215, 1084; 1H NMR (400 MHz, DMSO-d6) δ 6.75 (d, J = 2.16 Hz, 1H), 6.81–6.84 (dd, J = 8.48, 2.24 Hz, 1H), 7.59–7.68 (m, 5H), 8.20 (s, 1H), 10.65 (s, 1H); 13C NMR (DMSO-d6) δ 102.22 (CH), 112.37 (C), 113.98 (CH), 121.35 (C), 121.75 (C), 130.60 (CH), 130.76 (2 × CH), 131.57 (2 × CH), 134.77 (C), 141.86 (CH), 155.48 (C), 160.33 (C), 161.95 (C); ESI-MS (m/z) 317 (M + H)+; HRMS (ESI) calculated for C15H10BrO3 (M + H)+ 316.9813, found 316.9806.
7-Hydroxy-3-phenyl-2H-chromen-2-one (3r)19. Yield 78% as light yellow solid: mp 180–182 °C; FT-IR (KBr, cm−1) 3400, 3019, 1611, 1215, 1069; 1H NMR (400 MHz, DMSO-d6) δ 6.76 (d, J = 2.12 Hz, 1H), 6.81–6.84 (dd, J = 8.48, 2.28 Hz, 1H), 7.36–7.46 (m, 3H), 7.60 (d, J = 8.52 Hz, 1H), 7.68–7.70 (m, 2H), 8.15 (s, 1H), 10.59 (s, 1H); 13C NMR (DMSO-d6) δ 102.19 (CH), 112.47 (C), 113.86 (CH), 122.66 (C), 128.47 (CH), 128.63 (2 × CH), 128.73 (2 × CH), 130.46 (CH), 135.58 (C), 141.58 (CH), 155.40 (C), 160.54 (C), 161.72 (C); ESI-MS (m/z) 239 (M + H)+; HRMS (ESI) calculated for C15H11O3 (M + H)+ 239.0708, found 239.0738.
7-Hydroxy-3-(p-tolyl)-2H-chromen-2-one (3s)34. Yield 88% as light yellow solid: mp 242–244 °C; FT-IR (KBr, cm−1) 3400, 3019, 1602, 1215, 1084; 1H NMR (400 MHz, DMSO-d6) δ 2.34 (s, 3H), 6.74 (d, J = 2.24 Hz, 1H), 6.80–6.83 (dd, J = 8.48, 2.28 Hz, 1H), 7.24 (d, J = 7.96 Hz, 2H), 7.58–7.60 (dd, J = 8.52, 2.08 Hz, 3H), 8.11 (s, 1H), 10.56 (s, 1H); 13C NMR (DMSO-d6) δ 21.26 (CH3), 102.16 (CH), 112.51 (C), 113.81 (CH), 122.57 (C), 128.56 (2 × CH), 129.21 (2 × CH), 130.33 (CH), 132.65 (C), 137.90 (C), 140.91 (CH), 155.27 (C), 160.58 (C), 161.56 (C); ESI-MS (m/z) 253 (M + H)+; HRMS (ESI) calculated for C16H13O3 (M + H)+ 253.0865, found 253.0846.
3-(3,4-Dimethylphenyl)-7-hydroxy-2H-chromen-2-one (3t). Yield 82% as light brown solid: mp 210–212 °C; FT-IR (KBr, cm−1) 3399, 3019, 1602, 1215, 1084; 1H NMR (400 MHz, DMSO-d6) δ 2.25 (s, 3H), 2.26 (s, 3H), 6.74 (d, J = 2.12 Hz, 1H), 6.79–6.82 (dd, J = 8.48, 2.24 Hz, 1H), 7.18 (d, J = 7.88 Hz, 1H), 7.41–7.47 (m, 2H), 7.59 (d, J = 8.52 Hz, 1H), 8.09 (s, 1H), 10.56 (s, 1H); 13C NMR (DMSO-d6) δ 19.61 (CH3), 19.93 (CH3), 102.15 (CH), 112.53 (C), 113.79 (CH), 122.69 (C), 126.12 (CH), 129.62 (CH), 129.71 (CH), 130.28 (CH), 133.00 (C), 136.34 (C), 136.66 (C), 140.80 (CH), 155.24 (C), 160.57 (C), 161.50 (C); ESI-MS (m/z) 267 (M + H)+; HRMS (ESI) calculated for C17H15O3 (M + H)+ 267.1021, found 267.1018.
7-Hydroxy-3-(4-methoxyphenyl)-2H-chromen-2-one (3u)35. Yield 85% as light brown solid: mp 220–222 °C; FT-IR (KBr, cm−1) 3408, 3019, 1611, 1215, 1069; 1H NMR (400 MHz, DMSO-d6) δ 3.79 (s, 3H), 6.74 (d, J = 2.16 Hz, 1H), 6.79–6.82 (dd, J = 8.48, 2.28 Hz, 1H), 6.98–7.01 (m, 2H), 7.59 (d, J = 8.56 Hz, 1H), 7.63–7.67 (m, 2H), 8.08 (s, 1H), 10.53 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 55.65 (CH3), 102.15 (CH), 112.58 (C), 113.77 (CH), 114.08 (2 × CH), 122.32 (C), 127.78 (C), 129.96 (2 × CH), 130.17 (CH), 140.19 (CH), 155.12 (C), 159.62 (C), 160.68 (C), 161.37 (C); ESI-MS (m/z) 269 (M + H)+; HRMS (ESI) calculated for C16H13O4 (M + H)+ 269.0814, found 269.0773.
3-(4-Chlorophenyl)-7-methoxy-2H-chromen-2-one (3v)33. Yield 58% as white solid: mp 158–160 °C; FT-IR (KBr, cm−1) 3020, 1613, 1215, 1076; 1H NMR (400 MHz, CDCl3) δ 3.89 (s, 3H), 6.86–6.89 (m, 2H), 7.39–7.45 (m, 3H), 7.63–7.66 (m, 2H), 7.76 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 55.84 (CH3), 100.47 (CH), 112.97 (CH), 113.18 (C), 123.58 (C), 128.63 (2 × CH), 128.94 (CH), 129.69 (2 × CH), 133.44 (C), 134.45 (C), 140.08 (CH), 155.41 (C), 160.66 (C), 162.87 (C); ESI-MS (m/z) 287 (M + H)+; HRMS (ESI) calculated for C16H12ClO3 (M + H)+ 287.0475, found 287.0473.
7-Methoxy-3-(p-tolyl)-2H-chromen-2-one (3w)19. Yield 51% as white solid: mp 105–107 °C; FT-IR (KBr, cm−1) 3019, 1618, 1215, 1069; 1H NMR (400 MHz, CDCl3) δ 2.42 (s, 3H), 3.91 (s, 3H), 6.88–6.90 (m, 2H), 7.26–7.28 (m, 2H), 7.43–7.46 (m, 1H), 7.60–7.62 (m, 2H), 7.76 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.26 (CH3), 55.79 (CH3), 100.43 (CH), 112.73 (CH), 113.46 (C), 124.86 (C), 128.27 (2 × CH), 128.73 (CH), 129.13 (2 × CH), 132.14 (C), 138.44 (C), 139.39 (CH), 155.23 (C), 161.00 (C), 162.49 (C); ESI-MS (m/z) 267 (M + H)+; HRMS (ESI) calculated for C17H15O3 (M + H)+ 267.1021, found 267.1010.
6-Chloro-3-(p-tolyl)-2H-chromen-2-one (3y)36. Yield 67% as white solid: mp 176–178 °C; FT-IR (KBr, cm−1) 3019, 1621, 1230, 1071; 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H), 7.28–7.34 (m, 3H), 7.47–7.49 (dd, J = 8.76, 2.4 Hz, 1H), 7.54 (d, J = 2.36 Hz, 1H), 7.62 (d, J = 8.12 Hz, 1H), 7.72 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.32 (CH3), 117.85 (CH), 120.83 (C), 126.97 (CH), 128.42 (2 × CH), 129.27 (2 × CH), 129.50 (C), 129.67 (C), 131.06 (CH), 131.35 (C), 137.72 (CH), 139.38 (C), 151.78 (C), 160.09 (C); ESI-MS (m/z) 271 (M + H)+; HRMS (ESI) calculated for C16H12ClO3 (M + H)+ 271.0526, found 271.0522.
6-Bromo-3-(4-methoxyphenyl)-2H-chromen-2-one (3z)37. Yield 52% as white solid: mp 197–199 °C; FT-IR (KBr, cm−1) 3019, 1615, 1228, 1067; 1H NMR (400 MHz, CDCl3) δ 3.89 (s, 3H), 7.01 (d, J = 8.80 Hz, 2H), 7.28–7.29 (brd, 1H), 7.59–7.62 (dd, J = 8.71, 2.12 Hz, 1H), 7.68–7.70 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 55.41 (CH3), 114.03 (2 × CH), 116.98 (C), 118.12 (CH), 121.43 (C), 126.57 (C), 129.04 (C), 129.91 (C & 2 × CH), 133.69 (CH), 136.83 (C), 152.11 (C), 160.47 (C); ESI-MS (m/z) 331 (M + H)+; HRMS (ESI) calculated for C16H12BrO3 (M + H)+ 330.9970, found 330.9971.
3-(4-Chlorophenyl)quinolin-2(1H)-one (3za)38. Yield 65% as white solid: mp 252–254 °C; FT-IR (KBr, cm−1) 3410, 3019, 1656; 1H NMR (400 MHz, DMSO-d6) δ 7.18–7.22 (m, 1H), 7.34 (d, J = 8.2 Hz, 1H), 7.48–7.53 (m, 3H), 7.73 (d, J = 7.16 Hz, 1H), 7.80–7.83 (m, 2H), 8.16 (s, 1H), 11.99 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 115.20 (CH), 119.91 (C), 122.45 (CH), 128.40 (2 × CH), 128.69 (CH), 130.54 (C), 130.90 (3 × CH), 132.97 (C), 135.50 (C), 138.36 (CH), 138.91 (C), 161.33 (C); ESI-MS (m/z) 236 (M + H)+; HRMS (ESI) calculated for C15H11ClNO (M + H)+ 256.0529, found 256.0525.
4-Methyl-3-phenylquinolin-2(1H)-one (3zb)39. Yield 52% as white solid: mp 240–242 °C; FT-IR (KBr, cm−1) 3408, 3019, 1646; 1H NMR (400 MHz, DMSO-d6) δ 2.25 (s, 3H), 7.19–7.25 (m, 3H), 7.33–7.38 (m, 2H), 7.41–7.45 (m, 2H), 7.49–7.53 (m, 1H), 7.77–7.79 (dd, J = 8.12, 0.96 Hz, 1H), 11.78 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 17.02 (CH3), 115.60 (CH), 120.36 (C), 122.25 (CH), 125.70 (CH), 127.62 (CH), 128.29 (2 × CH), 130.45 (CH), 130.74 (2 × CH), 132.46 (C), 136.76 (C), 138.27 (C), 143.71 (C), 161.47 (C); ESI-MS (m/z) 236 (M + H)+; HRMS (ESI) calculated for C16H14NO (M + H)+ 236.1075, found 236.1068.
3-(7-Chloroquinolin-4-yl)-7-hydroxy-2H-chromen-2-one (4). Yield 51% as light yellow solid: mp 225–230 °C; FT-IR (KBr, cm−1) 3409, 3019, 1644, 1215, 1069; 1H NMR (400 MHz, DMSO-d6) δ 6.85–6.98 (m, 2H), 7.61–7.66 (m, 3H), 7.97 (d, J = 9.12 Hz, 1H), 8.17 (d, J = 14.68 Hz, 2H), 9.01 (d, J = 3.84 Hz, 1H), 10.80 (brd, 1H); 13C NMR (100 MHz, DMSO-d6) δ 102.53 (CH), 111.97 (C), 114.06 (CH), 120.02 (C), 123.26 (CH), 125.47 (C), 127.76 (CH), 128.36 (CH), 128.64 (CH), 130.85 (CH), 134.60 (C), 142.71 (C), 145.11 (CH), 148.74 (C), 152.01 (CH), 156.23 (C), 160.18 (C), 162.45 (C); ESI-MS (m/z) 324 (M + H)+; HRMS (ESI) calculated for C18H11ClNO3 (M + H)+ 324.0427, found 324.0429.
3-Phenylpyridin-2(1H)-one (6a). Yield 76% as white solid: mp 197–199 °C; FT-IR (KBr, cm−1) 3402, 3019, 1641; 1H NMR (400 MHz, DMSO-d6) δ 6.29 (t, J = 6.68 Hz, 1H), 7.28–7.31 (m, 1H), 7.36–7.39 (m, 3H), 7.62–7.64 (dd, J = 6.88, 2.0 Hz, 1H), 7.69–7.72 (m, 2H), 11.77 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 105.85 (CH), 127.66 (CH), 128.32 (2 × CH), 128.58 (2 × CH), 130.43 (C), 135.15 (CH), 137.27 (C), 139.16 (CH), 161.70 (C); ESI-MS (m/z) 172 (M + H)+; HRMS (ESI) calculated for C11H10NO (M + H)+ 172.0762, found 172.0756.
3-(4-Chlorophenyl)pyridin-2(1H)-one (6b). Yield 78% as white solid: mp 182–184 °C; FT-IR (KBr, cm−1) 3409, 3019, 1631; 1H NMR (400 MHz, DMSO-d6) δ 6.29 (t, J = 6.72 Hz, 1H), 7.39–7.44 (m, 3H), 7.67–7.69 (dd, J = 6.96, 2.08 Hz, 1H), 7.78 (d, J = 8.6 Hz, 2H), 11.84 (s, 1H); 13C NMR (100 MHz, DMSO-d6): δ 105.90 (CH), 128.31 (2 × CH), 128.88 (C), 130.27 (2 × CH), 132.27 (C), 135.61 (CH), 136.01 (C), 139.37 (CH), 161.52 (C); ESI-MS (m/z) 206 (M + H)+; HRMS (ESI) calculated for C11H9ClNO (M + H)+ 206.0373, found 206.0369.
3-(4-Bromophenyl)pyridin-2(1H)-one (6c). Yield 82% as light brown solid: mp 245–247 °C; FT-IR (KBr, cm−1) 3412, 3020, 1652; 1H NMR (400 MHz, DMSO-d6) δ 6.29 (t, J = 6.8 Hz, 1H), 7.39–7.42 (dd, J = 6.4, 2.04 Hz, 1H), 7.54–7.58 (m, 2H), 7.67–7.73 (m, 3H), 11.84 (s, 1H): 13C NMR (100 MHz, DMSO-d6) δ 106.00 (CH), 120.88 (C), 128.93 (C), 130.61 (2 × CH), 131.26 (2 × CH), 135.63 (CH), 136.37 (C), 139.43 (CH), 161.49 (C), ESI-MS (m/z) 250 (M + H)+; HRMS (ESI) calculated for C11H9BrNO (M + H)+ 249.9868, found 249.9860.
3-(4-Fluorophenyl)pyridin-2(1H)-one (6d). Yield 68% as white solid: mp 218–220 °C; FT-IR (KBr, cm−1) 3418, 3019, 1632; 1H NMR (400 MHz, DMSO-d6) δ 6.29 (t, J = 6.68 Hz, 1H), 7.17–7.22 (m, 2H), 7.37–7.39 (dd, J = 6.4, 2.0 Hz, 1H), 7.63–7.65 (dd, J = 6.92, 2.0 Hz, 1H), 7.75–7.79 (m, 2H), 11.80 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 105.88 (CH), 115.21 (d, J = 21.03 Hz, 2 × CH), 129.28 (C), 130.60 (d, J = 7.89 Hz, 2 × CH), 133.57 (C), 135.21 (CH), 139.10 (CH), 161.65 (C), 163.11 (d, J = 242.77 Hz, C–F); ESI-MS (m/z) 190 (M + H)+; HRMS (ESI) calculated for C11H9FNO (M + H)+ 190.0668, found 190.0667.
3-(p-Tolyl)pyridin-2(1H)-one (6e)21. Yield 71% as light yellow solid: mp 191–194 °C; FT-IR (KBr, cm−1) 3412, 3019, 1636; 1H NMR (400 MHz, DMSO-d6) δ 2.31 (s, 3H), 6.27 (t, J = 6.76 Hz, 1H), 7.18 (d, J = 7.92 Hz, 2H), 7.34–7.36 (dd, J = 6.4, 2.08 Hz, 1H), 7.59–7.63 (m, 3H), 11.72 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 21.25 (CH3), 105.84 (CH), 128.42 (2 × CH), 128.90 (2 × CH), 130.36 (C), 134.36 (C), 134.77 (CH), 136.91 (C), 138.62 (CH), 161.75 (C); ESI-MS (m/z) 186 (M + H)+; HRMS (ESI) calculated for C12H12NO (M + H)+ 186.0919, found 186.0913.
1-Methyl-3-phenylpyridin-2(1H)-one (6f)21. Yield 72% as white solid: mp 127–129 °C; FT-IR (KBr, cm−1) 3019, 1641; 1H NMR (400 MHz, CDCl3) δ 3.61 (s, 3H), 6.25 (t, J = 6.8 Hz, 1H), 7.29–7.34 (m, 2H), 7.37–7.41 (m, 2H), 7.47–7.49 (dd, J = 6.88, 2.0 Hz, 1H), 7.67–7.70 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 38.20 (CH3), 105.82 (CH), 127.65 (CH), 128.09 (2 × CH), 128.61 (2 × CH), 131.64 (C), 136.85 (C), 137.41 (CH), 137.59 (CH), 161.95 (C), ESI-MS (m/z) 186 (M + H)+; HRMS (ESI) calculated for C12H12NO (M + H)+ 186.0919, found 186.0907.
1-Methyl-3-phenylpyridin-2(1H)-one (6g)21. Yield 77% as white solid: mp 135–137 °C; FT-IR (KBr, cm−1) 3019, 1646; 1H NMR (400 MHz, CDCl3) δ 3.61 (s, 3H), 6.25 (t, J = 6.84 Hz, 1H), 7.31–7.33 (dd, J = 6.68, 2.0 Hz, 1H), 7.36 (d, J = 8.64 Hz, 2H), 7.46–7.48 (dd, J = 7.0, 2.04 Hz, 1H), 7.65 (d, J = 8.64 Hz, 2H): 13C NMR (100 MHz, CDCl3) δ 38.25 (CH3), 105.84 (CH), 128.25 (2 × CH), 129.89 (2 × CH), 130.34 (C), 133.52 (C), 135.23 (CH), 137.57 (CH), 137.76 (C), 161.74 (C), ESI-MS (m/z) 206 (M + H)+; HRMS (ESI) calculated for C12H11ClNO (M + H)+ 220.0529, found 220.0517.
3-(4-Bromophenyl)-1-methylpyridin-2(1H)-one (6h)14. Yield 74% as light brown solid: mp 144–146 °C; FT-IR (KBr, cm−1) 3021, 1656; 1H NMR (400 MHz, CDCl3) δ 3.61 (s, 3H), 6.25 (t, J = 6.88 Hz, 1H), 7.31–7.33 (dd, J = 6.68, 2.0 Hz, 1H), 7.46–7.48 (dd, J = 7.0, 2.04 Hz, 1H), 7.49–7.53 (m, 2H), 7.57–7.60 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 38.27 (CH3), 105.86 (CH), 121.77 (C), 130.21 (2 × CH), 130.37 (C), 131.22 (2 × CH), 135.70 (C), 137.57 (CH), 137.80 (CH), 161.68 (C); ESI-MS (m/z) 264 (M + H)+; HRMS (ESI) calculated for C12H11BrNO (M + H)+ 264.0024, found 264.0020.
1-Methyl-3-(p-tolyl)pyridin-2(1H)-one (6i)21. Yield 73% as light yellow solid: mp 136–138 °C; FT-IR (KBr, cm−1) 3019, 1652; 1H NMR (400 MHz, DMSO-d6) δ 2.32 (s, 3H), 3.49 (s, 3H), 6.29 (t, J = 6.84 Hz, 1H), 7.18 (d, J = 7.92 Hz, 2H), 7.55–7.59 (m, 3H), 7.69–7.71 (dd, J = 6.68, 2.04 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 21.24 (CH3), 37.91 (CH3), 105.59 (CH), 128.61 (2 × CH), 128.90 (2 × CH), 129.67 (C), 134.55 (C), 136.96 (C), 137.75 (CH), 139.30 (CH), 161.36 (C), ESI-MS (m/z) 200 (M + H)+; HRMS (ESI) calculated for C13H14NO (M + H)+ 200.1075, found 200.1067.
3-(4-Methoxyphenyl)-1-methylpyridin-2(1H)-one (6j)21. Yield 76% as white solid: mp 110–112 °C; FT-IR (KBr, cm−1) 3020, 1657; 1H NMR (400 MHz, CDCl3) δ 3.45 (s, 3H), 3.68 (s, 3H), 6.07 (t, J = 6.8 Hz, 1H), 6.76–6.79 (m, 2H), 7.09–7.12 (m, 1H), 7.28–7.30 (dd, J = 6.96, 2.24 Hz, 1H), 7.49–7.52 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 38.20 (CH3), 55.32 (CH3), 105.90 (CH), 113.56 (2 × CH), 129.30 (C), 129.79 (2 × CH), 131.23 (C), 136.66 (2 × CH), 139.79 (CH), 159.25 (C), 162.09 (C); ESI-MS (m/z) 216 (M + H)+; HRMS (ESI) calculated for C13H14NO2 (M + H)+ 216.1025, found 216.1012.
4-((2,2,6,6-Tetramethylpiperidin-1-yl)oxy)benzonitrile (7)40. Yield 64% as white solid: mp 78–81 °C; FT-IR (KBr, cm−1) 3019, 2231, 1591; 1H NMR (400 MHz, CDCl3) δ 0.99 (s, 6H), 1.25 (s, 6H), 1.44–1.49 (m, 1H), 1.59–1.66 (m, 5H), 7.28 (s, 2H), 7.54 (d, J = 9.12 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.90 (CH2), 20.46 (4 × CH3), 32.31 (CH2), 39.69 (CH2), 60.76 (2 × C), 103.21 (C), 114.89 (2 × CH), 119.61 (C), 133.52 (2 × CH), 166.91 (C).
Acknowledgements
P. C., M. R. and S. S. are thankful to the Council of Scientific and Industrial Research (CSIR), New Delhi, for Fellowship, Mr Anoop K. Srivastava for providing technical support, SAIF-CDRI Lucknow for providing spectral and analytical data. This work was supported by CSIR network project “THUNDER” (BSC0102). This is CDRI communication no. 9144.
Notes and references
-
(a) R. Narayan, K. Matcha and A. P. Antonchick, Chem.–Eur. J., 2015, 21, 1 CrossRef PubMed;
(b) F. Tinnis, E. Stridfeldt, H. Lundberg, H. Adolfsson and B. Olofsson, Org. Lett., 2015, 17, 2688 CrossRef CAS PubMed;
(c) Q.-L. Xu, H. Gao, M. Yousufuddin, D. H. Ess and L. Kurti, J. Am. Chem. Soc., 2013, 135, 14048 CrossRef CAS PubMed;
(d) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed;
(e) T. L. Chan, Y. Wu, P. Y. Choy and F. Y. Kwong, Chem.–Eur. J., 2013, 19, 15802 CrossRef CAS PubMed.
-
(a) A. M. Berman, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2010, 75, 7863 CrossRef CAS PubMed;
(b) A. Carrer, J.-D. Brion, S. Messaoudi and M. Alami, Org. Lett., 2013, 15, 5606 CrossRef CAS PubMed;
(c) J. Kwak, M. Kim and S. Chang, J. Am. Chem. Soc., 2011, 133, 3780 CrossRef CAS PubMed;
(d) L. Huang, Q. Li, C. Wang and C. Qi, J. Org. Chem., 2013, 78, 3030 CrossRef CAS PubMed;
(e) F. Jafarpour, S. Rahiminejadan and H. Hazrati, J. Org. Chem., 2010, 75, 3109 CrossRef CAS PubMed;
(f) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed;
(g) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed;
(h) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed;
(i) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
-
(a) D. P. Hari, P. Schroll and B. Konig, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef CAS PubMed;
(b) D. A. Nagib and D. W. C. MacMillan, Nature., 2011, 480, 224 CrossRef CAS PubMed;
(c) W. Du, J. Lai, L. Tian, X. Xie, X. She and S. Tang, Chem. Commun., 2014, 15, 14017 RSC.
-
(a) S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673 CrossRef CAS PubMed;
(b) E. Shirakawa, K.-I. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537 CrossRef CAS PubMed;
(c) C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044 CrossRef CAS PubMed;
(d) A. Dickschat and A. Studer, Org. Lett., 2010, 12, 3972 CrossRef CAS PubMed;
(e) S. Castro, J. J. Fernandez, R. Vicente, F. J. Fananas and F. Rodriguez, Chem. Commun., 2012, 48, 9089 RSC;
(f) C. Galli, Chem. Rev., 1988, 88, 765 CrossRef CAS.
-
(a) J. Hofmann, H. Jasch and M. R. Heinrich, J. Org. Chem., 2014, 79, 2314 CrossRef CAS PubMed;
(b) P. Patil, A. Nimonkar and K. G. Akamanchi, J. Org. Chem., 2014, 79, 2331 CrossRef CAS PubMed;
(c) M. Ravi, P. Chauhan, R. Kant, S. K. Shukla and P. P. Yadav, J. Org. Chem., 2015, 80, 5369 CrossRef CAS PubMed.
-
(a) H. R. El-Seedi, J. Nat. Prod., 2007, 17, 118 CrossRef PubMed;
(b) J. M. Narvaez-Mastache, F. Novillo and G. Delgado, Phytochemistry, 2008, 69, 451 CrossRef CAS PubMed;
(c) L. You, R. An, X. Wang and Y. Li, Bioorg. Med. Chem. Lett., 2010, 20, 7426 CrossRef CAS PubMed;
(d) M. J. Matos, D. Vina, E. Quezada, C. Picciau, G. Delogu, F. Orallo, L. Santana and E. Uriarte, Bioorg. Med. Chem. Lett., 2009, 19, 3268 CrossRef CAS PubMed;
(e) L. Piazzi, A. Cavalli, F. Belluti, A. Bisi, S. Gobbi, S. Rizzo, M. Bartolini, V. Andrisano, M. Recanatini and A. Rampa, J. Med. Chem., 2007, 50, 4250 CrossRef CAS PubMed;
(f) T. Matsui, T. Sugiura, H. Nakai, S. Iguchi, S. Shigeoka, Y. Odagaki, H. Takada, Y. Nagao, Y. Ushio, K. Ohmoto, H. Iwamura, S. Yamazaki, Y. Arai and M. Kawamura, J. Med. Chem., 1992, 35, 3307 CrossRef CAS PubMed.
-
(a) M. Catto, O. Nicolotti, F. Leonetti, A. Carotti, A. D. Favia, R. Soto-Otero, E. Mendez-Alvarez and A. Carotti, J. Med. Chem., 2006, 49, 4912 CrossRef CAS PubMed;
(b) M. J. Matos, C. Teran, Y. Perez.-Castillo, E. Uriarte, L. Santana and D. Vina, J. Med. Chem., 2011, 54, 7127 CrossRef CAS PubMed.
-
(a) L. Piazzi, A. Rampa, A. Bisi, S. Gobbi, F. Belluti, A. Cavalli, M. Bartolini, V. Andrisano, P. Valenti and M. Recanatini, J. Med. Chem., 2003, 46, 2279 CrossRef CAS PubMed;
(b) K. V. Sashidhara, R. K. Modukuri, P. Jadiya, K. B. Rao, T. Sharma, R. Haque, D. K. Singh, D. Banerjee, M. I. Siddiqi and A. Nazir, ACS Med. Chem. Lett., 2014, 5, 1099 CrossRef CAS PubMed.
-
(a) E. B. B. Ong, N. Watanabe, A. Saito, Y. Futamura, K. H. A. E. Galil, A. Koito, N. Najimudin and H. Osada, J. Biol. Chem., 2011, 286, 14049 CrossRef CAS PubMed;
(b) D. Olmedo, R. Sancho, L. M. Bedoya, J. L. Lopez-Perez, E. D. Olmo, E. Munoz, J. Alcami, M. P. Gupta and A. S. Feliciano, Molecules, 2012, 17, 9245 CrossRef CAS PubMed.
-
(a) C. Wang, C. Wu, J. Zhu, R. H. Miller and Y. Wang, J. Med. Chem., 2011, 54, 2331 CrossRef CAS PubMed;
(b) K. G. Reddie, W. H. Humphries, C. P. Bain, C. K. Payne, M. L. Kemp and N. Murthy, Org. Lett., 2012, 14, 680 CrossRef CAS PubMed.
-
(a) S. Hibi, K. Ueno, S. Nagato, K. Kawano, K. Ito, Y. Norimine, O. Takenaka, T. Hanada and M. Yonaga, J. Med. Chem., 2012, 55, 10584 CrossRef CAS PubMed;
(b) P. Wu and Y. Hu, Med. Chem. Commun., 2012, 3, 1337 RSC;
(c) A. A. Cumaraswamy, A. Todic, D. Resetca, M. D. Minden and P. T. Gunning, Med. Chem. Commun., 2012, 3, 22 RSC.
-
(a) Y. Li, Z. Qi, H. Wang, X. Fu and C. Duan, J. Org. Chem., 2012, 77, 2053 CrossRef CAS PubMed;
(b) M. Khoobi, M. Alipour, S. Zarei, F. Jafarpour and A. Shafiee, Chem. Commun., 2012, 48, 2985 RSC;
(c) T. Ithara and F. Ouseto, Synthesis, 1984, 488 CrossRef.
- M. Kojima, K. Oisaki and M. Kanai, Chem. Commun., 2015, 51, 9718 RSC.
- A. Nakatani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 1377 CrossRef CAS PubMed.
-
(a) K. V. Sashidhara, G. R. Palnati, S. R. Avula and A. Kumar, Synlett, 2012, 23, 611 CrossRef CAS;
(b) A. V. Klinin, A. J. M. Da Silva, C. C. Lopes, R. S. C. Lopes and V. Sneicker, Tetrahedron Lett., 1998, 39, 4995 CrossRef.
- H. Zhao, B. Yan, L. B. Peterson and B. S. J. Blagg, ACS Med. Chem. Lett., 2012, 3, 327 CrossRef CAS PubMed.
- F. Jafarpour, S. Zarei, M. B. A. Olia, N. Jalalimanesh and S. Rahiminejadan, J. Org. Chem., 2013, 78, 2957 CrossRef CAS PubMed.
- F. Jafarpour, M. B. A. Olia and H. Hazrati, Adv. Synth. Catal., 2013, 355, 3407 CrossRef CAS.
- F. Jafarpour, H. Hazrati, N. Mohasselyazdi, M. Khoobi and A. Shafiee, Chem. Commun., 2013, 49, 10935 RSC.
- Y. Liang, J. Gloudeman and S. F. Wnuk, J. Org. Chem., 2014, 79, 4094 CrossRef CAS PubMed.
- A. Modak, S. Rana and D. Maiti, J. Org. Chem., 2015, 80, 296 CrossRef CAS PubMed.
-
(a) P. Chauhan, M. Ravi, S. Singh, K. S. R. Raju, V. Bajpai, B. Kumar, Wahajuddin and P. P. Yadav, RSC Adv., 2014, 4, 43336 CAS;
(b) S. Singh, P. Chauhan, M. Ravi, I. Taneja, Wahajuddin and P. P. Yadav, RSC Adv., 2015, 5, 61876 RSC.
-
(a) P.-K. C. Huang and E. M. Kosower, J. Am. Chem. Soc., 1968, 90, 2354 CrossRef CAS;
(b) P.-K. C. Huang and E. M. Kosower, J. Am. Chem. Soc., 1968, 90, 2362 CrossRef CAS;
(c) M. R. Smith III and G. L. Hill, J. Am. Chem. Soc., 1988, 110, 4066 CrossRef.
- H. Kawai, S. Okusu, Z. Yuan, E. Tokunga, A. Yamano, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2013, 52, 2221 CrossRef CAS PubMed.
- J.-B. Liu, L. Nie, H. Yan, L.-H. Jiang, J. Weng and G. Lu, Org. Biomol. Chem., 2013, 11, 8014 CAS.
- D. P. Curran and A. I. Killer, J. Am. Chem. Soc., 2006, 128, 13706 CrossRef CAS PubMed.
- L. Li, Y.-L. Zhao, Q. Wang, T. Lin and Q. Liu, Org. Lett., 2015, 17, 370 CrossRef CAS PubMed.
- Y. Jiang, W. Chen and W. Lu, Tetrahedron, 2013, 69, 3669 CrossRef CAS.
- P. Hanson, P. W. Lovenich, S. C. Rowell, P. H. Walton and A. W. Timms, J. Chem. Soc., Perkin Trans. 2, 1999, 49 RSC.
- E. Yoneda, T. Sugioka, K. Hirao, S.-W. Zhang and S. Takahashi, J. Chem. Soc., Perkin Trans. 1, 1998, 477 RSC.
- S. P. Bondarenko, E. V. Podobii, M. S. Frasinyuk, V. I. Vinogradova and V. P. Khilya, Chem. Nat. Compd., 2014, 50, 420 CrossRef CAS.
- M. M. Garazd, Y. L. Garazd, A. S. Ogorodniichuk and V. P. Khilya, Chem. Nat. Compd., 2009, 45, 158 CrossRef CAS.
- D. I. Sherris and J. Plain, US Pat., 0197567 A1, 2007.
- N. P. Buu-Hoi, B. Ekert and R. Royer, J. Org. Chem., 1954, 19, 1548 CrossRef CAS.
- Y. Zhang, Y. Xu, S. Tan, L. Xu and X. Qian, Tetrahedron Lett., 2012, 53, 6881 CrossRef CAS.
- H. Zeng and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 13862 CrossRef CAS PubMed.
- E. Quezada, G. Delogu, C. Picciau, L. Santana, G. Podda, F. Borges, V. Garcia-Morales, D. Vina and F. Orallo, Molecules, 2010, 15, 270 CrossRef CAS PubMed.
- V. A. Mamedov, V. L. Mamedova, S. F. Kadyrova, G. Z. Khikmatona, A. T. Gubaidullin, I. K. Rizvanov and S. K. Latypov, Tetrahedron, 2015, 71, 2670 CrossRef CAS.
- P. J. Manley and M. T. Bilodeau, Org. Lett., 2004, 6, 2433 CrossRef CAS PubMed.
- Y. Li, W. Xie and X. Jiang, Chem.–Eur. J., 2015, 21, 16059 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, data and NMR spectra of all the final compounds. See DOI: 10.1039/c5ra20954d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.