
Facile preparation and properties of multifunctional polyacetylene via highly efficient click chemistry
Fayin Zhanga,
Shanyi Guang*b,
Fuyou Kea,
Jing Lia and
Hongyao Xu*a
aThe State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China. E-mail: hongyaoxu@163.com
bCollege of Chemistry and Bioengineering, Donghua University, Shanghai 201620, China
First published on 22nd December 2015
Abstract
A series of novel multifunctional polyacetylenes bearing oxadiazole and azo groups as molecular pendants were designed and synthesized via highly effective click chemistry. The resultant polymers showed good solubility in common organic solvents. The structure and properties of the resulting polymer were characterized and evaluated by FT-IR, 1H-NMR, UV-vis, TGA and FL analyses. The results show that the thermal stability of the synthesized functional polyacetylene was significantly increased when compared to single polyacetylene due to the jacket effect between the functional groups and backbone. The influence mechanism of the oxadiazole and azo groups was also discussed in detail. Simultaneously, the optical properties of the functional polyacetylene were investigated. It was found that the optical properties of the resulting polyacetylene were affected by the oxadiazole and azo groups. For P5–P7, the intensity of the absorption peak at ca. 452 nm, belonging to the azo group, decreased gradually as the content of M1 was reduced. In contrast, the intensity of the absorption peak at ca. 312 nm, belonging to the oxadiazole group, increased as the content of M2 increased. Due to azo group isomerization under the photo-thermal conditions, quenching of the fluorescence occurred in the range of molecules for M1, P2 and P3.
Introduction
Polyacetylene is an extensively conjugated polymer that shows good conductivity and large third-order non-linear optical susceptibilities1–3 because of the large π-electron conjugation along the alternating double-bond backbone. However, the strong π–π stacking interactions between the main chains of polyacetylene often result in intractability and instability in air, which have significantly limited the scope of its potential applications.4 The existence of two hydrogen atoms in its repeat unit provides ample opportunities to decorate the backbone with appropriate substituents.5 Therefore, many researchers have tried their best to change the substituents in the molecular structure to produce novel functional polyacetylene derivatives. Some mono-substituted or multi-substituted functional polyacetylenes have been successfully prepared. In our group, the previous work was mainly focused on the molecular design and relationship between the molecular structure and properties such as non-linear optical (NLO) properties. A series of polyacetylenes containing azobenzene or stilbene derivatives were synthesized and investigated. The different pendants with various conjugation lengths and jointed group structures have a greater influence on the optical limiting, NLO and solubility properties.6–13 In addition, it was found that the resultant polyacetylenes with aromatic azobenzene groups have good thermal stability due to the “jacket effect”. When compared with mono-substituted polyacetylene, multi-substituted poly-acetylene is superior in performance. In general, varieties of substituted polyacetylenes with novel functionalities, such as electro-optic activity,14–17 bioactivity18,19 and image sensing,20,21 were synthesized through a polymerization reaction using different functional acetylene monomers and exhibited many unique advantages with respect to the archetypal polyacetylene.However, some important factors such as catalyst and monomer reactivity have a great influence in the polymerization reaction process. Due to the different reactivity of the copolymerized monomer and steric hindrance, the content of each composition and structure are difficult to control effectively. Therefore, functionalization after the polymerization reaction is a rapid method to synthesize diverse materials. With the advent of click chemistry, it provides a convenient way to prepare multifunctional polymers after the polymerization reaction. Click chemistry has been growing exponentially since its introduction in 2001 due to mild reaction conditions, easily available reagents and more importantly, its high efficiency with respect to traditional chemical reactions.22,23 Moreover, it is widely used in the synthesis of DNA, supermolecules, dendritic molecules and functional polymers in recent years.24–26 Recently, some multifunctional polymers have been successfully synthesized via click chemistry and have been reported in the literature.27–32 However, the synthesis of multi-functional polyacetylene using this method has rarely been reported. Yang's group33 synthesized thermo-sensitive and amphiphilic polymer brushes based on helical polyacetylene by means of click chemistry, which was believed to have potential applications in chiral materials and smart materials. Tang and co-workers34–36 focused on the post-functionalization of disubstituted polyacetylenes via click chemistry. However, the polyacetylene prepared had only one functional group as a pendent in this work, which cannot meet the requirement of practical applications.
In this study, we successfully synthesized three functional polyacetylenes with higher thermal stability using non-linear optical azo and electron transport oxadiazole chromophore moieties as pendants via efficient click chemistry. Simultaneously, the relationship between the pendant and optical properties was explored in detail.
Results and discussion
Click synthesis and structural characterization
The five target functional polyacetylenes containing triazole and azobenzene groups were prepared according to Scheme 1. The synthesis details are available in the Experimental section. The click reaction using copper sulfate and sodium ascorbic acid as catalyst was used in the preparation of P3–P7 in an almost quantitative yield. The molecular structure of all the intermediates and objective polymers were characterized using standard spectroscopic methods and satisfactory spectroscopic analytical data.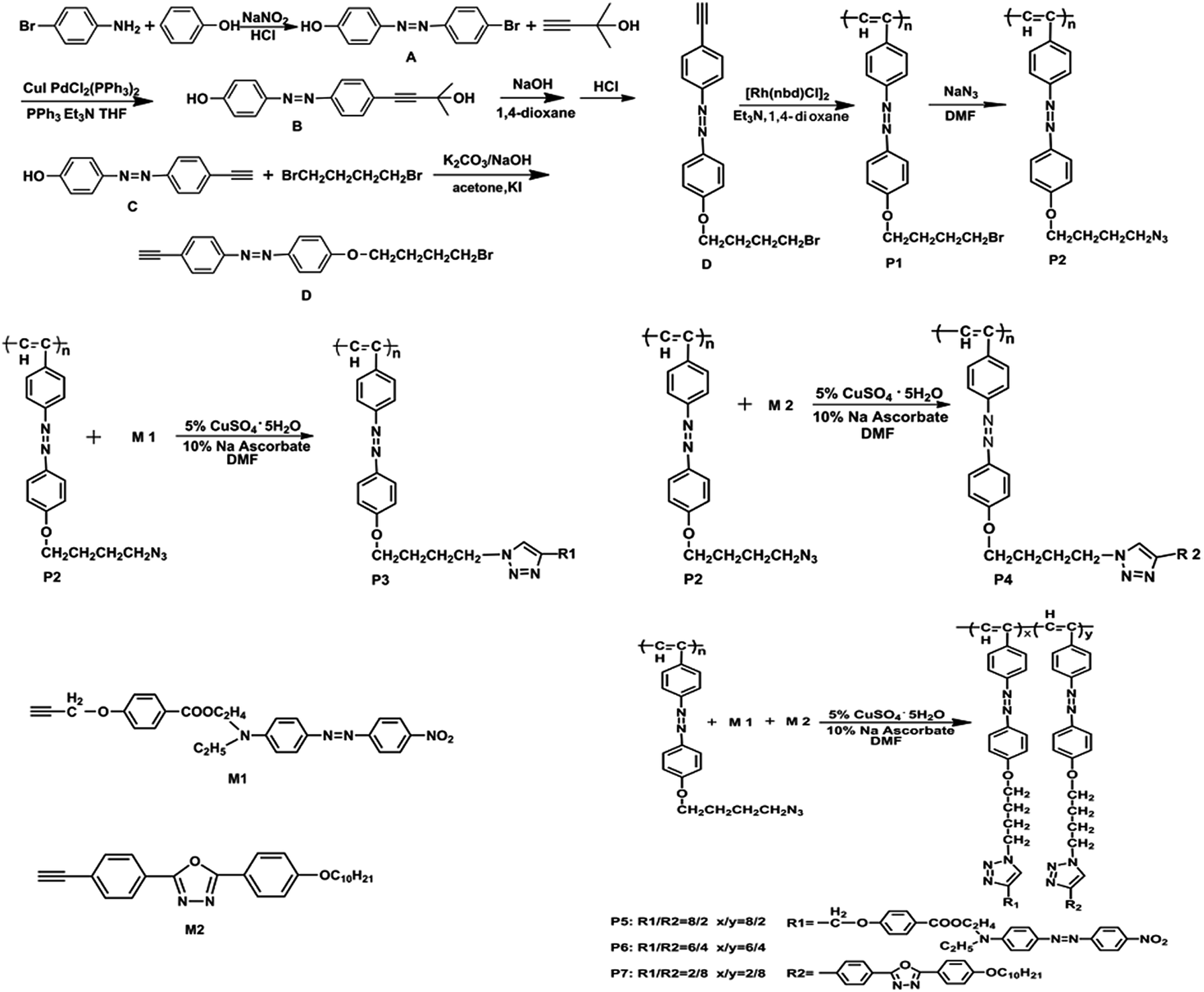 | ||
| Scheme 1 Synthetic route. | ||
As can be seen from the Fig. 1(a), monomer M1 clearly exhibits two characteristic absorptions at ca. 2126 cm−1 and 3263 cm−1, corresponding to the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– and C–H stretching vibration absorptions. Moreover, these peaks completely disappeared in the spectra of P2 and P3. The strong peak at 2096 cm−1 corresponding to the –N3 vibration absorption was observed in P2. However, due to the reaction between the azide group and alkynyl, two characteristic absorption peaks located at 1717 cm−1 and 1655 cm−1 reveal the generated nitro group and triazole ring, respectively. This indicates that the click reaction occurred between the azide group and alkynyl in P3.
C– and C–H stretching vibration absorptions. Moreover, these peaks completely disappeared in the spectra of P2 and P3. The strong peak at 2096 cm−1 corresponding to the –N3 vibration absorption was observed in P2. However, due to the reaction between the azide group and alkynyl, two characteristic absorption peaks located at 1717 cm−1 and 1655 cm−1 reveal the generated nitro group and triazole ring, respectively. This indicates that the click reaction occurred between the azide group and alkynyl in P3.
 | ||
| Fig. 1 The FTIR spectra of (a) M1, P2, P3 (b) M2, P2, P4 (c) M1, M2, P2, P5. | ||
From Fig. 1(b), the peaks at 2106 cm−1 and 3273 cm−1 correspond to the –CC– and C–H stretching vibration absorptions in M2, but the intensity of the –CC– vibration peak was very weak in the spectrum and they completely disappeared in P2 and P4. The stretching vibration at ca. 2100 cm−1 of the azidobenzene derivatives can be assigned to the strong –N3 absorption. It was transformed into a triazole ring absorption peak at ca. 1655 cm−1 in P4 after the click reaction. It demonstrates that P4 was successfully synthesized.
From Fig. 1(c), M1 and M2 have a strong characteristic absorption peak at ca. 3273 cm−1 and a very weak absorption peak at ca. 2106 cm−1, which correspond to single substituted alkynyl CH and –CC– stretching vibration absorptions, respectively. However, they disappeared in P2 and P5. A new absorption peak of the nitro group at ca. 1717 cm−1 was generated in P5. P2 has a strong stretching vibration absorption, which corresponds to the azide group at ca. 2100 cm−1 that disappeared in P5 replaced by a triazole ring absorption peak at ca. 1650 cm−1. This proved that the –CC– and –N3 reacted successfully in the click reaction. In addition, the peaks in P5 located at ca. 2928 cm−1, ca. 2856 cm−1, ca. 1608 cm−1, ca. 1256 cm−1 and ca. 834 cm−1 correspond to –CH3, –CH2, benzene ring –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– group, Ar-O– and –Ar– stretching vibration absorptions.
C– group, Ar-O– and –Ar– stretching vibration absorptions.
1H-NMR analysis
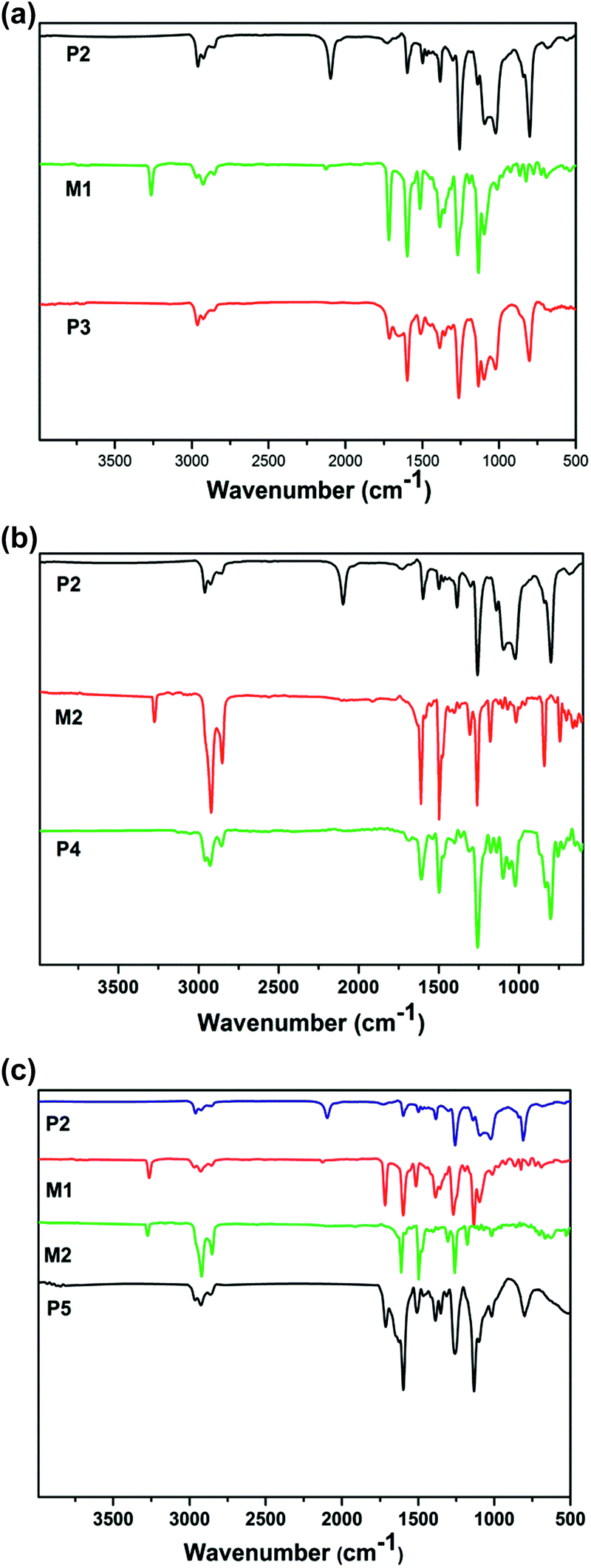
Fig. 2 shows the 1H-NMR spectra of the monomers and polymers in chloroform-d. From Fig. 2(a), four characteristic peaks were newly generated in P3 when compared with P2. The proton absorption peaks at δ = 5.49 ppm, δ = 6.63–6.83 ppm, δ = 4.46 ppm and δ = 0.85–0.87 ppm correspond to the connecting –CH2– protons between the triazole ring and ether bond, Ar-H, –O–CH2– and –CH3, respectively, which support the objective that P3 was successfully synthesized. From Fig. 2(b), the spectrum clearly shows that some of the new proton absorption peaks were exhibited in P4 when compared with P2. The signals at δ = 0.88 ppm, δ = 1.25–1.44 ppm and δ = 4.49 ppm in the P4 spectrum are assigned to the end methyl protons, the long alkyl (–CH2–) protons at the end of oxadiazole and the methylene protons connecting with the triazole ring, respectively. This indicates that P4 was successfully prepared. From Fig. 2(c), some new characteristic peaks were observed in P5, P6 and P7. The δ = 5.49 ppm, δ = 6.63–6.83 ppm, δ = 4.46 ppm, δ = 0.85–0.87 ppm, δ = 1.25–1.44 ppm and δ = 4.49 ppm peaks correspond to the connecting –CH2– proton peak between the triazole ring and ether bond, Ar-H, –O–CH2–, –CH3, the long alkyl (–CH2–) protons at the end of oxadiazole and the methylene protons connecting with the triazole ring, respectively. Importantly, a new peak corresponding to the resonances of the triazole ring was observed at δ = 7.6 in the spectrum of the polymers containing a triazole ring.37 In addition, from the spectrum of P5, P6 and P7, it was found that the area of the absorption peak at δ = 6.63–6.83 ppm and δ = 5.49 ppm belonging to Ar-H and the –CH2– proton peak between the triazole ring and ether bond decreased gradually due to the reduction in the content of the M1 monomer in the polymers.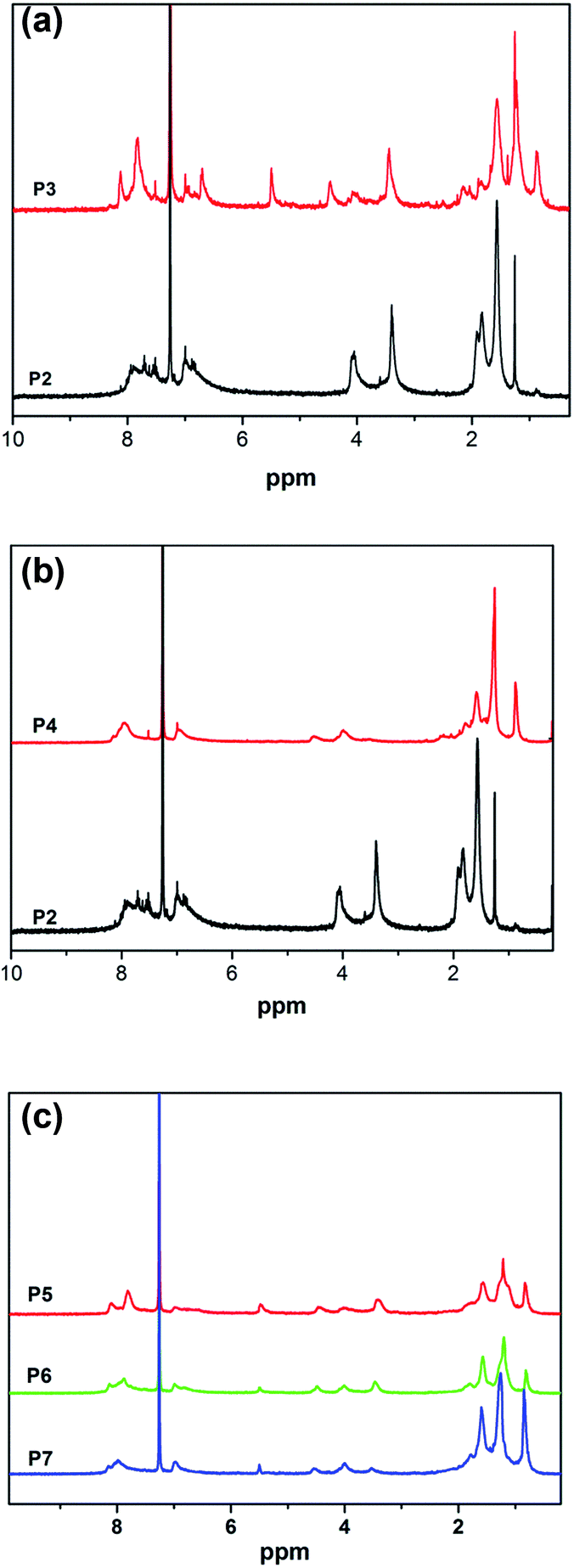 | ||
| Fig. 2 The 1H-NMR spectra of (a) P2, P3, (b) P2, P4 and (c) P5, P6, P7. | ||
UV-vis spectroscopic analysis
The UV spectra of the monomers in THF (1 × 10−3 mol L−1) were obtained on a Shimadzu UV-265 spectrometer and shown in Fig. 3(a). As shown in Fig. 3(a), the monomers M1 and M2 show strong absorption peaks at ca. 452 nm and ca. 312 nm, respectively, which are assigned to π–π* electronic transitions of their large conjugated structure and oxadiazole chromophore unit.38 In addition, a weak absorption peak located at ca. 287 nm was observed in M1, which was attributed to the benzene ring. P2 displays a strong absorption peak at ca. 364 nm, which was assigned to the π–π* electronic transition of the azobenzene group. For P3, the absorption peaks at ca. 287 nm and ca. 452 nm were the same as M1. However, it was surprisingly found that the absorption peak at ca. 369 nm has a 5 nm red shift when compared with P2, which may result from the superposition of the azobenzene group between M1 and P2. For P4, when M1 was replaced with M2, the absorption peak at ca. 364 nm was the same as P2, the maximum absorption peak at ca. 323 nm shows a 11 nm red shift in comparison with that of M2, which was attributed to the following reasons: the strong electron-withdrawing properties of the newly generated triazole ring after the click reaction enhances the electron mobility in P4. Moreover, a larger conjugate plane structure was formed between the rigid oxadiazole group and triazole ring, and the degree of conjugation was increased simultaneously. The UV spectra of the polymers P5–P7 in THF (0.01 mg mL−1) were obtained as shown in Fig. 3(b). As it can be seen from the spectra, the absorption peaks of P5–P7 are located at ca. 312 nm, ca. 360 nm and ca. 452 nm, respectively. It was found that the intensity of the absorption peak at ca. 452 nm was decreased gradually from P5 to P7, which was a result of the reduction in the content of M1 (the absorption peak at ca. 452 nm) in polymers P5–P7. Simultaneously, the intensity of the absorption peak at 312 nm was increased gradually as the content of M2 (the absorption peak at ca. 312 nm) increased from P5 to P7. | ||
| Fig. 3 The UV-vis spectra of (a) M1, M2, P2, P3, P4 and (b) P5, P6, P7. | ||
PL spectroscopic analysis
In order to understand the influence of the functional groups on the luminous performance, solution PL spectra were obtained and the results are displayed in Fig. 4. Due to azo group isomerization under photo-thermal conditions, quenching of the fluorescence occurred within the molecule; therefore, the monomer M1, P2 and P3 have no fluorescence properties. The PL spectra of M2 and P4 in DMF (1.0 × 10−4 mol L−1) were acquired. The emission peaks of M2 and P4 are located at ca. 376 nm and ca. 379 nm, respectively. It was found that the P4 emission peak has a 3 nm red shift with respect to M2, which resulted from the increase in the conjugation plane between the oxadiazole group and triazole ring after the click reaction. However, the fluorescence intensity of P4 decreased to a large extent when compared with M2. This was because the side chain of P4 has an azo group and its absorption peak was located at ca. 360 nm, so a large part of the fluorescence inspired by the oxadiazole group was assimilated by the azo group causing the fluorescent strength of P4 to decrease to a great degree. From Fig. 4(b), the PL spectra of the polymers P5–P7 in DMF (0.01 mg mL−1) were obtained. As can be seen from the spectra, P5–P7 have a maximum emission peak at ca. 379 nm and the intensity of the peak was increased gradually as the content of M2 increased.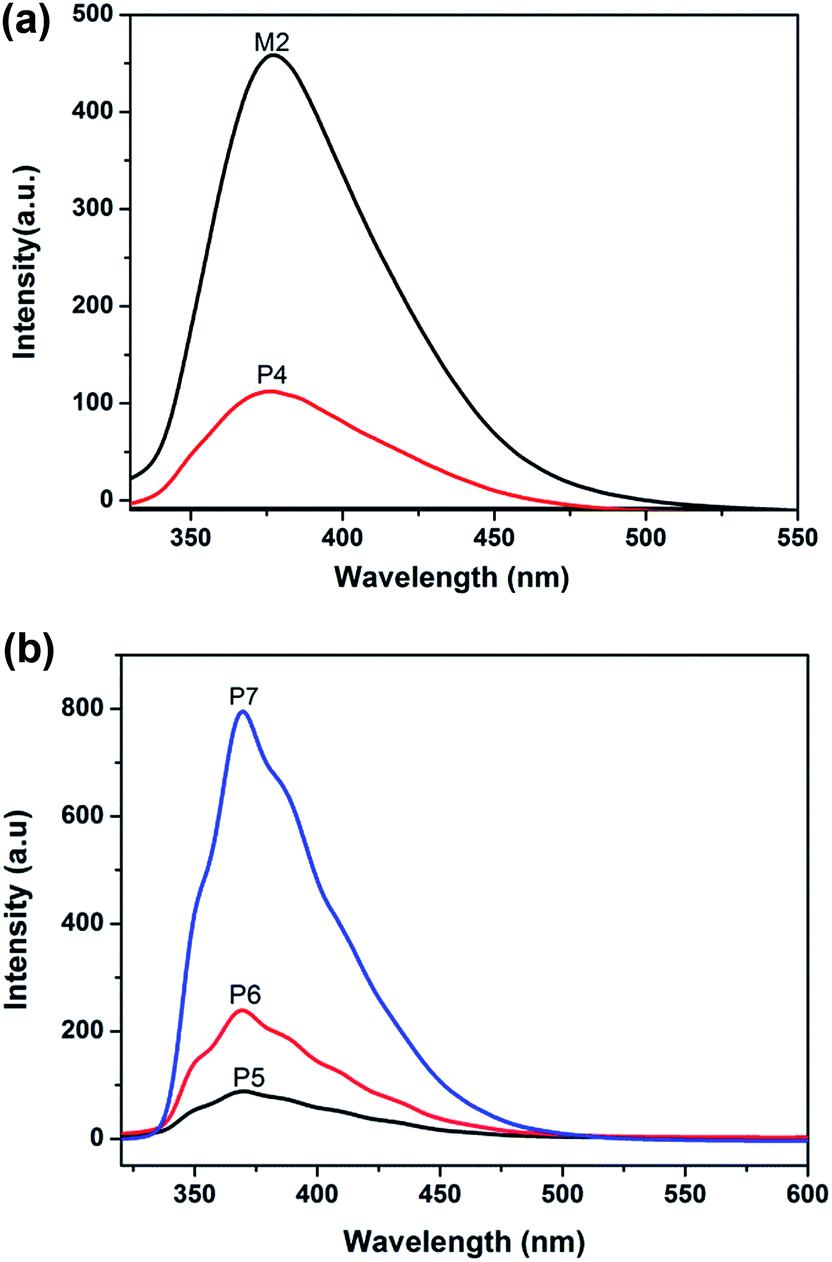 | ||
| Fig. 4 The PL spectra of (a) M2, P4 and (b) P5, P6, P7. | ||
Thermal properties
The thermal stability of the resulting monomers and polymers was evaluated using thermogravimetric analysis (TGA) under a nitrogen atmosphere. Their TGA thermograms are shown in Fig. 5. From Fig. 5(a), it can be seen that the thermal decomposition temperatures (Td, 5 wt% loss) of P3 and P1 are 281.3 °C and 265.9 °C, respectively. It means that P3 has a better thermal stability than of P1, which may be due to a protective “jacket” effect that shields the polyacetylene backbone through a strong electron interaction within the azo groups.39 Tang's group previously found that the alternating double-bond back-bone of the polymers may be surrounded by a rigid “jacket” formed through the intra- and interchain molecular interactions of the mesogenic groups, shielding the main chains of the polymer from thermal attack.40–42 Simultaneously, the protective “jacket” effect of the modified polymer containing a longer branch chain was more obvious after the click reaction. For P4, as it can be seen that the thermal decomposition temperature (Td, 5 wt% loss) was 372.9 °C. The reason why the thermal stability was improved more greatly than P1 and P3 may be attributed to two reasons. On the one hand, the backbone was surrounded by polar azo groups and oxadiazole groups, and a strong electronic interaction occurs within the groups so that the backbone was protected from the effects of heat, which is the so-called “jacket” effect. On the other hand, due to the stronger electronic interactions found with the oxadiazole group than the azobenzene group, the thermal stability of the oxadiazole group was better than the azobenzene group.43 From Fig. 5(b), it can be seen clearly that degradation temperature (Td, 5 wt% loss) of P5–P7 are 283.7 °C, 312.4 °C and 349.9 °C. The mechanism of thermal stability enhancement may be attributed to the reasons mentioned above. Simultaneously, the degradation temperatures of P5–P7 were improved significantly as the oxadiazole content increased and the polymer residues of combustion were increased from 31.83% (P5) to 58.01% (P7). From the spectra of Fig. 5(b), the rate of degradation decreased gradually from P5 to P7. This was because the azobenzene group is an optically active functional group and can undergo a cis–trans isomerization transition under thermal conditions. Therefore, the thermal isomerization of the azobenzene group first occurs and then the degradation of azobenzene occurs. With the reduction in the azobenzene group content in polymers P5–P7, the rate of degradation was increased gradually from P5 to P7. This was manifested clearly in the region of 300–500 °C.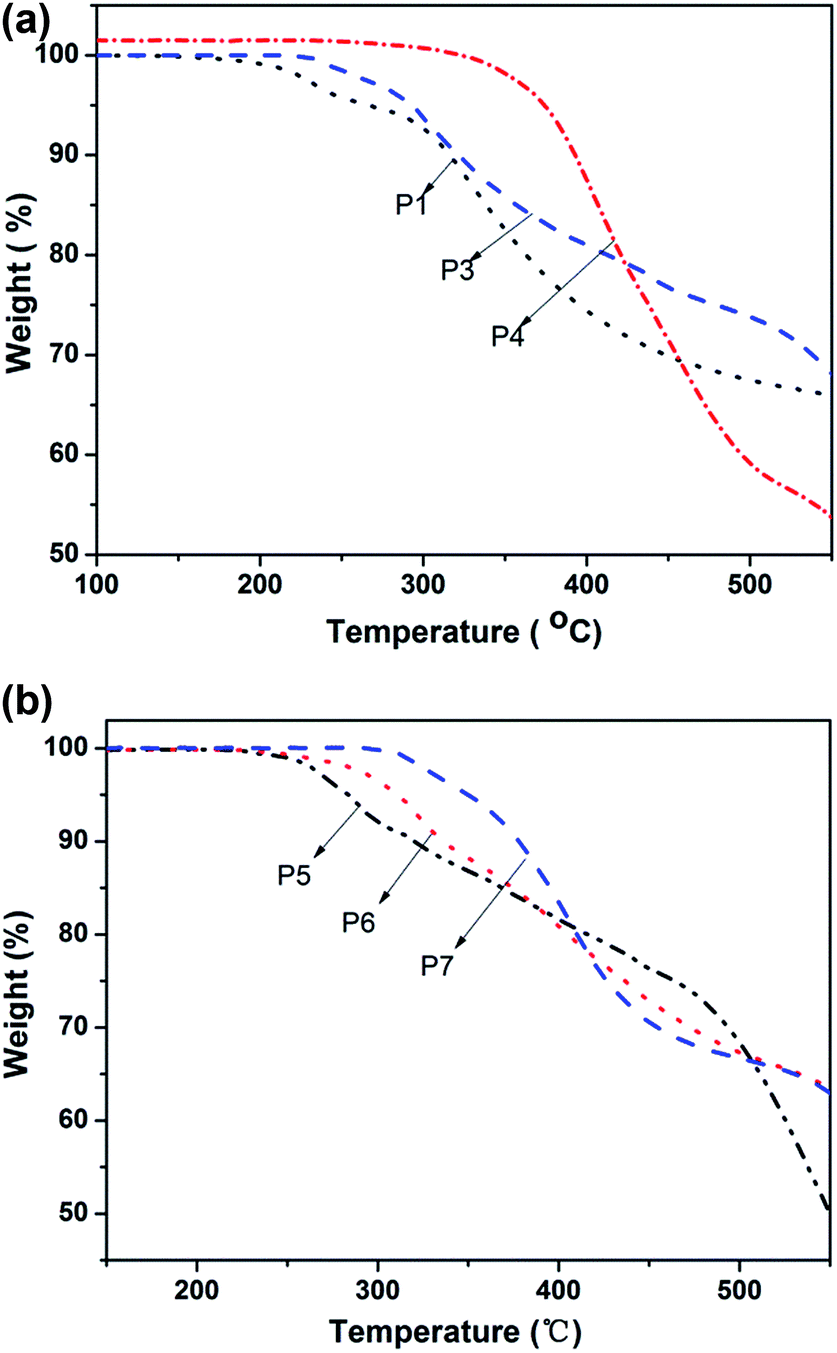 | ||
| Fig. 5 The TGA spectra of (a) M2, P4 and (b) P5, P6, P7. | ||
Experimental section
Materials
Cuprous iodide, dimethyl sulfoxide, 1,4-dibromobutane, n-hexane, N,N-dimethyl formamide, triethylamine, 4-bromoaniline, triphenylphosphine, sodium hydroxide, sodium and anhydrous magnesium sulfate were supplied from Shanghai Chemical Reagent Company. Palladium acetate, 2-methyl-3-butyl acetylene-2-alcohol and triphenyl phosphorus dichloride palladium were purchased from Alfa Aesar. All commercial reagents were used directly without any further purification except some solvents, which were distilled prior to use.Instrumentation
FTIR spectra were obtained using a KBr disk on a Nicolet NEXUS 8700 FTIR spectrometer. 1H-NMR spectra were obtained on a Bruker DMX-400 spectrometer using chloroform-d (CDCl3) as the NMR solvent. The optical properties of the samples were measured using a Shimadzu UV-265 spectrometer with a 1 cm quartz cell. TGA (thermogravimetric analysis) was performed on a Perkin-Elmer TGA under an N2 atmosphere at a heating rate of 10 °C min−1. Elemental analyses were performed using a CHNOS Elemental Analyzer.Preparation of the monomers and polymers
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :petroleum ether = 1:6) to afford a red solid powder in 85.8% yield. FT-IR (KBr) cm−1: 3438 (–OH), 1584 (NN), 835 (–Ar–), 543 (Ar-Br). 1H-NMR (CDCl3): δ (ppm) = 6.93 (2H, m, J = 8.8 Hz, 4-H), δ 7.62 (2H, m, J = 8.7 Hz, 3-H), δ 7.75 (2H, m, J = 20 Hz, 2-H), δ 7.86 (2H, m, J = 8.8 Hz, 1-H), δ 10.38 (1H, s, 7-H).:petroleum ether = 1:1) to afford a red solid powder in 60% yield. FT-IR (KBr) cm−1: 2978, 2923, 2851 (–CH3), 2228 (CC), 1589 (NN), 844 (–Ar–). 1H-NMR (DMSO): δ (ppm) = 1.18–1.48 (6H, m, –(CH3)2), δ 7.55 (2H, d, J = 8.3 Hz, 2-H), δ 7.81 (2H, m, J = 6.6 Hz, 3-H), δ 7.78 (2H, d, J = 4.3 Hz, 4-H), δ 6.95 (2H, d, J = 8.7 Hz, 5-H), δ 5.54 (1H, s, –OH), δ 10.38 (1H, s, Ar-OH).:petroleum ether = 1:6) to afford a red solid powder in 80% yield. FT-IR (KBr) cm−1: 3264 (CC–H), 1589 (NN), 845 (–Ar–). 1H-NMR (DMSO): δ (ppm) = 4.42 (1H, s, –CCH), δ 7.66 (2H, t, J = 6.7 Hz, 2-H), δ 7.82 (2H, t, J = 4.5 Hz, 3-H), δ 7.81 (2H, d, J = 2.3 Hz, 4-H), δ 6.95 (2H, m, J = 14.8 Hz, 5-H), δ 10.41 (1H, s, –OH).:petroleum ether = 1:6 or 1:10) to obtain a red solid powder in 83% yield. FT-IR (KBr) cm−1: 3279 (CC–H); 2952, 2933, 2869 (–CH3; –CH2–), 2108 (CC), 1582 (NN), 1247 (Ar-O), 844 (–Ar–). 1H-NMR (CDCl3): δ (ppm) = 3.25 (1H, s, –CCH), δ 7.65 (2H, m, J = 8.5 Hz, 2-H), δ 7.95 (2H, m, J = 9 Hz, 3-H), δ 7.87 (2H, m, J = 8.6 Hz, 4-H), δ 7.04 (2H, m, J = 9 Hz, 5-H), δ 4.13 (2H, t, J = 12 Hz, –O–CH2), δ 2.02–2.16 (4H, m, –CH2–CH2–), δ 3.55 (2H, t, J = 13 Hz, Br–CH2–).N), 1253 (Ar-O), 839 (–Ar–). 1H-NMR (CDCl3): δ (ppm) = 7.94–7.52 (Ar-H), 7.02–6.93 (Ar-H), 4.1–4.06 (–OCH2–), 3.51–3.5 (–CH2Br), 2.17–1.85 (–CH2–).N), 1253 (Ar-O). 1H-NMR (CDCl3), δ (ppm) = 7.94–7.52 (Ar-H), 7.02–6.93 (Ar-H), 4.1–4.06 (–OCH2–), 3.41–3.5 (–CH2N3), 2.17–1.85 (–CH2–).N), 1268 (Ar-O), 801 (–Ar–). 1H-NMR (CDCl3): δ (ppm) = 8.12–7.51 (Ar-H), 7.18–6.93 (Ar-H), 6.83–6.63 (Ar-H), 5.49 (–O–CH2–), 4.46 (–COO–CH2–C–), 4.1–4.06 (–OCH2–), 3.41–3.5 (–CH2N3), 1.78–1.22 (–CH2–), 0.87–0.85 (–CH3). Mw = 1.51 × 104, Mw/Mn = 2.2.N), 1258 (Ar-O), 841 (–Ar–). 1H-NMR (CDCl3) δ (ppm) = 8.03–7.51 (Ar-H), 6.99–6.97 (Ar-H), 4.61–4.4 (–CH2N3), 4.1–4.06 (–OCH2–), 2.04–1.25 (–CH2–), 0.88 (–CH3). Mw = 2.67 × 104, Mw/Mn = 1.03.:2, 6:4 and 2:8, which correspond to polymer 5, polymer 6 and polymer 7. Polymer 5, polymer 6 and polymer 7 were obtained as deep red solids. FT-IR (KBr) cm−1: 2955–2854 (–CH3; –CH2–), 1717 (–NO2), 1598 (NN), 1260 (Ar-O), 801 (–Ar–). 1H-NMR (CDCl3): δ (ppm) = 8.12–7.51 (Ar-H), 7.18–6.93 (Ar-H), 6.83–6.63 (Ar-H), 5.49 (–O–CH2–), 4.46 (–COO–CH2–C–), 4.1–4.06 (–OCH2–), 3.41–3.5 (–CH2N3), 2.15–1.22 (–CH2–), 0.87–0.85 (–CH3).
:petroleum ether = 1:6) to afford a red solid powder in 85.8% yield. FT-IR (KBr) cm−1: 3438 (–OH), 1584 (NN), 835 (–Ar–), 543 (Ar-Br). 1H-NMR (CDCl3): δ (ppm) = 6.93 (2H, m, J = 8.8 Hz, 4-H), δ 7.62 (2H, m, J = 8.7 Hz, 3-H), δ 7.75 (2H, m, J = 20 Hz, 2-H), δ 7.86 (2H, m, J = 8.8 Hz, 1-H), δ 10.38 (1H, s, 7-H).:petroleum ether = 1:1) to afford a red solid powder in 60% yield. FT-IR (KBr) cm−1: 2978, 2923, 2851 (–CH3), 2228 (CC), 1589 (NN), 844 (–Ar–). 1H-NMR (DMSO): δ (ppm) = 1.18–1.48 (6H, m, –(CH3)2), δ 7.55 (2H, d, J = 8.3 Hz, 2-H), δ 7.81 (2H, m, J = 6.6 Hz, 3-H), δ 7.78 (2H, d, J = 4.3 Hz, 4-H), δ 6.95 (2H, d, J = 8.7 Hz, 5-H), δ 5.54 (1H, s, –OH), δ 10.38 (1H, s, Ar-OH).:petroleum ether = 1:6) to afford a red solid powder in 80% yield. FT-IR (KBr) cm−1: 3264 (CC–H), 1589 (NN), 845 (–Ar–). 1H-NMR (DMSO): δ (ppm) = 4.42 (1H, s, –CCH), δ 7.66 (2H, t, J = 6.7 Hz, 2-H), δ 7.82 (2H, t, J = 4.5 Hz, 3-H), δ 7.81 (2H, d, J = 2.3 Hz, 4-H), δ 6.95 (2H, m, J = 14.8 Hz, 5-H), δ 10.41 (1H, s, –OH).:petroleum ether = 1:6 or 1:10) to obtain a red solid powder in 83% yield. FT-IR (KBr) cm−1: 3279 (CC–H); 2952, 2933, 2869 (–CH3; –CH2–), 2108 (CC), 1582 (NN), 1247 (Ar-O), 844 (–Ar–). 1H-NMR (CDCl3): δ (ppm) = 3.25 (1H, s, –CCH), δ 7.65 (2H, m, J = 8.5 Hz, 2-H), δ 7.95 (2H, m, J = 9 Hz, 3-H), δ 7.87 (2H, m, J = 8.6 Hz, 4-H), δ 7.04 (2H, m, J = 9 Hz, 5-H), δ 4.13 (2H, t, J = 12 Hz, –O–CH2), δ 2.02–2.16 (4H, m, –CH2–CH2–), δ 3.55 (2H, t, J = 13 Hz, Br–CH2–).N), 1253 (Ar-O), 839 (–Ar–). 1H-NMR (CDCl3): δ (ppm) = 7.94–7.52 (Ar-H), 7.02–6.93 (Ar-H), 4.1–4.06 (–OCH2–), 3.51–3.5 (–CH2Br), 2.17–1.85 (–CH2–).N), 1253 (Ar-O). 1H-NMR (CDCl3), δ (ppm) = 7.94–7.52 (Ar-H), 7.02–6.93 (Ar-H), 4.1–4.06 (–OCH2–), 3.41–3.5 (–CH2N3), 2.17–1.85 (–CH2–).N), 1268 (Ar-O), 801 (–Ar–). 1H-NMR (CDCl3): δ (ppm) = 8.12–7.51 (Ar-H), 7.18–6.93 (Ar-H), 6.83–6.63 (Ar-H), 5.49 (–O–CH2–), 4.46 (–COO–CH2–C–), 4.1–4.06 (–OCH2–), 3.41–3.5 (–CH2N3), 1.78–1.22 (–CH2–), 0.87–0.85 (–CH3). Mw = 1.51 × 104, Mw/Mn = 2.2.N), 1258 (Ar-O), 841 (–Ar–). 1H-NMR (CDCl3) δ (ppm) = 8.03–7.51 (Ar-H), 6.99–6.97 (Ar-H), 4.61–4.4 (–CH2N3), 4.1–4.06 (–OCH2–), 2.04–1.25 (–CH2–), 0.88 (–CH3). Mw = 2.67 × 104, Mw/Mn = 1.03.:2, 6:4 and 2:8, which correspond to polymer 5, polymer 6 and polymer 7. Polymer 5, polymer 6 and polymer 7 were obtained as deep red solids. FT-IR (KBr) cm−1: 2955–2854 (–CH3; –CH2–), 1717 (–NO2), 1598 (NN), 1260 (Ar-O), 801 (–Ar–). 1H-NMR (CDCl3): δ (ppm) = 8.12–7.51 (Ar-H), 7.18–6.93 (Ar-H), 6.83–6.63 (Ar-H), 5.49 (–O–CH2–), 4.46 (–COO–CH2–C–), 4.1–4.06 (–OCH2–), 3.41–3.5 (–CH2N3), 2.15–1.22 (–CH2–), 0.87–0.85 (–CH3).Conclusion
Five novel multifunctional polyacetylenes with oxadiazole and azo groups were designed and fabricated using copper sulfate and sodium ascorbic acid catalysts via a highly efficient click reaction. The results show that the incorporation of the non-linear optical azo molecule and electron transport oxadiazole chromophore molecule into the side chain of polyacetylene not only endowed the multifunctional polyacetylenes with good solubility, but also improved the thermal stability, which may be attributed to the ‘jacket effect’ of the pendants, resulting from strong intra- and inter-molecular electronic interactions between the polarized azo groups and oxadiazole groups. This study provides an effective and accurate method for preparing soluble functional polyacetylenes with characteristic optical properties and high thermal stability.Acknowledgements
This research was financially supported by the National Natural Science Fund of China (Grant No. 21471030, 21271040 and 21171034), the Shanghai Municipal Natural Science Foundation for Youths (No. 12ZR144100) and the “Chen Guang” project (No. 12CG37) supported by Shanghai Municipal Education Commission and Shanghai Education Development Foundation.Notes and references
- F. Kajzar, S. Etemad, G. L. Baker and J. Messier, Solid State Commun., 1987, 63, 1117 CrossRef.
- W. S. Fann, S. Benson, J. M. J. Madey, S. Etemad, G. L. Baker and F. Kajzar, Phys. Rev. Lett., 1989, 62, 1492–1495 CrossRef CAS PubMed.
- A. Takahashi, Synth. Met., 1997, 85, 1087–1088 CrossRef CAS.
- D. Neher, A. Kaltbeitzel, A. Wolf, C. Bubeck and G. Wegner, J. Phys. D: Appl. Phys., 1991, 24, 1193–1202 CrossRef CAS.
- W. Y. L. Jacky and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745–754 CrossRef PubMed.
- S. C. Yin, H. Y. Xu, X. Y. Su, X. Y. Su, W. Lei, Y. L. Song and B. Z. Tang, Dyes Pigm., 2007, 75, 675–680 CrossRef CAS.
- S. C. Yin, H. Y. Xu, W. F. Shi, L. Bao, Y. C. Gao, Y. L. Song and B. Z. Tang, Dyes Pigm., 2007, 72, 119–123 CrossRef.
- S. Y. Guang, S. C. Yin, H. Y. Xu, W. J. Zhu, Y. C. Gao and Y. L. Song, Dyes Pigm., 2007, 73, 285–291 CrossRef CAS.
- X. Y. Su, H. Y. Xu, Q. Z. Guo, S. Y. Guang, J. Y. Yang and Y. L. Song, J. Polym. Sci., Part A: Polym. Chem., 2006, 46, 4529–4541 CrossRef.
- X. Wang, S. Y. Guang, H. Y. Xu, X. Y. Su, X. H. Zhu and C. Li, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1406–1414 CrossRef CAS.
- X. Y. Su, H. Y. Xu, J. Y. Yang, N. B. Lin and Y. L. Song, Polymer, 2008, 49, 3722–3730 CrossRef CAS.
- H. Y. Xu, S. C. Yin, W. J. Zhu, Y. L. Song and B. Z. Tang, Polymer, 2006, 47, 6986–6992 CrossRef CAS.
- H. Y. Xu, X. Wang and J. C. Wu, Chin. Chem. Lett., 2008, 19, 141–145 CrossRef CAS.
- F. Sanda, T. Nakai, N. Kobayashi and T. Masuda, Macromolecules, 2004, 37, 2703–2708 CrossRef CAS.
- J. W. Y. Lam, Y. Dong, H. S. Kwork and B. Z. Tang, Macromolecules, 2006, 47, 6551–6559 Search PubMed.
- J. Qu, Y. Suzuki, M. Shiotsuki and F. Sanda, Polymer, 2007, 48, 4628–4636 CrossRef CAS.
- C. C. W. Law, J. W. Y. Lam, A. Qin, Y. Dong, H. S. Kwork and B. Z. Tang, Polymer, 2006, 47, 6642–6651 CrossRef CAS.
- K. K. L. Cheuk, J. W. Y. Lam, B. S. Li, Y. Xie and B. Z. Tang, Macromolecules, 2007, 40, 2633–2642 CrossRef CAS.
- H. B. Wolfgang, K. Christian, J. S. Christoph and F. Gernot, Macromolecules, 2005, 38, 9405–9410 CrossRef.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, Chem.–Eur. J., 2006, 12, 6776–6786 CrossRef PubMed.
- D. A. Ossipov and J. Hilborn, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS.
- J. Bolley, E. Guenin, N. Lievre, M. Lecouvey, M. Soussan, Y. Lalatonne and L. Motte, Langmuir, 2013, 29, 14639–14647 CrossRef CAS PubMed.
- D. L. Thorek and D. R. Elias, Mol. Imaging, 2009, 8, 221–229 CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- B. C. Englert, S. Bakbak and U. H. F. Bunz, Macromolecules, 2005, 38, 5686–5877 CrossRef.
- Y. Xu, Y. Suzuki, T. Ishizuka, C. D. Xiao, X. Liu, T. Hayashi and M. Komiyama, Bioorg. Med. Chem., 2014, 22, 4419–4421 CrossRef CAS PubMed.
- T. Smyth, K. Petrova, N. M. Payton, I. Persaud, J. S. Redzic, M. W. Gruner and P. Smith-Jones, Bioconjugate Chem., 2014, 25, 1777–1784 CrossRef CAS PubMed.
- H. S. Zhao, E. Heusler, G. Jones, L. H. Li and V. Werner, J. Struct. Biol., 2014, 186, 420–430 CrossRef CAS PubMed.
- C. Hu, X. Guo, Y. H. Jing, J. Chen, C. Zhang and J. J. Huang, J. Appl. Polym. Sci., 2014 DOI:10.1002/app.40636.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, Chem.–Eur. J., 2006, 12, 6776–6786 CrossRef PubMed.
- D. A. Ossipov and J. Hilborn, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS.
- X. Y. Wang, A. Kimyonok and M. Weck, Chem. Commun., 2006, 3933–3935 RSC.
- L. Ding, C. N. Chen, J. P. Deng and W. T. Yang, Polym. Bull., 2012, 69, 1023–1040 CrossRef CAS.
- L. Tong, A. J. Qin, Y. Mao, J. Z. Sun and B. Z. Tang, Sci. China: Chem., 2011, 54, 1948–1954 CrossRef CAS.
- Q. Zeng, Z. A. Li, Z. Li, C. Ye, J. G. Qin and B. Z. Tang, Macromolecules, 2007, 40, 5634–5637 CrossRef CAS.
- C. K. W. Jim, A. J. Qin, J. W. Y. Lam and B. Z. Tang, J. Inorg. Organomet. Polym. Mater., 2007, 17, 289–293 CrossRef CAS.
- C. K. W. Jim, A. J. Qin, J. W. Y. Lam and B. Z. Tang, J. Inorg. Organomet. Polym. Mater., 2007, 17, 289–293 CrossRef CAS.
- S. H. Jin, M. Y. Kim and J. Y. Kim, J. Am. Chem. Soc., 2004, 126, 2474–2480 CrossRef CAS PubMed.
- M. Teraguchi and T. Masuda, Macromolecules, 2000, 33, 240–242 CrossRef CAS.
- B. Z. Tang, X. X. Kong, X. H. Wan, H. Peng and W. Y. Lam, Macromolecules, 1998, 31, 2419–2432 CrossRef CAS.
- B. Z. Tang, X. Kong, X. Wan and X. D. Feng, Macromolecules, 1997, 30, 5620–5626 CrossRef CAS.
- J. W. Y. Lam, Y. P. Dong, K. K. L. Cheuk and B. Z. Tang, Macromolecules, 2002, 35, 1229–1240 CrossRef CAS.
- X. Wang, S. Y. Guang, H. Y. Xu, X. Y. Su, J. Y. Yang, Y. L. Song, N. B. Lin and X. Y. Liu, J. Mater. Chem., 2008, 18, 4204–4209 RSC.
| This journal is © The Royal Society of Chemistry 2016 |