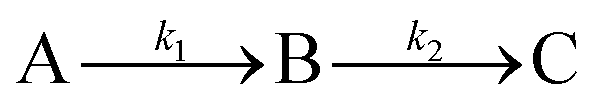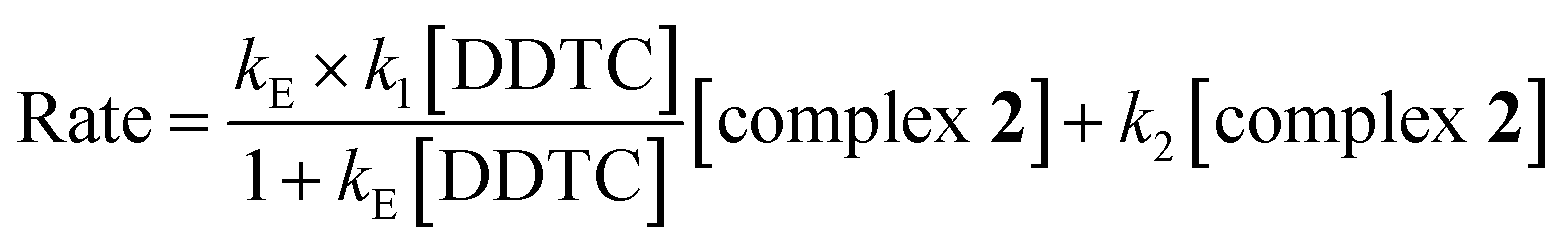DOI:
10.1039/C5RA21161A
(Paper)
RSC Adv., 2016,
6, 18288-18299
An experimental and theoretical approach on the kinetics and mechanism for the formation of a four-membered (S, S) chelated Pt(II) complex†
Received
12th October 2015
, Accepted 3rd February 2016
First published on 4th February 2016
Abstract
The interaction of a chemoprotective agent, diethyldithiocarbamate (DDTC), with cis-[Pt(pic)(H2O)2]2+, 2 (where pic = 2-aminomethylpyridine), in aqueous medium was investigated experimentally and theoretically. The equilibrium constant (KE) for the formation of the outer sphere association complex and the rate constants for both steps have been evaluated experimentally, and were compared with the theoretically determined values. The activation parameters (ΔHǂ, ΔSǂ and ΔGǂ) for both the steps were calculated and an associative mechanism is proposed. The bonding modes of the product [Pt(pic)(DDTC)]+, 3, were confirmed by spectroscopic measurements and supported by Time Dependent Density Functional Theory (TD-DFT) and Natural Bond Orbital (NBO) calculations. Penta coordinated platinum transition state geometries for both the steps were fully optimized and confirmed by frequency analysis and an intrinsic reaction coordinate method molecular docking study has been performed to understand the binding interaction of complex 2 with B-DNA. The anticancer properties of 2 and 3 were investigated on a HeLa cell line, and they showed remarkable activity of about 70% compared to cisplatin at 50 μM concentration.
Introduction
Cisplatin is the most widely used drug for the treatment of cancer among the commercially available Pt(II)-based chemotherapeutic agents.1–5 The clinical use of cisplatin is curbed by its inactivity against several types of cancer, acquired resistance in long term treatment along with high toxicity to normal cells. Consequently, researchers are aiming to synthesise safer Pt(II)-based anticancer drugs with reduced toxicity and wider application. One of the recognised causes for the development of resistance is reduced transport of drugs across the cell membrane. Y. Kidani and co-workers6 noted that cellular resistance of cisplatin can be overcome by changing the ammine ligands to 1,2-diaminocyclohexane (DACH). As a result, certain planned amines were used to synthesize Pt(II) complexes7,8 and good results were found. It is therefore of much interest to investigate how variation in the structures of amine ligands affect the toxicity of platinum complexes. We have chosen 2-aminomethylpyridine (pic) as the carrier ligand in the Pt(II) complex [Pt(pic)(H2O)2]2+ 2 due to its σ-donor and π-acceptor properties, which make it suitable for substitution by biomolecules and kinetic investigations. To tune the activity of Pt(II)-based anticancer drugs, a new strategy has been developed to design novel Pt(II) complexes9–12 with N- and S-containing ligands.
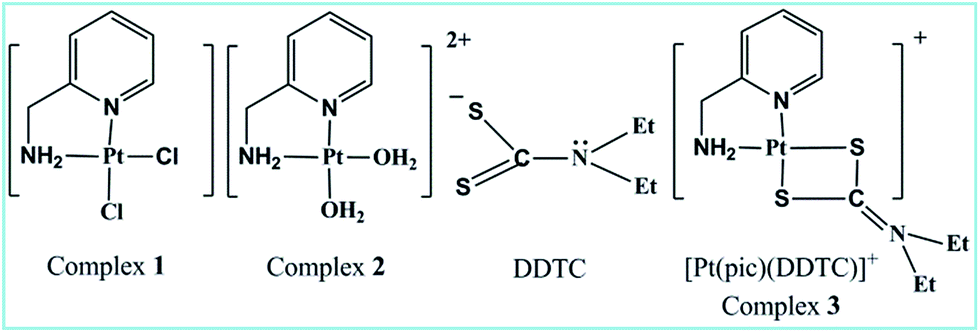
Dithiocarbamate or its substituted derivative diethyldithiocarbamate (DDTC) has versatile uses in the area of bio-inorganic chemistry as a rescue agent13,14 against cisplatin toxicity. Another fascinating aspect of the bidentate (S, S) chelating ligand DDTC is its high selectivity to protect from gastrointestinal, renal and bone marrow toxicity induced by Pt(II)-based anticancer drugs without inhibition of the latter’s antitumor effect.15–18 Few Pt(II) and Pd(II) dithiocarbamate complexes have been reported for their remarkable cytotoxic properties, lower cross-resistance and minimum nephrotoxicity19 compared to cisplatin. Binding of Pt(II) to dithiocarbamate sulfur tends to be irreversible in contrast to thio-ether sulfur20–22 and results in a stable adduct, which however does not interfere with the tumoricidal Pt–DNA interaction. Thus, the protective action of dithiocarbamate against the toxicity of cisplatin seems to be the formation of its stable platinum dithiocarbamate complexes. In vitro kinetic and mechanistic studies on complex 2 with a few thiols23–25 have already been investigated. Combined experimental and theoretical investigation on the kinetics and mechanism of the substitution reaction of 2 with DDTC has not yet been studied in detail. Herein, we have endeavoured to correlate the experimental and theoretical kinetic data for the reaction. The kinetic study aims to understand the drug reservoir mechanism between DDTC and complex 2. Cell-based assays and DNA binding studies were done to evaluate the anticancer properties of the complexes (Fig. 1).
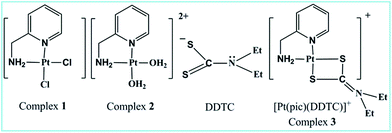 |
| | Fig. 1 Structure of ligand and complexes. | |
Experimental
Instrumentation
Elemental analysis of the complexes was carried out on a Perkin-Elmer 2400 series-II CHNS/O Analyser. Conductivity measurements were performed with a Systronics conductivity meter (Model 308), where the cell constant was calibrated with standard buffer solution. The pH measurements were done with the help of a EUTECH digital pH meter (pH Tutor) with an accuracy of ±0.01 U, calibrated with a standard phosphate buffer solution (KH2PO4/Na2HPO4). Fourier transform infrared (FTIR) spectra of the complexes were recorded on a Nicolet-iS-10 spectrometer (KBr disk, in the range of 4000–400 cm−1). The kinetic measurements were conducted at pH 4.0 on a Shimadzu UV 1800 spectrophotometer attached to a thermoelectric cell temperature controller (Model TCC-100, accuracy ±0.1 °C). 1H NMR spectra were recorded with a Bruker Avance-400 (400 MHz) spectrometer using D2O or DMSO-d6 as the solvent. ESI mass spectra were carried out on a Waters Q-TOF Micro YA263 Mass spectrometer in water. The pKa values of complex 2 were determined by a Metrohm 888 Titrando titro-processor. A Stat Fax™® 2100 Microplate (USA) Enzyme-linked immunosorbent assay (ELISA) plate reader was used for the MTT assay.
Reagents and methods
All chemicals used for the kinetic and bioactivity study were of the highest purity available. The starting compounds K2PtCl4 (99%), 2-aminomethylpyridine (pic) (97%), and sodium diethyldithiocarbamate (NaDDTC) were purchased from Sigma Aldrich. AgClO4 (97%), AgNO3 (99%), K2HPO4 and NaClO4 were purchased from Merck. All these reagents were used without further purification. For kinetic purposes, double distilled water was used to prepare all kinds of solutions and molecular biology grade water was used for the bioactivity study. All solvents used were of analytical grade. Cisplatin (Cis-gland) was purchased from Gland Pharma Limited. Culture medium (DMEM) and FBS were purchased from Hi-Media.
Synthesis of complexes 1–3
Complex 1 was prepared according to the literature method26 with some modification. K2PtCl4 (250 mg, 0.6023 mmol) was dissolved in 7.5 mL water and heated at 40–45 °C followed by dropwise addition of an aqueous solution of 2-aminomethyl pyridine (pic) (65 mg, 0.6023 mmol). The reaction mixture was heated at 50–60 °C with constant stirring for two hours. A greenish yellow coloured precipitate was obtained and the solution became colourless after a few hours of stirring indicating the completion of the reaction. The precipitate was filtered and subsequently washed with water, acetone and diethyl ether. It was then dried in a desiccator until a constant weight was obtained (yield = 175 mg, 78%). Elemental analysis (%): C6H8N2Cl2Pt (calc. in parentheses): C 18.8 (19.2), H 2.0 (2.1), N 7.4 (7.5) and Cl 9.5 (9.7).
The diaqua complex 2 was prepared in solution using the method of Hay and Basak.27 The chloro complex was converted into the diaqua analogue in solution by adding two equivalents of AgClO4. The solution was kept in the dark overnight, and the AgCl precipitate was removed by filtration through a 0.1 μm pore size membrane filter. Great care was taken to ensure that the resulting solution was free of Ag+ ions and that the chloro complex had been converted completely into the corresponding diaqua species. For the bioactivity study, another aliquot of the diaqua [Pt(pic)(H2O)2](NO3)2 complex 2 solution was prepared from [Pt(pic)Cl2] as the NO3− salt using AgNO3 solution (the same method as for AgClO4).
Job’s method of continuous variation was carried out to confirm the metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand (M:L) ratio for the reaction between complex 2 with DDTC at pH 4.0. Complex 2 and the ligand were mixed according to their probable ratios, like 1:1, 1:2, 2:1 etc. and heated to about 50 °C for a few hours and the absorbances were measured until a constant maximum was reached. The exact M:L ratio was found with proper calculation and shows a bell shaped curve when absorbance vs. [M]/[M] + [L] was plotted (ESI Fig. S1†). The development of a characteristic peak in the product complex 3 at 262 nm was monitored as a function of time at different fixed temperatures. Complex 2 and the ligand DDTC were mixed in a 1:1 molar ratio at pH 4.0 in accordance with Job’s result to obtain the solid product by slow evaporation.
:ligand (M:L) ratio for the reaction between complex 2 with DDTC at pH 4.0. Complex 2 and the ligand were mixed according to their probable ratios, like 1:1, 1:2, 2:1 etc. and heated to about 50 °C for a few hours and the absorbances were measured until a constant maximum was reached. The exact M:L ratio was found with proper calculation and shows a bell shaped curve when absorbance vs. [M]/[M] + [L] was plotted (ESI Fig. S1†). The development of a characteristic peak in the product complex 3 at 262 nm was monitored as a function of time at different fixed temperatures. Complex 2 and the ligand DDTC were mixed in a 1:1 molar ratio at pH 4.0 in accordance with Job’s result to obtain the solid product by slow evaporation.
Synthesis safety note
Metal perchlorate salts should be handled carefully as these are explosive and hazardous upon strong heating. Appropriate safety precautions were taken while handling the salt. No explosion event was encountered in the present study.
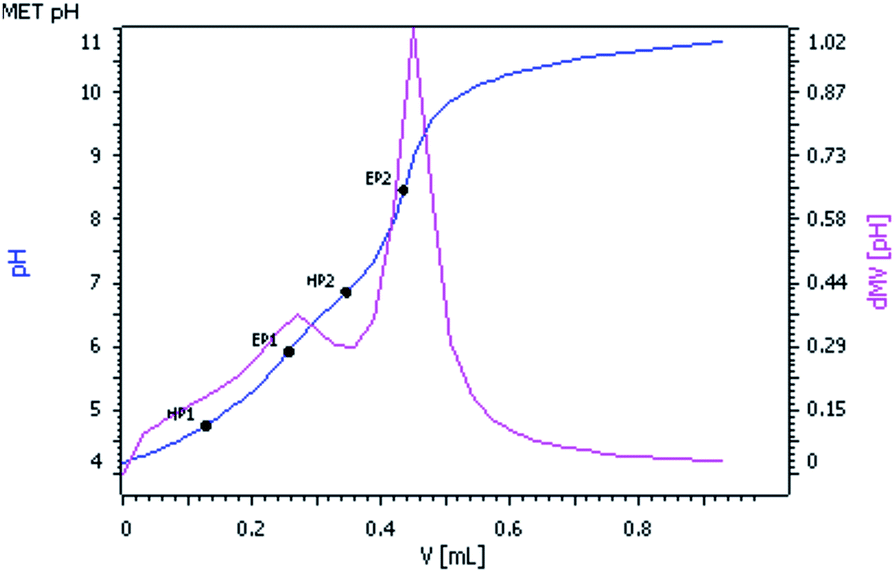
pKa value determination of complex 2
The pKa values of complex 2 were determined by the potentiometric method using the tiamo 2.4 software package. The electrode and titro processor were calibrated with standard buffer solutions prepared according to NBS specifications. pH versus volume titration (Fig. 2) of complex 2 (2.05 × 10−3 M) with standard 0.05 M KOH solution was done at a constant ionic strength (0.1 M NaClO4) and at 25 °C. pH was plotted against the volume of base consumed. The relationship pH − p[H] = 0.05 was observed. Equivalent points EP1 and EP2 were observed in the titration curve corresponding to the pKa values HP1 and HP2. The pKa1 and pKa2 values of complex 2 were found to be 4.76 and 6.83 respectively, which are comparable with the reported pKa values of the almost similar system [Pt(MAMP)(H2O)2]2+.7
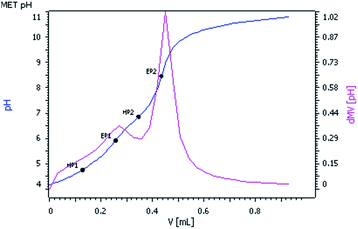 |
| | Fig. 2 pH vs. volume (mL) of 0.05 M KOH solution. | |
Spectroscopic study
Electronic absorption spectrum of 1 in DMSO, λmax (ε/M−1 cm−1): 308 nm (5012). Selected IR frequencies (ESI Fig. S2†) (KBr disk, cm−1): 3200–3170(b), 3110(s), 2924(s), 1616(s), 1559(m), 1449(m), 1288(s), 1162(m) and 425(s). 1H NMR (400 MHz, DMSO-d6, δ in ppm, J in Hz) of 1 (ESI Fig. S3†): δ 8.91 (d, J = 6.9, 1Ha), δ 8.25 (t, J = 1.9, 8.4, 1Hc), δ 7.81 (d, J = 10.8, 1Hd), δ 7.46 (t, J = 9.9, 7.6, 1Hb), δ 6.8 (s, 2Hg), δ 4.40 (t, J = 7.5, 7.5, 2Hf), δ 3.3 (water in DMSO), and δ 2.49 (solvent DMSO).
13C NMR of 1 (ESI Fig. S4†) (400 MHz, δ in ppm, DMSO-d6): δ Ce 150.20, Ca 141.64, Cc 124.74, Cd 122.9, Cb 41.56 and Cf 39.52.
Electronic spectrum of DDTC substituted product 3 displays bands at different λmax values in aqueous medium with hypsochromic as well as hyperchromic shift as compared to complex 2 (ESI Fig. S5†).
The important bands in the IR spectra of 3 (ESI Fig. S6†) are: 3400–3380(br) cm−1, 2974(br) cm−1, 2823(m) cm−1, 1684(s) cm−1, 1637(s) cm−1, 1541 cm−1, 1522 cm−1, 1285–1080(br), 940(s) cm−1, 627(s) cm−1 and 476(s) cm−1.
1H NMR of 3 (ESI Fig. S7†) (400 MHz, DMSO-d6, δ in ppm, J in Hz) of complex 3: δ 8.5 (d, J = 6.4, 1Ha), δ 8.29 (t, J = 2.8, 2.0, 1Hc), δ 7.74 (t, J = 7.6, 8.0, 1Hd), 7.56 (d, J = 6.4, 1Hb), 4.40 (d, J = 5.6, 2Hf), 2.92 (m, 4Hx & x′), and 1.15 (t, 6Hy & y′).
The important peaks of the IR spectrum of the free ligand DDTC (ESI Fig. S8†) are: 3500–3200(br) cm−1, 2978(s) cm−1, 2923(s) cm−1, 2075(br) cm−1, 1671(s) cm−1, 1617(s) cm−1, 1590(s) cm−1, 1434(s) cm−1, 1262(s) cm−1, 1204(s) cm−1, 1131 cm−1, 1063 cm−1, 936 cm−1 and 911 cm−1.
The ESI mass spectrum of product complex 3 (ESI Fig. S9†) shows a molecular ion peak m/z at 451.08 (100%) in water.
Kinetic investigation
The usual mixing technique was followed and pseudo-first-order reaction conditions were employed throughout the kinetic runs for the reactions. The progress of the reaction was followed by measuring the increase in absorbance for DDTC with complex 2 at 310 nm, where the spectral differences between complex 2 and the product complex 3 are at a maximum. The pH and ionic strength were maintained at 4.0 and 0.1 mol dm−3 (with NaClO4) respectively. The method of Weyh and Hamm28 was adopted to calculate the rate constants for both the kinetic studies. The rate data, represented as an average of duplicate runs, were reproducible to within ±4%.
Computational details
All calculations were performed with the Gaussian09 (ref. 29) programme. For optimisation of complex 3 in both the gas and aqueous phases, we have used three density functional theory methods. The first is the gradient-corrected DFT level using the three parameter fit of exchange and correlation functionals of Becke (B3LYP)30 which includes the correlation functional of Lee, Yang and Parr (LYP).31 The second is MPW1PW91 (ref. 32 and 33) which utilizes a modification of the gradient-corrected Perdew–Wang exchange functional, PW91, combined with the non-local Perdew–Wang correlation functional and the last one is PBEPBE.34,35 The relativistic effective core potential (ECP) and associated valence double ξ (zeta) basis set of Hay and Wadt36,37 (LANL2DZ) were employed for Pt. This comprises of electrons in the 6s, 6p, and 5d orbitals. The standard split valence basis set 6-31G(d)38,39 was applied for carbon, oxygen, nitrogen, hydrogen and sulfur. For the studied reaction, all the stationary points located on the PES were characterized as minima (all positive frequencies) or first-order transition states (one imaginary frequency) through harmonic frequency calculations at the B3LYP/6-31G(d)/LANL2DZ level of theory. The vibrational normal modes have been assigned on the basis of the potential energy distribution (PED) calculated with the use of the VEDA program.40
For every transition state, the corresponding structures were confirmed by the intrinsic reaction coordinate (IRC) method41,42 implemented in Gaussian 09. Thermal contribution of the energetic properties was also considered at the standard state, namely, at 298.15 K and 1 atm. The rate constants (k) were calculated within the transition state theory according to the Eyring equation:43
| | |
k(T) = (KBT/h)e−ΔGǂ/KT
| (1) |
where
T is the absolute temperature,
kB is the Boltzmann constant, and
h is Planck’s constant. Δ
Gǂ is the free energy of activation for each step. The standard concentration (
c0 = 1 mol dm
−3) was considered in the theoretical calculations.
Natural bond orbital (NBO) analysis44 at the B3LYP/LANL2DZ/6-31G(d) level was calculated to understand the orbital interactions and charge delocalization during the course of reaction. In the NBO analysis, NBOs are first defined for each covalent bond, lone pair, and anti-bonding orbital using the molecular orbitals obtained by quantum chemical calculations, and the orbital interaction energies are consequently calculated for all possible interactions between electron-donor Lewis-type NBOs (i), and acceptor non-Lewis NBOs (j). For each electron-donor NBO (i) and acceptor NBO (j), the stabilization energy Eij associated with the delocalization i → j is estimated by eqn (2):
| | |
Eij = qif(i,j)2/εj − εi
| (2) |
where
qi is the donor orbital occupancy,
εi and
εj are the orbital energies and
Eij is the off-diagonal NBO Fock matrix element.
45,46
Molecular docking of complex 2 with B-DNA (PDB ID:1BNA) was done using HEX 8.0.0 software.47 HEX calculates DNA–complex binding, using the knowledge of 3D shape only by assuming that the complex is rigid and that it superposes pairs of molecules using the knowledge of 3D shape only. It uses a spherical polar Fourier correlation to accelerate the calculations. In order to run the docking protocol in HEX, first the DNA and ligand in PDB format were loaded. By clicking on the control option in the main menu, docking was selected and activated. The parameters which were used in the docking process are listed in the ESI, Table S1.† The results obtained after the docking process were analysed. The E-total values of the complex against DNA were calculated. To validate the procedure and results, we ran the same protocol three times and obtained almost the same results. RMSD values were calculated and the RMSD is lower than 2 Å.
Bioactivity
Anticancer properties, cells and culture conditions. HeLa cells were used for investigation of the anticancer properties of the complexes. The cells were cultured in DMEM containing 10% FBS and 1% of penicillin/streptomycin (50 IU mL−1 and 500 mg L−1) and cultured for two days at 37 °C in a 5% CO2 incubator.
In vitro cytotoxicity assay. Cytotoxicity was investigated by MTT colorimetric assay.48,49 Approximately 10000–12000 cells were added to each well of a 96-well plate containing DMEM medium. The cells were treated with the desired concentrations (5–50 μM) of complex 2 and 3. Cisplatin (5–50 μM) was used as the positive control50 and the untreated cells were used as the negative control. The plates were incubated for 24 hours. 20 μL MTT (5 mg mL−1 in PBS) solution was then added to each well and incubation was done for another 3 hours. 150 μL of DMSO was added to each well to dissolve the blue formazan product. The absorbance of this product was measured at 540 nm, using an ELISA plate reader.51
Microscopic observation of complex-treated HeLa cells. The HeLa cells were allowed to grow in 100 mm culture plates containing DMEM with 10% FBS and an appropriate amount of penicillin/streptomycin. To acquire around 70% confluence, they were incubated at 37 °C in a 5% CO2 incubator. These cells were treated with a 10 μM concentration of complex 2, 3 and cisplatin, and incubated for 12 hours. The negative control plate was also maintained, where the cells remained untreated. These plates were washed with 1× PBS and used to capture images by an inverted microscope with 100× magnification (Ziess Axio Observer.Z1).
Results and discussion
Spectroscopic analyses
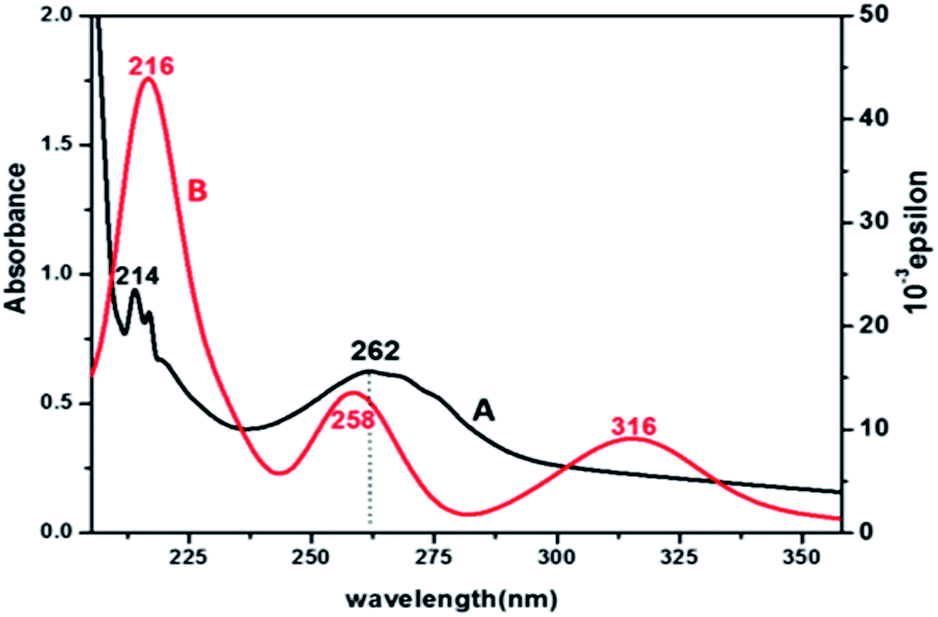
Electronic spectra and TD-DFT. The UV-vis spectrum of product 3 (Fig. 3) shows that there are three absorption peaks at 214, 262 and 310 nm, which are due to π–π* and n–π* transitions in different MOs of the ligands. Complex 3 also shows a hyperchromic and slight red shift as compared to complex 2. To evaluate the nature of the orbitals involved in the transitions related to the experimental observations, a theoretical approach Time Dependent Density Functional Theory (TD-DFT) computational method was applied. The theoretical calculations predict that each molecular orbital is formed by the combination of a large number of atomic orbitals. The bands in the simulated spectrum appear at 216, 258 and 316 nm. The experimental and theoretical λmax values corresponding to the bands observed for the electronic transitions are given in Table 1, which support the experimental results. The major contribution of the molecular orbitals involved in the electronic transitions is shown in the ESI, Fig. S10.†
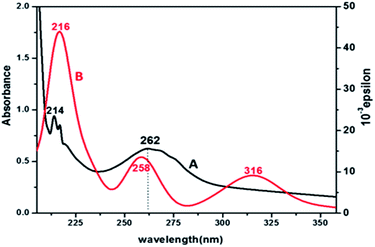 |
| | Fig. 3 UV-vis (A, black) and simulated TD-DFT (B, red) spectra of complex 3. | |
Table 1 TD-DFT and experimental band position of [Pt(pic)(DDTC)]
| Experimental band (ε dm3 mol−1 cm−1) |
TD-DFT peaks (oscillator strength) |
Major contribution |
| 310 |
316 (0.1103) |
H−1 → LUMO (86%) H−1 → L+1 (5%) |
| 262 |
258 (0.1673) |
H−1 → L+3 (91%) |
| 214 |
216 (0.005) |
HOMO → L+7 (66%), H−7 → L+2 (11%), H−7 → L+3 (14%) |
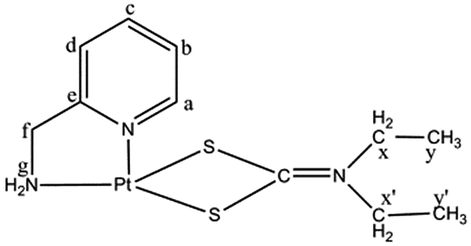
FTIR spectra. The ligand DDTC has three prospective donor centres (S, S and N) and FTIR has been used to confirm the bonding nature of the ligand in complex 3. The important bands in the FTIR spectrum of 3 have been assigned by comparison with the spectral data of DDTC and complex 2. The IR peak at 2075 cm−1 in DDTC has shifted to 2025 cm−1 in 3. This is most probably due to the partial multiple bond nature between carbon and sulfur (–S![[partial double bond, bottom dashed]](https://www.rsc.org/images/entities/char_e00f.gif) CNEt2), which is quite similar to the behaviour of SCN− when coordinated to Pt(II). The peak is broad, possibly due to delocalisation of charge between both the sulfur donor centres (Scheme 1) and their coordination to the metal centre. A Pt–S stretch should appear below 400 cm−1 and was obtained theoretically at 317 cm−1. A strong band at 1541 cm−1 and a medium band at 940 cm−1 can be assigned to the (CN) and (CS) partial double bond str. frequency respectively. The position of these bands in complex 3 reveals that DDTC binds to Pt(II) as a bidentate ligand through the sulfur centres.
CNEt2), which is quite similar to the behaviour of SCN− when coordinated to Pt(II). The peak is broad, possibly due to delocalisation of charge between both the sulfur donor centres (Scheme 1) and their coordination to the metal centre. A Pt–S stretch should appear below 400 cm−1 and was obtained theoretically at 317 cm−1. A strong band at 1541 cm−1 and a medium band at 940 cm−1 can be assigned to the (CN) and (CS) partial double bond str. frequency respectively. The position of these bands in complex 3 reveals that DDTC binds to Pt(II) as a bidentate ligand through the sulfur centres.
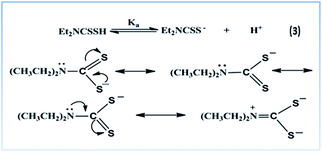 |
| | Scheme 1 Resonance structures of diethyldithiocarbamate (DDTC). | |
1H NMR spectra. The δ values of the aromatic protons in 3 are consistent with those in complex 2. The chemical shift position of the ethyl protons is also unaffected when compared to that in DDTC, which indicates that the amine nitrogen is not a coordinating site to the metal.
ESI mass spectra. The product complex 3 obtained from the reaction mixture of a 1:1 molar ratio of complex 2 and DDTC shows a molecular ion peak m/z at 451.08, which corresponds to the exact molecular mass of product 3 as suggested in the mechanism.
Conductivity measurement
Conductivity measurement of complex 3 in conductivity water shows 1:1 electrolytes, which clearly indicates that the complex is carrying one unit of positive charge.
Kinetic study (experimental)
The pKa value52 of the ligand DDTC is 3.37 at 25 °C, thus at pH 4.0, the ligand DDTC exists in its anionic form which is involved in the kinetic process. Both the sulfur centres are reactive due to negative charge delocalisation between them as shown in Scheme 1.
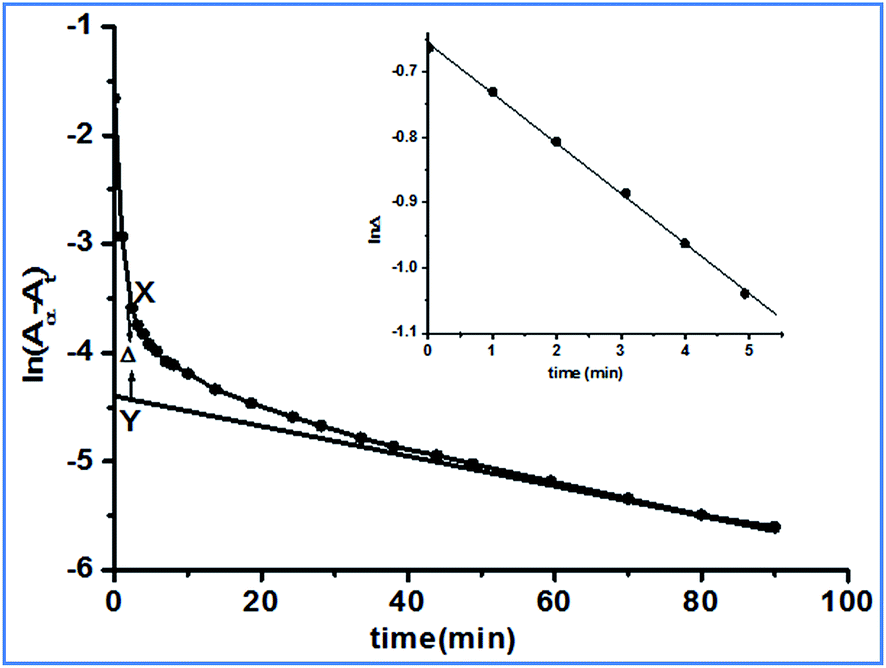
The pKa values of complex 2 were obtained at 4.76 and 6.83. However, the pKa1 value was found to be different from the previously reported26 value (5.82). The pKa values were determined using tiamo software, which is more reliable and accurate than the formerly used methods. We can assume that at pH 4.0 the reactant complex 2 exists in its diaqua form. At a particular temperature, pH 4.0 and a fixed concentration of complex 2, the plots of ln(Aα − At) versus time (where Aα and At are the absorbances at infinite time, i.e. after the completion of the reaction and at time t) for various ligand concentrations were curved at the initial stage and subsequently were of a constant slope (Fig. 4). This indicates that the reaction proceeds through a two-step consecutive process and we suggest that in the first step one aqua ligand is replaced in 2 by one sulfur atom of DDTC−. The second step is slower, where another aqua molecule is substituted by the other sulfur atom of the dithiocarbamate group and ring closure takes place.
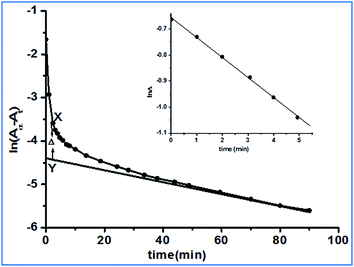 |
| | Fig. 4 A typical kinetic plot of ln(Aα − At) versus time: [complex 2] = 2.43 × 10−4 mol dm−3; [DDTC] = 2.43 × 10−3 mol dm−3; temperature = 25 °C. Inset: plot of lnΔ versus time. | |
The rate constants of the reaction for such a two-step process can be evaluated by assuming the following:
where A is the diaqua complex
2, B is the sulfur bonded one aqua ligand substituted intermediate, and C is the final product
3. Formation of C from B is predominant after some time has passed,
i.e. when the reaction is complete.
Evaluation of k1
The first step substitution process, the A → B step, is dependent on the incoming ligand concentration. At each temperature, the rate constant k1(obs) values were evaluated using the method of Weyh and Hamm28 from the plots of lnΔ (the variation of Δ is shown in Fig. 4, inset) versus time (where t is small) and are collected in the ESI, Table S2.† Using the usual consecutive rate law:| | |
(Aα − At) = a1exp(−k1(obs)t) + a2exp(−k2(obs)t)
| (4) |
where a1 and a2 are constants that depend on the rate constant and extinction coefficient, the values of [(Aα − At) − a2exp(−k2(obs)t)] were obtained from the parameter X–Y at different times.| | |
lnΔ = constant − k1(obs)t
| (5) |
where Δ is the difference between the observed and extrapolated part of the linear portion of (Aα − At) versus time (when t is small). Hence, k1(obs) is derived from the slope of the plot of lnΔ versus t (correction coefficient is 0.9879).
The same procedure was followed in the range of ligand concentration from 2.43 × 10−3 mol dm−3 to 7.29 × 10−3 mol dm−3, at a constant complex 2 concentration (2.43 × 10−4 mol dm−3) and at different temperatures (25, 30, 35, 40 and 45 °C).
The reaction rate increases with the increase in ligand concentration up to a limiting rate (ESI Fig. S11†). The dependency of the k1(obs) values on ligand concentration can be explained in terms of rapid formation of the inner sphere association complex between the reactant complex 2 and DDTC in the step A → B. This is followed by inner sphere association through sulfur coordination via displacement of one water molecule resulting in the intermediate B. The following Scheme 2 can be proposed for the reaction:
 |
| | Scheme 2 | |
Based on the above equations, a rate expression (6) can be derived for the A → B step:
| | |
d[B]/dt = k1KE[2][DDTC]/(1 + KE[DDTC])
| (6) |
| | |
d[B]/dt = k1(obs)[2]T
| (7) |
where the subscript T stands for the total concentration of the complex (
2), thus:
| | |
1/k1(obs) = 1/k1 + 1/k1KE[DDTC]
| (8) |
where
k1 is the rate constant for the formation of B and
KE is the outer sphere association equilibrium constant. A plot of 1/
k1(obs) versus 1/[DDTC] was found to be linear (ESI Fig. S12
†) with an intercept of 1/
k1 and a slope of 1/
k1KE. This equation is applicable to all the temperatures under the kinetic study. The first step rate constant
k1 and
KE values were obtained from the intercept (1/
k1) and from the slope-to-intercept ratios (1/
k1KE). The
k1 and
KE values (
Table 2) thus obtained are dependent on the ligand concentration.
Table 2 Rate constants for the reaction of 2 with DDTC at pH 4.0 and with 0.1 M NaClO4
| DDTC |
| Temp. (°C) |
103k1 (M−1 s−1) |
KE1 (dm3 mol−1) |
105k2 (s−1) |
| 25 |
3.33 ± 0.08 |
261 ± 0.13 |
2.15 ± 0.01 |
| 30 |
4.54 ± 0.05 |
300 ± 0.22 |
2.80 ± 0.03 |
| 35 |
6.66 ± 0.12 |
349 ± 0.16 |
3.79 ± 0.03 |
| 40 |
9.09 ± 0.09 |
398 ± 0.21 |
4.94 ± 0.01 |
| 45 |
14.28 ± 0.05 |
448 ± 0.32 |
6.45 ± 0.03 |
Evaluation of k2
The second step B → C involves displacement of another water molecule accompanied by ring closure through another sulfur atom of the dithiocarbamate group of DDTC (Scheme 2). According to the HSAB principle, formation of a Pt(II)–sulfur four-membered ring is more favourable than a platinum–nitrogen four-membered chelate. In the literature, few four-membered metal chelates have been reported with soft metal ions like Pt(II)53 and Pd(II)54,55 where dithiocarbamate moieties use both of their S-donor centres to chelate the metal ion. Consequently, we expect that DDTC would show a similar type of bonding nature with Pt(II) in complex 3 and the same has been confirmed from spectroscopic data.
At each temperature, the k2(obs) values were calculated from the limiting linear portion (when t is large) of the plot of ln(Aα − At) versus t (Fig. 4) and the values are collected in the ESI, Table S3.† Unlike k1, the k2 values were found to be independent of ligand concentration at all the temperatures studied. The derived rate equation of the reaction based on kinetic observations is as follows:
| |
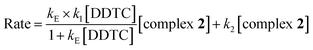 | (9) |
where
KE is the outer sphere association equilibrium constant.
Effect of temperature
The reaction was studied at five different temperatures at different ligand concentrations and the substitution rate constants for both the steps (k1 and k2) are given in Table 2. The activation parameters calculated from the Eyring plots (ESI Fig. S13a and b†) (R2 for k1 is 0.9795 and R2 for k2 is 0.9865) are given in Table 3. The Gibbs free energy of activation was derived from the thermodynamic relation ΔGǂ = ΔHǂ − TΔSǂ. The activation parameters ΔHǂ and ΔSǂ are compared with analogous systems in the ESI, Table S4.†
Table 3 Experimental activation parameters for the reaction of complex 2 with DDTC
| System |
Step 1 |
ΔH1ǂ (kJ mol−1) |
ΔS1ǂ (J K mol−1) |
ΔG1ǂ (kJ mol−1) |
| Complex 2/DDTC |
53.87 ± 1.46 |
−107.43 ± 1.13 |
85.83 ± 1.80 |
| Step 2 |
ΔH2ǂ (kJ mol−1) |
ΔS2ǂ (J K mol−1) |
ΔG2ǂ (kJ mol−1) |
| 40.98 ± 1.17 |
−193.67 ± 1.81 |
100.18 ± 1.71 |
Computational study
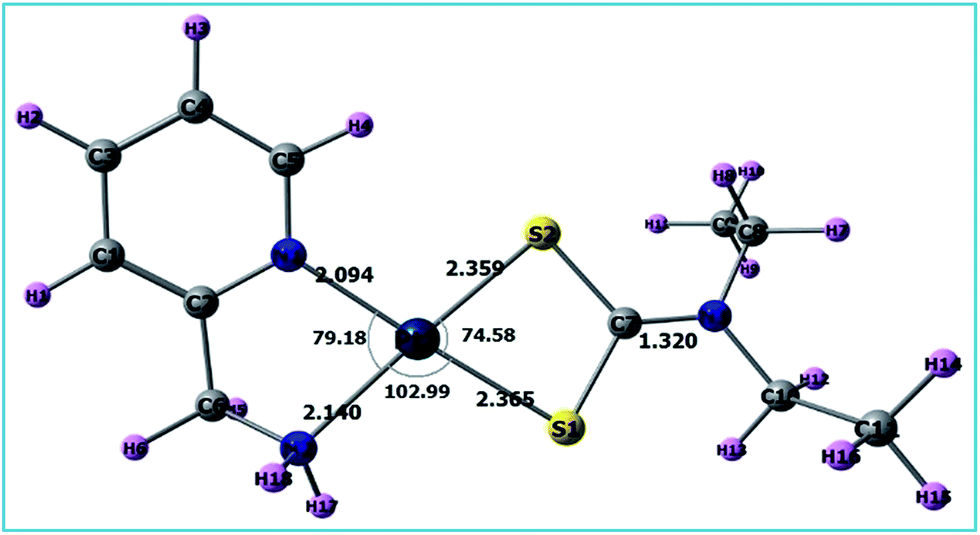
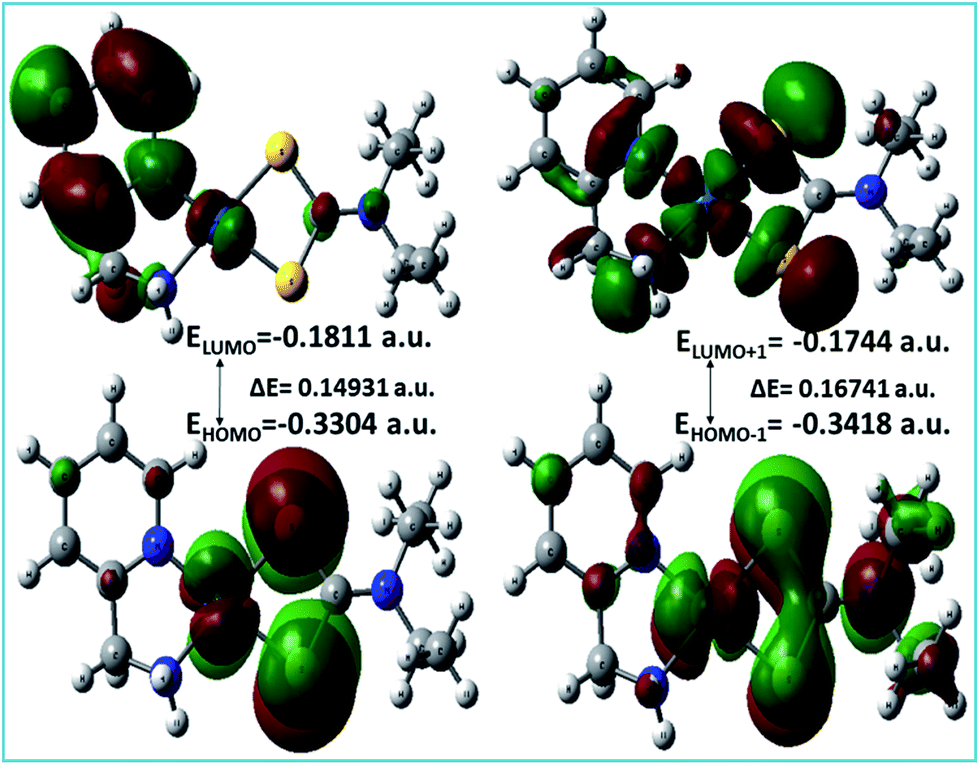
A list of bond distances (Å) and angles (degrees) obtained from the optimized geometry of complex 3 (Fig. 5) is presented in Table 4 and compared with the reported data of closely related structures.56–58 The energies of the most significant orbitals (HOMO, LUMO, HOMO−1, and LUMO+1) of complex 3 and their energy differences are shown in Fig. 6.
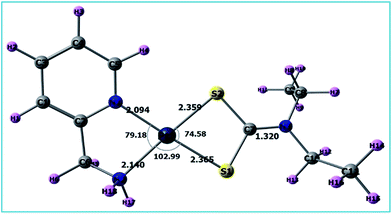 |
| | Fig. 5 Optimized structure of complex 3. | |
Table 4 Important bond lengths (Å) and bond angles (°) of the final product [Pt(pic)(DDTC)] using different protocols in the gas and aqueous phases
| Method |
B3LYP |
MPW1PW91 |
PBEPBE |
Ref. 52–54 |
| Medium |
Gas |
Aqueous |
Gas |
Aqueous |
Gas |
Aqueous |
| Pt–N1 |
2.094 |
2.085 |
2.066 |
2.059 |
2.070 |
2.065 |
2.026(3) |
| Pt–N2 |
2.140 |
2.120 |
2.110 |
2.089 |
2.130 |
2.112 |
2.015(3) |
| Pt–S1 |
2.365 |
2.377 |
2.340 |
2.349 |
2.356 |
2.364 |
2.356(3) |
| Pt–S2 |
2.359 |
2.377 |
2.334 |
2.351 |
2.348 |
2.363 |
2.349(3) |
| C7–N3 |
1.320 |
1.320 |
1.314 |
1.315 |
1.330 |
1.330 |
— |
| N1–Pt–N2 |
79.18 |
79.42 |
79.60 |
79.90 |
79.64 |
79.84 |
— |
| N1–Pt–S2 |
103.25 |
103.44 |
102.95 |
103.31 |
102.82 |
102.07 |
— |
| S1–Pt–S2 |
74.58 |
74.28 |
74.89 |
74.64 |
74.88 |
74.64 |
— |
| N2–Pt–S1 |
102.99 |
102.86 |
102.56 |
102.16 |
102.67 |
102.45 |
— |
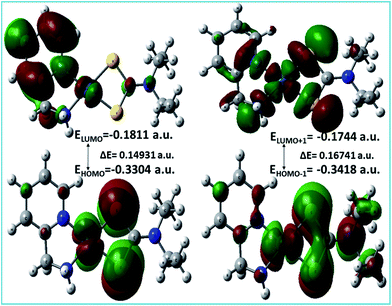 |
| | Fig. 6 HOMO and LUMO and their energy difference of complex 3. | |
Kinetic investigation by DFT
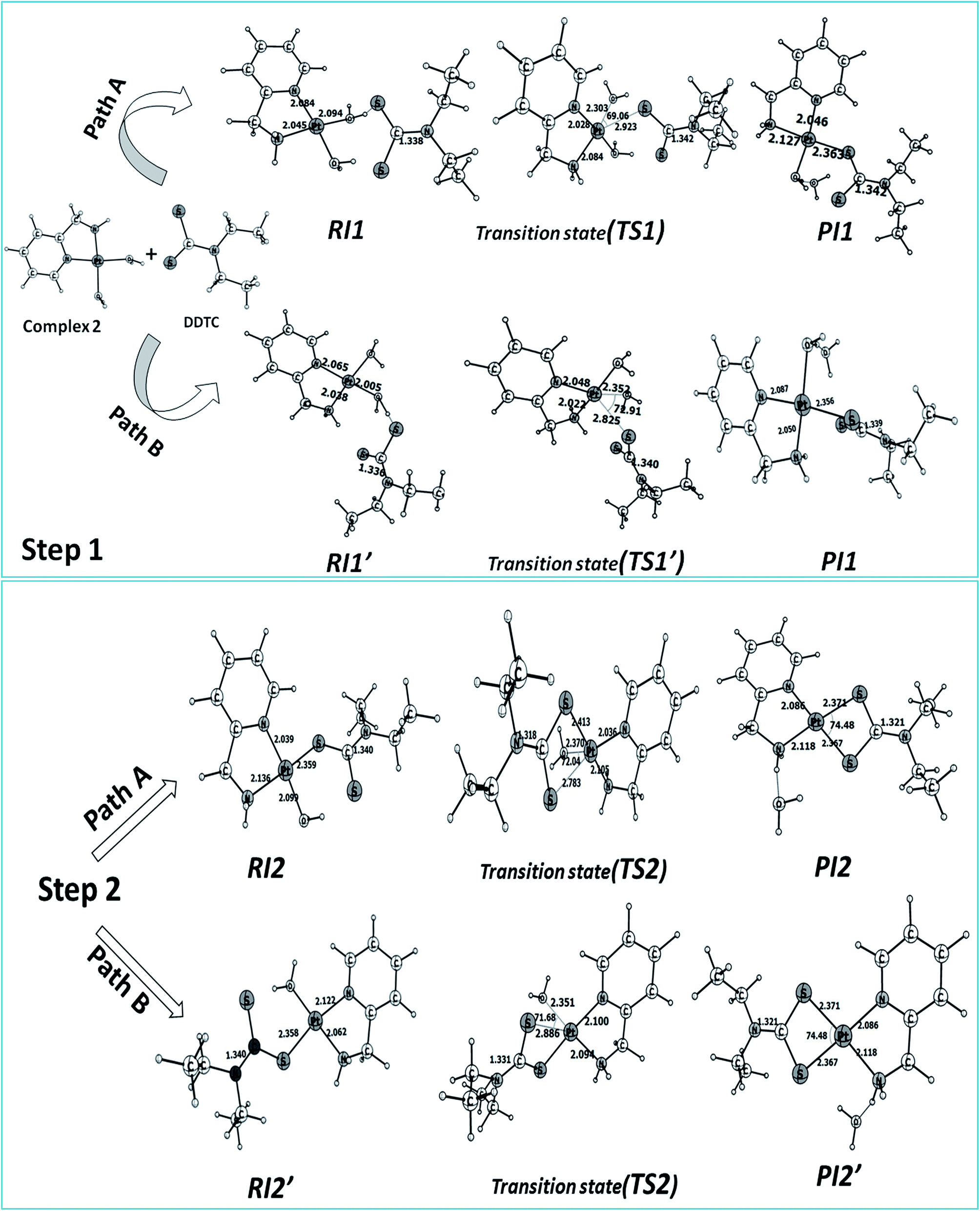
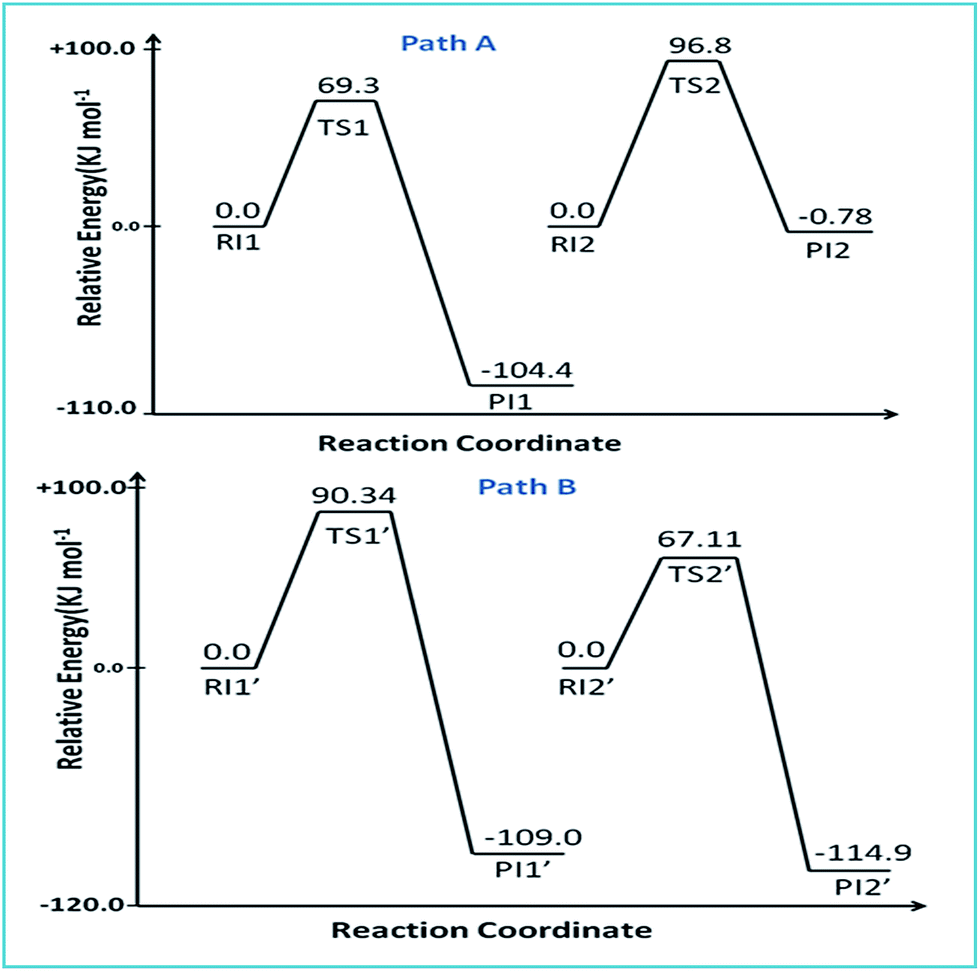
The reaction of complex 2 with DDTC is a second-order nucleophilic substitution (SN2) process. In a general SN2 reaction path, five stationary states e.g., the reactant (R), reactant intermediate (RI), TS, product intermediate (PI) and product (P) are found.59,60 The reaction may proceed in two different paths (A and B) both having two steps. In path A, the aqua molecule trans to the ammine (NH2) group of the carrier ligand pic leaves first through a trigonal bipyramidal (tbp) transition state resulting in a mono aqua [Pt(pic)(H2O)–S–DDTC] complex (B). In the second step, the aqua molecule trans to the pyridine nitrogen leaves the platinum coordination zone resulting in the product complex 3. This is also true for path B, where the aqua molecule trans to the pyridine nitrogen leaves the coordination zone first through tbp to the mono aqua complex [Pt(pic)(H2O)–S–DDTC] complex (B′). The next step is formation of the (S, S) coordinated complex (C). All the intermediate structures found in this reaction are stabilized through H bonds since the interaction with the Pt(II) atom is negligible (Pt–S distances being above 3.365 Å as compared to 2.356 Å.57,58 All stationary states (Fig. 7), i.e. the four reactant intermediates (RI1, RI1′, RI2 and RI2′), transition states (TS1, TS1′, TS2, and TS2′) and four product intermediates (PI1, PI1′, PI2 and PI2′), located on the PES are confirmed by frequency analysis. The coordinates of all the stationary states and negative frequencies of the corresponding first-order saddle point are shown in the ESI, Table S5.† The total energy profile diagrams (energies are in kJ mol−1) for the studied reaction in the different possible paths (A and B) are shown in Fig. 8. Rate constants and activation parameters for the reaction calculated for both the possible paths (A and B) are shown in Table 5, in which the relative energy change in path A is reasonable compared to path B. The theoretically calculated first step rate constant (k1 = 4.43 M−1 s−1) is found to be higher than the experimentally determined first step rate constant (k1 = 3.33 × 10−3 M−1 s−1). It might be explained on the basis of the fact that the outer sphere association equilibrium (KE) constant and first step reaction rate constant are not distinguishable or separable in the computational method. The association equilibrium (KE) constant and actual first step rate constant (k1) are collectively shown as a theoretical first step rate constant, which is greater than the experimental rate constant at 25 °C. In the case of the second step, the theoretical rate constant (k2 = 0.68 × 10−5 s−1) is closer to the experimental value (k2 = 2.15 × 10−5 s−1) at 25 °C. As a result, the theoretically calculated and experimentally determined rate constants are reasonably comparable.
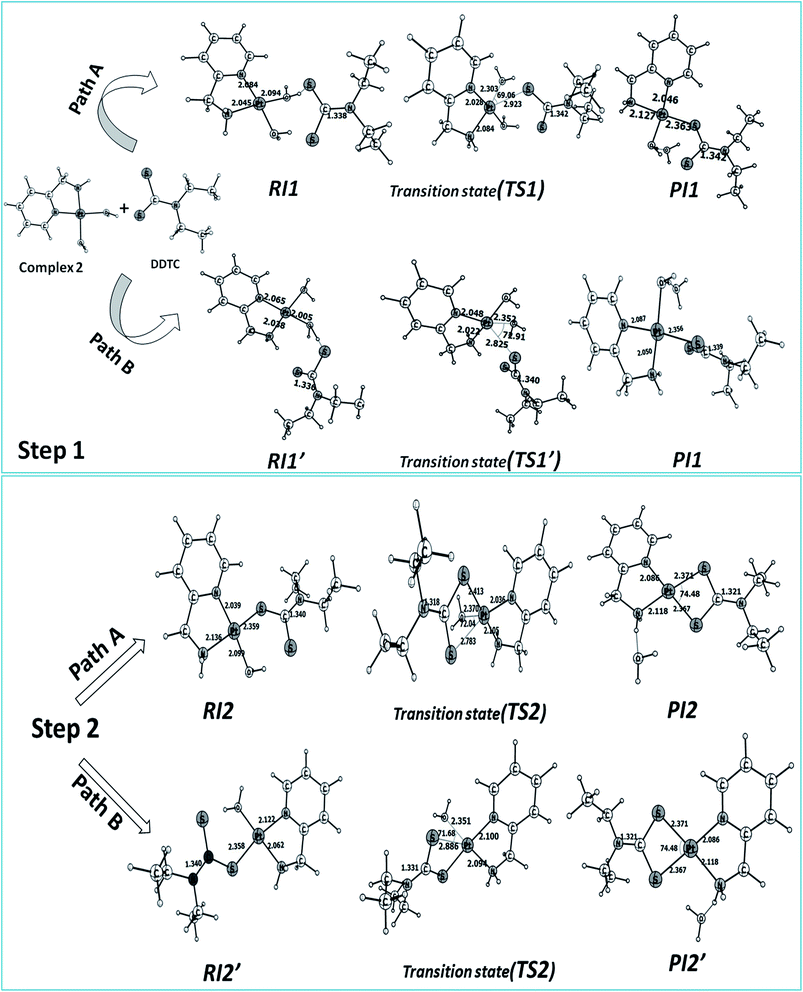 |
| | Fig. 7 Optimized structures and selected structural parameters for both the steps of the substitution reaction on complex 2 with DDTC for paths A and B. Distance and bond angles are in Å and degrees (°) respectively. | |
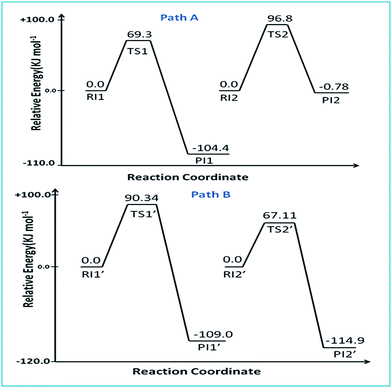 |
| | Fig. 8 Energy profile diagram for paths A and B. | |
Table 5 Activation free energy and rate constants based on the computational study using the B3LYP/6-31g(d)/LANL2DZ protocol
| Complex 2/DDTC |
Path A |
Path B |
| ΔGǂ (kJ mol−1) |
Rate (M−1 s−1) |
ΔGǂ (kJ mol−1) |
Rate (s−1) |
| First step |
69.3 |
4.43 |
90.34 |
8.82 × 10−4 |
| Second step |
96.8 |
0.68 × 10−5 |
67.11 |
10.86 |
Natural bond orbital (NBO) analysis
The coordinating sites to Pt(II) were confirmed by natural bond orbital (NBO) analysis, calculated at the B3LYP/LANL2DZ/6-31G(d) level. NBO analysis provides detailed insight into the nature of electronic structure and bonding in the probable complexes 3 and 3′ with DDTC by considering61 chelation through (S, S) or (S, N) respectively in the substitution reaction. The calculated atomic charge and electronic configurations for complex 3 (S, S) (Fig. 5) and the other probable complex 3′ (S, N) (ESI Fig. S14†) are listed in Table 6. The formal charge of Pt in both complexes 3 and 3′ is +2.0 a.u. The calculated natural charge on Pt(II) in complex 3 (0.1553 a.u.) is lower than that in 3′ (0.31241 a.u.) which implies that there is more charge donation to the Pt(II) centre from (S, S) in 3 than from (S, N) in 3′. Moreover, the formal charge on both (S, S) in complex 3 (0.05890 and 0.05940 a.u.) is less than that on (S, N) in complex 3′ (0.10555 and −0.49922 a.u.). The formation of complex 3 through (S, S) coordination can hence be confirmed by considering better charge transfer from the donor centres (S, S) to Pt(II) rather than (S, N), which is in concurrence with the spectroscopic results and also the SHAB principle.
Table 6 Charges and electronic configuration for the complexes 3 (S, S) and 3′ (S, N)
| Complex 3 (S, S) |
Complex 3′ (S, N) |
| Atom |
Charge |
Natural electronic configuration |
Charge |
Natural electronic configuration |
| Pt |
0.15537 |
[core]6S(0.58)5d(8.91)6p(0.34)6d(0.02) |
0.31241 |
[core]6S(0.58)5d(8.83)6p(0.27)6d(0.02) |
| N1 |
−0.45201 |
[core]2S(1.28)2p(4.15)3p(0.01) |
−0.44872 |
[core]2S(1.28)2p(4.15)3p(0.01) |
| N2 |
−0.82678 |
[core]2S(1.40)2p(4.41)3p(0.01) |
−0.84424 |
[core]2S(1.40)2p(4.42)3p(0.01) |
| S1 |
0.05890 |
[core]3S(1.71)3p(4.20)3d(0.02)4p(0.01) |
0.16600 |
[core]3S(1.70)3p(4.10)3d(0.02)4p(0.01) |
| S2 |
0.05940 |
[core]3S(1.71)3p(4.20)3d(0.02)4p(0.01) |
0.10555 |
[core]3S(1.74)3p(4.13)4S(0.01)3d(0.01)4p(0.01) |
| N3 |
−0.38469 |
[core]2S(1.41)2p(4.41)3p(0.02) |
−0.49922 |
[core]2S(1.32)2p(4.17)3p(0.01) |
Molecular docking with B-DNA
Molecular docking studies of complex 2 with the DNA duplex of sequence d(CGCGAATTCGCG)2 dodecamer (PDB ID:1BNA) were performed in order to predict the chosen binding site. Complex 2 is flexible enough to adopt a conformation which is complementary to the minor groove62 of the DNA (Fig. 9). The almost planar structure of complex 2 fits into the minor groove of B-DNA in a parallel way with respect to the DNA backbone. The platinum complex forms two hydrogen bonds with the guanine (2.8 Å) and adenine (1.9 Å) bases of the 3′ to 5′ polynucleotide chain in DNA. The resulting binding energy of the docked metal complex was found to be −34.77 kcal mol−1.
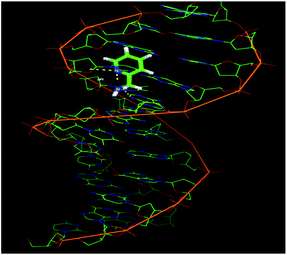 |
| | Fig. 9 Molecular docking model illustrating the interaction between complex 2 and B-DNA. | |
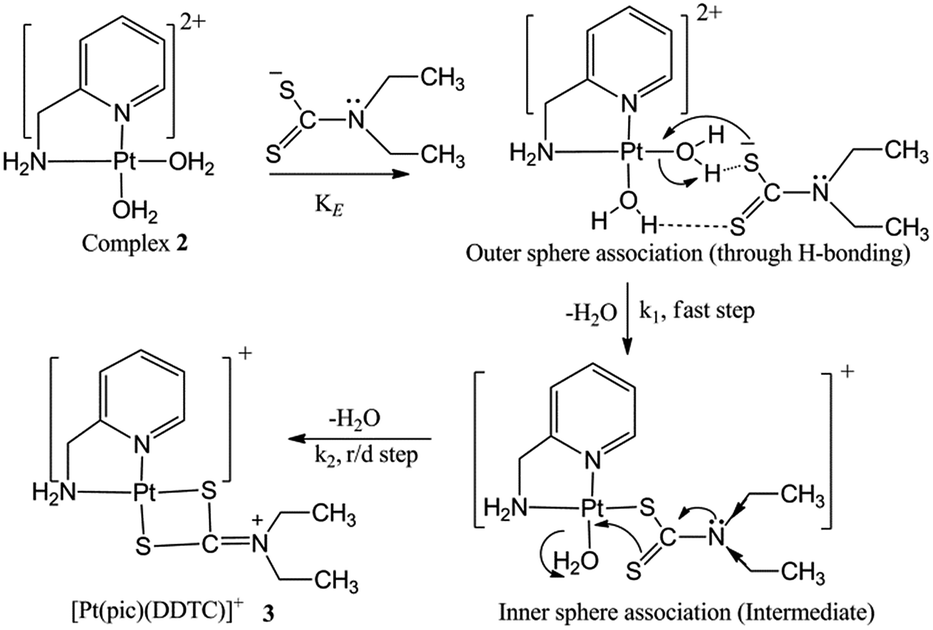
Reaction mechanism
The experimental kinetic study shows a rapid equilibrium due to outer sphere association between complex 2 and the ligand DDTC. DDTC substitutes two water molecules of complex 2 and gives the substituted product 3 through (S, S) coordination. The leaving group H2O trans to the primary amine is substituted first rather than the one trans to the pyridine nitrogen due to its more labile nature. The second step is slower than the first, and is also independent of the ligand concentration. The four-membered Pt–S bonded complex is quite unnatural in Pt(II) chemistry and should be unstable. However, here it is stabilised by simultaneous electromeric and inductive effects of the ethyl groups attached to the nitrogen in DDTC. A 1:1 metal–ligand ratio obtained from Job’s method of continuous variation also corroborates with our proposition. The IR spectrum of 3 also confirms that DDTC behaves as a bidentate ligand (Scheme 3). Theoretical investigations suggest that the reaction proceeds through path A instead of path B. A theoretical study has been performed to verify our proposed mechanism, which is established based on experimental work and good agreement was found between them.
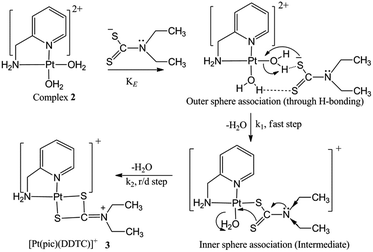 |
| | Scheme 3 Proposed mechanism for the reaction of DDTC with complex 2 at pH 4.0. | |
Bioactivity
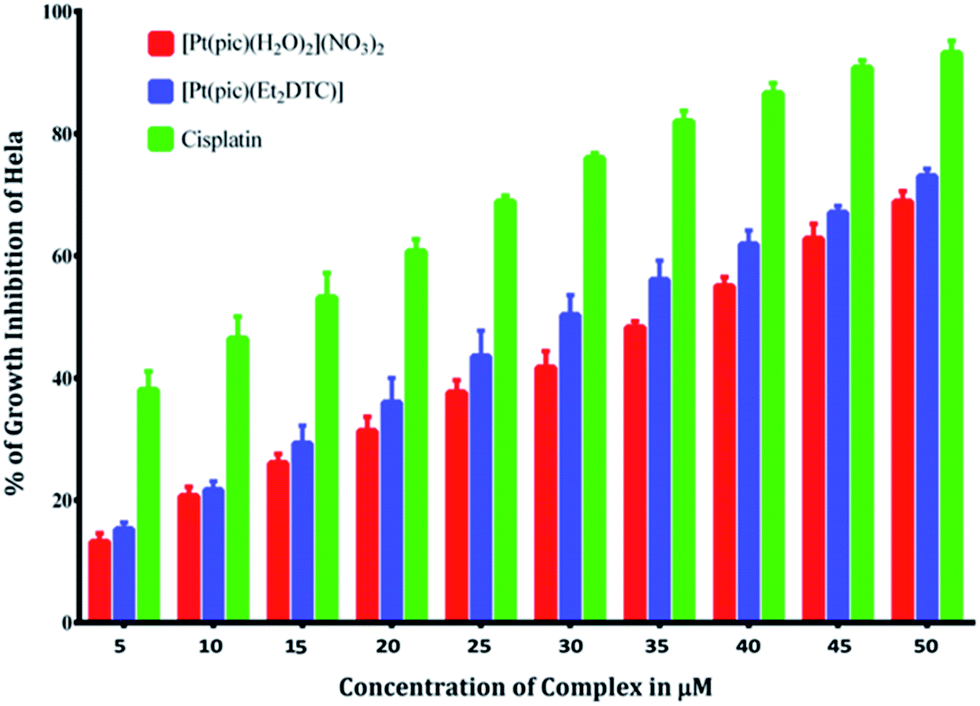
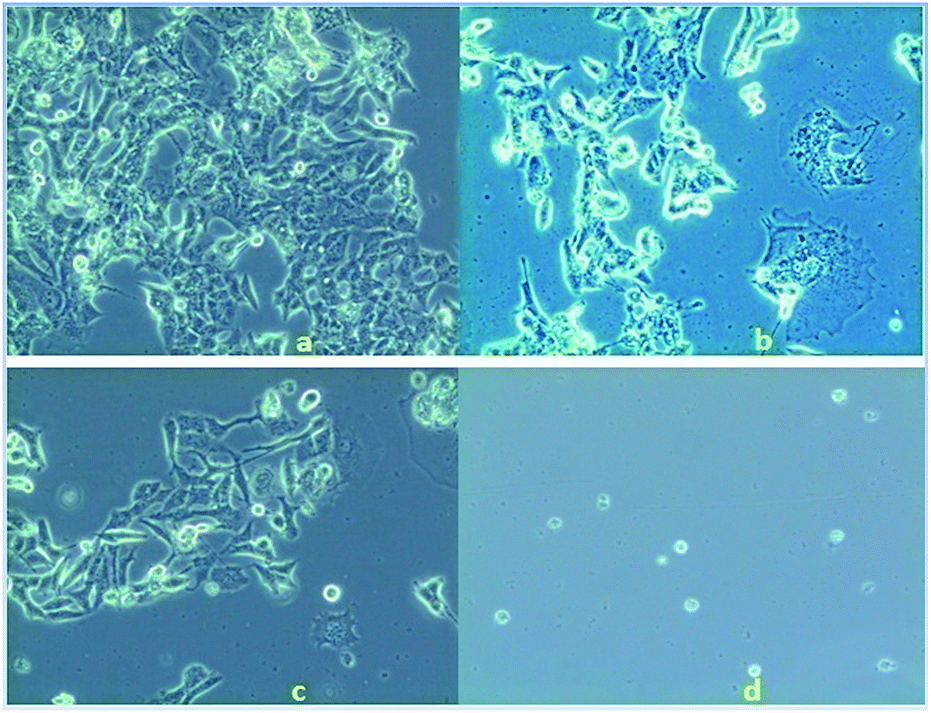
Anticancer property and IC50 value of complexes 2 and 3. Inhibition of cell proliferation was investigated by incubating HeLa cells with various concentrations (5.0–50 μM) of complex 2, 3 and cisplatin by comparing with the negative control (untreated cells) under the same experimental conditions. A graphical plot (Fig. 10) of the percentage of growth inhibition of the HeLa cells versus the complex concentrations reveals that the rate of growth inhibition increases with the increasing concentrations of the complexes. This indicates that the complexes show potential inhibition of HeLa cell proliferation in a concentration-dependent treatment. Complexes 2 and 3 thus have potent anticancer activity comparable with that of the established standard metallodrug, cisplatin. The half maximal inhibitory concentration (IC50) values of complexes 2 and 3 were calculated and found to be 36 ± 0.6 and 30 ± 0.4 μM respectively, as compared to 12 μM of cisplatin42 for HeLa cells. The microscopic images of HeLa cells treated with a 10 μM concentration of complex 2, 3 and cisplatin show adequate cellular growth inhibition in the order of (d) > (c) > (b) as compared with the untreated cells (a) (Fig. 11).
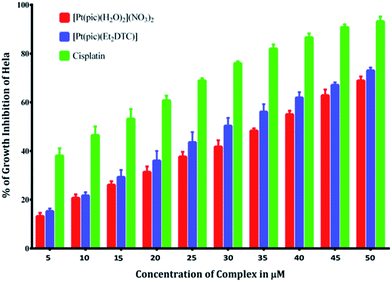 |
| | Fig. 10 % of growth inhibition of HeLa cells in the presence of [Pt(pic)(H2O)2](NO3)2 and the substituted complex [Pt(pic)(DDTC)] NO3 from 5 μM to 50 μM concentration compared with cisplatin. | |
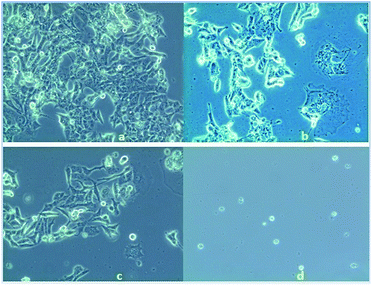 |
| | Fig. 11 Microscopic view of (a) untreated HeLa cells, (b) treated with complex 2 (10 μM), (c) treated with complex 3 (10 μM) and (d) treated with cisplatin (10 μM). | |
The result of the MTT assay and the microscopic images depict that complex 3 has better anticancer properties than complex 2. The higher cell growth inhibition activity observed for complex 3 might be explained on the basis of its highly strained four-membered ring structure.
Conclusions
Complex 2 reacts with DDTC at pH 4.0 and proceeds through two consecutive steps, to form complex 3. The rate constants (k1 and k2) of the two step-process were evaluated experimentally and were comparable to the theoretically calculated ones. Among the three potential donor centres (S, S and N) of DDTC, only the (S, S) donor pair chelates the metal and forms a stable four-membered complex 3, which is unusual in Pt(II) chemistry. DFT study and the simulated TD-DFT spectrum of complex 3 also support the formation of the four-membered complex. NBO analysis also clearly reveals that the (S, S) donor pair of DDTC chelates the metal rather than the (S, N) donor pair. The plausible associative mechanism is confirmed by theoretical investigation. The activation parameters (low ΔH1ǂ and ΔH2ǂ values and negative ΔS1ǂ and ΔS2ǂ values) for the first and second steps suggest an associative mode of activation for the substitution process, with a good degree of ligand participation in the transition state.
Four reactant intermediates (RI1, RI1′, RI2 and RI2′) and four product intermediates (PI1, PI1′, PI2 and PI2′) are observed corresponding to the first and second step of reaction for both paths A and B. Molecular docking of the Pt(II) complex 2 with B-DNA reveals minor groove binding with a total binding free energy of −34.77 kcal mol−1. Complexes 2 and 3 have acceptable water solubility and are stable in aqueous solution. A potent in vitro anticancer activity (∼70% as compared to cisplatin) against HeLa cells is observed at higher concentrations for complexes 2 and 3. Hence, there is a fair chance to employ these Pt(pic) complexes to treat tumour proliferations after clinical trials.
Acknowledgements
The authors are thankful to the National Institute of Technology Durgapur-713209 and Department of Science and Technology (DST), Government of India (Project No.: SB/EMEQ-028/2013) for providing the necessary assistance and financial support for carrying out this work. We would like to thank the referees for their valuable comments and suggestions.
References
- B. Rosenberg, L. V. Camp and T. Krigas, Nature, 1965, 205, 698 CrossRef CAS PubMed.
- B. Rosenberg, L. V. Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385 CrossRef CAS PubMed.
- B. Lippert, Cisplatin: Chemistry and biochemistry of a leading anticancer drug, Wiley, Zürich, 1999 Search PubMed.
- P. J. Sadler, Adv. Inorg. Chem., 1991, 36, 1 CrossRef CAS.
- M. J. Abrams and B. A. Murrer, Science, 1993, 261, 725 CAS.
- Y. Kidani, R. Kizu, M. Miyazaki, M. Noji, A. Matsuzawa, Y. Takeda, N. Akiyama and M. Eriguchi, Platinum and Other Metal Coordination Compounds in Cancer Chemotherapy, ed. H. M. Pinedo and J. H. Schornagel, Plenum Press, New York, 1996, p. 43 Search PubMed.
- S. Mukherjee, V. P. Reddy B., I. Mitra, R. N. Saha, J. C. Bose K., S. Reddy D., W. Linert and S. C. Moi, RSC Adv., 2015, 5, 76987 RSC.
- T. Shoeib and B. L. Sharp, Metalomics, 2012, 4, 1308 RSC.
- C. Makedonas, C. A. Mitsopoulou, F. J. Lahoz and A. I. Balana, Inorg. Chem., 2003, 42(26), 8853 CrossRef CAS PubMed.
- C. Makedonas and C. A. Mitsopoulou, Inorg. Chim. Acta, 2007, 360(14), 3997 CrossRef CAS.
- S. D. Cummings and R. Eisenberg, Inorg. Chem., 1995, 34(8), 2007 CrossRef CAS.
- C. Makedonas and C. A. Mitsopoulou, Eur. J. Inorg. Chem., 2006, 3, 590 CrossRef.
- R. Mital, N. Jain and T. S. Seivastava, Inorg. Chim. Acta, 1989, 166, 135 CrossRef CAS.
- A. Gringeri, P. C. Keng and R. F. Borch, Cancer Res., 1988, 5708 CAS.
- M. A. Zemaitis and F. E. Greene, Toxicol. Appl. Pharmacol., 1979, 48(2), 343 CrossRef CAS PubMed.
- R. Mital, N. Jain and T. S. Srivastava, Inorg. Chim. Acta, 1989, 166(1), 135 CrossRef CAS.
- N. Manav, A. K. Mishra and N. K. Kaushik, Spectrochim. Acta, Part A, 2006, 65(1), 32 CrossRef CAS PubMed.
- G. Faraglia, S. Sitran and D. Montagner, Inorg. Chim. Acta, 2005, 358(4), 971 CrossRef CAS.
- C. Marzano, A. Trevisoan, L. Giovagnini and D. Fregona, Toxicol. In Vitro, 2002, 16, 413 CrossRef CAS PubMed.
- S. J. Berners-Price and P. W. Kuchel, J. Inorg. Biochem., 1990, 38, 327 CrossRef CAS PubMed.
- T. Ishikawa, C. D. Wright and H. Ishizuka, J. Biol.Chem., 1994, 269, 29085 CAS.
- N. Jain and T. S. Srivastava, Inorg. Chim. Acta, 1987, 128(2), 151 CrossRef CAS.
- K. Misra, G. K. Ghosh, I. Mitra, S. Mukherjee, V. P. Reddy B, W. Linert, B. Misini, J. C. Bose K, S. Mukhopadhyay and S. C. Moi, RSC Adv., 2015, 5, 12454 RSC.
- K. Misra, I. Mitra, G. K. Ghosh, S. Mukherjee, B. Misini, W. Linert and S. C. Moi, Trans Met. Chem., 2014, 39, 789 CrossRef CAS.
- A. Samanta, G. K. Ghosh, I. Mitra, S. Mukherjee, J. C. Bose K, S. Mukhopadhyay, W. Linert and S. C. Moi, RSC Adv., 2014, 4, 43516 RSC.
- N. Summa, W. Schiessl, R. Puchta, N. van, E. Hommes and R. van Eldik, Inorg. Chem., 2006, 45(7), 2948 CrossRef CAS PubMed.
- R. W. Hay and A. K. Basak, J. Chem. Soc., Dalton Trans., 1982, 1819 RSC.
- J. A. Weyh and R. E. Hamm, Inorg. Chem., 1969, 8, 2298 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- J. P. Perdew, Electronic Structure of Solids ‘91, ed. P. Ziesche and H. Eschrig, Akademie, Berlin, 1991 Search PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284 CrossRef.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS.
- W. Hehre, J. R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- M. H. Jamroz, Vibrational Energy Distribution Analysis VEDA 4, Warsaw, 2004–2010, http://smmg.nazwa.pl/index.php/software-michal-jamroz/sowtware-veda.html Search PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS.
- K. A. Conors, Chemical Kinetics–The Study of Reaction Rate in Solution, Wiley, New York, 1990, p. 200 Search PubMed.
- D. E. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, Version 3.1 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- A. Ebrahimia, F. Deyhimib and H. Roohi, J. Mol. Struct.: THEOCHEM, 2003, 626, 223 CrossRef.
- G. Macindoe, L. Mavridis, V. Venkatraman, M. D. Devignes and D. W. Ritchie, Nucleic Acids Res., 2010, 38, 445 CrossRef PubMed.
- R. I. Freshney, Culture of animal cells: A manual of basic technique, Wiley-Liss, NY, 5th edn, 1987, p. 200 Search PubMed.
- R. W. Masters, Animal cell culture: Cytotoxicity and viability assays, 3rd edn, 2000, vol. 23, p. 202 Search PubMed.
- D. Garmann, A. Warnecke, G. V. Kalayda, F. Kratz and U. Jaehde, J. Controlled Release, 2008, 131, 100 CrossRef CAS PubMed.
- Y. C. Sasaki and A. Passaniti, BioTechniques, 1998, 24, 1038 Search PubMed.
- K. I. Aspila, S. J. Joris and C. L. Chakrabarti, J. Phys. Chem., 1970, 74, 3625 CrossRef CAS.
- S. K. Mukhopadhyay, Transition Met. Chem., 2008, 33, 739 CrossRef CAS.
- S. Mallik, B. Bera, S. Mondal, P. Karmakar, A. Mandal and A. K. Ghosh, J. Chem. Sci., 2011, 123(3), 311 CrossRef.
- V. Alverdi, L. Giovagnini, C. Marzano, R. Seraglia, F. Bettio, S. Sitran, R. Graziani and D. Fregona, J. Inorg. Biochem., 2004, 98, 1117 CrossRef CAS PubMed.
- D. Li and D. Liu, Cryst. Res. Technol., 2004, 39(4), 359 CrossRef CAS.
- A. N. Skvortsov, A. N. Reznikov, D. A. de Vekki, A. I. Stash, V. K. Belsky, V. N. Spevak and N. K. Skvortsov, Inorg. Chim. Acta, 2006, 359, 1031 CrossRef CAS.
- E. Colacio, R. Cuesta, M. Ghazi, M. A. Huertas, J. M. Moreno and A. Navarrete, Inorg. Chem., 1997, 36, 1652 CrossRef CAS PubMed.
- S. Roy, P. Mondal, P. S. Sengupta, D. Dhak, R. C. Santra, S. Das and P. S. Guin, Dalton Trans., 2015, 44, 5428 RSC.
- T. Zimmerman, M. Zeizinger and J. V. Burda, J. Inorg. Biochem., 2005, 99, 2184 CrossRef PubMed.
- J. R. Dilworth and J. R. Miller, J. Chem. Educ., 1991, 68(9), 788 CrossRef CAS.
- K. Suntharalingam, O. Mendoza, A. A. Duarte, D. J. Mann and R. Vilar, Metallomics, 2013, 5, 514 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21161a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.