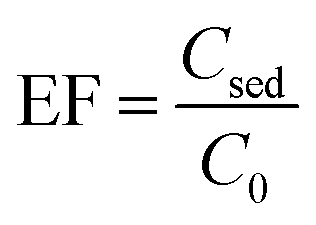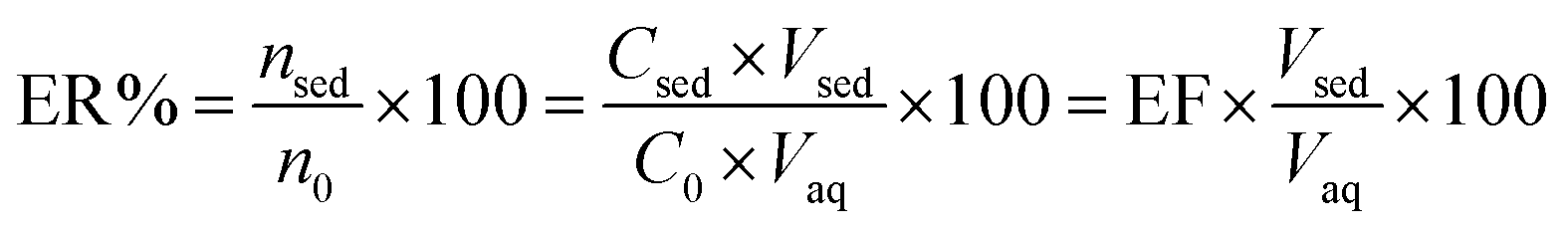DOI:
10.1039/C5RA23191D
(Paper)
RSC Adv., 2016,
6, 14927-14936
Development of a new version of homogenous liquid–liquid extraction based on an acid–base reaction: application for extraction and preconcentration of aryloxyphenoxy-propionate pesticides from fruit juice and vegetable samples
Received
4th November 2015
, Accepted 27th January 2016
First published on 29th January 2016
Abstract
In this paper, a new version of homogeneous liquid–liquid extraction based on an acid–base reaction combined with dispersive liquid–liquid microextraction followed by gas chromatography-flame ionization detection has been developed for the extraction and determination of aryloxyphenoxy-propionate pesticides in fruit juice and vegetable samples. In this method, initially a basic solvent (amine) is mixed with acidified deionized water spiked with the analytes, which leads to the formation of a homogenous solution. Afterward, the homogenous solution is transferred into a narrow bore tube containing the mixture of sodium chloride and sodium carbonate. As the solution is passed through the filled portion of the tube, fine droplets of the organic solvent (amine) are released and move through the aqueous phase to the top of the tube and form a separated phase in which the target analytes are extracted. In the second step, the collected organic phase is removed and mixed with a preconcentration solvent and injected into acidified deionized water for more enrichment of the analytes. Effective parameters controlling the performance of the proposed method, such as type and volume of extraction solvent and flow rate in homogenous liquid–liquid extraction step and type and volume of preconcentration solvent, ionic strength and centrifugation rate and time in the dispersive liquid–liquid extraction step were optimized. Under the optimum extraction conditions, the method showed low limits of detection and quantification within the ranges of 0.12–0.40 and 0.40–1.3 μg L−1, respectively. Relative standard deviations were in the range of 3–5% (n = 6, C = 50 or 100 μg L−1). Comparison of the proposed method with the existing reports in the literature demonstrates that it is simple, rapid, selective, and sensitive, and has high enrichment factors and low detection limits for determining aryloxyphenoxy-propionate pesticides in the studied samples.
1. Introduction
Aryloxyphenoxy-propionate pesticides are regarded as selective and systemic herbicides that are absorbed by plants. They are used in many agriculture productions like barley, wheat, wild oat, soybean, cotton, tomato, potato, onion, garlic, etc. to control annual and perennial grasses.1–4 Despite their many merits, pesticides are some of the most toxic, environmentally stable, and mobile substances in the environment. Pesticide residues, which remain after an agricultural treatment, may penetrate plant tissues and may be found in the pulp and the juice of the fruits.5
Among the analytical techniques used in analysis of pesticide residues, the most important ones are gas chromatography (GC) with element specific detectors such as the nitrogen phosphorus detector (NPD), flame photometric detector (FPD), electron capture detector (ECD), and mass spectrometry (MS),6,7 and liquid chromatography (LC) with ultraviolet (UV) and MS detectors.8,9 Although the above-mentioned methods are sensitive instrumental techniques of analysis, another step before the analysis, known as sample preparation, is still required.
A variety of sample preparation methods have been reported for the extraction of pesticides in fruit juice and vegetable matrices. Liquid–liquid extraction (LLE)10–12 and solid phase extraction (SPE)13,14 have been applied to the extraction of pesticides from juice samples. However, LLE is time consuming and requires large volumes of toxic organic solvents. SPE uses much less solvent than LLE but the cartridge needs pretreatment and can be relatively expensive and labor intensive. Another extraction method is homogeneous liquid–liquid extraction (HLLE) that extracts the desired solutes existing in a homogeneous aqueous solution into a water-immiscible phase formed by each kind of phase separation phenomenon.15–18
Nowadays, to overcome the mentioned problem of the extraction methods, several microextraction techniques by reducing organic solvent consumption and simplifying the sample preparation techniques have been reported as the alternatives to conventional sample preparation procedures, such as solid phase microextraction (SPME),19,20 and liquid phase microextraction (LPME).21,22 SPME by combining extraction and preconcentration in a single step, is an effective and solvent-free technique. However, SPME fibers are relatively expensive, generally fragile, and have a limited lifetime, especially for some direct immersion extraction from complex sample matrices. Moreover, SPME has been reported to suffer from sample carry-over problems.23 With the advantages of being economical, effective, and solvent-minimized, LPME has gained considerable applicability to various compounds and has been developed in a variety of configurations and modes, such as single drop microextraction (SDME),24 headspace LPME,25 dynamic LPME,26 hollow fiber protected LPME,27–29 continuous flow LPME,30 and solvent bar microextraction.31,32 As a relatively new mode of LPME, dispersive liquid–liquid microextraction (DLLME) was first introduced in 2006 by Assadi et al.33 In this method, a solvent system containing a mixture of an extraction solvent and a disperser solvent is rapidly injected into an aqueous sample. The advantages of DLLME are good repeatability, low volume of extraction solvent, reduced extraction time, high extraction recovery (ER) and high enrichment factor (EF). This technique has been widely used to determine different compounds in various samples.34–36
The aim of this work was to develop a new version of HLLE based on an acid–base reaction performed in a narrow bore tube and its combination with DLLME for the extraction and high preconcentration of aryloxyphenoxy-propionate pesticides from fruit juice and vegetable samples. To the best of our knowledge, for the first time, this version of HLLE is developed and applied for the analysis of aryloxyphenoxy-propionate pesticide residues in the samples without dilution except for garlic.
2. Materials and methods
2.1. Reagents and solutions
All pesticides including clodinafop-propargyl, haloxyfop-R-methyl, fenoxaprop-P-ethyl, and diclofop-methyl had a purity > 98% and were kindly provided by GYAH Corporation (Karadj, Iran). Di-iso-butylamine, tripropylamine, chloroform, acetone, 1,2-dibromoethane (1,2-DBE), and carbon tetrachloride were purchased from Merck (Darmstadt, Germany). Sodium chloride and sodium carbonate were also supplied from Merck. 1,1,2,2-Tetrachloroethane (1,1,2,2-TCE) and tributylamine were from Janssen (Beers, Belgium) and Fluka (Buchs, Switzerland), respectively. Deionized water was from Ghazi Company (Tabriz, Iran). A mixture stock solution of the selected pesticides was prepared in acetone with a concentration of 1000 mg L−1 (each pesticide). This solution was stored at 4 °C in a refrigerator until use. Working solutions were prepared daily by diluting the stock solution with deionized water. A mixture standard solution of the analytes (1000 mg L−1 of each pesticide) in 1,2-DBE (extraction solvent) was prepared and directly injected into the separation system each day (three times) in order to evaluate the instrumental system quality and to calculate EFs and ERs of the analytes.
2.2. Samples
Fresh onion, tomato, melon, strawberry, cucumber, and garlic were purchased from local markets (Tabriz, Iran). Grape, apple and sour cherry juice samples from different brands were also supplied from local supermarkets. The samples were stored at 4 °C until use. The vegetable samples were cut into small pieces and their juices were extracted by a juice extractor. The juice samples were centrifuged at 700 × g for 5 min and 30 mL of the supernatant was collected in a 50 mL centrifuge tube. Garlic juice was diluted at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 with deionized water before analysis, while the other samples were used without dilution.
:3 with deionized water before analysis, while the other samples were used without dilution.
2.3. Instrumentation
Quantitative analysis of the selected pesticides was performed on a Shimadzu 2014 gas chromatograph (Kyoto, Japan) equipped with a split/splitless injector operated at 300 °C in a splitless/split mode (sampling time, 1 min, and split ratio of 1:5) and a flame ionization detector (FID). Helium (99.999%, Gulf Cryo, United Arab Emirates) was used as the carrier gas at a linear velocity of 30 cm s−1 and make up gas at a flow rate of 30 mL min−1. Chromatographic separation was achieved on an RTX-1 capillary column (30 m × 0.25 mm i.d., and film thickness 0.25 μm) (100% dimethyl polysiloxane) (Restek, USA). The column oven temperature was initially held at 80 °C for 2 min, then raised to 300 °C at a rate of 12 °C min−1, and held at 300 °C for 4 min. The FID temperature was maintained at 300 °C. For FID hydrogen gas was generated with a hydrogen generator (OPGU – 1500S, Shimadzu, Japan) at a flow rate of 40 mL min−1. The flow rate of air for FID was 300 mL min−1. Gas chromatography-mass spectrometry (GC-MS) analysis was carried out on an Agilent 7890A–5975C apparatus (Agilent Technologies, CA, USA). MS operational conditions were: electron ionization (EI) at 70 eV; ionic source temperature, 250 °C; transfer line temperature, 260 °C; mass range, m/z 55–350; acquisition rate, 20 Hz; and detector voltage, −1700 V. Library searching was performed using the commercial NIST library. The separation was performed on an HP-5 MS capillary column (30 m × 0.25 mm i.d., and film thickness of 0.25 μm). The carrier gas was helium at a flow rate of 1.0 mL min−1. The column oven temperature programming was the same as used in GC-FID analysis mentioned above.
2.4. Procedure
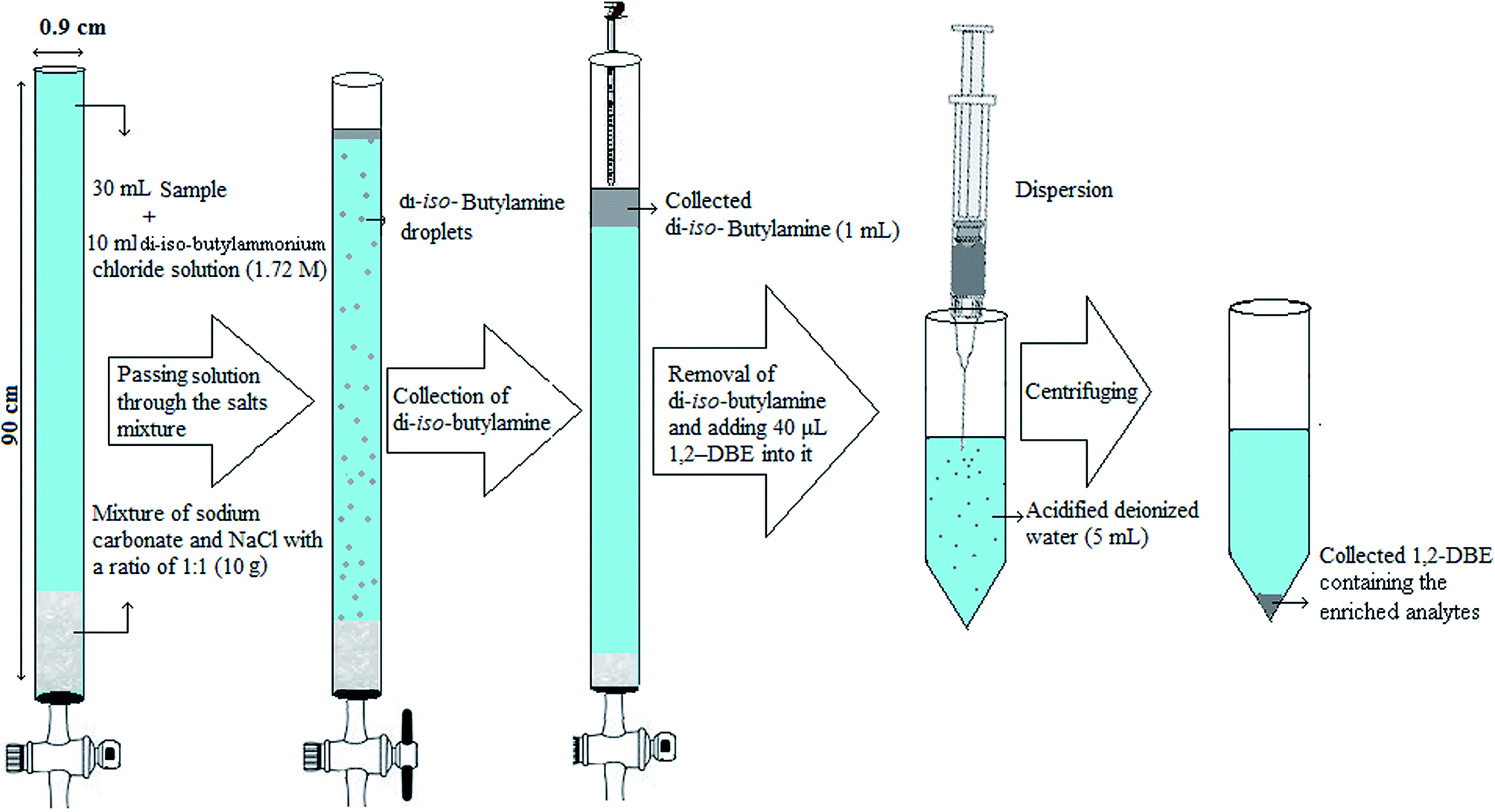
Initially, the end of a narrow bore tube (90 × 0.9 cm i.d.) was connected to a stopcock and then 10 g mixture of NaCl and sodium carbonate with a ratio of 1:1 (w/w) was filled into the tube. Then, 7 mL hydrochloric acid solution (2.5 M) was mixed with 3 mL di-iso-butylamine and gently shaken with hand to obtain a clear solution. Afterward, this homogenous solution was mixed with 30 mL deionized water spiked with the selected pesticides at a concentration of 300 μg L−1 (each pesticide). In the following, the solution was pulled into the tube and allowed to pass at a flow rate of 1.5 mL min−1. By passing the solution through the column, di-iso-butylamine was generated by the reaction of di-iso-butylammonium ions and sodium carbonate. In the following the fine droplets of organic phase (di-iso-butylamine) formed at the interface of solution and solid sodium carbonate, moved through the aqueous phase to top of the tube and floated on the surface of aqueous phase as a separated layer due to low density of di-iso-butylamine with respect to water. After passing 10 mL solution the stopcock was closed. During passing the fine droplets of the organic phase through the aqueous phase, the analytes were extracted into the organic phase. In this step, volume of the collected organic phase was 1.0 ± 0.1 mL. In the continuation, the upper phase was carefully removed by a pipette and mixed with 50 μL 1,2-DBE. Then the mixture was rapidly injected into 5 mL deionized water containing 7.5%, w/v, NaCl and 0.3 M acetic acid placed in a 10 mL glass centrifuge tube with conical bottom. A cloudy solution was resulted from dispersion of the tiny droplets of 1,2-DBE into the aqueous solution and the pesticides were extracted into 1,2-DBE. The mixture was then centrifuged for 4 min at 700 × g, which led to settle down the dispersed droplets of the extractant in bottom of the tube. After centrifuging, 10 ± 0.5 μL of the settled organic phase was obtained. Finally an aliquot (1 μL) of the organic phase was removed and injected into the separation system for analysis. Extraction and preconcentration procedure is schematically shown in Scheme 1.
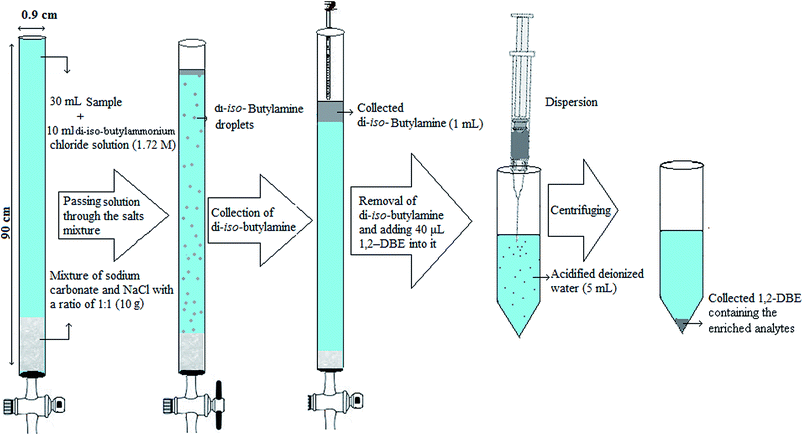 |
| | Scheme 1 Extraction and preconcentration procedure. | |
2.5. Calculation of EF and ER
To evaluate the performance of the proposed method, EF and ER were calculated using eqn (1) and (2)| |
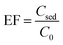 | (1) |
where Csed and C0 are concentration of the analyte in the sedimented phase in DLLME step and the initial concentration of the analytes in aqueous solution, respectively. Csed was calculated by comparison of peak areas obtained for injection of the sedimented phase after performing the method with those obtained for direct injection of pesticides standard solution prepared in 1,2-DBE.
ER is defined as the percentage of the total analyte amount (n0) which is extracted into the sedimented phase (nsed).
| |
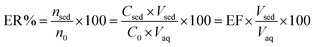 | (2) |
where
Vsed and
Vaq are the volumes of sedimented phase and sample solution, respectively.
3. Results and discussion
In order to reach a high extraction efficiency, the influence of different parameters such as type and volume of extraction solvent, and flow rate in HLLE step and type and volume of preconcentration solvent, ionic strength, and centrifugation rate and time in DLLME step were investigated.
3.1. Optimization of HLLE step
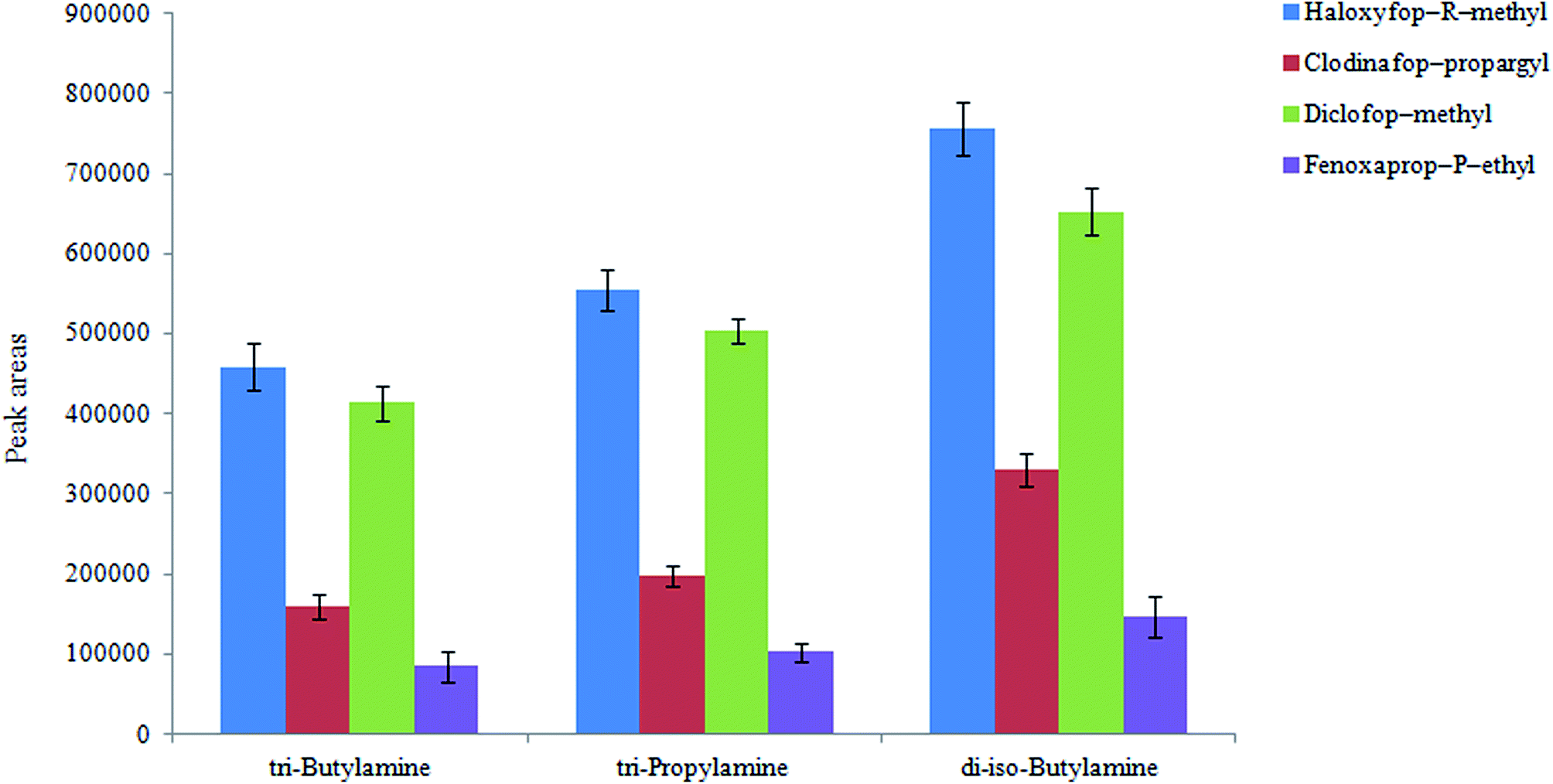
3.1.1. Selection of extraction solvent. Selecting an appropriate extraction solvent is critical in all extraction methods. In the present work, the extraction solvent has a double role: (i) as an extractant in HLLE step, and (ii) as a disperser in the next preconcentration step (DLLME). The extraction solvent must satisfy the following requirements: low water solubility, high affinity for the analytes, low density compared to water, and it forms a single phase with water at low pHs as its protonated form. On the basis of these considerations, di-iso-butylamine, tributylamine, and tripropylamine (3.5 mL in each case) were tested as the extraction solvent in HLLE step and they were mixed with 6.5 mL hydrochloric acid solution. It is noted that, in each case the concentration of hydrochloric acid was chosen different to obtain a neutral solution after mixing with the extraction solvents mentioned above (2.0, 1.4 and 1.8 M for di-iso-butylamine, tributylamine, and tripropylamine, respectively). In all cases, it was allowed that 1.0 mL of the organic phase was collected on the surface of aqueous phase by passing different volumes of aqueous phase through the tube. This volume is used in the following DLLME step. One mL was selected for the collected organic phase volume because it will be used as a disperser solvent in the DLLME step. It is mentioned that in most DLLME techniques 1.0 mL disperser solvent has been used for 5 mL aqueous solution in the published papers in literature. The obtained results are shown in Fig. 1. As it can be seen di-iso-butylamine is a suitable extraction solvent for extraction of the selected pesticides from aqueous solution. Therefore it was used in further experiments.
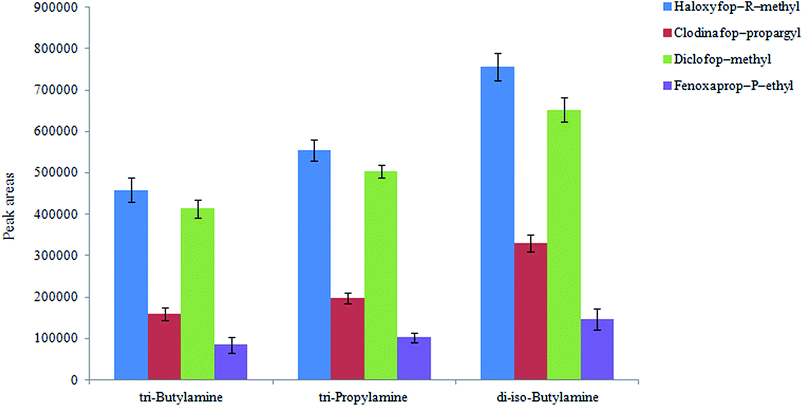 |
| | Fig. 1 Selection of extraction solvent in HLLE. Conditions: aqueous solution, 30 mL deionized water spiked with 300 μg L−1 of each pesticide; extraction solvent volume, 3.5 mL mixed with 7 mL HCl (2.5 M); flow rate 1 mL min−1; mixture of sodium carbonate and NaCl (1:1, w/w) filled in tube, 10 g; extraction solvent in DLLME, 1,2-DBE (50 μL); aqueous phase in DLLME, 5 mL deionized water; and centrifuge rate and time, 1006 × g and 6 min, respectively. The error bars indicate the minimum and maximum of three determinations. | |
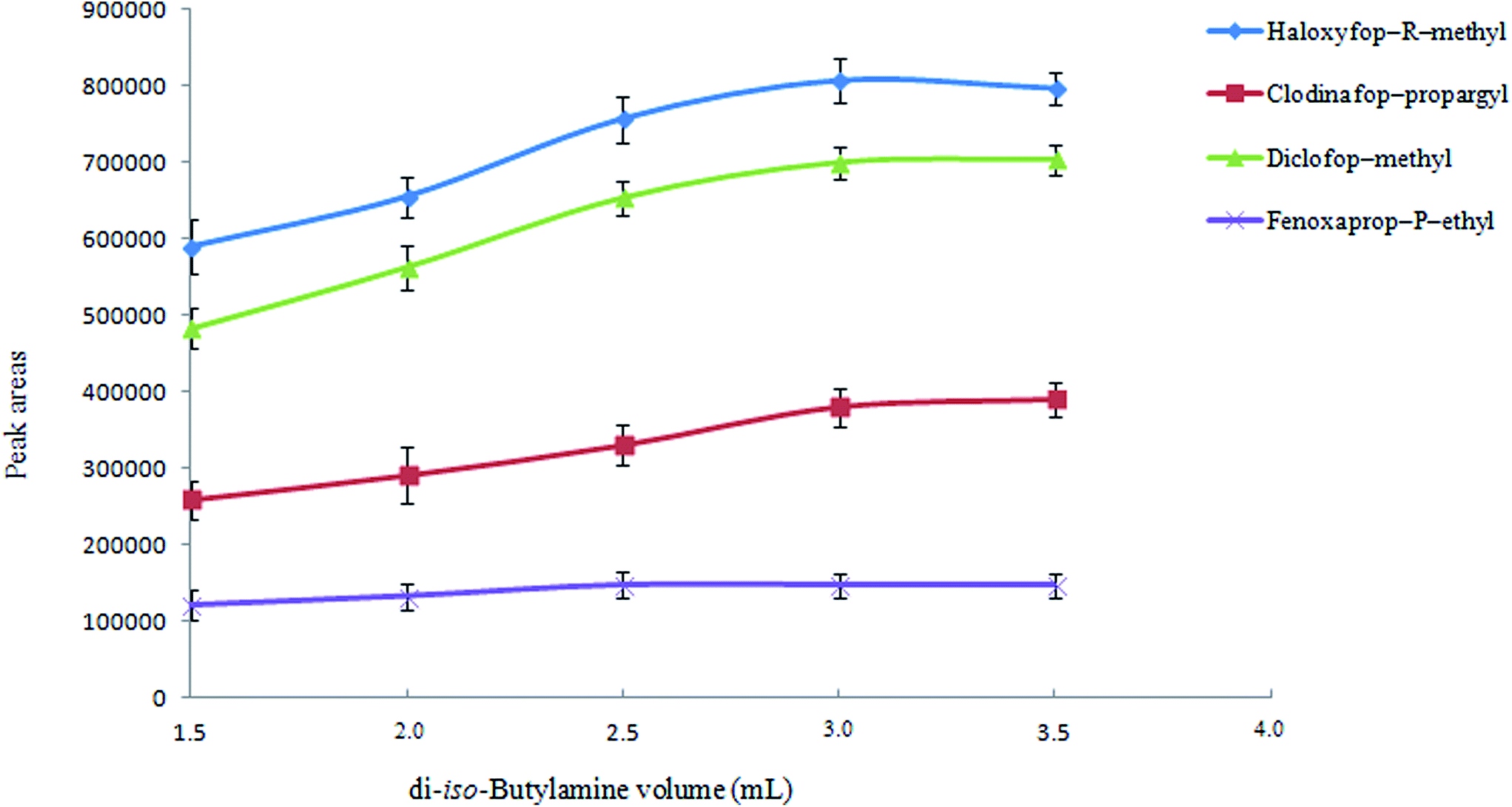
3.1.2. Selection of extraction solvent volume. The volume of extraction solvent is a crucial parameter that has a significant effect on the extraction efficiency. It is predictable that extraction efficiency is increased by increasing percent of di-iso-butylammonium (as di-iso-butylammonium ions) in aqueous solution. To examine the effect of extraction solvent volume on the extraction efficiency, different volumes of di-iso-butylamine (1.5, 2.0, 2.5, 3.0, and 3.5 mL) were investigated. It is remarkable that by increasing the volume of di-iso-butylamine, the concentration of hydrochloric acid solution was increased to obtain a homogenous solution (0.8, 1.1, 1.4, 1.7, and 2.0 M for 1.5, 2.0, 2.5, 3.0, and 3.5 mL, respectively). Fig. 2 shows that by increasing di-iso-butylamine volume, analytical signals increase till 3 mL and then remain constant. Therefore, 3 mL di-iso-butylamine was selected as an extraction solvent volume for the further studies.
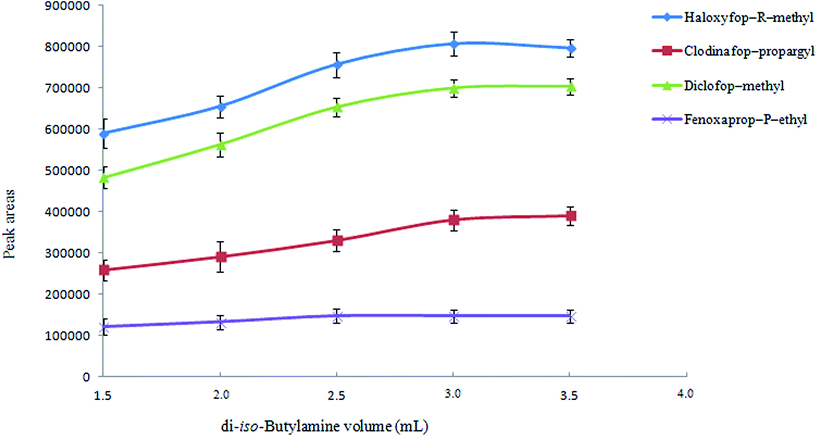 |
| | Fig. 2 Study of di-iso-butylamine volume in HLLE. Conditions: the same as used in Fig. 1, except di-iso-butylamine was used as the extraction solvent. | |
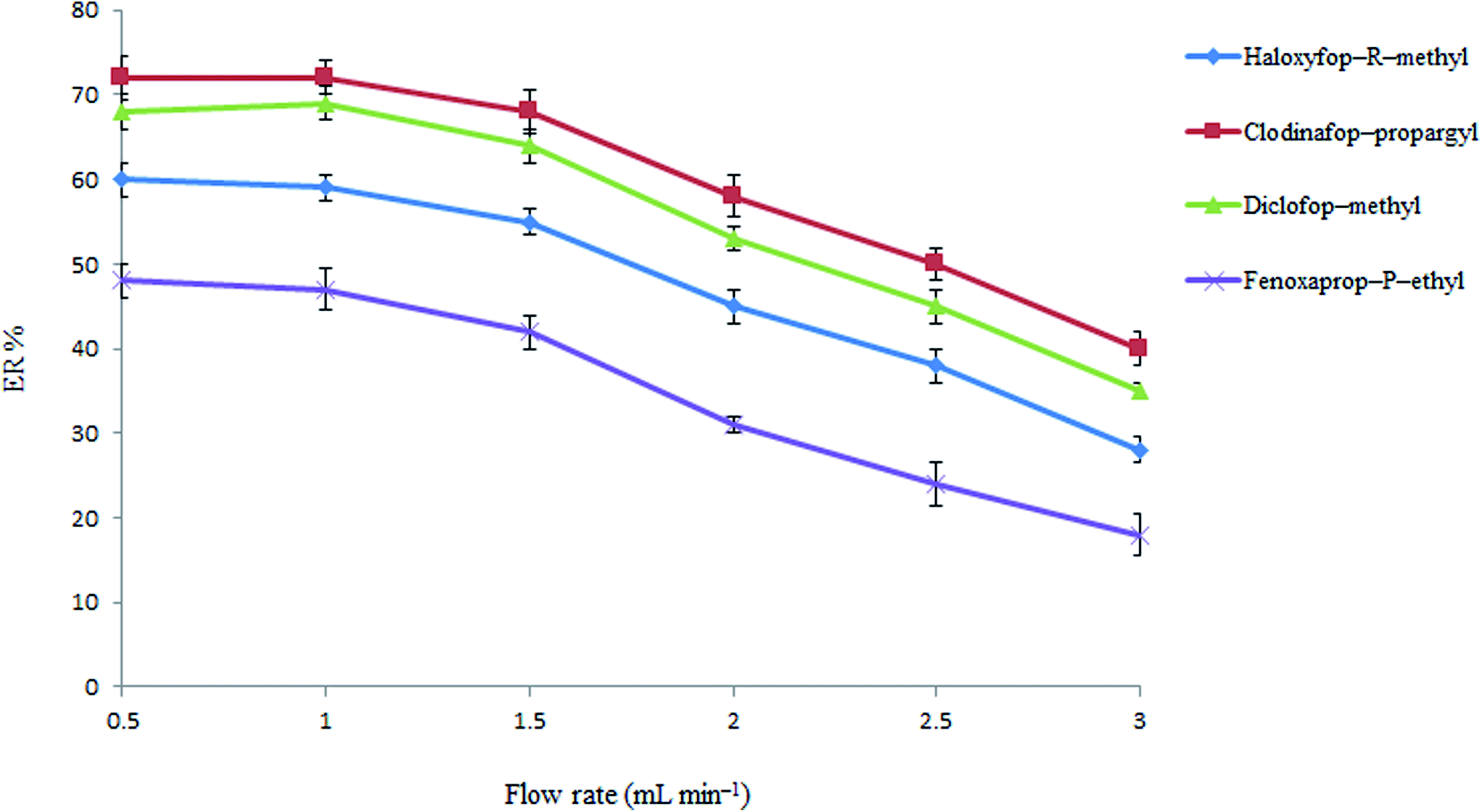
3.1.3. Flow rate study. The sample flow rate through the tube filled with sodium carbonate and NaCl mixture is one of the most important parameters which allows ER of the presented method to be improved by controlling rate and amount of the produced di-iso-butylamine from di-iso-butylammonium ions. Therefore, the influence of sample flow rate on the extraction efficiency of the analytes was investigated in the range of 0.5–3 mL min−1 by passing 10 mL of the solution through tube. The volumes of 0.3, 0.5, 0.7, 0.9, 1.0 and 1.0 mL di-iso-butylamine were collected at flow rates of 3.0, 2.5, 2.0, 1.5, 1.0, and 0.5 mL min−1, respectively. It should be noted that in those cases which the collected volumes was less than 1.0 mL, the collected organic phase was diluted with pure di-iso-butylamine to 1.0 mL before performing the following DLLME step. The results in Fig. 3 show that analytical signals are nearly constant up to 1.5 mL min−1 and then decrease at higher flow rates. This indicates that an inadequate extraction of the analytes from the aqueous phase is obtained at flow rates higher than 1.5 mL min−1. This behavior can be explained because the amount of the produced di-iso-butylamine is decreased at high flow rates and it leads to a decrease in efficiency. Therefore, a flow rate of 1.5 mL min−1 was chosen for the further analysis.
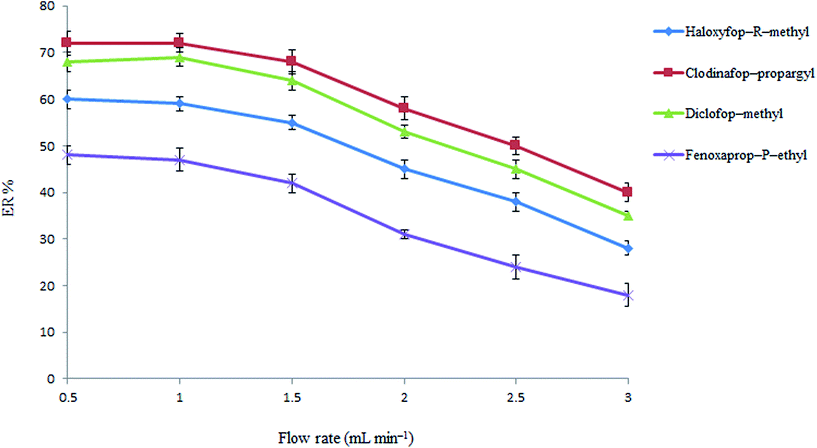 |
| | Fig. 3 Effect of flow rate in HLLE. Conditions: the same as used in Fig. 2, except di-iso-butylamine volume which was 3 mL. | |
3.2. Optimization of parameters in DLLME step
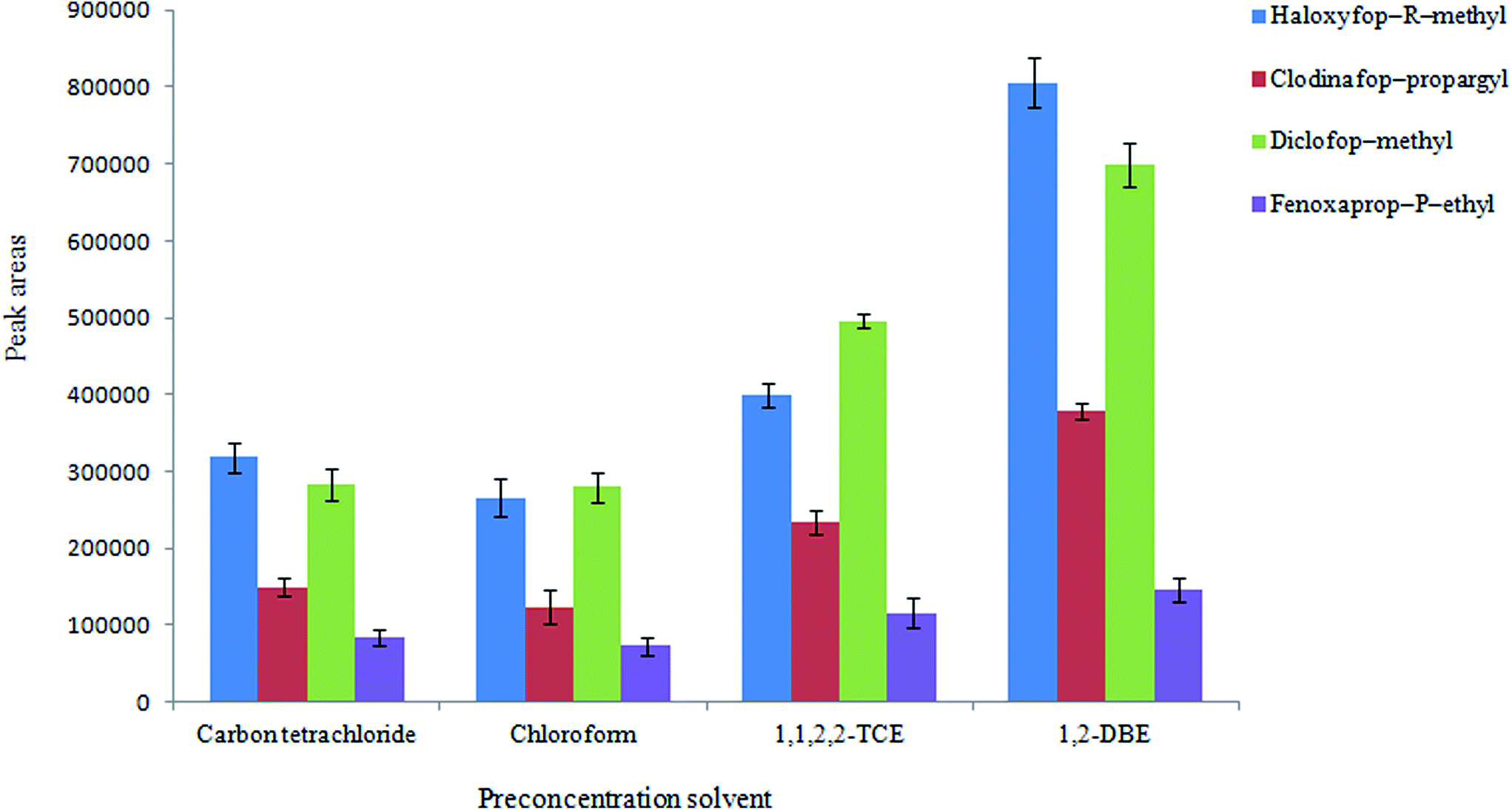
3.2.1. Selection of preconcentration solvent. In this study a DLLME step is performed after HLLE step to reduce volume of the organic phase from mL-level to a few μL. This step leads to the analytes are concentrated more in the extractant (preconcentration solvent). The choice of extraction solvent is very important for all DLLME processes in order to maximize the extraction efficiency. In this case, it should have low solubility in water and high extraction capability for the target analytes and it is immiscible with the disperser solvent (extraction solvent in HLLE step e.g. di-iso-butylamine). Four solvents including 1,2-DBE, 1,1,2,2-TCE, chloroform, and carbon tetrachloride were examined as the preconcentration solvent. To achieve a constant volume of the sedimented phase, some experiments were carried out using 1 mL di-iso-butylamine (obtained from the first step) including various volumes of the mentioned solvents. Using 28 μL of 1,1,2,2-TCE, 38 μL of carbon tetrachloride, 40 μL of 1,2-DBE, and 80 μL of chloroform, the sedimented phase volume was 10 ± 0.5 μL. Ten μL was selected as the sedimented organic phase volume owing to good repeatability of the method and to facilitate its removal. The results (Fig. 4) show that 1,2-DBE can efficiently extract the desired analytes among the tested solvents. So it was selected as a preconcentration solvent in other experiments.
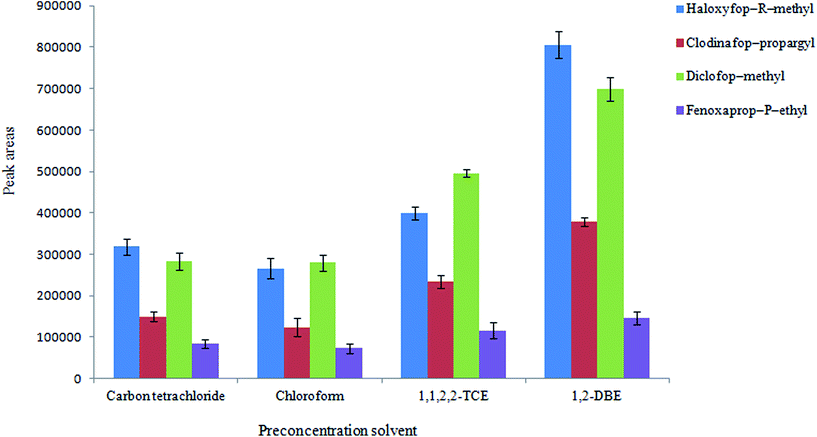 |
| | Fig. 4 Selection of preconcentration solvent in DLLME conditions: the same as used in Fig. 3, except flow rate which was 1.5 mL min−1. | |
3.2.2. Volume of the preconcentration solvent. The volume of extraction solvent (preconcentration solvent) plays an important role in DLLME efficiency. To obtain the optimum volume of the preconcentration solvent different volumes of 1,2-DBE (40, 50, 60, and 70 μL) were examined. The obtained results showed that analytical signals decreased when 1,2-DBE volume increased from 40 to 70 μL due to dilution effect. At low volumes of 1,2-DBE, as expected, higher EFs can be attainable. It is worth mentioned that low volumes of extraction solvent (e.g., below 40 μL) led to difficulty in collecting the sedimented phase by a microsyringe. Additionally, the repeatability of results reduced when low volumes of the extraction solvent were used. In consideration of a suitable balance of the analytical signals and ease of collecting the extract, 40 μL of 1,2-DBE was applied for the subsequent experiments.
3.2.3. Influence of ionic strength. Commonly salt addition is used in LLE and SPME procedures for increasing the ionic strength of aqueous phase which leads to enhancing analyte partition into the adsorbent and organic phase in SPME and LLE, respectively. In the present study, investigation on ionic strength effect was performed by adding various amounts of NaCl (0–10%, w/v) into aqueous phase, whereas the other experimental conditions (except preconcentration solvent volume) were kept constant. It is noted that addition of NaCl increased volume of the sedimented phase due to decreasing in solubility of the preconcentration solvent in aqueous solution compared to deionized water. The obtained results showed that the extraction efficiency increased with increasing NaCl concentration up to 7.5%, w/v, and then remained almost constant at higher concentrations. Based on these results, NaCl (7.5%, w/v) was added to aqueous solution in further studies.
3.2.4. Centrifugation time and rate. Centrifugation is an important step for separating organic phase from aqueous solution in the proposed method, and it could affect the volume of the settled phase. In order to attain the best extraction efficiency, centrifugation time and rate were studied in the ranges of 2–8 min and 112–1006 × g, respectively. The experimental results showed that the best performance was obtained at ≥4 min and ≥700 × g for centrifuging time and rate, respectively. Therefore, 4 min and 700 × g were chosen as the optimized centrifugation time and rate, respectively.
3.3. Quantitative aspects
Under the optimized conditions, the performance of the HLLE-DLLME-GC-FID method with regard to the linearity, limit of detection (LOD), limit of quantification (LOQ), ER, EF, and repeatability was investigated to validate the proposed method. The results obtained are given in Table 1. As can be seen, all calibration curves exhibit good linearity with coefficients of determination (r2) > 0.997. The relative standard deviations (RSDs) are below 6% for all analytes, showing good repeatability of the method. The ERs and EFs are in the ranges of 53–77% and 2120–3080, respectively. The LODs and LOQs for the selected pesticides, calculated at signal-to-noise (S/N) ratios of 3 and 10, are in the ranges of 0.12–0.40 and 0.40–1.3 μg L−1, respectively. Good repeatability, high EFs and ERs, and low LODs and LOQs are main advantages of the proposed method.
Table 1 Quantitative features of the proposed method for the selected pesticides
| Analyte |
LODa |
LOQb |
LRc |
r2d |
RSDe% |
ER ± SDf |
EF ± SDg |
| Intra-day |
Inter-day |
| Limit of detection (S/N = 3) (μg L−1). Limit of quantification (S/N = 10) (μg L−1). Linear range (μg L−1). Coefficient of determination. Relative standard deviation (n = 6, C = 50 μg L−1) for intra-day and (n = 4, C = 50 μg L−1) for inter-day. Extraction recovery ± standard deviation (n = 3). Enrichment factor ± standard deviation (n = 3). |
| Haloxyfop-R-methyl |
0.12 |
0.40 |
0.5–500 |
0.999 |
3 |
3 |
68 ± 1 |
2720 ± 40 |
| Clodinafop-propargyl |
0.20 |
0.66 |
1–500 |
0.997 |
5 |
6 |
77 ± 3 |
3080 ± 120 |
| Diclofop-methyl |
0.14 |
0.46 |
0.5–500 |
0.998 |
3 |
3 |
75 ± 2 |
3000 ± 80 |
| Fenoxaprop-P-ethyl |
0.40 |
1.3 |
1.5–500 |
0.999 |
3 |
4 |
53 ± 2 |
2120 ± 80 |
3.4. Comparison of the proposed method with other approaches
The performance of the proposed method in extraction and determination of aryloxyphenoxy-propionate pesticides in fruit juice and vegetable samples was compared with those of the other methods, and the results are summarized in Table 2. The proposed method has the following advantages over the other mentioned methods. Firstly, this method has high EFs compared to the other methods published for the determination of selected pesticides. Secondly, the RSD% of the proposed method are better than other methods. Overall the method can be considered as a simple, fast, and efficient sample preparation method. Moreover, it seems that this technique can be used for preconcentration of the acidic compounds in aqueous samples and extended to other applications.
Table 2 Comparison of the presented method with other methods used in preconcentration and determination of the studied pesticides
| Pesticide |
Sample |
RSDa (%) |
LODb |
EFc |
Method |
Ref. |
| Relative standard deviation. Limit of detection (μg L−1). Enrichment factor. Immersed solvent microextraction-gas chromatography-mass spectrometry. Sequential dispersive liquid–liquid microextraction-high performance liquid chromatography-ultraviolet detector. Single drop microextraction-gas chromatography-mass spectrometry. |
| Haloxyfop-P-methyl |
Aqueous sample |
4 |
0.05 |
272 |
ISME-GC-MSd |
2 |
| Clodinafop-propargyl |
6 |
0.15 |
317 |
| Fenoxaprop-P-ethyl |
7 |
0.15 |
403 |
| Haloxyfop-R-methyl |
Water sample |
3 |
4.35 |
171 |
SDLLME-HPLC-UVe |
37 |
| Fenoxaprop-P-ethyl |
4 |
1.50 |
173 |
| Clodinafop-propargyl |
Aqueous sample |
9 |
0.02 |
415 |
SDME-GC-MSf |
38 |
| Fenoxaprop-P-ethyle |
10 |
0.05 |
287 |
| Haloxyfop-R-methyl |
Fruit juices and vegetables |
3 |
0.12 |
2720 |
HLLE-DLLME-GC-FID |
This method |
| Clodinafop-propargyl |
5 |
0.20 |
3080 |
| Diclofop-methyl |
3 |
0.14 |
3000 |
| Fenoxaprop-P-ethyl |
3 |
0.40 |
2120 |
3.5. Real samples analysis
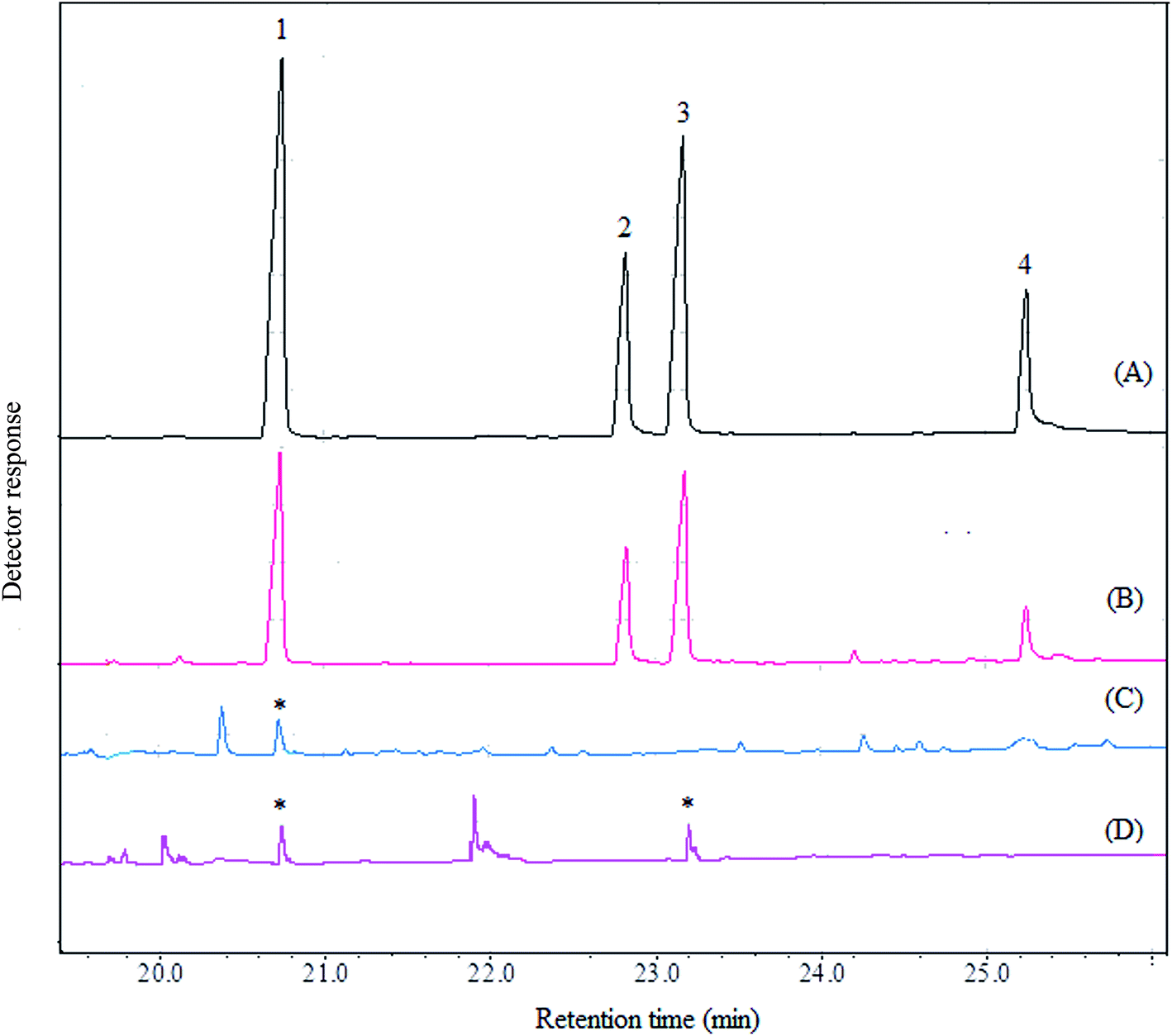
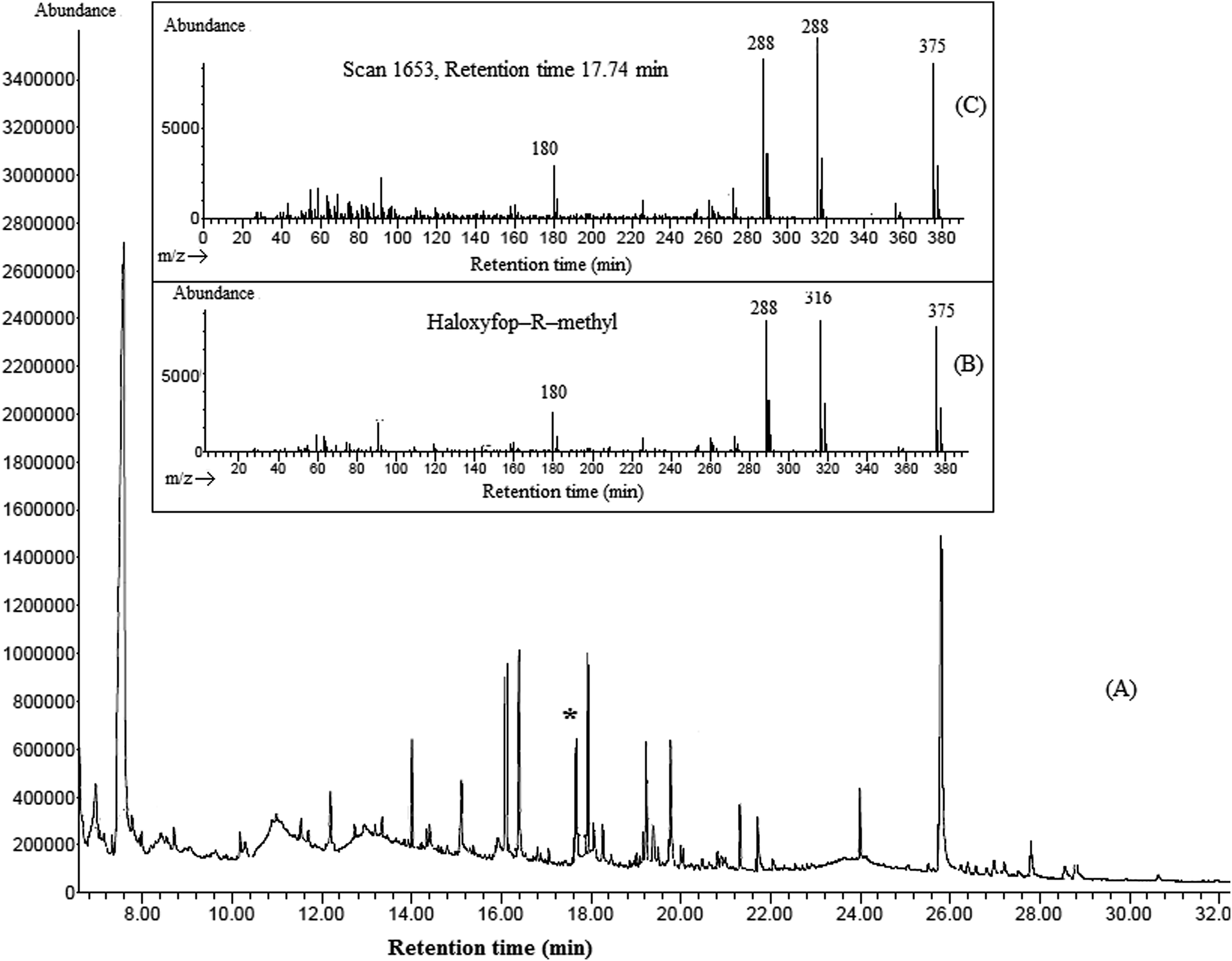
In order to evaluate the applicability of the proposed method, it was used in determination of four aryloxyphenoxy-propionate pesticides in fruit juice and vegetable samples including apple, grape, sour cherry, strawberry, garlic, melon, onion, cucumber, and tomato. Fig. 5 shows the typical GC-FID chromatograms of a standard solution at a concentration of 1000 mg L−1 in 1,2-DBE (each pesticide), deionized water spiked with 250 μg L−1 of each pesticide, garlic, and onion after performing the proposed method, expect chromatogram (A) in which 1 μL of the standard solution was directly injected into the separation system. The results show that haloxyfop-R-methyl and diclofop-methyl were found in onion at the concentrations of 49 ± 3 and 35 ± 3 μg L−1 (n = 3), respectively. Haloxyfop-R-methyl was also found in garlic at a concentration of 18 ± 2 μg L−1 (n = 3). Other samples were free of the studied pesticides. In order to more identify the compounds eluted in the retention times of the analytes in GC-FID chromatograms, the garlic and onion samples were also injected into GC-MS after performing the proposed sample preparation method on them. Total ions current (TIC) chromatogram and mass data for the compound eluted in the retention time of haloxyfop-R-methyl in garlic are given in Fig. 6. The presence of haloxyfop-R-methyl in the garlic was confirmed by comparison of mass spectrum of haloxyfop-R-methyl and scan 1653 (retention time 17.74 min). Also the obtained GC-MS data for onion sample (not shown here) verified the presence of haloxyfop-R-methyl and diclofop-methyl in that sample. The relative recoveries obtained for the analytes in samples in comparison with the results obtained for deionized water spiked at the same three concentration (30, 150, and 500 μg L−1 of each pesticide) are summarized in Table 3. According to the obtained results, matrices of the samples have no significant effect on the performance of the proposed method. It is noted that, garlic juice sample was diluted at a ratio of 1:3 with deionized water to reduce its matrix effect significantly. The results demonstrate that all obtained relative recoveries are between 82 and 99%.
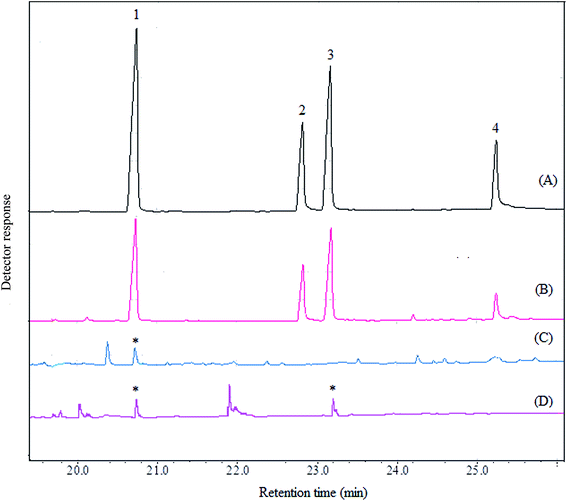 |
| | Fig. 5 Typical GC-FID chromatograms of: (A) standard solution (1000 mg L−1, each analyte in 1,2-DBE), (B) deionized water spiked with 250 μg L−1 of each pesticide, (C) garlic, and (D) onion. In all cases except chromatogram (A) the method was performed on the samples and 1 μL of the sedimented phase was injected into GC-FID. In chromatogram (A) 1 μL of the standard solution was directly injected into the separation system. Peaks identification: 1, haloxyfop-R-methyl; 2, clodinafop-propargyl; 3, diclofop-methyl; and 4, fenoxaprop-P-ethyl. | |
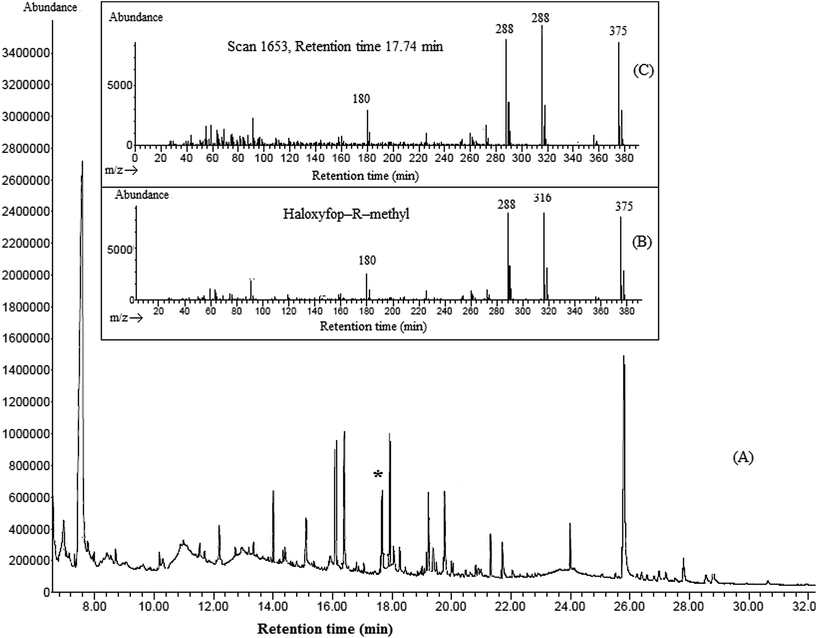 |
| | Fig. 6 Typical GC-TIC-MS chromatogram of garlic (A), mass spectra of haloxyfop-R-methyl (B), and scan of 1653 (retention time 17.74 min) (C). | |
Table 3 Study of matrix effect in fruit juice and vegetable samples. Analytes' contents of the samples were subtracted. All samples were used without dilution, except garlic which was diluted with deionized water at a ratio of 1:3
| Analyte |
Mean relative recovery ± standard deviation (n = 3) |
| Strawberry |
Garlic |
Apple |
Melon |
Onion |
Grape |
Sour cherry |
Tomato |
Cucumber |
| All samples were spiked with each analyte at a concentration of 30 μg L−1 |
| Haloxyfop-R-methyl |
94 ± 4 |
90 ± 2 |
97 ± 2 |
98 ± 3 |
88 ± 3 |
95 ± 3 |
94 ± 1 |
98 ± 2 |
94 ± 4 |
| Clodinafop-propargyl |
91 ± 2 |
82 ± 1 |
99 ± 3 |
95 ± 2 |
87 ± 4 |
93 ± 2 |
91 ± 2 |
96 ± 1 |
97 ± 1 |
| Diclofop-methyl |
92 ± 3 |
89 ± 3 |
97 ± 3 |
97 ± 2 |
90 ± 3 |
94 ± 1 |
90 ± 4 |
97 ± 1 |
95 ± 4 |
| Fenoxaprop-P-ethyl |
91 ± 1 |
85 ± 3 |
95 ± 2 |
96 ± 3 |
89 ± 4 |
91 ± 3 |
92 ± 3 |
94 ± 1 |
93 ± 3 |
|
| All samples were spiked with each analyte at a concentration of 150 μg L−1 |
| Haloxyfop-R-methyl |
94 ± 2 |
92 ± 2 |
98 ± 2 |
97 ± 3 |
89 ± 2 |
96 ± 2 |
95 ± 3 |
99 ± 2 |
95 ± 3 |
| Clodinafop-propargyl |
92 ± 1 |
83 ± 2 |
98 ± 2 |
95 ± 4 |
88 ± 3 |
92 ± 2 |
90 ± 3 |
96 ± 1 |
98 ± 2 |
| Diclofop-methyl |
94 ± 4 |
89 ± 4 |
97 ± 3 |
97 ± 4 |
90 ± 5 |
95 ± 3 |
91 ± 4 |
97 ± 3 |
96 ± 5 |
| Fenoxaprop-P-ethyl |
92 ± 3 |
88 ± 2 |
96 ± 4 |
97 ± 4 |
90 ± 3 |
93 ± 1 |
92 ± 2 |
95 ± 1 |
95 ± 2 |
|
| All samples were spiked with each analyte at a concentration of 500 μg L−1 |
| Haloxyfop-R-methyl |
95 ± 3 |
92 ± 2 |
98 ± 2 |
97 ± 2 |
91 ± 2 |
97 ± 1 |
96 ± 2 |
99 ± 2 |
96 ± 3 |
| Clodinafop-propargyl |
93 ± 1 |
86 ± 2 |
99 ± 1 |
96 ± 2 |
89 ± 4 |
93 ± 2 |
92 ± 2 |
97 ± 1 |
98 ± 3 |
| Diclofop-methyl |
94 ± 3 |
91 ± 4 |
97 ± 3 |
97 ± 2 |
91 ± 4 |
95 ± 3 |
92 ± 4 |
97 ± 2 |
98 ± 4 |
| Fenoxaprop-P-ethyl |
94 ± 2 |
90 ± 2 |
95 ± 3 |
98 ± 2 |
92 ± 2 |
94 ± 2 |
91 ± 2 |
96 ± 2 |
99 ± 2 |
4. Conclusion
In the present study, a new version of HLLE technique based on an acid–base reaction and its combination with DLLME followed by GC-FID analysis was developed for the extraction, preconcentration, and determination of aryloxyphenoxy-propionate pesticides in different samples. It was demonstrated that the proposed method is a fast, simple, and effective sample preparation method. Moreover, high EFs of the method afforded low LODs for the selected pesticides, making it possible to analyze the studied pesticides in the selected samples with convenience and sensitivity.
Abbreviations
| HLLE | Homogenous liquid–liquid extraction |
| DLLME | Dispersive liquid–liquid microextraction |
| EF | Enrichment factor |
| ER | Extraction recovery |
| FID | Flame ionization detector |
| FPD | Flame photometric detector |
| HPLC | High-performance liquid chromatography |
| GC | Gas chromatography |
| LLE | Liquid–liquid extraction |
| LOD | Limit of detection |
| LPME | Liquid phase microextraction |
| LOQ | Limit of quantification |
| MS | Mass spectrometry |
| NPD | Nitrogen phosphorous detector |
| RSD | Relative standard deviation |
| SPE | Solid phase extraction |
| SDME | Single drop microextraction |
| SPME | Solid phase microextraction |
| UV | Ultraviolet |
Acknowledgements
The authors thank the Research Council of University of Tabriz for financial support.
References
- L. Lucini and G. Pietro Molinari, Pest Manage. Sci., 2010, 66, 621–626 CAS.
- H. Bagheri, A. Es'haghi and A. Es-haghi, Int. J. Environ. Anal. Chem., 2013, 93, 450–460 CrossRef CAS.
- S. Roy and S. B. Singh, J. Chromatogr. A, 2005, 1065, 199–206 CrossRef CAS PubMed.
- H. Bao, S. Fang, Z. Liu, H. Shi, Y. Ye and M. Wang, J. Agric. Food Chem., 2010, 58, 8167–8170 CrossRef CAS PubMed.
- J. Wang, W. Chow, D. Leung and J. Chang, J. Agric. Food Chem., 2012, 60, 12088–12104 CrossRef CAS PubMed.
- J. W. Wong, K. Zhang, D. G. Hayward and C. Kai-Meng, in Mass spectrometry in food Safety, 2011, pp. 131–172 Search PubMed.
- J. W. Wong, K. Zhang, K. Tech, D. G. Hayward, C. M. Makovi, A. J. Krynitsky, F. J. Schenck, K. Banerjee, S. Dasgupta and D. Brown, J. Agric. Food Chem., 2010, 58, 5868–5883 CrossRef CAS PubMed.
- M. Gómez-Ramos, C. Ferrer, O. Malato, A. Agüera and A. Fernández-Alba, J. Chromatogr. A, 2013, 1287, 24–37 CrossRef PubMed.
- W. Guan, C. Li, X. Liu, S. Zhou and Y. Ma, Food Addit. Contam., Part A, 2014, 31, 250–261 CrossRef CAS PubMed.
- E. Orejuela and M. Silva, J. Chromatogr. A, 2003, 1007, 197–201 CrossRef CAS PubMed.
- N. Kolbe and J. T. Andersson, J. Agric. Food Chem., 2006, 54, 5736–5741 CrossRef CAS PubMed.
- A. Sannino, Rapid Commun. Mass Spectrom., 2007, 21, 2079–2086 CrossRef CAS PubMed.
- B. Albero, C. Sánchez-Brunete and J. L. Tadeo, Talanta, 2005, 66, 917–924 CrossRef CAS PubMed.
- S. Topuz, G. Özhan and B. Alpertunga, Food Control, 2005, 16, 87–92 CrossRef CAS.
- S. Igarashi, N. Ide and Y. Takagai, Anal. Chim. Acta, 2000, 424, 263–269 CrossRef CAS.
- K. Murata, Y. Yokoyama and S. Ikeda, Anal. Chem., 1972, 44, 805–810 CrossRef CAS PubMed.
- A. Koltsakidou, C. K. Zacharis and K. Fytianos, J. Chromatogr. A, 2015, 1377, 46–54 CrossRef CAS PubMed.
- M. A. Farajzadeh, M. Bahram, S. Zorita and B. G. Mehr, J. Hazard. Mater., 2009, 161, 1535–1543 CrossRef CAS PubMed.
- S. Cortés-Aguado, N. Sánchez-Morito, F. Arrebola, A. G. Frenich and J. M. Vidal, Food Chem., 2008, 107, 1314–1325 CrossRef.
- A. L. S. Simplıício and L. S. V. Boas, J. Chromatogr. A, 1999, 833, 35–42 CrossRef.
- D. A. Lambropoulou and T. A. Albanis, J. Biochem. Biophys. Methods, 2007, 70, 195–228 CrossRef CAS PubMed.
- M. I. Pinto, G. Sontag, R. Bernardino and J. Noronha, Microchem. J., 2010, 96, 225–237 CrossRef CAS.
- Y. Yang, D. J. Miller and S. B. Hawthorne, J. Chromatogr. A, 1998, 800, 257–266 CrossRef CAS PubMed.
- J. M. Kokosa, TrAC, Trends Anal. Chem., 2015, 71, 194–204 CrossRef CAS.
- J. Zhang, T. Su and H. K. Lee, Anal. Chem., 2005, 77, 1988–1992 CrossRef CAS PubMed.
- Y. Wang, Y. C. Kwok, Y. He and H. K. Lee, Anal. Chem., 1998, 70, 4610–4614 CrossRef CAS PubMed.
- G. Shen and H. K. Lee, Anal. Chem., 2002, 74, 648–654 CrossRef CAS PubMed.
- C. Basheer, A. Jayaraman, M. K. Kee, S. Valiyaveettil and H. K. Lee, J. Chromatogr. A, 2005, 1100, 137–143 CrossRef CAS PubMed.
- N. Ratola, A. Alves, N. Kalogerakis and E. Psillakis, Anal. Chim. Acta, 2008, 618, 70–78 CrossRef CAS PubMed.
- W. Liu and H. K. Lee, Anal. Chem., 2000, 72, 4462–4467 CrossRef CAS PubMed.
- L. Wang, L. Wang, J. Chen, W. Du, G. Fan and X. Lu, J. Chromatogr. A, 2012, 1256, 9–14 CrossRef CAS PubMed.
- W. Liu, L. Zhang, L. Fan, Z. Lin, Y. Cai, Z. Wei and G. Chen, J. Chromatogr. A, 2012, 1233, 1–7 CrossRef CAS PubMed.
- M. Rezaee, Y. Assadi, M.-R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- S. Rastegarzadeh, N. Pourreza and A. Larki, Anal. Methods, 2014, 6, 3500–3505 RSC.
- M. M. Zahedi, M. Rahimi-Nasrabadi, S. M. Pourmortazavi, G. R. F. Koohbijari, J. Shamsi and M. Payravi, Microchim. Acta, 2012, 179, 57–64 CrossRef CAS.
- Z. Gao and X. Ma, Anal. Chim. Acta, 2011, 702, 50–55 CrossRef CAS PubMed.
- S. Li, P. Gao, J. Zhang, Y. Li, B. Peng, H. Gao and W. Zhou, J. Sep. Sci., 2012, 35, 3389–3395 CrossRef CAS PubMed.
- H. Bagheri, M. Dehghan, A. Es'haghi and M. Naderi, Anal. Methods, 2013, 5, 4846–4851 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.