
Theoretical study on the tailored side-chain architecture of benzil-like voltage stabilizers for enhanced dielectric strength of cross-linked polyethylene
Hui Zhang*a,
Yan Shanga,
Hong Zhaoa,
Xuan Wanga,
Baozhong Hana and
Zesheng Lib
aKey Laboratory of Engineering Dielectrics and Its Application of Ministry of Education & College of Chemical and Environmental Engineering, Harbin University of Science and Technology, Harbin 150080, P. R. China. E-mail: hust_zhanghui11@hotmail.com; hbzhlj@163.com; Fax: +86-451-86392708; Fax: +86-451-86391615
bKey Laboratory of Cluster Science of Ministry of Education & School of Chemistry, Beijing Institute of Technology, Beijing, 100081, P. R. China. E-mail: zeshengli@bit.edu.cn; Fax: +86-10-68918670
First published on 13th January 2016
Abstract
A theoretical investigation on the mechanism of the tailored side-chain architecture of benzil-like voltage stabilizers for enhanced dielectric electrical breakdown strength of cross-linked polyethylene at the atomic and molecular levels is accomplished. The HOMO–LUMO energy gaps, the ionization potentials, and the electron affinities at the ground states of a series of benzil-like molecules are obtained at the B3LYP/6-311+G(d,p) level. The 8 isomerization reactions at the S0 state, including the harmonic vibration frequencies of the equilibrium geometries and the minimum energy paths (MEP) found using the intrinsic reaction coordinate (IRC) theory, are obtained at the same level. The substituent group effect, functional group effect, and electronic effect of the different heteroatoms (O, N, S) in substituent groups have been evaluated. The results show that benzil-like molecules, which have much smaller HOMO–LUMO energy gaps, much lower ionization potentials, and much larger electron affinities than those of aliphatic chains, can increase the electrical breakdown strength effectively as voltage stabilizers in cross-linked polyethylene. This result is consistent with Jarvid’s suggestions (Journal of Polymer Science, Part B: Polymer Physics, 2014, 52(16): 1047–1054).
1. Introduction
Cross-linked polyethylene (XLPE) with excellent dielectric properties, heat resistance, and chemical corrosion resistance has become one of the most widely applied insulation materials for high voltage cables up to 500 kV in real applications. The pre-breakdown degradation phenomenon known as electrical treeing initiates at points of high and divergent electric fields and is associated with partial discharges and insulation failure in power cables.1,2 The initial electron may be created by electrode emission under the local high electric field in XLPE insulated high-voltage cables, such as the inside Schottky effect from insulation, the Schottky effect from the electrode, or collision ionization. The initial electron which gains enough kinetic energy under the electric field in insulation is known as a hot electron. Afterwards, a bound electron is knocked free from an atom through a collision with a hot electron, creating a new carrier. If a chain reaction forms during this process, the carrier concentration increases sharply, resulting in the electron avalanche effect and the loss of insulating properties of the material. This phenomenon is known as electric breakdown. If the electron avalanche mentioned above is limited to a local area, the C–C bond of the polyethylene chain will be attacked, initiating a partial degradation known as electric treeing. Accumulation of the damage can cause dielectric breakdown as soon as an electrical tree channel has bridged and it leads to thorough damage of the insulation matrix. Some organic additives, such as polycyclic aromatic compounds or those with benzophenone-like structures, can serve as so-called voltage stabilizers to effectively increase the resistance to electrical treeing.3–8 This may be the next solution for XLPE insulating compounds for insulation of power cables exceeding 500 kV. Recently, our research group employed both experimental and computational methods to study the effect of acetophenone as a voltage stabilizer. The addition of acetophenone, a prototypical aromatic ketone compound, leads to a 50% increase in the alternate current (AC) electrical breakdown strength of XLPE.9 Using the theoretical studies, we foremost elucidated that keto–enol tautomerism of aromatic carbonyl compounds is one of the important factors for inhibiting initiation and propagation of electrical treeing in polyethylene, and proposed mechanisms for increasing the electrical breakdown strength of XLPE with aromatic carbonyl compounds as voltage stabilizers in 2013.10–12 However, the compatibility of acetophenone with the XLPE matrix is poor, and it is easy for it to migrate out of the polymeric matrix, leading to worse performance of PE insulation materials. In 2012, a benzil-type compound with a larger alkoxy chain was evaluated as a voltage stabilizer in a super-clean XLPE by Jarvid and his co-workers.7 As a result, the electrical treeing inception level was effectively raised, and the compatibility with the PE matrix was also significantly improved. After that, the synthesis and physico-chemical properties of seven benzil-type voltage stabilizers were also reported by Jarvid and his co-workers.8 These additives substantially enhance the dielectric strength of the insulating material under high-voltage alternating current conditions. The dielectric strength, 70% higher than the reference XLPE, was improved by the addition of voltage stabilizers with short (methyl) side chains linked to the benzil core via an ester or tertiary amine group.8This inspired us to see that it is essential to investigate the relationship between the electrical character of voltage stabilizers and their molecular structures and the mechanism of PE electrical breakdown strength increments at atomic and molecular levels.13 These proposed mechanisms are crucial to understand the molecular functioning of voltage stabilizers in XLPE composites in preventing the degradation of the polymer matrix. In this paper, we aim at providing a systematic investigation on the tailored side-chain architecture of benzil voltage stabilizers for enhanced dielectric strength of XLPE. The behavior of benzil-like molecules in preventing the treeing of XLPE composite materials would be proposed on the basis of the theoretical study, which would be useful for the rational molecular design and synthesis of the desired voltage stabilizers to enhance the transmission efficiency of tomorrow’s power grids in a real commercial application.
The different substituent groups with the heteroatoms O, N and S have been chosen in this study to evaluate the substituent effect, functional group effect, and electronic effect. The role of the voltage stabilizers in increasing the electrical breakdown strength of PE is discussed by focusing on the three effects. The molecular formulae, the names and corresponding abbreviations of the studied molecules are listed in Table 1. In this work, n-hexane (Pe6) is selected as a model molecule of PE; acetophenone (Ap), benzophenone (Bz), and benzil (Ben) are selected as reference molecules.
| Molecular formula | Molecular name | Ab. |
|---|---|---|
 |
n-Hexane | Pe6 |
 |
Acetophenone | Ap |
 |
Benzophenone | Bz |
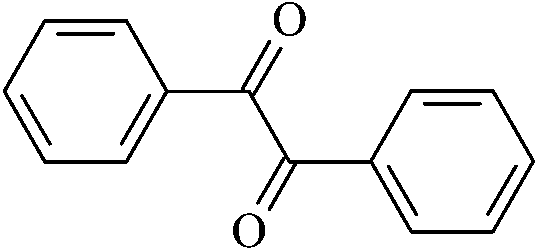 |
Benzil | Ben |
 |
4,4′-Dimethoxybenzil | Et1 |
 |
4,4′-Dioctyloxybenzil | Et8 |
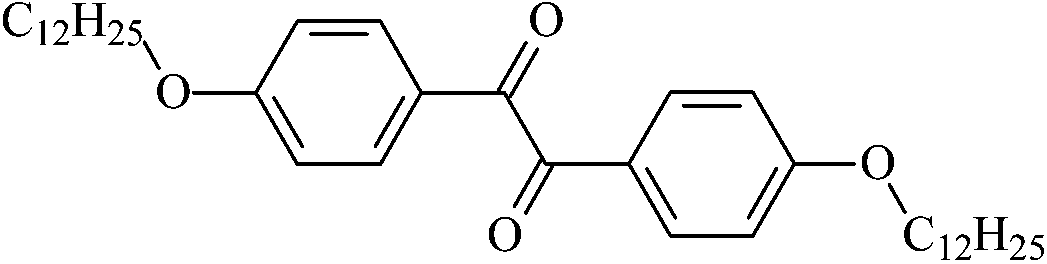 |
4,4′-Didodecyloxybenzil | Et12 |
 |
4,4′-Diacetoxybenzil | Es2 |
 |
4,4′-Dioctanoyloxybenzil | Es8 |
 |
4,4′-Didodecanoyloxybenzil | Es12 |
 |
4,4′-Dicarboxylbenzil | Dc1 |
 |
4,4′-Dimethoxycarbonylbenzil | Dc2 |
 |
4,4′-Dioctyloxycarbonylbenzil | Dc8 |
 |
4,4′-Didodecyloxycarbonylbenzil | Dc12 |
 |
4,4′-Diaminocarbonylbenzil | An0 |
 |
4,4′-Bis(N-methylaminocarbonyl)benzil | An1 |
 |
4,4′-Bis(N-octylaminocarbonyl)benzil | An8 |
 |
4,4′-Bis(N-dodecylaminocarbonyl)benzil | An12 |
 |
4,4′-Bis(N,N-dimethylaminocarbonyl)benzil | Nc1 |
 |
4,4′-Bis(N,N-dioctylaminocarbonyl)benzil | Nc8 |
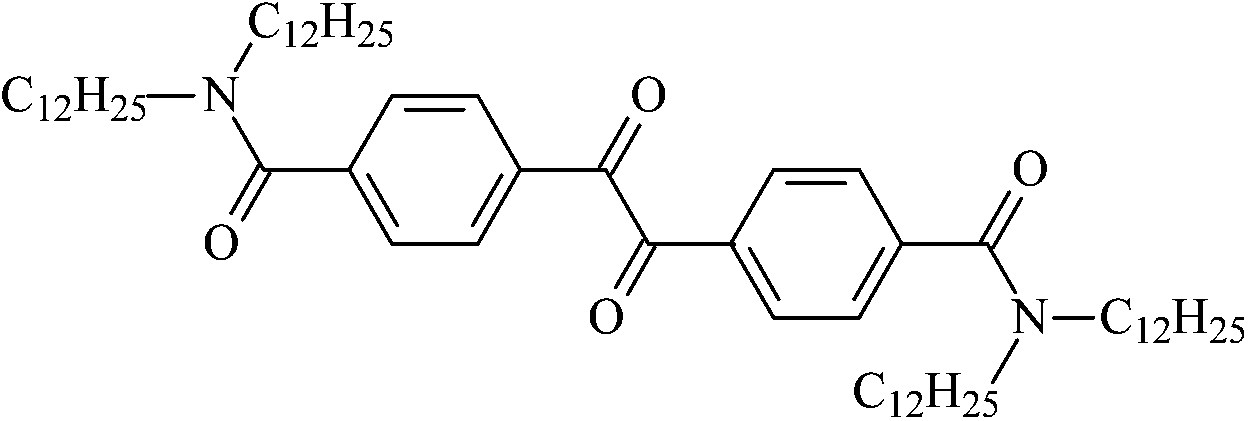 |
4,4′-Bis(N,N-didodecylaminocarbonyl)benzil | Nc12 |
 |
4,4′-Diaminobenzil | Am0 |
 |
4,4′-Bis(N,N-dimethylamino)benzil | Am1 |
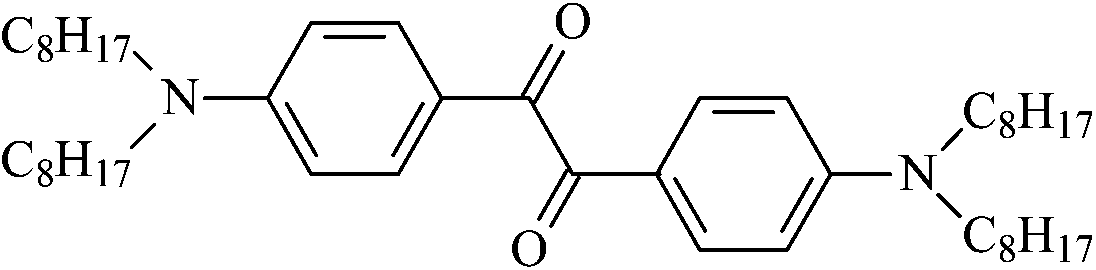 |
4,4′-Bis(N,N-dioctylamino)benzil | Am8 |
 |
4,4′-Bis(N,N-didodecylamino)benzil | Am12 |
 |
4,4′-Diacetamidobenzil | Ad2 |
 |
4,4′-Dioctanoylamidobenzil | Ad8 |
 |
4,4′-Didodecanoylamidobenzil | Ad12 |
 |
4,4′-Dithiobenzil | Th0 |
 |
4,4′-Dimethylthiobenzil | Th1 |
 |
4,4′-Dioctylthiobenzil | Th8 |
 |
4,4′-Didodecylthiobenzil | Th12 |
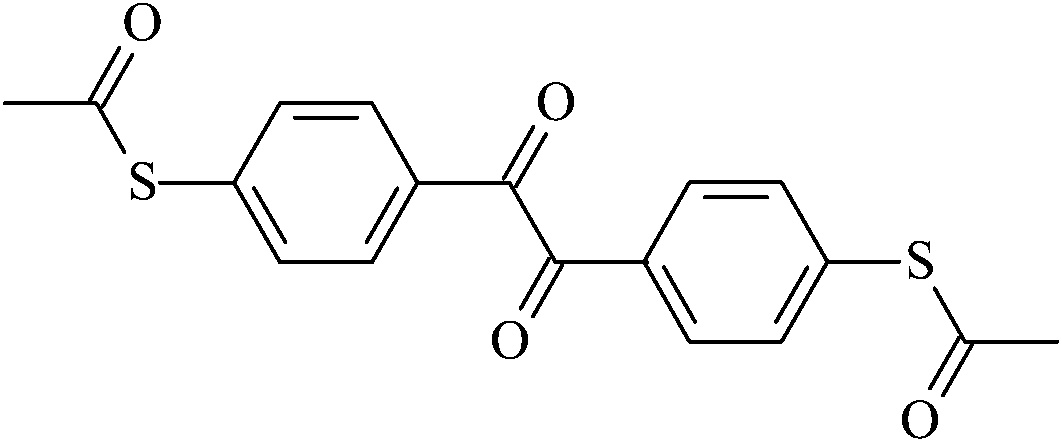 |
4,4′-Diacetylthiobenzil | To2 |
 |
4,4′-Dioctanoylthiobenzil | To8 |
 |
4,4′-Didodecanoylthiobenzil | To12 |
 |
4,4′-Dithioacetylthiobenzil | Tt2 |
 |
4,4′-Dithiooctanoylthiobenzil | Tt8 |
 |
4,4′-Dithiododecanoylthiobenzil | Tt12 |
 |
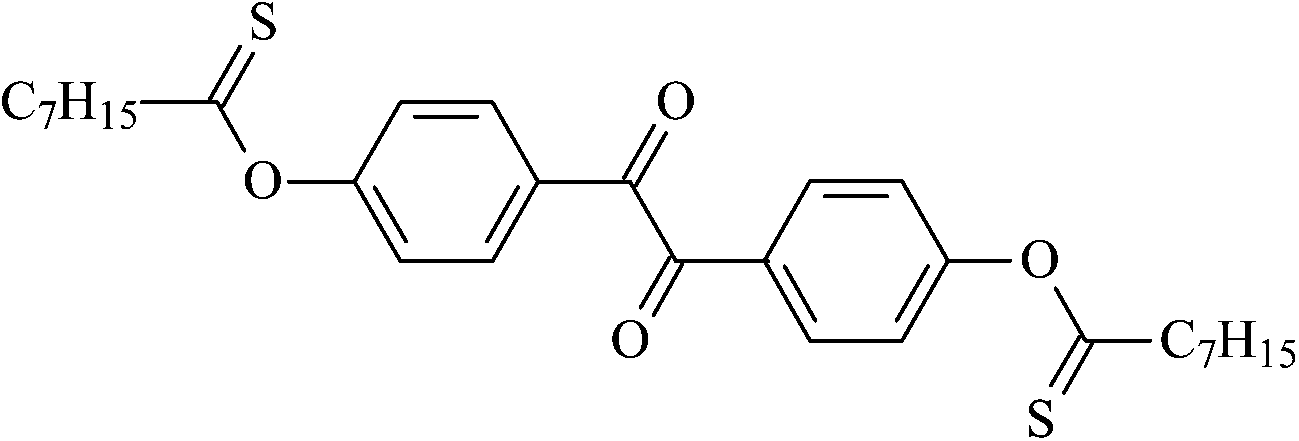
4,4′-Dithioacetoxybenzil | Oz2 |
 |
4,4′-Dithiooctanoyloxybenzil | Oz8 |
 |
4,4′-Dithiododecanoyloxybenzil | Oz12 |
2. Computational methods
The equilibrium geometries of all the stationary points of the studied molecules at the ground state S0 were optimized and frequencies were obtained using the B3LYP14–17 functional with the 6-311+G(d,p) basis set in this work. The B3LYP functional and the 6-311+G(d,p) basis set were confirmed to be suitable for the current study since the computational values of the adiabatic ionization potentials IP(a) and the electron affinities EA(a) at this level are in the most agreement with the corresponding experimental values in our previous paper.18 Based on these optimized geometries, the energy gaps (Eg) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), ionization potentials (IPs), and electron affinities (EAs) were obtained. The equilibrium geometries and frequencies of reactants, transition states, and products of 8 isomerization reactions at S0 were calculated at the same level. The minimum energy path (MEP) was obtained using intrinsic reaction coordinate (IRC) theory with a gradient step-size of 0.05 (amu)1/2 bohr. Then, the first and second energy derivatives were obtained to calculate the curvature of the reaction path and the generalized vibrational frequencies along the reaction path. The relevant schematic formulae can be defined as follows,| IP(v) = E+(M) − E(M) |
| IP(a) = E+(M+) − E(M) |
| EA(v) = E(M) − E−(M) |
| EA(a) = E(M) − E−(M−) |
 | ||
| Fig. 1 Schematic description of geometric coordinate modifications and energy changes. | ||
3. Results and discussion
3.1. Stationary point geometries
The optimized geometric structures of the transition states of 8 isomerization reactions at the B3LYP/6-311+G(d,p) level are presented in Fig. 2. Optimized bond lengths of breaking and forming bonds for the 8 transition states, the corresponding reactant C–H, O–H, and N–H bonds, and the product O–H and S–H bonds, and the calculated harmonic vibrational frequencies corresponding to the stretching modes at the same level are listed in Table 2 with acetophenone as the reference. All the transition states are confirmed to have and only have one imaginary frequency corresponding to the stretching modes of the coupling breaking and forming bonds by normal-mode analysis. The other harmonic vibrational frequencies of the neutral and ionic states of the studied molecules are confirmed to be real by normal-mode analysis. The transition states are abbreviated as TS, and reaction channels are represented as R. In Table 2, it can be seen that in the transition state of the isomerization reaction RDc1, the lengths of the broken and formed O–H bonds are equal, indicating that the isomerization reaction RDc1 proceeds via a symmetrical barrier. Other transition state structures of the studied isomerization reactions have a common character, the elongation of the breaking bond is larger than that of the forming bond, and they are all product-like, i.e., those reaction pathways will proceed via “late” transition states, which is consistent with Hammond’s postulate,20 applied for an endothermic reaction.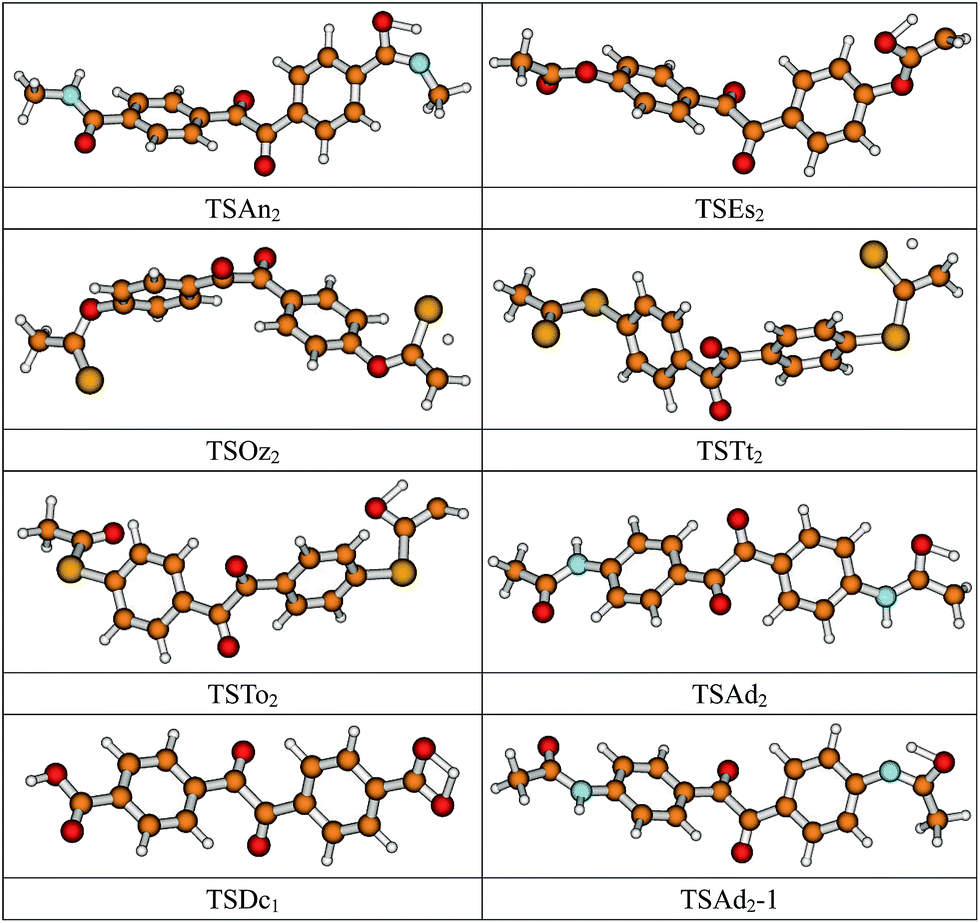 | ||
| Fig. 2 Optimized transition state geometric structures of the studied molecules at the B3LYP/6-311+G(d,p) level. | ||
| Ab. | Molecular formula | LR | Lb/f | Lp | Freq. |
|---|---|---|---|---|---|
| Ap |  |
1.089 | 1.491/1.276 | 0.963 | 2175i |
| Es2 | 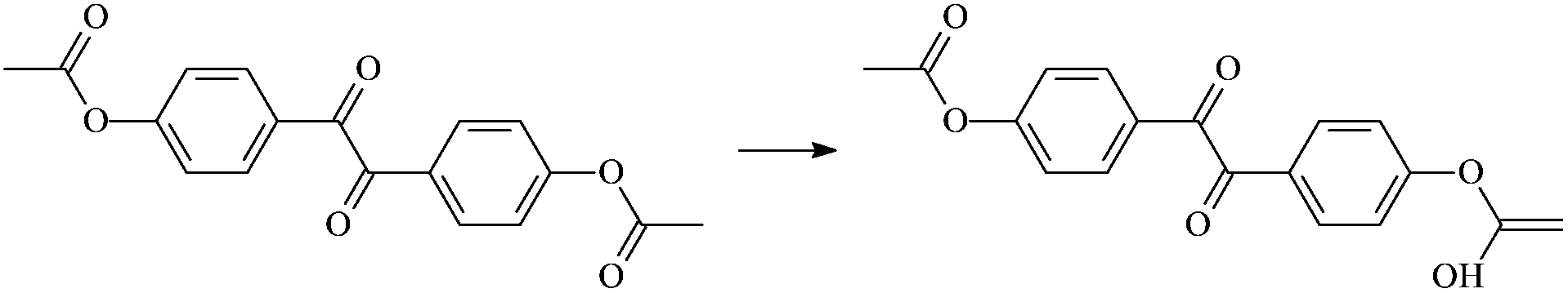 |
1.088 | 1.524/1.263 | 0.964 | 2099i |
| Dc1 | 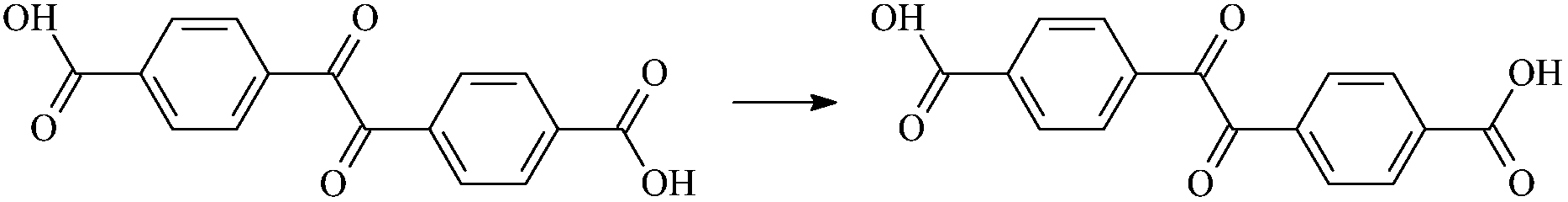 |
0.969 | 1.305/1.305 | 0.969 | 1962i |
| An1 |  |
1.007 | 1.342/1.308 | 0.969 | 1932i |
| Ad2 |  |
1.091 | 1.529/1.245 | 0.964 | 2003i |
| Ad2-1 | 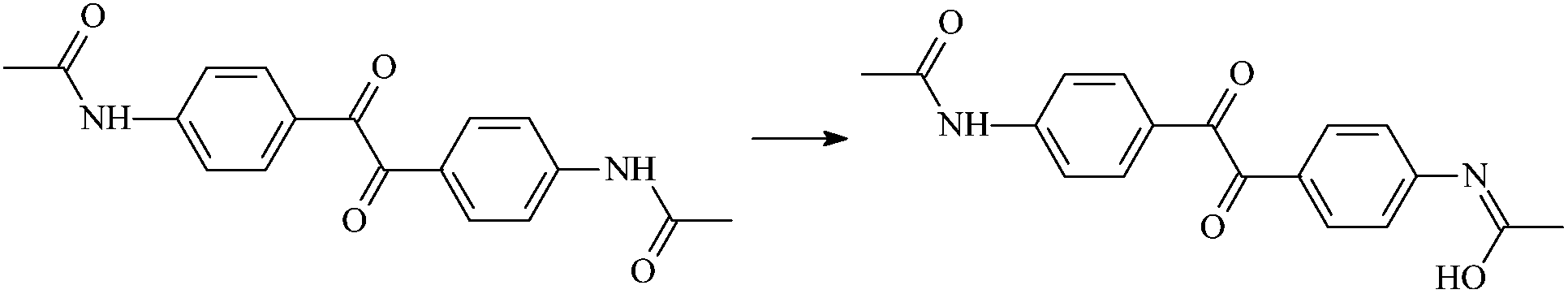 |
1.013 | 1.338/1.308 | 0.969 | 1911i |
| To2 |  |
1.094 | 1.521/1.267 | 0.966 | 2112i |
| Tt2 | 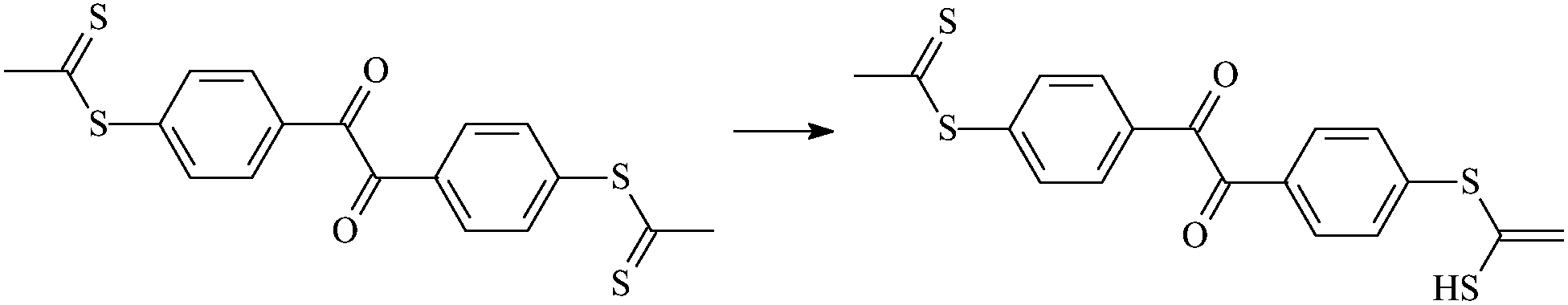 |
1.095 | 1.524/1.666 | 1.351 | 1894i |
| Oz2 | 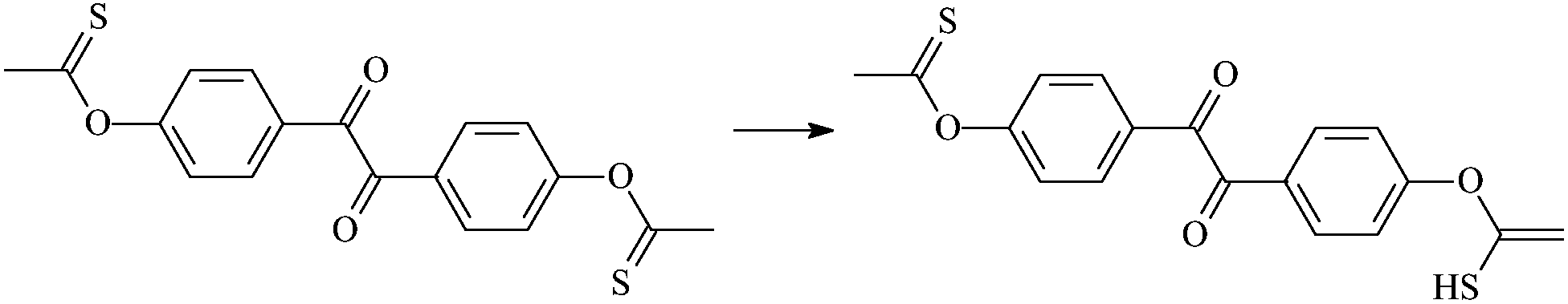 |
1.086 | 1.523/1.665 | 1.348 | 1854i |
3.2. Energies: frontier MOs, IPs and EAs
Both the ionization potentials (IPs) and the electron affinities (EAs) of the molecules are important parameters to characterize their reduction and oxidation abilities, respectively. The calculated values of the vertical and adiabatic IPs, EAs at the B3LYP/6-311+G(d,p) level and the corresponding experimental data21 (in brackets) are listed in Table 3, as well as the calculated HOMO–LUMO energy gap (Eg). To clarify quantitatively the influences of different substituent effects on their HOMOs and LUMOs, Pe6, Ap, Bz, and Ben were investigated as reference molecules.| Ab. | Molecular formula | Eg | IP(a) | IP(v) | EA(a) | EA(v) |
|---|---|---|---|---|---|---|
| Pe6 |  |
8.76 | 9.76 (9.97) | 10.80 | −1.20 | −1.21 |
| Ap |  |
5.20 | 8.95 (9.10 ± 0.10) | 9.19 | 0.33 (0.33) | 0.093 |
| Bz |  |
4.90 | 8.64 | 8.67 | 0.73 | 0.50 |
| Ben | 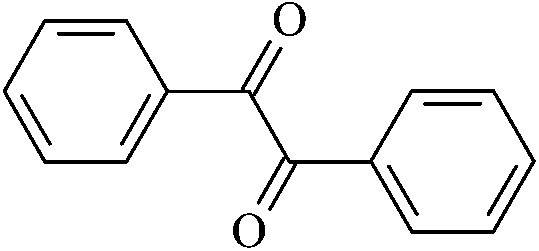 |
4.23 | 8.24 | 8.53 | 1.53 | 1.02 |
| Et1 |  |
4.22 | 7.74 | 7.92 | 1.31 | 0.75 |
| Et8 |  |
4.22 | 7.60 | 7.81 | 1.28 | 0.71 |
| Et12 |  |
4.22 | 7.60 | 7.77 | 1.28 | 0.70 |
| Es2 |  |
4.20 | 8.05 | 8.41 | 1.70 | 1.16 |
| Es8 |  |
4.20 | 7.98 | 8.32 | 1.68 | 1.14 |
| Es12 |  |
4.20 | 7.97 | 8.25 | 1.68 | 1.14 |
| Dc1 |  |
3.98 | 8.55 | 8.87 | 2.21 | 1.84 |
| Dc2 |  |
4.02 | 8.39 | 8.71 | 2.07 | 1.69 |
| Dc8 |  |
4.02 | 8.30 | 8.56 | 2.04 | 1.65 |
| Dc12 |  |
4.02 | 8.27 | 8.43 | 2.04 | 1.65 |
| An0 | 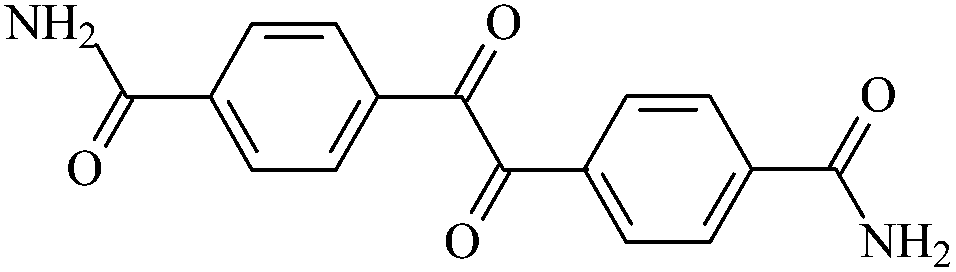 |
4.08 | 8.42 | 8.66 | 2.05 | 1.61 |
| An1 | 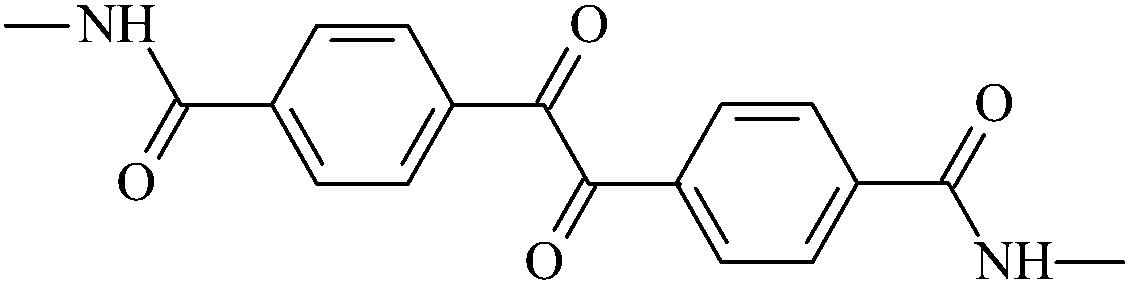 |
4.09 | 8.31 | 8.48 | 1.97 | 1.53 |
| An8 |  |
4.09 | 8.23 | 8.33 | 1.95 | 1.49 |
| An12 |  |
4.09 | 8.21 | 8.27 | 1.95 | 1.49 |
| Nc1 |  |
4.17 | 8.12 | 8.18 | 1.87 | 1.35 |
| Nc8 | 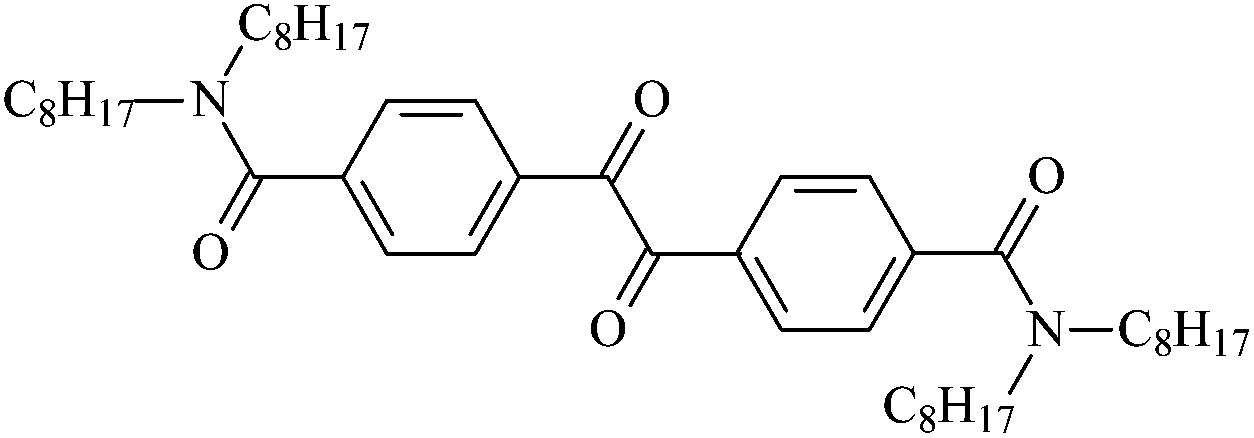 |
4.16 | 7.78 | 7.86 | 1.82 | 1.29 |
| Nc12 |  |
4.16 | 7.76 | 7.84 | 1.82 | 1.26 |
| Am0 |  |
4.11 | 7.30 | 7.48 | 1.11 | 0.50 |
| Am1 |  |
3.86 | 6.81 | 6.88 | 1.02 | 0.38 |
| Am8 |  |
3.81 | 6.54 | 6.59 | 0.94 | 0.38 |
| Am12 |  |
3.77 | 6.53 | 6.57 | 0.94 | 0.38 |
| Ad2 |  |
4.09 | 7.77 | 7.99 | 1.57 | 1.12 |
| Ad8 |  |
4.09 | 7.76 | 7.89 | 1.56 | 1.10 |
| Ad12 |  |
4.09 | 7.72 | 7.86 | 1.56 | 1.10 |
| Th0 | 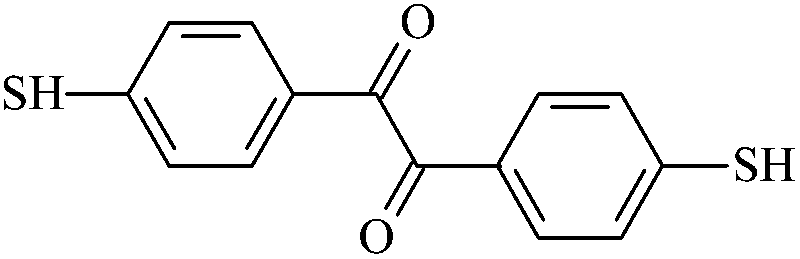 |
4.01 | 7.90 | 7.96 | 1.70 | 1.17 |
| Th1 |  |
3.82 | 7.50 | 7.55 | 1.64 | 1.04 |
| Th8 |  |
3.80 | 7.32 | 7.37 | 1.64 | 0.99 |
| Th12 |  |
3.80 | 7.31 | 7.37 | 1.64 | 0.99 |
| To2 | 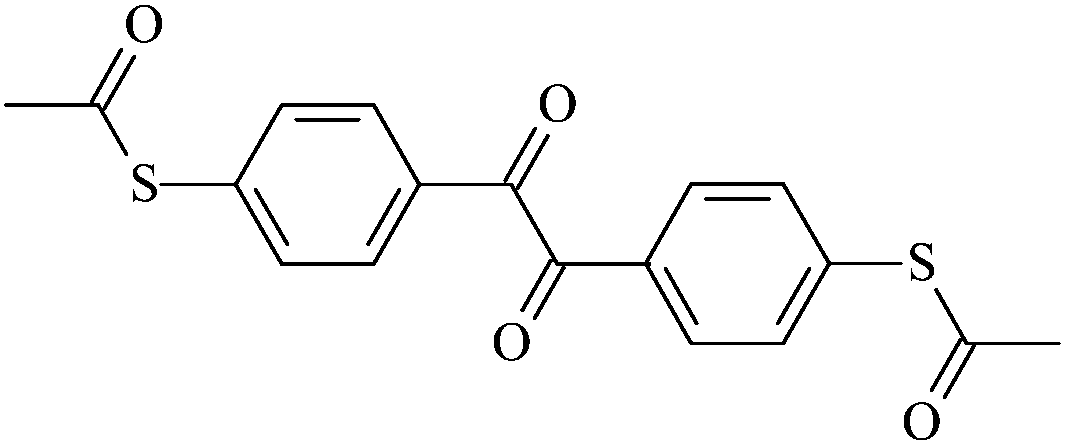 |
4.11 | 8.00 | 8.25 | 1.88 | 1.38 |
| To8 |  |
4.09 | 7.89 | 8.11 | 1.86 | 1.37 |
| To12 | 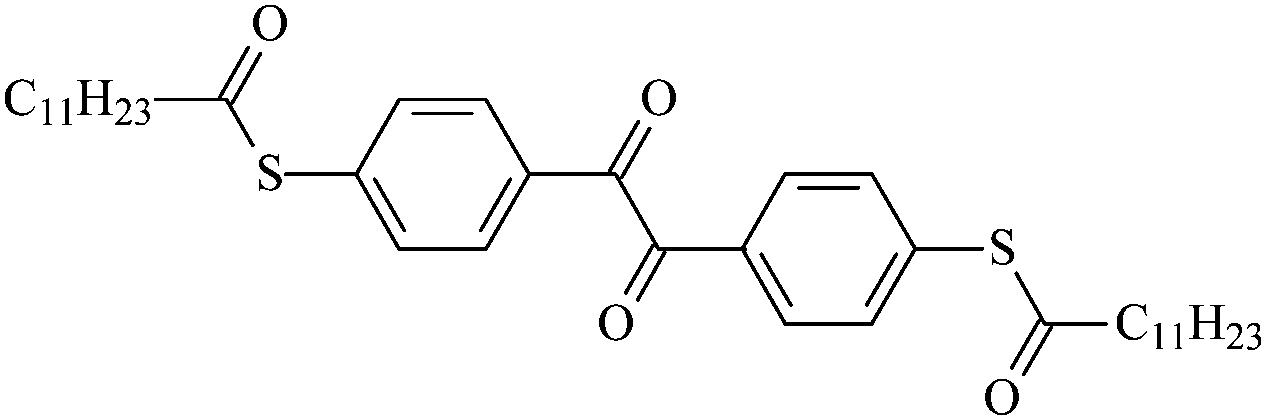 |
4.09 | 7.87 | 8.09 | 1.86 | 1.37 |
| Tt2 | 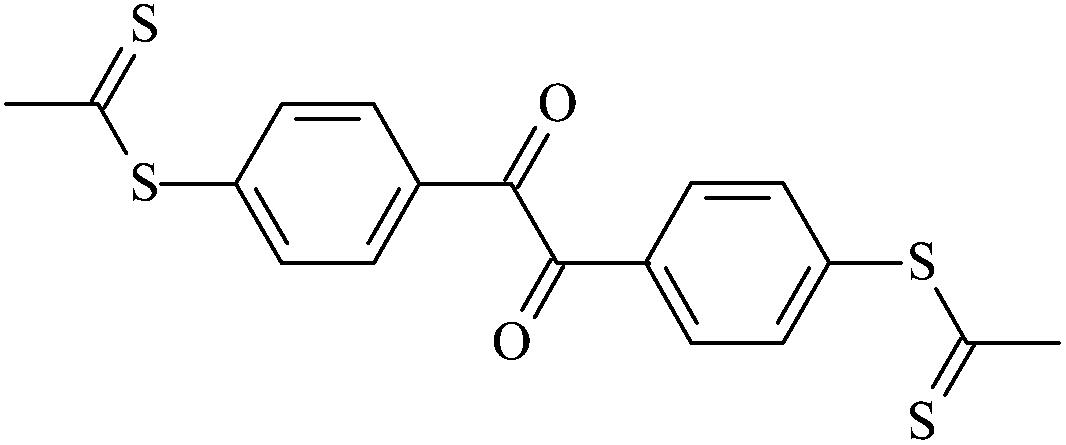 |
3.36 | 7.58 | 7.66 | 2.04 | 1.62 |
| Tt8 |  |
3.35 | 7.44 | 7.52 | 2.02 | 1.59 |
| Tt12 |  |
3.32 | 7.43 | 7.50 | 2.02 | 1.59 |
| Oz2 |  |
3.78 | 7.79 | 7.90 | 1.84 | 1.30 |
| Oz8 |  |
3.78 | 7.66 | 7.79 | 1.80 | 1.29 |
| Oz12 | 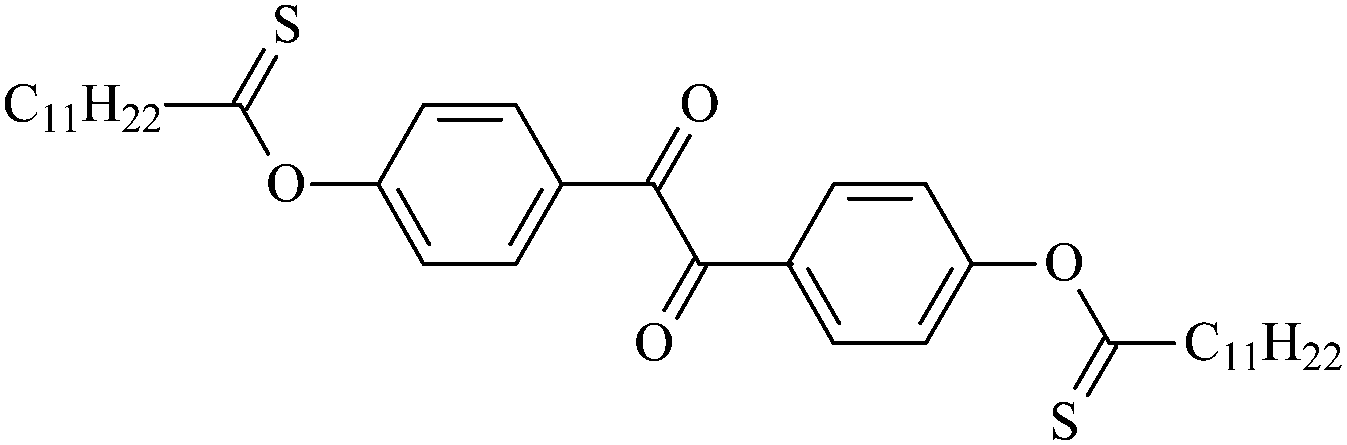 |
3.77 | 7.66 | 7.79 | 1.80 | 1.29 |
The varied trends of the ionization potentials and electron affinities are similar to those of the negative values of the HOMO and LUMO energies, respectively. Comparing with an aliphatic chain (Pe6), the introduction of carbonyl, phenyl, and heteroatom groups into the molecules is efficacious at decreasing the Eg.13 These groups are propitious for electronic dissociation because of their high HOMO energy and hence small ionization potential in terms of Koopmans’ theorem; in addition, they are also favorable for accepting electrons when the molecules possess low LUMO energy and hence large electron affinity energies. For instance, IP(a) (Am12, 6.53 eV) < IP(a) (Ap, 8.95 eV) < IP(a) (Pe, 9.76 eV); EA(a) (Dc1, 0.73 eV) > EA(a) (Ap, 0.33 eV) > EA(a) (Pe6, −1.20 eV). These benzil-like molecules possess a lower IP(v) than Pe6, so they can give rise to collision ionization before the polyethylene chain has been ionized. This can prevent degradation of the polymer matrix as voltage stabilizers, which is consistent with Jarvid’s suggestion.8
From Table 3, it is obvious that the introduction of carbonyl and phenyl groups in Bz and Ben molecules can decrease the values of Eg compared with Ap, because their conjugated system extends. The π–π–π and π–π–π–π conjugated systems are formed in Bz and Ben, respectively, resulting in the electronegativity of C in the phenyl ring (sp2) being larger than that in the alkyl group (sp3). Therefore, the abilities of π-electron delocalization and electron trapping are stronger than those of Ap (π–π). Bz and Ben molecules possess lower barriers to accepting electrons because of their low LUMO energy levels. For Bz, as the two benzene rings link to a carbonyl group directly, the steric hindrance is big, causing the two benzene rings to not be planar, and not providing the optimal π-orbital overlap. For Ben, as the two benzene rings link with different carbonyls, the steric hindrance decreases evidently, the degree of coplanarity increases, and the conjugation effect is increased, so Eg (Ben) < Eg (Bz). It can be seen that the contribution of the carbonyl group is larger in decreasing the Eg and increasing the EA in the Ben molecule than that of the phenyl group, because the electron withdrawing inductive effect of the carbonyl group is stronger than that of the phenyl group. Thus, Eg (Ben) < Eg (Bz) < Eg (Ap); EA(a) (Ap, 0.33 eV) < EA(a) (Bz, 0.73 eV) < EA(a) (Ben, 1.53 eV). The EA of benzil-like molecules increases gradually when –Z,  , and
, and  groups are linked to the para-position of Ben, so they are in the order EA(–Z) < EA(
groups are linked to the para-position of Ben, so they are in the order EA(–Z) < EA(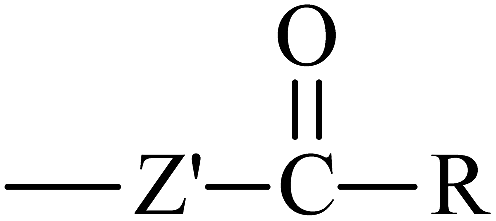 ) < EA(
) < EA(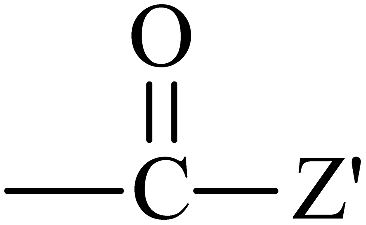 ), where Z = OH, OR, NH2, NHR, NR2, SH, and SR; Z′ = O and NH. EA(Dc1) is the largest.
), where Z = OH, OR, NH2, NHR, NR2, SH, and SR; Z′ = O and NH. EA(Dc1) is the largest.
Comparing with Ap (π–π conjugated system), not only a π–π–π–π conjugated system between carbonyl groups and benzene rings is formed in Et and Am, but also a p–π conjugated system is formed, as the electron-donating groups –OR or –NR2 are linked to the benzene ring, stretching the conjugated system. It can be concluded that the Eg decreases, for example Eg (Et12, 4.22 eV) < Eg (Ap, 5.20 eV); Eg (Am12, 3.77) < Eg (Ap). The calculated results have been well explained by Jarvid, Englund, and their co-workers2,7,8 that the introduction of a carbonyl or alkoxy group into benzophenone molecules is an exciting molecular design because they can effectively decrease the Eg value and increase the compatibility with polymer matrices. The energy of electronic transition becomes smaller as the Eg decreases. In addition, the –OR or –NR2 groups in Et or Am exhibit inductive electron-donating effects, which makes the electron densities of the benzene ring larger than those of Ben, resulting in the weaker ability to accept electrons. For instance, EA(a) (Et12, 1.28 eV) < EA(a) (Ben, 1.53 eV) and EA(a) (Am12, 0.94 eV) < EA(a) (Ben). There are two carbonyl groups in Ben, and one carbonyl group in Bz. The electron density in the benzene ring in Bz is larger than that of Ben, so EA (Bz, 0.73 eV) < EA (Ben). The Eg decreases evidently with the increasing conjugated effect, the N in the amino group is the best one that conjugated with a benzene ring among the studied molecules, thus Eg (Ad) < Eg (Es); Eg (Am) < Eg (Et).
From Table 3, the results indicate that the introduction of electron-withdrawing groups  (Z = OH, OR, NH2, NHR, and NR2) can increase the ability of accepting electrons, and the EA increases markedly. For example, EA(a) (Ben, 1.53 eV) < EA(a) (Dc1, 2.21 eV); EA(a) (Ben) < EA(a) (An0). When –SR,
(Z = OH, OR, NH2, NHR, and NR2) can increase the ability of accepting electrons, and the EA increases markedly. For example, EA(a) (Ben, 1.53 eV) < EA(a) (Dc1, 2.21 eV); EA(a) (Ben) < EA(a) (An0). When –SR, 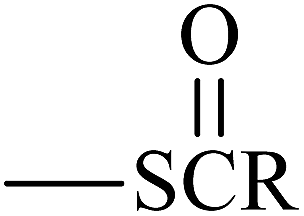 , and
, and  groups are linked to the para-position of Ben, the Eg of the benzil-like molecules decreases and the EA increases gradually. Because the radius of the S atom is larger than that of the O atom, the space of delocalization has expanded. Thus, EA(a) (Th) < EA(a) (To) ≈ EA(a) (Oz) < EA(a) (Tt).
groups are linked to the para-position of Ben, the Eg of the benzil-like molecules decreases and the EA increases gradually. Because the radius of the S atom is larger than that of the O atom, the space of delocalization has expanded. Thus, EA(a) (Th) < EA(a) (To) ≈ EA(a) (Oz) < EA(a) (Tt).
3.3. Energetics
The reaction enthalpies at 298 K (ΔH0298) and the potential barrier heights (ΔETS) with zero-point energy (ZPE) corrections for the 8 isomerization reactions obtained based on the electronic structure calculations at the B3LYP/6-311+G(d,p) level are listed in Table 4. The calculated ΔETS of acetophenone at the B3LYP/6-311+G(d,p) level is 62.31 kcal mol−1, which has been reported in our previous paper.11 The mechanisms have been firstly proposed on the basis of our theoretical results10,11 that voltage stabilizers can trap hot electrons, facilitate electronic transition, and restore the aliphatic cation radical. In addition, keto–enol and valence bond isomerization is suggested to occur when acetophenone is used as a voltage stabilizer.| Reaction equation | ΔETS + ZPE | ΔH0298 | |
|---|---|---|---|
| RAp |  |
62.31 | 13.81 |
| REs2 |  |
71.53 | 26.38 |
| RDc1 | 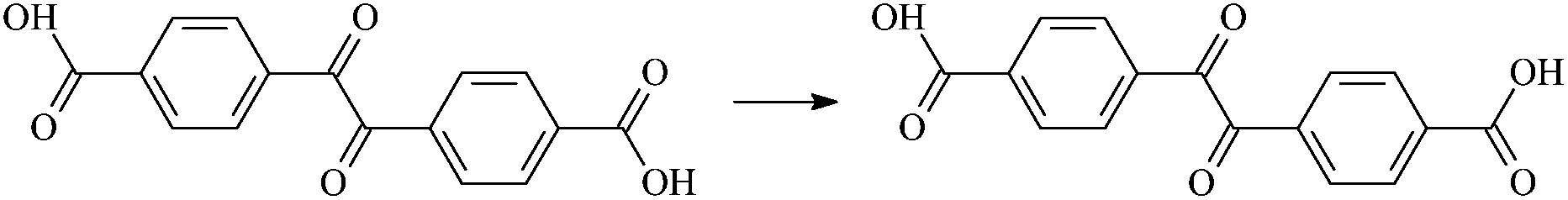 |
32.76 | 0.01 |
| RAn1 | 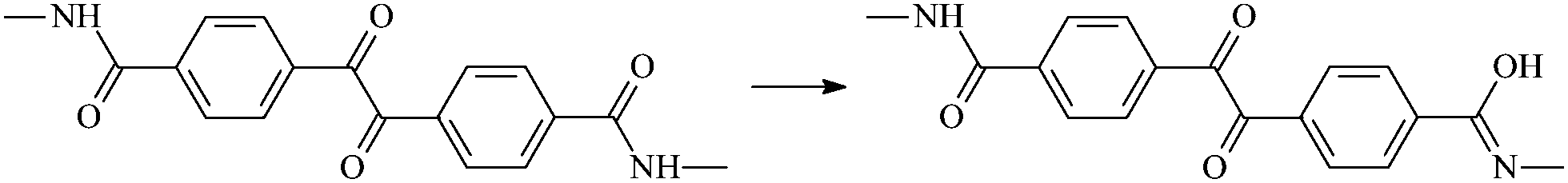 |
44.07 | 16.35 |
| RAd2 | 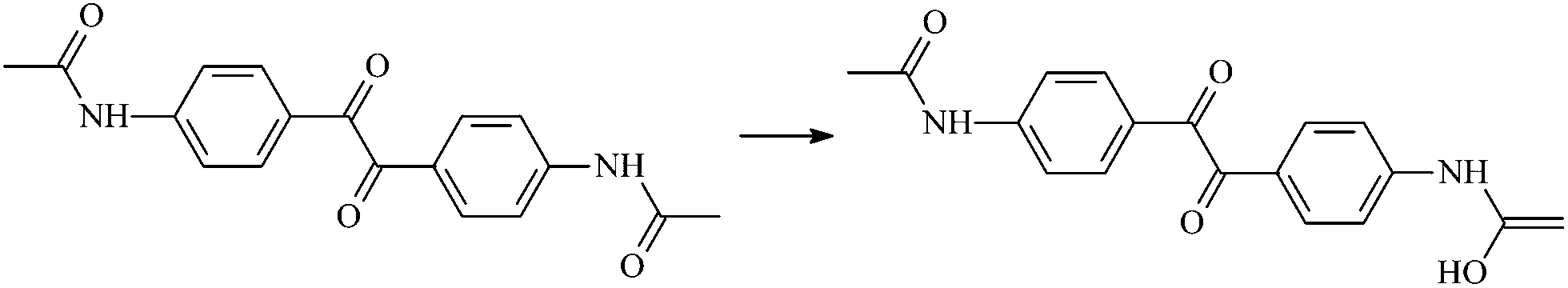 |
64.38 | 25.74 |
| RAd2-1 | 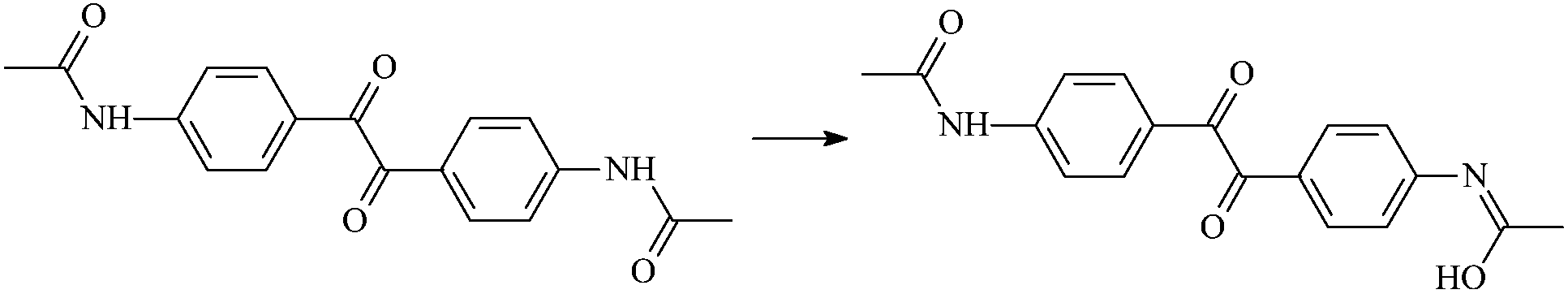 |
37.16 | 8.05 |
| RTo2 |  |
67.66 | 21.86 |
| RTt2 | 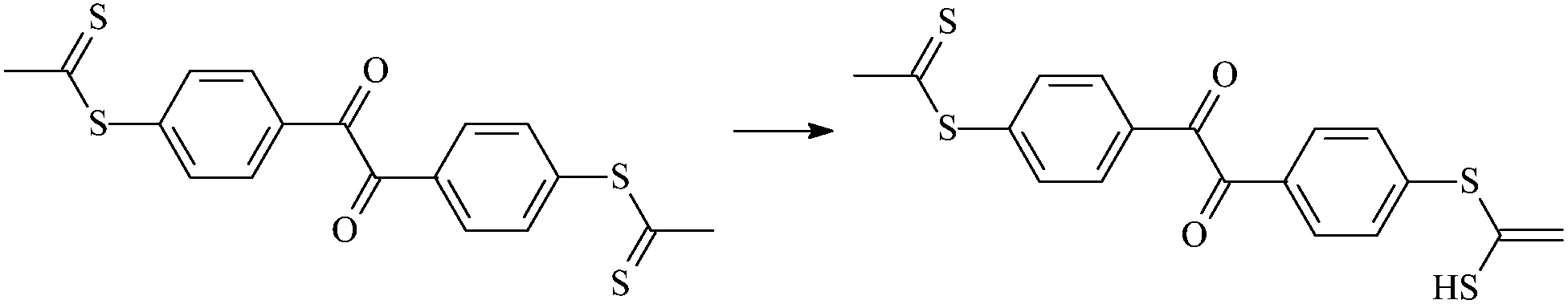 |
54.89 | 9.75 |
| ROz2 |  |
57.24 | 13.84 |
The 8 isomerization reactions have energy barriers of both the forward and reverse reactions. Comparing RAp with RDc1, the –CH3 in the acetyl group of Ap has been replaced by –OH. The H in the –OH of the Dc1 molecule is more active than that in –CH3 due to the larger electronegativity of O than of C. Therefore, the H in –OH dissociates more easily and leads to a more facile carboxyl isomerization reaction with lower energy barriers. In addition, when –CH3 is replaced by –OH, it exhibits an inductive electron withdrawing effect and electron donating conjugated effect at the same time, while the latter is stronger than the former, resulting in a good electron donating effect at last. This further facilitates the transfer of π electrons to the O in –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, which in turn exerts a stronger attraction to the H in the acetyl group, and thus the isomerization reaction occurs in an easier fashion. Similarly, when –CH3 is replaced by –NHR, its isomerization reaction energy barriers are lower than that of Ap. The electronegativity is in the order C < N < O, and the corresponding reaction activity of the H atom is in the order –CH < –NH < –OH, too. The above results indicate that the studied aromatic carbonyl and carboxyl molecules can undergo isomerization reactions, absorbing and transferring the energy of hot electrons when hot electrons are injected into the polyethylene compound. A lesser amount of energy (about 32.76–71.53 kcal mol−1) is needed to complete the isomerization reaction, which is much lower than that of the carbon–carbon single bond energy (the average bond energy is 82.95 kcal mol−1) in XLPE. It means that if the isomerization reaction energy barrier of any molecule is higher than 82.95 kcal mol−1, the reaction process is not competitive to the C–C bond breaking, and then those molecules cannot be used as voltage stabilizer adulterated to XLPE material. In principle, the studied benzil-like molecules could be effective as voltage stabilizers for increasing the breakdown strength of insulating XLPE materials.
O, which in turn exerts a stronger attraction to the H in the acetyl group, and thus the isomerization reaction occurs in an easier fashion. Similarly, when –CH3 is replaced by –NHR, its isomerization reaction energy barriers are lower than that of Ap. The electronegativity is in the order C < N < O, and the corresponding reaction activity of the H atom is in the order –CH < –NH < –OH, too. The above results indicate that the studied aromatic carbonyl and carboxyl molecules can undergo isomerization reactions, absorbing and transferring the energy of hot electrons when hot electrons are injected into the polyethylene compound. A lesser amount of energy (about 32.76–71.53 kcal mol−1) is needed to complete the isomerization reaction, which is much lower than that of the carbon–carbon single bond energy (the average bond energy is 82.95 kcal mol−1) in XLPE. It means that if the isomerization reaction energy barrier of any molecule is higher than 82.95 kcal mol−1, the reaction process is not competitive to the C–C bond breaking, and then those molecules cannot be used as voltage stabilizer adulterated to XLPE material. In principle, the studied benzil-like molecules could be effective as voltage stabilizers for increasing the breakdown strength of insulating XLPE materials.
4. Conclusion
Comparing with aliphatic compounds, we conjecture that the studied benzil-like molecules with heteroatom (O, N and S) groups as voltage stabilizers can: (1) effectively improve the ability of trapping hot electrons (larger electronegativity) and decrease the kinetic energy of the hot electrons (so as to not have enough energy to break the C–C bonds of polyethylene and prevent degradation of the polymer matrix); (2) obtain the energy of hot electrons and dissipate it through isomerization reactions (the energies of isomerization reactions are lower than that of C–C single bond breakdown in XLPE; (3) prevent the hot electrons from bombarding the C–C bonds of the XLPE matrix and electronic transition (lower HOMO–LUMO energy gaps); and (4) transform the XLPE aliphatic cation to a relatively stable aromatic cation and restore the partially injured aliphatic cation to prevent the degradation of the insulation matrix (as aromatic cations possess much stronger abilities of π-electron delocalization than aliphatic cations).With the increase in electronegativity of the substituent group, the value of EA increases, the hot electron in the XLPE insulated material has been bound, and benzil-like molecules can not only decrease electronic mobility, but also reduce electronic conductance when they are used as voltage stabilizers. This is favorable for improving the insulating properties of the material. At the same time, the probabilities of electronic collision ionization and C–C bond cleavages are also decreased. In this way, benzil-like molecules can effectively inhibit the initiation and propagation of polyethylene electrical treeing and simultaneously strengthen the alternate electric breakdown that polyethylene can endure. From the macro-point of view, as a consequence, the insulating XLPE material exhibits an elevated AC breakdown strength. With the increase in the conjugation effect, the value of the Eg decreases. Further work to account for reaction energy barriers, enthalpies and products of the isomerization of negative molecular ions under electron injection, together with the following electronic transfer, as well as electronic mobility and interface energy etc. of benzil-like molecules as voltage stabilizers in XLPE is under way.
Acknowledgements
We thank Professor Tierui Zhang (Key Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry (TIPC), Chinese Academy of Sciences (CAS), Beijing 100190, China) for his fruitful discussions and for checking the English. This work is supported by the National Basic Research Program of China (2012CB723308), the National Natural Science Foundation of China (51337002 and 50977019), the Doctoral Foundation by the Ministry of Education of China (20112303110005), the Science Foundation for Distinguished Young Scholar of Heilongjiang Province (JC201206).References
- A. C. Ashcraft, R. M. Eichhorn and R. G. Shaw, Laboratory Studies of Treeing in Solid Dielectrics and Voltage Stabilization of Polyethylene, Conf. Rec. IEEE Int. Symp. Electr. Insul., 1976, 6 Search PubMed.
- M. Jarvid, A. Johansson, V. Englund, A. Lundin, S. Gubanski, C. Müller and M. R. Andersson, High Electron Affinity: A Guiding Criterion for Voltage Stabilizer Design, J. Mater. Chem. A, 2015, 3, 7273–7286 CAS.
- Y. Yamano and H. Endoh, Increase in Breakdown Strength of PE Film by Additives of Azocompounds, IEEE Trans. Dielectr. Electr. Insul., 1998, 5, 270–275 CrossRef CAS.
- Y. Yamano, Roles of Polycyclic Compounds in Increasing Breakdown Strength of LDPE Film, IEEE Trans. Dielectr. Electr. Insul., 2006, 13, 773–781 CrossRef CAS.
- Y. Yamano and M. Iizuka, IEEE Trans. Dielectr. Electr. Insul., 2009, 16(1), 189–198 CrossRef CAS.
- V. Englund, R. Huuva and S. M. Gubanski, IEEE Trans. Dielectr. Electr. Insul., 2009, 16(5), 1455–1460 CrossRef CAS.
- M. Jarvid, A. Johansson, V. Englund, S. Gubanski and M. R. Andersson, Electrical Tree Inhibition by Voltage Stabilizers, Annual Repot Conference on Electrical Insulation and Dielectric Phenomena, October 2012, pp. 605–608 Search PubMed.
- M. Jarvid, A. Johansson, J. M. Bjuggren, H. Wutzel, V. Englund, S. Gubanski, C. Müller and M. R. Andersson, Tailored Side-Chain Architecture of Benzil Voltage Stabilizers for Enhanced Dielectric Strength of Cross-Linked Polyethylene, J. Polym. Sci., Part B: Polym. Phys., 2014, 52(16), 1047–1054 CrossRef CAS.
- H. Zhang, Y. Shang, H. Zhao, B. Z. Han and Z. S. Li, Study of the Effect of Valence Bond Isomerizations on Electrical Breakdown by Adding Acetophenone to Polyethylene as Voltage Stabilizers, Comput. Theor. Chem., 2015, 1062, 99–104 CrossRef CAS.
- H. Zhang, Y. Shang, H. Zhao, B. Z. Han and Z. S. Li, Mechanisms on Inhibition of Polyethylene Electrical Tree Aging: A Theoretical Study, J. Mol. Model., 2013, 19, 3035–3044 CrossRef CAS PubMed.
- H. Zhang, Y. Shang, H. Zhao, B. Z. Han and Z. S. Li, Mechanisms on Electrical Breakdown Strength Increment of Polyethylene by Acetophenone and its Analogues Addition: A Theoretical Study, J. Mol. Model., 2013, 19, 4477–4485 CrossRef CAS PubMed.
- H. Zhang, Y. Shang, X. Wang, H. Zhao, B. Z. Han and Z. S. Li, Mechanisms on Electrical Breakdown Strength Increment of Polyethylene by Aromatic Carbonyl Compounds Addition: A Theoretical Study, J. Mol. Model., 2013, 19, 5429–5438 CrossRef CAS PubMed.
- H. Zhang, H. Zhao, X. Wang, Y. Shang, B. Z. Han and Z. S. Li, Theoretical Study on the Mechanism of Polyethylene Electrical Breakdown Strength Increment by the Addition of Voltage Stabilizers, J. Mol. Model., 2014, 20, 2211–2222 CrossRef PubMed.
- T. N. Truong, W. T. Duncan and R. L. Bell, Chemical Applications of Density-Functional Theory, American Chemical Society, Washington, DC, 1996, p. 85 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle–Salvetti correlation energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- A. D. Becke, Density-functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- H. Zhang, Y. Shang, M. X. Li, H. Zhao, X. Wang and B. Z. Han, Theoretical study on the radical reaction mechanism in the cross-linking process of polyethylene, RSC Adv., 2015, 5, 90343–90353 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Revision A.02, Wallingford, CT, 2009 Search PubMed.
- G. S. Hammond, A Correlation of Reaction Rates, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- S. G. Lias and J. E. Bartmess, in NIST ChemistryWebBook, NIST Standard Reference Database Number 69, 2016 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |