
In situ transmission electron microscopy study of the electrochemical sodiation process for a single CuO nanowire electrode†
Liqiang Zhang‡
ab,
Yuecun Wang‡c,
Degang Xie‡c,
Yushu Tangab,
Chunyang Wud,
Lishan Cuiab,
Yongfeng Li*a,
Xiaohui Ning*c and
Zhiwei Shanc
aState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Changping, Beijing 102249, China. E-mail: yfli@cup.edu.cn
bDepartment of Materials Science and Engineering, China University of Petroleum, Beijing 102249, China
cThe Center for Advancing Materials Performance from the Nanoscale (CAMP-Nano), Xi An Jiao Tong University, Xian, 710049, China. E-mail: xiaohuining@mail.xjtu.edu.cn
dSchool of Materials Science and Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, China
First published on 12th January 2016
Abstract
In this work, an in situ transmission electron microscopy (TEM) study of the electrochemically driven sodiation and desodiation of a CuO nanowire (NW) was performed. Upon sodiation, Na ions first reacted with CuO, yielding a mixture of Cu, Cu2O, and Na2O, and then some Cu2O was subsequently reduced into nanocrystal Cu. The final sodiation product was nanocrystalline Cu mixed with Na2O and Cu2O. Upon extraction of Na+, the nanocrystalline Cu first oxidized into Cu2O and finally transformed back to nanocrystalline CuO. The volume of the CuO NW was found to expand markedly during the first sodiation, but the morphology of the NW remained unchanged during the following cycles. The mechanism by which high-performance CuO NW anodes are used for rechargeable Na ion batteries was analyzed by carrying out an in situ TEM technique based on our results, and this study may be beneficial for designing the optimal structure of the CuO anode, improving its cycle performance, and making CuO more feasible for Na ion batteries.
Introduction
The forecasted increasing demand for lithium, with its expanding electric vehicle and grid applications, suggests a looming lithium shortage. Thus, increasing numbers of researchers are seeking new chemistries with monovalent (K+, Na+) or divalent (Mg2+ and Ca2+) cations. Of these cations, the most appealing alternative to lithium metal is sodium, which has a high abundance in the earth's crust. Recently, research on Na ion batteries (NIBs) has attracted increasing attention.1–3 Many compounds such as transition metal oxides and sulfides have served as promising anode materials in NIBs.4–11 Of these compounds, CuO has attracted particularly considerable interest due to its low cost, chemical stability and nontoxic nature.12 CuO used to be extensively investigated as an anode material in lithium ion batteries (LIBs).13–15 CuO nanorod anodes for LIBs have been shown to exhibit an electrochemical capacity of 550 mA h g−1 after 100 cycles,14 which can be further improved by combining it with graphene.15 As in LIBs, the volume of the CuO anode in NIBs changes considerably upon Na+ insertion and extraction processes.16,17 In order to accommodate the volume changes and hence improve the capacity retention and cycling performance of CuO, many CuO nanoscale materials such as nanoparticles, nanowires (NWs), nanorods, and nanosheets have been designed and synthesized, and have shown better performance than bulk CuO.18–21 The one-dimensional CuO NW is considered to be able to provide an effective channel for electron transport and ion diffusion, leading to a better battery performance than provided by the other CuO nanomaterials.19,22,23 Yuan et al. proposed a strategy for scalable fabrication of flexible and porous CuO nanorod array anodes in NIBs, and these NIBs showed a high capacity of over 640 mA h g−1 and a good cycling stability even at a high current density.24 Wang et al. prepared porous CuO NW NIBs that in their investigation showed discharge capacities of 640 mA h g−1 in the first cycle and 303 mA h g−1 after 50 cycles.25 Although CuO NWs have been considered as high-performance anodes for rechargeable NIBs, the details of the electrochemical processes that occur in CuO NWs upon sodiation and desodiation are still unknown.Understanding the detailed microscopic mechanisms of the electrochemical reaction and material degradation is crucial for designing high-performance CuO anodes for NIBs. In this work, an in situ transmission electron microscopy (TEM) technique was applied to investigate the structural and chemical evolution of CuO NWs during Na+ insertion and extraction, and this investigation has provided an essential understanding of the electrochemical reaction mechanism of CuO NWs serving as anodes for NIBs.
Experimental section
The CuO NW was prepared via a thermal oxidation process. First, the copper foam substrate was cleaned in an aqueous 1.0 M HCl solution for ∼20 s, and then repeatedly rinsed with distilled water and dried under a nitrogen flow. Then, the copper foam substrate was placed in an aluminum boat and transferred into the center of a quartz tube. Here, it was immediately heated to 500 °C and kept for 4 h in air, after which many CuO NWs were found to have been synthesized on the surface of the copper foam.The in situ experiments investigating the electrochemically driven sodiation of the CuO NW were conducted in real time by using an all-solid nanobattery assembled inside the transmission electron microscope, and details of the nanobattery setup can be found in our previous publications.26,27 To confirm the negligible magnitude of the effects of the electron beam on the electrochemical reactions, both beam-on and blank experiments were performed. The in situ electrochemical tests and TEM imaging were carried out by using an in situ electrical testing system, Hysitron PI 95 H1H, inside the Hitachi H9500 TEM operated at 300 keV.
The electrochemical Na-storage properties of the CuO NWs were evaluated by galvanostatic cycling using CR2025-type coin cells. The electrode slurry was made by mixing 75 wt% active material (CuO NW/graphite), 15 wt% acetylene black and 10 wt% polyvinylidene fluoride (PVDF) in N-methyl pyrrolidone (NMP) with magnetic stirring for 2 h. The slurry was then coated onto Ni foam and dried at 100 °C in a vacuum overnight to make the working electrodes. The electrodes were then assembled into half cells in an Ar-filled glove box using an Na foil as the counter electrode and a Celgard 2300 polypropylene membrane as the separator. The electrolyte was 1 M NaPF6 dissolved in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in volume). The cells were charged and discharged at various current densities between 0.005 and 3.0 V vs. Na/Na+ on a Neware battery tester (Shenzhen, China). The specific capacity of CuO/G was calculated based on the total weight of the CuO NW and graphite. Cyclic voltammetry (CV) scanning was carried out on an Arbin BT2000 system between 0.005 and 3.0 V vs. Na/Na+ at various scan rates using three-electrode beaker cells with Na foil as both the counter and reference electrodes. All of the electrochemical measurements were carried out at room temperature.
:1 in volume). The cells were charged and discharged at various current densities between 0.005 and 3.0 V vs. Na/Na+ on a Neware battery tester (Shenzhen, China). The specific capacity of CuO/G was calculated based on the total weight of the CuO NW and graphite. Cyclic voltammetry (CV) scanning was carried out on an Arbin BT2000 system between 0.005 and 3.0 V vs. Na/Na+ at various scan rates using three-electrode beaker cells with Na foil as both the counter and reference electrodes. All of the electrochemical measurements were carried out at room temperature.
Results and discussion
Fig. 1(a) shows a schematic illustration of the CuO NW/Na+ nanobattery setup, which consisted of a few CuO NWs attached to an aluminum (Al) rod with conductive silver epoxy as the anode, metallic Na serving as the counter electrode, and the naturally grown sodium oxide (Na2O) and hydroxide (NaOH) on the surface of the metallic Na serving as the solid electrolyte. A potential of −3.5 V was applied to the CuO NW with respect to the Na counter electrode to initiate the sodiation, while the bias was reversed to facilitate desodiation. The CuO NWs were fabricated by the thermal oxidation of the copper foam substrate at 500 °C in air for 12 h, and the microscopic images of the Cu foam before and after the thermal oxidation process are shown in Fig. 1(b). The scanning electron microscope (SEM) image in Fig. 1(c) demonstrates that all of the CuO NWs grew uniformly and perpendicular to the surface of the Cu foam substrate. These NWs were observed to have lengths ranging from 2–20 μm and diameters of 30–200 nm. The high-resolution TEM image (Fig. 1(d)) and the electron diffraction pattern (EDP) of the CuO NW (Fig. 1(e)) demonstrated the pristine CuO NW to be of a single-crystalline phase. | ||
| Fig. 1 (a) Schematic illustration of the electrochemical device. The CuO NWs grown vertically on the surface of Cu foam was cleaned and glued to an Al rod using conducting epoxy. Bulk metallic Na (cathode) was scratched on the surface of a W tip. The naturally generated Na2O layer on the surface of the metallic Na served as the solid electrolyte. (b) Photographs of the Cu foam before and after heating for 12 h. (c) An SEM image of the Cu foam heated in air for 12 h. (d) A bright-field TEM image of a CuO NW and its HRTEM image are shown in the upper inset. (e) The EDP of the CuO NW. | ||
Fig. 2(a) shows the typical changes in the microstructure of an intrinsic CuO NW during the first sodiation process. When a potential of −3.5 V was applied, the sodiation (discharging) process of the NW occurred. As the reaction front (marked by the red arrows) propagated from (as shown in the figure) the left end to the right end of the CuO NW, the NW immediately elongated and expanded. The single crystalline pristine CuO NW was immediately transformed into a nanocrystal upon insertion of Na+ (Fig. 2(b)). Since the leading edge of the reaction front propagated longitudinally down the NW, the reaction front also proceeded progressively along the radial direction, i.e., from the surface to the center of CuO NW (Fig. S1, Movie S1†). EDPs for the pristine and sodiated CuO NWs confirmed that the single-crystalline CuO NW was first partially converted to Cu2O, and then decomposed into Cu (Fig. 2(c)). After the first discharge reaction, the contrast between a crystalline core and grey shell was clearly observed in the NW. The diameter of the crystalline core of the CuO NW decreased as the sodiation proceeded (Movie S1†). It took about a half an hour to discharge a NW with an initial length of 1.41 μm and diameter of 52 nm. After the first sodiation process, the CuO NW elongated by ∼20%, its diameter increased by ∼78%, and its total volume expanded by about 232%. The CuO NW clearly experienced a large radial expansion upon insertion of Na+. EDX line scans, at the region of the blue dashed line in Fig. 2(b), revealed the distribution of elements at the reaction front (Fig. 2(d)). Although elemental Cu and Na cannot form any alloy during the sodiation process, Na+ can still diffuse into the CuO lattice and form Na2O with the O2− of CuO; and then the Na2O in the CuO NW could further serve as the diffusion channel for Na+ transport. The maintenance of the integrity of the morphology of the CuO NW after the electrochemical reaction contributed to the stability of the battery. Had the CuO been a bulk material, its pulverization could not have been avoided upon the very large expansion of its volume. During the first sodiation, the NW generated a few nanocrystals dispersed in the amorphous matrix (the inset of Fig. 2(b)), and the EDP characterization showed diffraction rings mixed with the amorphous haloes, which could be indexed as Na2O and Cu mixed with Cu2O phases, which can be defined as Cu1−xCuxO1−(x/2). With the prolonged discharging, the diffraction spots related to CuO became weaker, and finally disappeared. The first discharging process has been shown to include three conversion reactions:24
| xNa+ + CuO + xe− → x/2Na2O + Cu1−xCuxO1−(x/2) | (1) |
| 2Cu1−xCuxO1−(x/2) + (2 − 2x)Na+ + (2 − 2x)e− → Cu2O + (1−x)Na2O | (2) |
| Cu2O + 2Na+ + 2e− → 2Cu + Na2O | (3) |
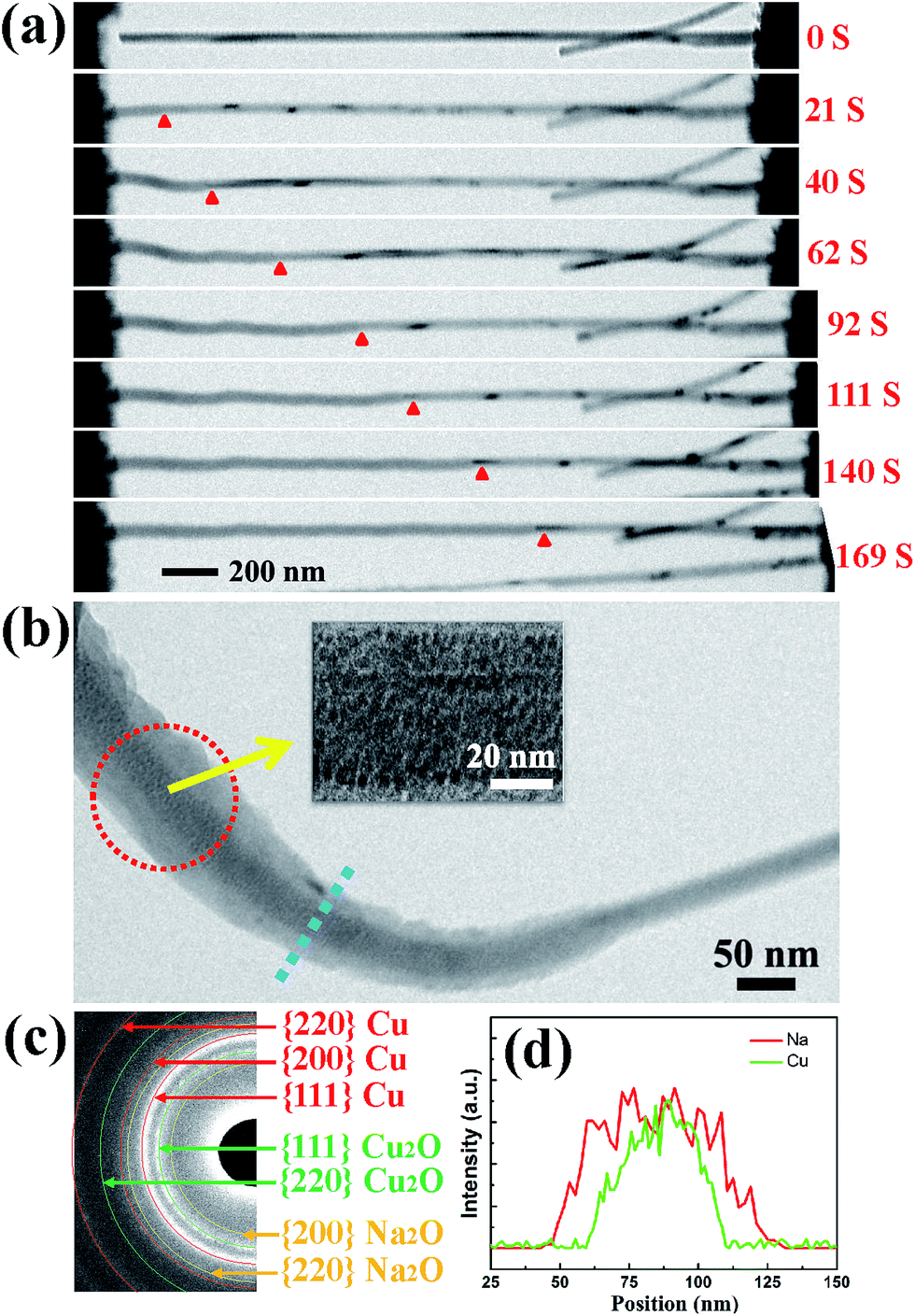 | ||
| Fig. 2 (a) A series of images showing the changes in the microstructure of an intrinsic CuO NW during the sodiation process. (b) A magnified image of the reaction front, with the inset demonstrating that the single crystal of CuO transformed into a nanocrystal upon sodiation. (c) An EDP showing that single crystalline CuO NW was converted to Cu2O and a Cu alloy. (d) EDX line scans of the region marked by the blue dashed line in (b) revealing the distribution of elements at the reaction front. | ||
The changes in the morphology of the CuO NW during the electrochemical cycling are shown in Fig. 3(a)–(d). The changes of the EDPs of the CuO NW undergoing sodiation/desodiation cycling are shown in Fig. 3(e)–(h). After the first discharging process, the pristine CuO NW became partially sodiated and transformed to a Cu phase mixed with Cu2O and Na2O (Fig. 3(e)), which was transformed back to the crystalline CuO in the following charging process (Fig. 3(f)). Note that the shape of the sodiated CuO NW remained unchanged during the desodiation, and there was a negligible decrease in the volume (Fig. 3(c)). In the following discharging process (Fig. 3(d)), the crystallized CuO transformed back to Cu, and Cu2O again (Fig. 3(g)), which was finally retransformed back to the crystallized CuO (Fig. 3(h)) in the following charging process as well. The CuO NW was found to undergo irreversible volume changes during the cycling processes, and the desodiated CuO NW could not return to its original state. In addition, the sodiated CuO became quite brittle after cycling, and broke suddenly after the second charging (Fig. S1†) due to a tiny vibration. The cycling performance of the CuO NW can be expressed as follows: decomposition of CuO into Cu2O, and then conversion into metallic Cu upon discharging; and reoxidation of Cu2O and Cu into CuO in the subsequent charging process.
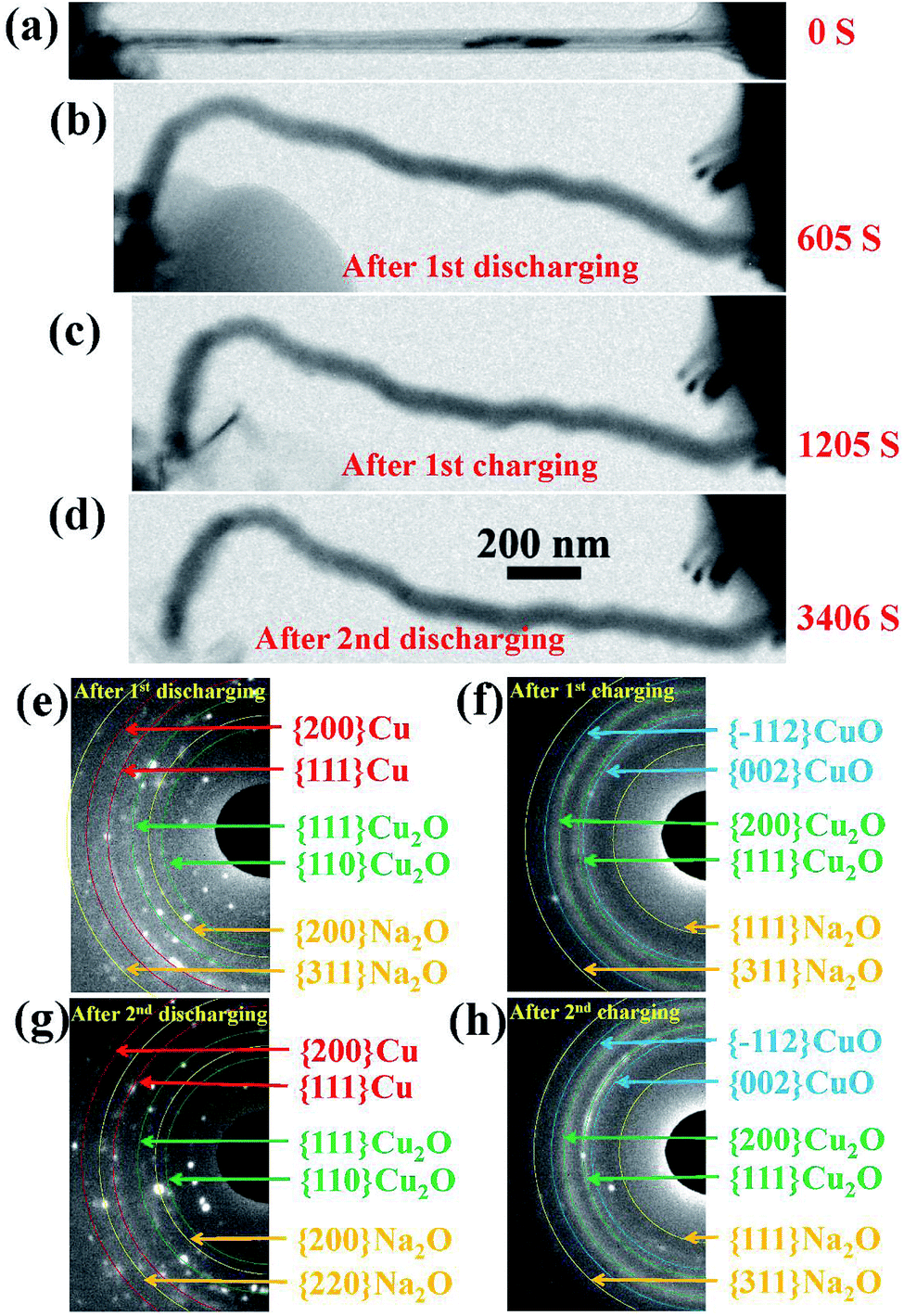 | ||
| Fig. 3 (a–d) Changes in the morphology of the CuO NW during cycling. (e–h) The changes of the EDPs of the CuO NW undergoing sodiation/desodiation cycling. | ||
In addition, galvanostatic discharge–charge cycling was performed to directly measure the capacity of the CuO NW electrode during sodiation and desodiation at a constant current rate of 200 mA g−1 in a coin cell. The cyclic voltammograms of the CuO NW electrode at a scan rate of 0.2 mV s−1 are shown in Fig. S2,† and they indicate that the CuO NW underwent multiple phase conversions during cycling. In the first discharging process, three reduction peaks at 1.3, 0.8 and 0.1 V were observed in the potential range of 0.01–3 V, which are attributed to the formation of Cu1−xCuxO1−(x/2), the generation of Cu2O and the final conversion into metallic Cu mixed with Na2O, respectively. In the following charging process, two oxidation peaks appeared at 1.3 and 2.1 V that can be indexed to the formation of Cu2O and restoration of CuO phase, respectively. All of the redox peaks reappeared quite consistently from one cycle to the next, except for a slight shift to higher potential, demonstrating good redox reversibility and structural stability.
Fig. 4 shows a schematic diagram of a CuO NW during the sodiation process. With the volume expansion, the reaction front is illustrated to migrate from the left end to the right end of the NW. The formation of Na2O on the surface is due to the surface diffusion and oxidization of Na+. In some carbon anodes without O2−, similar surface oxide phases (Li2O or Na2O) have also been observed under the in situ TEM electrochemical testing condition,28,29 which indicates that the electron beam may affect the formation of the surface oxide layer. Na+ is not able to extract O2− from CuO to form the surface shell as this process requires a very high input of energy. Instead, Na+ can diffuse into the CuO lattice and form Na2O with the O2− of CuO. The new generated Na2O phase in the sodiated CuO NW could serve as the diffusion channel for Na+ transport. As the migration of Na+, Cu2+ first becomes converted to Cu+, and finally to the metallic Cu. The displacement of the reaction front versus the reaction time is plotted in Fig. S3,† and the diameter of the NW was observed to be ∼40 nm. The nearly parabolic character implies the diffusion of Na+ over a long distance, which is significant in determining the battery performance. The different colors correspond to the different lengths of the NW. Based on the above results, the diffusivity rate of Na+ in the reacted amorphous sections was determined to be in the range of 2–9.3 nm s−1. The wide distribution of diffusivity values is reasonable here. In contrast to a crystal, the atomic structure in an amorphous region is not well defined. When subjected to different conditions, the amorphous NWs will form different structural features, such as different lengths, which can lead to different diffusion rates. Owing to the properties of CuO, Na+ can diffuse into the CuO lattice and the generated Na2O phase becomes diffusion channels for the transport of Na+, and this mechanism is quite different from that used by the Sn- or Si-based anode.30,31 The CuO NWs display no obvious changes in morphology during cycling (except the 1st discharging), so they have a potential to achieve long cycling lifetimes and good reversibility. The exterior Na2O shell on the surface of sodiated CuO NW is the solid electrolyte interface (SEI), which could consume the Na+ and lead to an irreversible capacity loss. In the future, investigators are expected to wrap an exterior protective layer (such as a carbon layer) on the surface of the CuO NW; such a wrap would allow the anode to freely expand while retaining good electrical conductivity and would prevent the cracking and pulverization of the CuO anodes. The above results show that significant challenges still need to be overcome before CuO anodes can be widely utilized in practical NIBs.
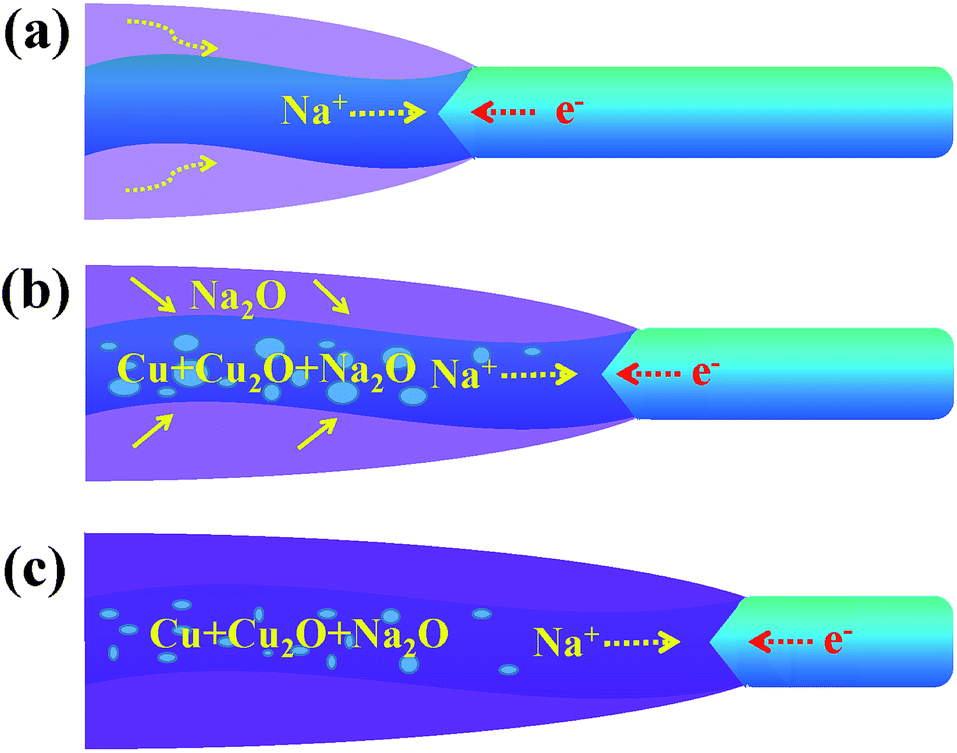 | ||
| Fig. 4 A schematic diagram for one part of CuO NW during the (a) start (b) middle and (c) end of the sodiation process. | ||
Conclusions
In summary, an in situ TEM has been performed to study the structural and phase changes of the CuO NWs during the sodiation and desodiation process. A very large increase in the volume of the NW was observed when a potential was applied to the nanobattery, and meanwhile the CuO NW was partially converted to Cu2O, and was finally decomposed into metallic Cu mixed together with Na2O. During the desodiation process, the reacted CuO did not display obvious changes in morphology. However, the phase transformation from Cu to Cu2O mixed with CuO was confirmed by the EDPs. The above results illustrate that there are significant challenges that need to be overcome before CuO anodes can be utilized in practical NIBs. For example, enough space should be reserved for the increase of the diameter of the CuO NW, and its elongation and distortion upon sodiation; providing such space could enhance the cycling lifetime and improve the reversibility of the battery. Moreover, the CuO NWs often become quite brittle and break easily during cycling, and thus more attention should be paid to improving the toughness of the NWs. Our work provides detailed information regarding the sodiation and desodiation processes of CuO NWs. And this information will potentially further our understanding of the microscopic electrochemical conversion mechanism and material degradation of CuO NWs applied in NIBs, and will also be of significance for designing other high-performance metal oxide anodes for NIBs.Acknowledgements
This work was financially supported by the Beijing Natural Science Foundation of China (no. 2144054), National Natural Science Foundation of China (no. 21322609, 21207144, 51401239, 11404138, 51401157), Key Program Project of National Natural Science Foundation of China (51231008), Science Foundation of China University of Petroleum, Beijing (no. 2462014QZDX01), the Key Project of Chinese Ministry of Education (313055), the National 973 program of China (2012CB619403), and Thousand Talents Program of China.Notes and references
- F. Klein, B. Jache, A. Bhide and P. Adelhelm, Phys. Chem. Chem. Phys., 2013, 15, 15876–15887 RSC.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- S. P. Ong, V. L. Chevrier, G. Hautier, A. Jain, C. Moore, S. Kim, X. Ma and G. Ceder, Energy Environ. Sci., 2011, 4, 3680–3688 CAS.
- D. Su and G. Wang, ACS Nano, 2013, 7, 11218–11226 CrossRef CAS PubMed.
- J. Park, J.-S. Kim, J.-W. Park, T.-H. Nam, K.-W. Kim, J.-H. Ahn, G. Wang and H.-J. Ahn, Electrochim. Acta, 2013, 92, 427–432 CrossRef CAS.
- D. Su, S. Dou and G. Wang, Chem. Commun., 2014, 50, 4192–4195 RSC.
- M. Mortazavi, C. Wang, J. Deng, V. B. Shenoy and N. V. Medhekar, J. Power Sources, 2014, 268, 279–286 CrossRef CAS.
- M. Gu, A. Kushima, Y. Shao, J.-G. Zhang, J. Liu, N. D. Browning, J. Li and C. Wang, Nano Lett., 2013, 13, 5203–5211 CrossRef CAS PubMed.
- P. Senguttuvan, G. Rousse, V. Seznec, J.-M. Tarascon and M. R. Palacín, Chem. Mater., 2011, 23, 4109–4111 CrossRef CAS.
- Y. Sun, L. Zhao, H. Pan, X. Lu, L. Gu, Y.-S. Hu, H. Li, M. Armand, Y. Ikuhara, L. Chen and X. Huang, Nat. Commun., 2013, 4, 1870 CrossRef PubMed.
- L. Xiao, Y. Cao, J. Xiao, W. Wang, L. Kovarik, Z. Nie and J. Liu, Chem. Commun., 2012, 48, 3321–3323 RSC.
- A. Li, H. Song, W. Wan, J. Zhou and X. Chen, Electrochim. Acta, 2014, 132, 42–48 CrossRef CAS.
- S. Ko, J.-I. Lee, H. S. Yang, S. Park and U. Jeong, Adv. Mater., 2012, 24, 4451–4456 CrossRef CAS PubMed.
- X. P. Gao, J. L. Bao, G. L. Pan, H. Y. Zhu, P. X. Huang, F. Wu and D. Y. Song, J. Phys. Chem. B, 2004, 108, 5547–5551 CrossRef CAS.
- Y. J. Mai, X. L. Wang, J. Y. Xiang, Y. Q. Qiao, D. Zhang, C. D. Gu and J. P. Tu, Electrochim. Acta, 2011, 56, 2306–2311 CrossRef CAS.
- Y. Liu, Y. Qiao, W. Zhang, P. Hu, C. Chen, Z. Li, L. Yuan, X. Hu and Y. Huang, J. Alloys Compd., 2014, 586, 208–215 CrossRef CAS.
- Q. Su, G. Du, J. Zhang, Y. Zhong, B. Xu, Y. Yang, S. Neupane and W. Li, ACS Nano, 2014, 8, 3620–3627 CrossRef CAS PubMed.
- A. Débart, L. Dupont, P. Poizot, J. B. Leriche and J. M. Tarascon, J. Electrochem. Soc., 2001, 148, A1266–A1274 CrossRef.
- X. Wang, D.-M. Tang, H. Li, W. Yi, T. Zhai, Y. Bando and D. Golberg, Chem. Commun., 2012, 48, 4812–4814 RSC.
- X. Lu, D. Zheng, T. Zhai, Z. Liu, Y. Huang, S. Xie and Y. Tong, Energy Environ. Sci., 2011, 4, 2915–2921 CAS.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
- F. Cheng, H. Wang, Z. Zhu, Y. Wang, T. Zhang, Z. Tao and J. Chen, Energy Environ. Sci., 2011, 4, 3668–3675 CAS.
- H. Ma, S. Zhang, W. Ji, Z. Tao and J. Chen, J. Am. Chem. Soc., 2008, 130, 5361–5367 CrossRef CAS PubMed.
- S. Yuan, X.-l. Huang, D.-l. Ma, H.-g. Wang, F.-z. Meng and X.-b. Zhang, Adv. Mater., 2014, 26, 2273–2279 CrossRef CAS PubMed.
- L. Wang, K. Zhang, Z. Hu, W. Duan, F. Cheng and J. Chen, Nano Res., 2014, 7, 199–208 CrossRef CAS.
- L. Q. Zhang, X. H. Liu, Y.-C. Perng, J. Cho, J. P. Chang, S. X. Mao, Z. Z. Ye and J. Y. Huang, Micron, 2012, 43, 1127–1133 CrossRef CAS PubMed.
- X. H. Liu, H. Zheng, L. Zhong, S. Huang, K. Karki, L. Q. Zhang, Y. Liu, A. Kushima, W. T. Liang, J. W. Wang, J.-H. Cho, E. Epstein, S. A. Dayeh, S. T. Picraux, T. Zhu, J. Li, J. P. Sullivan, J. Cumings, C. Wang, S. X. Mao, Z. Z. Ye, S. Zhang and J. Y. Huang, Nano Lett., 2011, 11, 3312–3318 CrossRef CAS PubMed.
- Y. Liu, F. Fan, J. Wang, Y. Liu, H. Chen, K. L. Jungjohann, Y. Xu, Y. Zhu, D. Bigio, T. Zhu and C. Wang, Nano Lett., 2014, 14, 3445–3452 CrossRef CAS PubMed.
- X. H. Liu, J. W. Wang, Y. Liu, H. Zheng, A. Kushima, S. Huang, T. Zhu, S. X. Mao, J. Li, S. Zhang, W. Lu, J. M. Tour and J. Y. Huang, Carbon, 2012, 50, 3836–3844 CrossRef CAS.
- J. W. Wang, X. H. Liu, S. X. Mao and J. Y. Huang, Nano Lett., 2012, 12, 5897–5902 CrossRef CAS PubMed.
- X. H. Liu, H. Zheng, L. Zhong, S. Huan, K. Karki, L. Q. Zhang, Y. Liu, A. Kushima, W. T. Liang, J. W. Wang, J. H. Cho, E. Epstein, S. A. Dayeh, S. T. Picraux, T. Zhu, J. Li, J. P. Sullivan, J. Cumings, C. S. Wang, S. X. Mao, Z. Z. Ye, S. L. Zhang and J. Y. Huang, Nano Lett., 2011, 11, 3312–3318 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The TEM image of CuO NW after cycling; a CV curve for CuO NW based NIB; plots of the reaction front migration distance vs. the time. See DOI: 10.1039/c5ra24086g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |