DOI:
10.1039/C5RA24624E
(Paper)
RSC Adv., 2016,
6, 15782-15787
Guanidine, amitrole and imidazole as nitrogen dopants for the synthesis of N-graphenes
Received
20th November 2015
, Accepted 31st January 2016
First published on 2nd February 2016
Abstract
Three N-containing organic compounds – guanidine, amitrole (3-amino-1,2,4-triazole) and imidazole were selected and evaluated as new nitrogen dopants for the preparation of N-graphene. A graphene oxide aqueous dispersion was subjected to hydrothermal treatment at 180 °C for 8 h in the presence of the selected N-compounds. The nitrogen contents in the resultant N-graphenes were as high as 13.4 at%, which is among the highest reported for graphene materials. The X-ray photoelectron spectroscopy revealed that pyridinic nitrogen was predominant in all of the N-graphenes, accounting for up to 47% of the total nitrogen content. Deoxygenation of graphene oxide under hydrothermal conditions was also improved through the use of the N-compounds. Possible reaction mechanisms between graphene oxide and each N-compound are proposed.
Introduction
During the last decade, graphene has garnered wide attention because of its unique properties, which include excellent electrical conductivity, high flexibility, good electrochemical stability and high surface area. In the last few years, the chemical modulation of graphene by doping with heteroatoms has been extensively investigated as a method to improve its performance in sensing1,2 and energy storage applications.2 Doping with nitrogen is a particularly well-adopted strategy because of the contribution of nitrogen p-electrons to the π-system, conferring enhanced conductivity and electrochemical performance to the graphene materials. The preparation of N-doped graphenes by chemical vapor deposition (CVD), nitrogen plasma, nitrogen-assisted arc discharge and high-temperature ammonization has been described in the literature.1–4 However, these techniques are not suitable for scale-up processes because of their complexity and high cost. In this regard, the hydrothermal treatment of graphene oxide in the presence of N-containing compounds such as ammonia,5 glucosamine,6 pyrrole,7 hydrazine,8 urea9 or aminoacids10 has recently been proposed as a simple and scalable procedure for the preparation of N-graphenes. This work is a preliminary report describing the preparation of N-graphenes by hydrothermal treatment of a GO aqueous dispersion in the presence of three organic N-containing compounds: guanidine (Gn), amitrole (Am) and imidazole (Im). To the best of our knowledge, this work represents the first time that N-enriched graphene materials have been synthesized using these compounds. Possible reaction mechanisms for the incorporation of nitrogen in the GO nanosheets for each N-dopant have been proposed.
Experimental
Synthesis of materials
A coal-tar pitch-based graphite supplied by INCAR-CSIC (Oviedo, Spain) was oxidized by a modified Hummer's method. Concentrated H2SO4 (48 ml) and NaNO3 (1 g) were added slowly with vigorous stirring to a 250 ml flask containing graphite powder (1 g). Then, KMnO4 (6 g) was added to the reaction mixture while the flask was held in an ice bath, followed by stirring for 3 h at 35 °C. Afterwards, 200 ml of H2O2 (3%) was added to the flask. Finally, the mixture was decanted, washed with Milli-Q water, and centrifuged several times until a pH-neutral supernatant was obtained. The resulting graphite oxide was treated with 5% HCl and then washed several times with Milli-Q water followed by centrifugation until the pH of the solution was neutral.11 After drying, 200 mg of graphite oxide was dispersed in 200 ml of water and ultrasonicated at 35 °C for 6 h, yielding a graphene oxide (GO) aqueous dispersion with a concentration of 1 mg ml−1. 2 g of each nitrogen organic compound (guanidine carbonate, amitrole or imidazole) was dissolved in 200 ml of GO suspension to reach the N-compound/GO mass ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
The chemical structures of the N-containing compounds are shown in Table 1. Hydrothermal treatments were performed in a steel-based home-made autoclave at 180 °C for 8 h. For comparison purposes, hydrothermal treatment was also applied to the GO dispersion without the addition of any reactant. After the reaction, the autoclave was allowed to cool to room temperature; the obtained black precipitates were centrifuged and washed in deionized water five times. Finally, the samples were washed with isopropanol and dried in an oven at 65 °C for 24 h. The N-doped reduced graphene oxides (rGOs) prepared using Gn, Am and Im were labeled as rGO-Gn, rGO-Am and rGO-Im, respectively. The reduced graphene oxide produced without the addition of any reactant was labeled as rGO.
Table 1 N-containing compounds used in the synthesis of N-graphenes
| Compound |
Molecule |
Symbol |
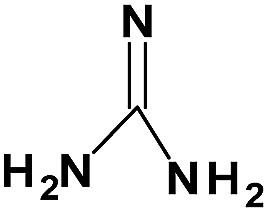
| Guanidine |
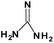 |
Gn |
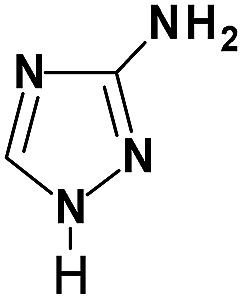
| Amitrole |
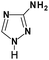 |
Am |
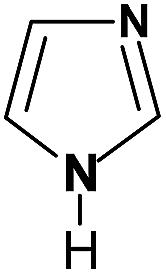
| Imidazole |
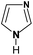 |
Im |
Characterization of materials
The microstructure and morphology of the materials were analyzed by field-emission scanning electron microscopy (FE-SEM, Merlin Zeiss) with an accelerating voltage of 3 kV, transmission electron microscopy (HRTEM, FEI Tecnai G2 20 X-TWIN) with an acceleration voltage of 200 kV and atomic force microscopy (AFM, Bruker, operating in tapping mode) measurements. The Fourier Transform Infrared (FTIR) spectra of the materials in the region of 4000–700 cm−1 were measured in a Bruker Vertex 70 spectrometer. Samples were analyzed in attenuated total reflectance mode (ATR) using the Pike MIRacle accessory equipped with a Ge crystal (Pike Technology). Prior to the measurements, the sample and detector compartment were evacuated in order to avoid the interference of carbon dioxide and water vapor. The final spectra were calculated as the mean of 256 measurements with a resolution of 2 cm−1. The X-ray photoelectron spectroscopy (XPS) analyses were carried out with a PHI 5000 VersaProbe (ULVAC-PHI) spectrometer. The sample charging was corrected using the C1s peak at 284.6 eV as an internal standard. The quantitative analysis of C1s, N1s and O1s spectra were performed using the CasaXPS software after Shirley background subtraction. The best peak fits were obtained using a Gaussian–Lorentzian (70/30) peak shape at the same FWHM for all fitted peaks. The surface atomic ratios were calculated from the ratio of the corresponding peak areas after correction with the theoretical sensitivity factors based on the Scofield's photoionization cross-sections. The C1s core level spectra were deconvoluted into four components corresponding to graphitic sp2 hybridized C (284.5 ± 0.1 eV), hydroxyl, epoxy groups and C–N linkages (286.5 ± 0.3 eV), quinones (287.6 ± 0.2 eV) and carboxylic groups (289 ± 0.4 eV).12–14 The N1s core level spectra of the N-doped samples were deconvoluted into five components corresponding to pyridinic-N (N6, 398.7 ± 0.3 eV), pyrrolic-N (N5, 400.3 ± 0.3 eV), quaternary-N (NQ, 401.4 ± 0.5 eV), amides/amines or lactams (NC, 399.7 ± 0.2 eV) and pyridine N-oxides (NX, 402–405 eV).15 The following oxygen moieties were identified by deconvolution of the O1s core level spectra: quinone groups (530.6 ± 0.1 eV), carbonyl and carboxyl (531.7 ± 0.1 eV), epoxy (532.8 ± 0.1 eV), and hydroxyl groups (533.7 ± 0.1 eV).6 The crystalline characteristics of the materials were studied by X-ray diffraction analyzer (XRD, Ultima IV Rigaku) equipped with a 2 kW X-ray tube (40 kV/30 mA) using Cu Kα2 radiation (λ = 1.54056 Å).
Results and discussion
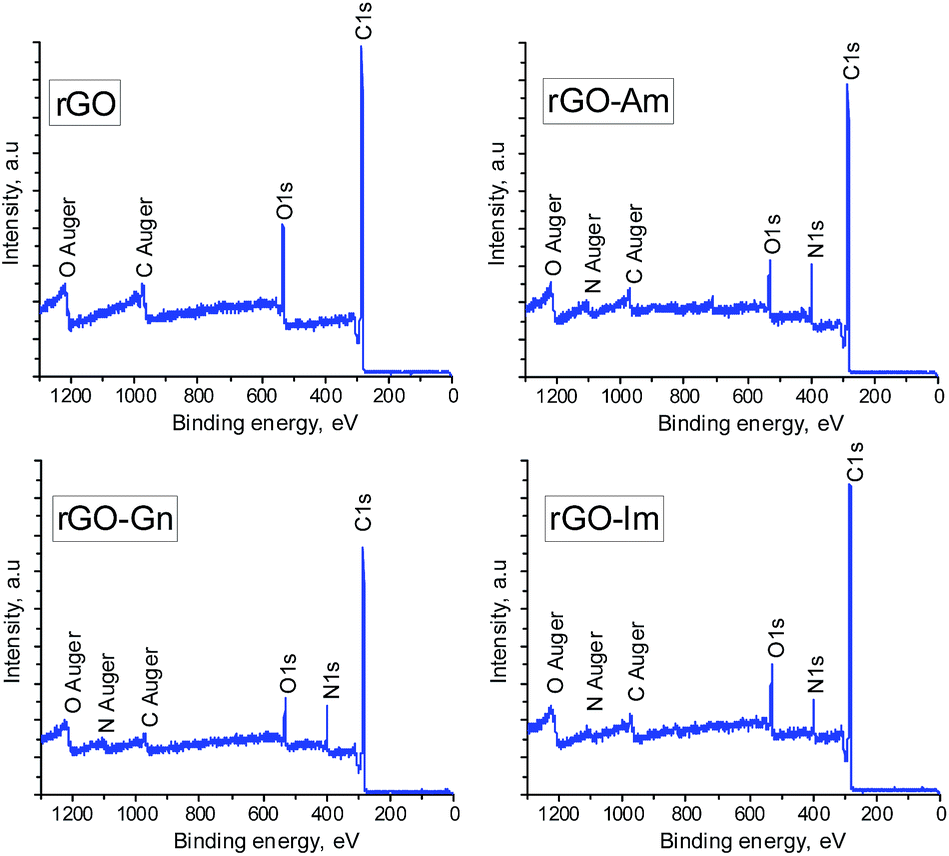
Fig. 1 presents the XPS survey spectra of the non-doped hydrothermally reduced graphene oxide (rGO) and the N-doped samples (rGO-Am, rGO-Gn and rGO-Im). The spectrum of rGO contains only the carbon and oxygen peaks. On the contrary, the presence of sharp N1s peaks in the XPS spectra of the N-doped samples clearly demonstrates the nitrogen doping by hydrothermal treatment of GO with N-dopants. The elemental compositions of GO and hydrothermally treated samples determined by XPS are presented in Table 2. GO displayed a typical high oxygen content of 37.2 at%,16 which decreased to 16.2 at% after hydrothermal treatment as a result of the acid-catalyzed deoxygenation promoted by water molecules.16 Importantly, the oxygen content of the samples prepared in the presence of the N-compounds was remarkably lower, in the range of 9.4 to 10.4 at%. In parallel to this enhanced removal of oxygen, the presence of N-containing compounds also promoted a substantial incorporation of nitrogen into the rGO nanosheets. Specifically, sample rGO-Am exhibited very high nitrogen content of 13.4 at%. To the best of our knowledge, this N content is among the highest reported for graphene-based materials.3 In most cases, the nitrogen content in N-enriched graphene materials reported by other authors does not exceed 10 at%, usually being in the range of 3–7 at% regardless of the methodology used for the nitrogen doping (Table 3).
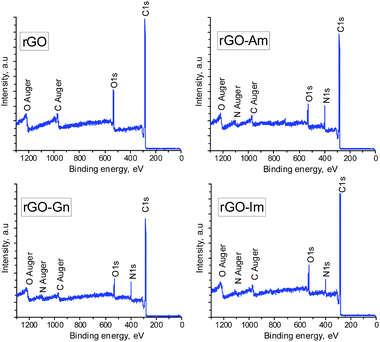 |
| | Fig. 1 XPS survey spectra of rGO and N-rGOs. | |
Table 2 Elemental composition (XPS) of GO and hydrothermally treated samples, at%
| Sample |
C |
N |
O |
| GO |
62.8 |
— |
37.2 |
| rGO |
83.8 |
— |
16.2 |
| rGO-Gn |
78.7 |
11.9 |
9.4 |
| rGO-Am |
77.2 |
13.4 |
9.4 |
| rGO-Im |
81.7 |
7.9 |
10.4 |
Table 3 Nitrogen contents of N-doped graphene materials prepared by different methods
| Dopant |
Synthesis method |
Synthesis conditions |
Nitrogen content [at%] |
Reference |
| Amitrole |
Hydrothermal |
GO/amitrole (1:10), 180 °C for 8 h |
13.4 |
This work |
| Guanidine |
Hydrothermal |
GO/guanidine (1:10), 180 °C for 8 h |
11.9 |
This work |
| Imidazole |
Hydrothermal |
GO/imidazole (1:10), 180 °C for 8 h |
7.9 |
This work |
| Urea |
Hydrothermal |
GO/urea (1:30), 160 °C for 3 h |
3.9–6.6 |
17 |
| NH4OH |
Hydrothermal |
GO/NH4OH (10:1), 180 °C for 12 h |
5.7–7.2 |
18 |
| (NH4)2CO3 |
Hydrothermal |
GO/(NH4)2CO3, (1:50)–(1:300), 110–140 °C for 10 h |
6.8–10.1 |
19 |
| Ethylenediamine |
Hydrothermal |
GO/ethylenediamine (1500:1), 180 °C for 12 h |
9.6 |
20 |
| Ammonium oxalate |
Hydrothermal |
GO/ammonium oxalate (1:8), 180 °C for 8 h |
6.8 |
21 |
| Hexamethylenetetramine |
Hydrothermal |
GO/hexamethylenetetramine (1:37), 180 °C for 12 h |
6.4–8.6 |
22 |
| Aminoacids |
Hydrothermal |
GO/aminoacids (1:2.5), 160 °C for 3 h |
1.0–8.9 |
10 |
| Urea |
Hydrothermal |
GO/urea (1:20.8), 180 °C for 3 h |
5.8 |
23 |
| NH3 |
Thermal annealing |
GRO, NH3, 800 °C for 5 min |
6.7 |
4 |
| NH3-Ar |
Thermal annealing |
GO, NH3-Ar, 300–1000 °C |
3–5 |
5 |
| NH3 |
Thermal annealing |
GO, NH3, 500–800 °C |
7.7–12.5 |
24 |
| Triazine |
CVD |
1,3,5-Triazine, H2-Ar, 500 °C |
2.1–5.6 |
25 |
| Monoethalonamine (ME) |
CVD |
ME, N2, 1000–1100 °C |
1.5–2.9 |
26 |
| NH3 |
Ball milling |
Graphite, NH3 |
4.5 |
27 |
| Melamine, urea |
Ball milling |
GO, melamine/urea |
7.5–9.3 |
28 |
| NH4OH |
Microwave heating |
GO, NH4OH, ethylene glycol, 160–180 °C, 10–30 min |
6–8.7 |
29 |
| NH3 |
Microwave heating |
GRO, NH3 |
4.0–5.5 |
30 |
| Urea |
Microwave heating |
GO, urea |
13–15 |
31 |
| o-Phenylenediamine |
Solvothermal |
GO, o-phenylenediamine, 180 °C for 12 h |
7.7 |
32 |
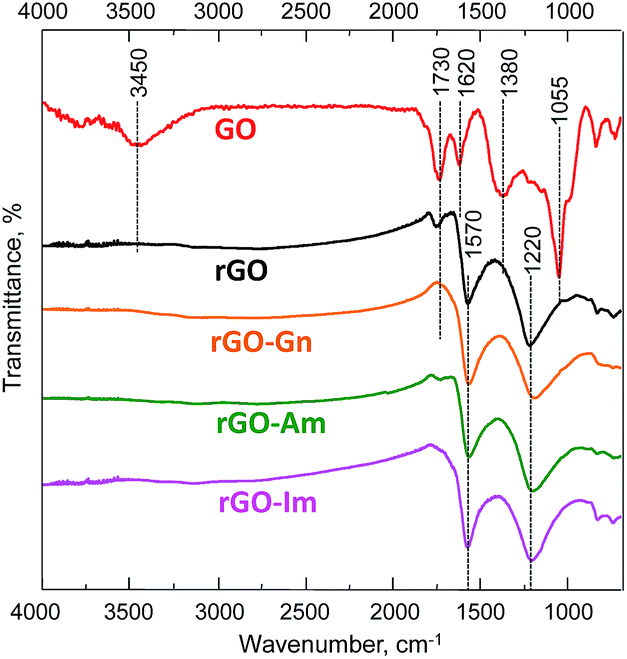
The nature of the oxygen and nitrogen functionalities of N-rGOs was investigated by FTIR. The FTIR spectrum of the initial GO (Fig. 2) confirmed its high oxidation degree. The characteristic peaks of hydroxyl (O–H) and carboxyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups appeared at 3450 and 1730 cm−1, respectively.33 A zone of overlapped bands in the range of 900–1450 cm−1 revealed the presence of C–O linkages in hydroxyl (1055 and 1380 cm−1) and epoxy (1220 cm−1) groups. The band at 1620 cm−1 was ascribed to the CC stretching vibration of unoxidized graphitic domains and the bending vibration in molecules of intercalated water. The intensity of all of the bands corresponding to oxygen functionalities decreased upon hydrothermal treatment. In particular, the band at 3400 cm−1 disappeared, indicating the successful removal of hydroxyl groups. A low-intensity band at 1720 cm−1 was still observed in the spectrum of rGO but not in those of the N-enriched samples, consistent with the lower reduction degree observed in this sample. Two main peaks were detected in the spectra of all of the rGOs. The peak at 1570 cm−1 was ascribed to the CC stretching and N–H bending vibrations, although the weak contribution of ketone groups could not be disregarded. The broad band at 1220 cm−1 was ascribed to the presence of residual quinone and epoxy groups. In previous studies the vibrational modes of C–N in N-graphene sheets were observed at 1030 and 1257 cm−1.5 In the current work, the peaks are not visible since they are being overlapped by the broad bands corresponding to oxygen linkages.
O) groups appeared at 3450 and 1730 cm−1, respectively.33 A zone of overlapped bands in the range of 900–1450 cm−1 revealed the presence of C–O linkages in hydroxyl (1055 and 1380 cm−1) and epoxy (1220 cm−1) groups. The band at 1620 cm−1 was ascribed to the CC stretching vibration of unoxidized graphitic domains and the bending vibration in molecules of intercalated water. The intensity of all of the bands corresponding to oxygen functionalities decreased upon hydrothermal treatment. In particular, the band at 3400 cm−1 disappeared, indicating the successful removal of hydroxyl groups. A low-intensity band at 1720 cm−1 was still observed in the spectrum of rGO but not in those of the N-enriched samples, consistent with the lower reduction degree observed in this sample. Two main peaks were detected in the spectra of all of the rGOs. The peak at 1570 cm−1 was ascribed to the CC stretching and N–H bending vibrations, although the weak contribution of ketone groups could not be disregarded. The broad band at 1220 cm−1 was ascribed to the presence of residual quinone and epoxy groups. In previous studies the vibrational modes of C–N in N-graphene sheets were observed at 1030 and 1257 cm−1.5 In the current work, the peaks are not visible since they are being overlapped by the broad bands corresponding to oxygen linkages.
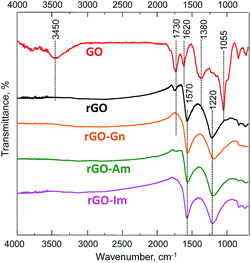 |
| | Fig. 2 FTIR-ATR spectra of GO, rGO and N-rGOs. | |
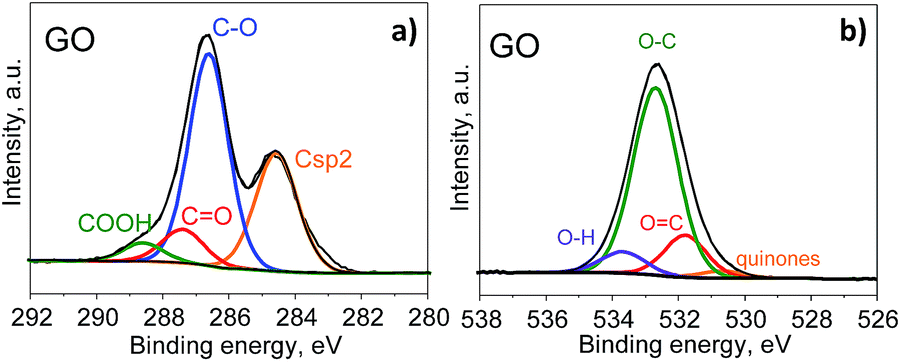
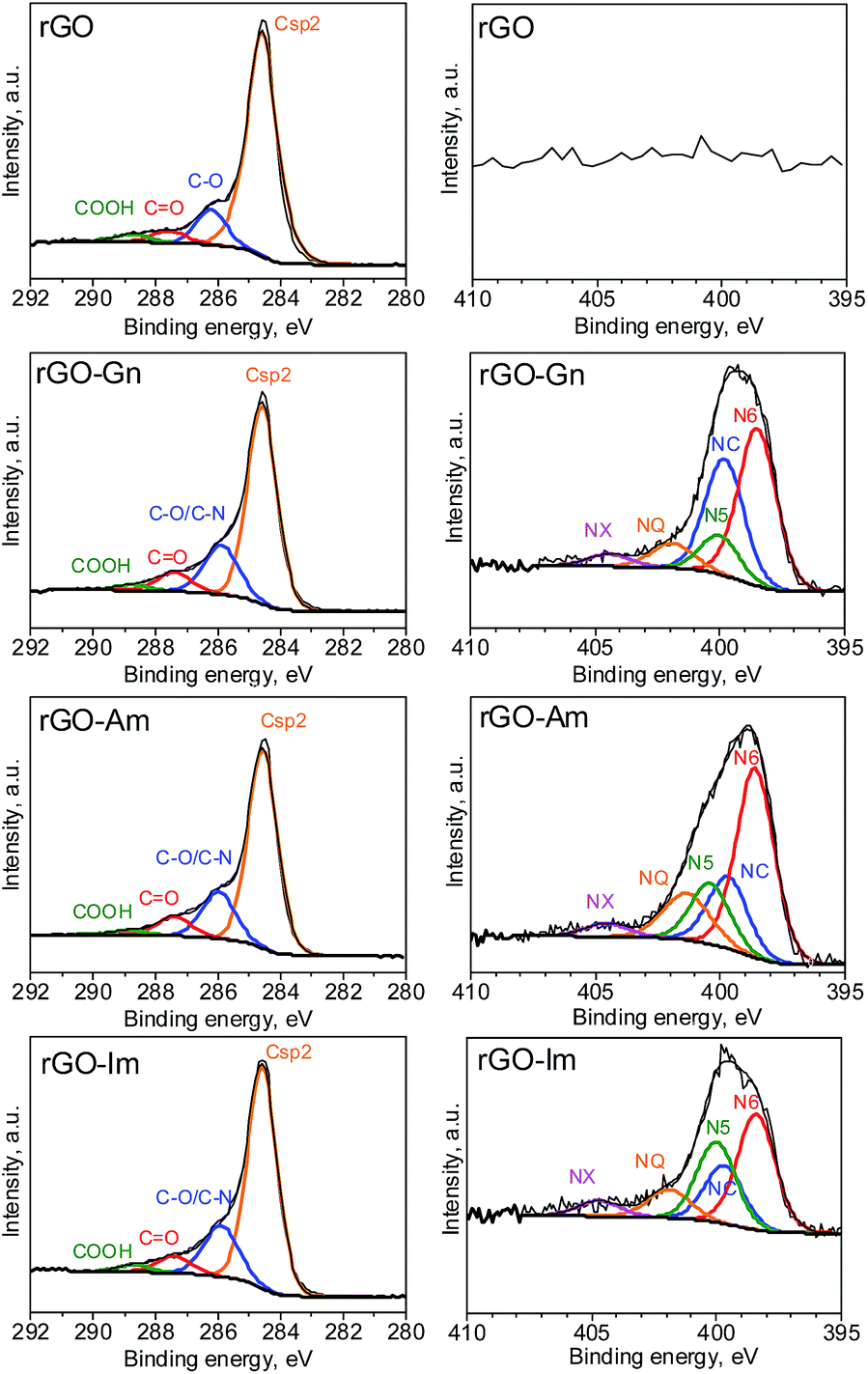
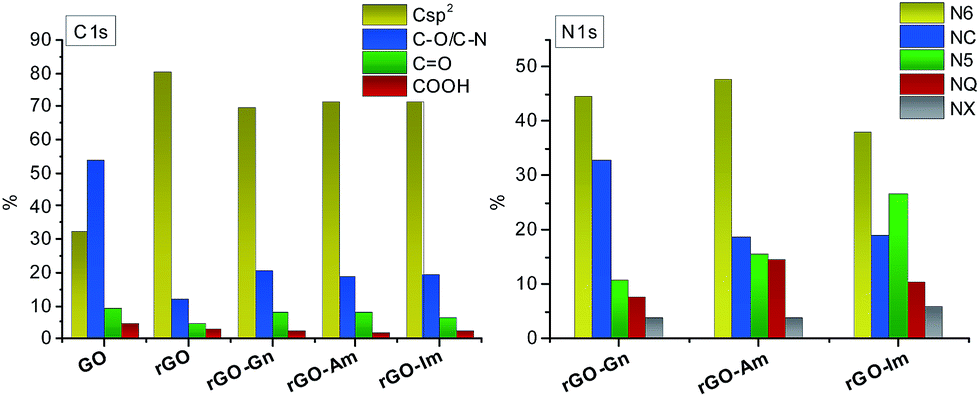
Further information about the oxygen and nitrogen functionalities was provided by deconvolution of the C1s, O1s and N1s core-level XPS spectra (Fig. 3 and 4). Fig. 5 presents the distribution of carbon and nitrogen linkages in the different graphene materials. GO exhibited a high contribution (44%) of hydroxyl groups and/or epoxy groups (Fig. 3a). Fitting of the O1s peak (Fig. 3b) revealed that the epoxy groups were predominant, whereas the contribution of hydroxyl groups was rather low (7%). On the basis of the C1s deconvolution for rGO and N-rGOs (Fig. 4), the successful removal of hydroxyl and epoxy groups during hydrothermal treatment was evident. The appearance of a well-defined and intense peak ascribed to carbon with sp2 hybridization suggests that the removal of oxygen moieties occurs simultaneously with the restoration of the C sp2 conjugation.
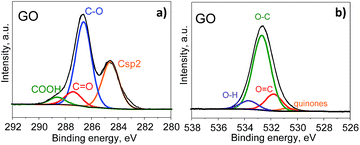 |
| | Fig. 3 C1s XPS (a) and O1s XPS (b) spectra of GO. | |
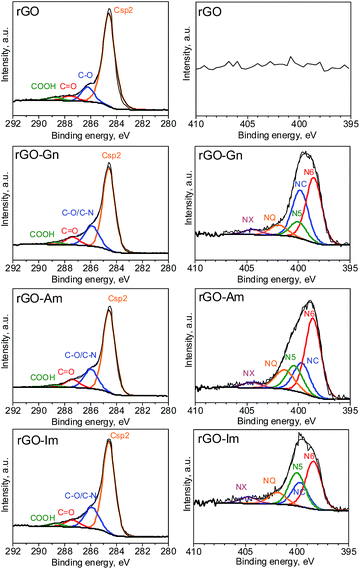 |
| | Fig. 4 C1s and N1s XPS spectra of rGO and N-rGOs. | |
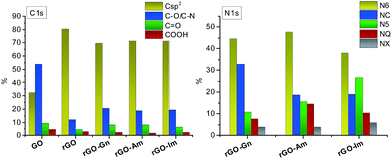 |
| | Fig. 5 Distribution of carbon and nitrogen linkages in GO and reduced samples. | |
Among the different nitrogen groups detected in the N-doped samples, pyridinic-N (N6) was predominant in all of the N-graphenes (Fig. 4). In particular, rGO-Am exhibited the highest content of pyridinic groups, with a contribution of 47% to the total nitrogen content (Fig. 5). rGO-Im was characterized by the highest content of pyrrolic-N (N5), with a contribution of 27%. rGO-Am and rGO-Im contained 14.4 and 10.5% quaternary-N (NQ), respectively, whereas rGO-Gn contained a higher proportion of amides/amines and lactams (NC), representing 33% of its total N content. A higher contribution of the peak at 286.5 eV (C–O/C–N groups) in the C1s core-level XPS spectra for N-rGOs compared with rGO confirms again a remarkable incorporation of nitrogen into the GO structure (Fig. 5).
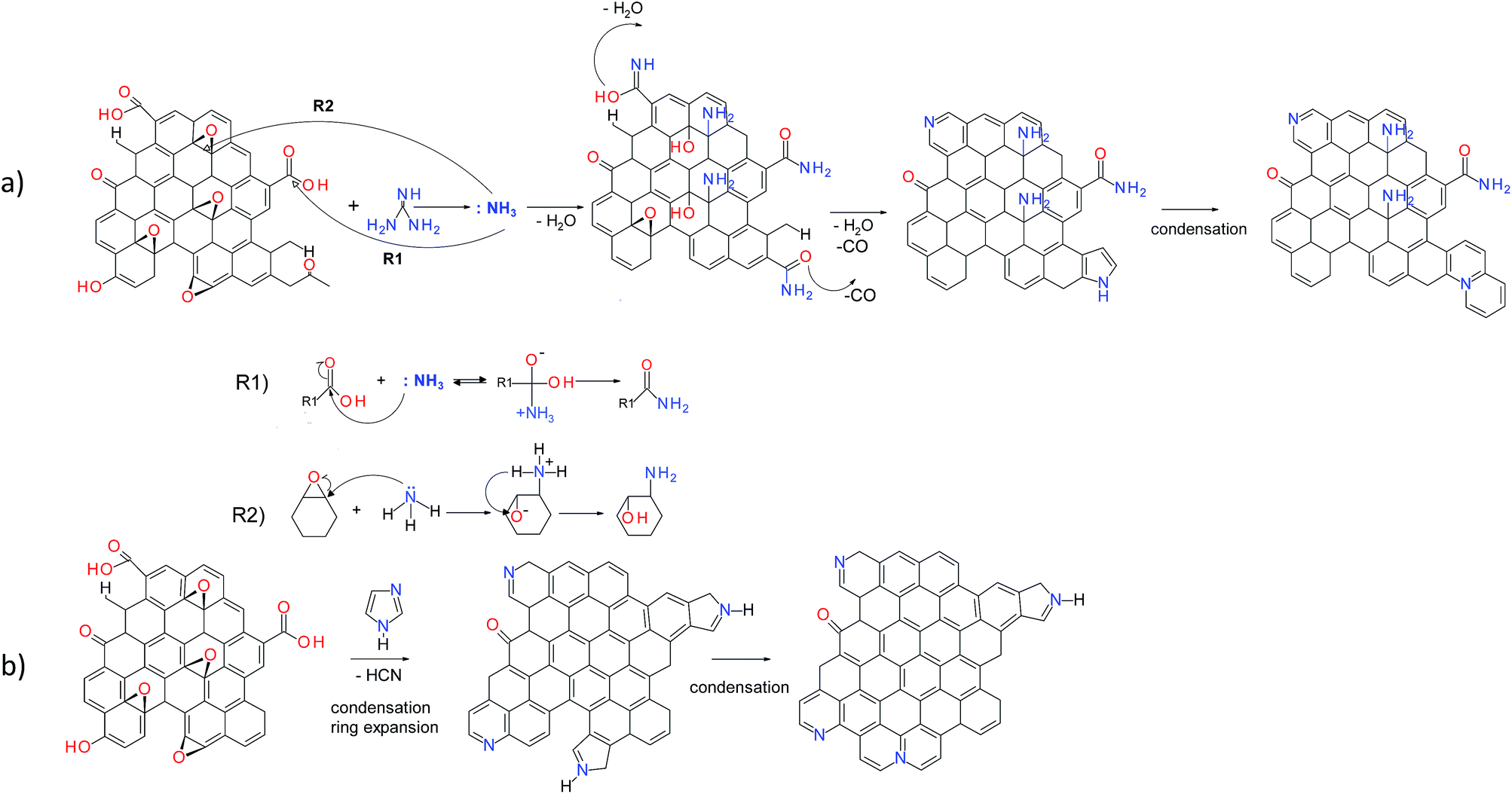
The possible reaction mechanisms for the incorporation of nitrogen in the GO nanosheets are proposed for different N-dopants based on the XPS and FTIR results (Fig. 6). The hydrothermal treatment of guanidine leads to the formation of ammonia.34 Further reactions involve the formation of amides via nucleophilic substitution of carboxyl groups by ammonia acting as Lewis acid (Fig. 6a, R1). In parallel the ammonia acts as the ring open-agent for the epoxides of GO, leading to the formation of amines (Fig. 6a, R2).35,36 During the hydrothermal treatment, parts of these functionalities can undergo dehydration or decarbonylation, giving rise to pyridine, pyridone and lactam groups or pyrroles, respectively.9
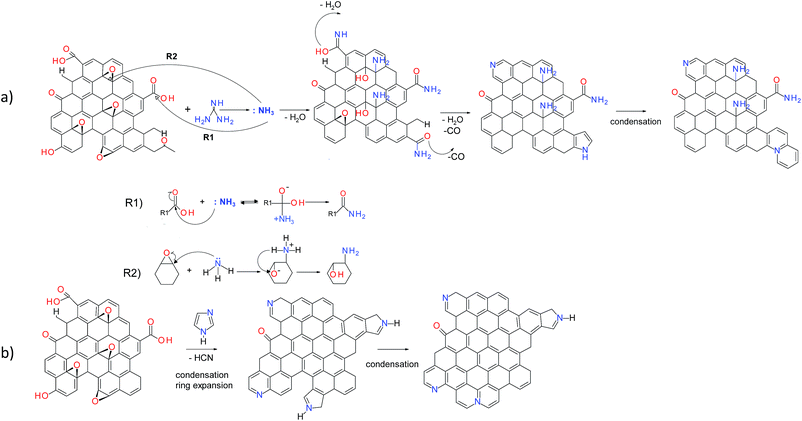 |
| | Fig. 6 Possible reaction pathways of GO with Gn (a) and Im (b). | |
Possible mechanisms of the reaction of GO with imidazole and amitrole involve the condensation of five-membered nitrogen-containing rings within the graphene network (Fig. 6b). Since the amitrole and imidazole can be considered as the analogues of the five-membered pyrrole, their further transformation likely follows the reactions occurring during thermal treatment of this compound. A part of the newly incorporated pyrrolic structures can undergo further ring expansion into pyridinic-N.37,38 Both pyrrolic- and pyridinic-N can subsequently condense into quaternary nitrogen. Because rGO-Am contains the largest amount of quaternary-N (Fig. 5), the presence of four nitrogen atoms in the amitrole molecule appears to favor the formation of condensed heteroaromatic structures.
The mechanism of the nitrogen doping affects the morphology of the resultant N-graphene materials as revealed by FE-SEM (Fig. 7a–c). rGO-Am and rGO-Im are predominantly composed of disordered and wrinkled graphene nanosheets (Fig. 7a and b). In contrast, rGO-Gn displays a more uniform structure with smoother surface (Fig. 7c). The differences in the morphologies of the samples can be explained based on the doping mechanism: guanidine decomposes to the ammonia which in turn reacts with the graphene oxide. Since the size of the ammonia molecule is relatively small, the flat and dense structure of the graphene layers is preserved to a higher extent. On the contrary, the five-membered heterocyclic rings of amitrole and imidazole molecules are substantially larger. Since the interaction of GO with both N-compounds involves the condensation of the dopant molecule with the graphene, this process leads to the formation of more disordered and wrinkled structures. The TEM observations revealed the presence of the lateral graphene nanosheets as the predominant building units of all the studied N-rGOs (Fig. 7d and e).
 |
| | Fig. 7 FE-SEM images of (a) rGO-Am, (b) rGO-Im, (c) rGO-Gn; TEM images of rGO-Am after being dispersed in DMF:H2O (1:1) (d, e). | |
Conclusions
We have shown for the first time the use of guanidine, amitrole and imidazole for the synthesis of N-graphenes. N-Doped graphene materials with very high nitrogen contents, in the range of 7.9–13.4 at%, were successfully prepared by the hydrothermal treatment of a GO dispersion in the presence of these N-containing compounds. Among the nitrogen functional groups formed, pyridinic nitrogen was most abundant in all the synthesized N-rGOs. The use of the N-dopants also promoted further oxygen removal during hydrothermal treatment, decreasing from 16.2 at% for rGO to 9.4 at% for N-graphenes. Further work focused on the advanced understanding of the chemical transformations during hydrothermal treatment of GO with selected N-dopants and optimization of the reaction conditions is currently in progress. Because of their very high nitrogen contents, where the N is, to a great extent, present in the form of heterocyclic groups as pyridinic- and pyrrolic-N, these N-graphenes could be promising for use in supercapacitors or electrochemical sensors.
Acknowledgements
The research leading to these results has received funding from the European Union's Research Fund for Coal and Steel (RFCS) research programme under grant agreement RFCR-CT-2013-00006.
References
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS.
- C. N. R. Rao, K. Gopalakrishnan and A. Govindaraj, Nano Today, 2014, 9, 324 CrossRef CAS.
- B. Xiong, Y. Zhou, R. O'Hayre and Z. Shao, Appl. Surf. Sci., 2013, 266, 433 CrossRef CAS.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939 CrossRef CAS PubMed.
- X. Fan, C. Yu, J. Yang, Z. Ling and J. Qiu, Carbon, 2014, 70, 130 CrossRef CAS.
- Y. Zhao, C. Hu, Y. Hu, H. Cheng, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2012, 51, 11371 CrossRef CAS PubMed.
- R. Wang, Y. Wang, C. Xu, J. Sun and L. Gao, RSC Adv., 2013, 3, 1194 RSC.
- L. Sun, L. Wang, C. Tian, Y. Xie, K. Shi, M. Li and H. Fu, RSC Adv., 2012, 2, 4498 RSC.
- T. Wang, L. Wang, D. Wu, W. Xia, H. Zhao and D. Jia, J. Mater. Chem. A, 2014, 2, 8352 CAS.
- N. Diez, A. Śliwak, S. Gryglewicz, B. Grzyb and G. Gryglewicz, RSC Adv., 2015, 5, 81831 RSC.
- S. Biniak, G. Szymański, J. Siedlewski and A. Świątkowski, Carbon, 1997, 35, 1799 CrossRef CAS.
- E. Lorenc-Grabowska, G. Gryglewicz and M. A. Diez, Fuel, 2013, 114, 235 CrossRef CAS.
- Q. Zhuo, Y. Zhang, Q. Du and C. Yan, J. Colloid Interface Sci., 2015, 457, 243 CrossRef CAS PubMed.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641 CrossRef CAS.
- S. A. Hasan, E. K. Tsekura, V. Sternhagen and M. Strømme, J. Chem. Phys. C, 2012, 116, 6530 CrossRef CAS.
- H. L. Guo, P. Su, X. Kang and S. K. Ning, J. Mater. Chem. A, 2013, 1, 2248 CAS.
- B. Jiang, C. Tian, L. Wang, L. Sun, C. Chen, X. Nong, Y. Qiao and H. Fu, Appl. Surf. Sci., 2012, 258, 3438 CrossRef CAS.
- H. Zhang, T. Kuila, N. H. Kim, D. S. Yu and J. H. Lee, Carbon, 2014, 69, 66 CrossRef CAS.
- P. Chen, J. J. Yang, S. S. Li, Z. Wang, T. Y. Xiao, Y. H. Qian and S. H. Yu, Nano Energy, 2013, 2, 249 CrossRef CAS.
- D. Wang, Y. Min, Y. Yu and B. Peng, J. Colloid Interface Sci., 2014, 417, 270 CrossRef CAS PubMed.
- J. W. Lee, J. M. Ko and J. D. Kim, Electrochim. Acta, 2012, 85, 459 CrossRef CAS.
- B. Zheng, J. Wang, F. B. Wang and X. H. Xia, Electrochem. Commun., 2013, 28, 24 CrossRef CAS.
- S. Sandoval, N. Kumar, J. O. Solé, A. Sundaresan, C. N. R. Rao, A. Fuertes and G. Tobias, Carbon, 2016, 96, 594 CrossRef CAS.
- Y. F. Lu, S. T. Lo, J. C. Lin, W. Zhang, J. Y. Lu, F. H. Liu, C. M. Tseng, Y. H. Lee, C. T. Liang and L. J. Li, ACS Nano, 2013, 7, 6522 CrossRef CAS PubMed.
- J. F. Bao, N. Kishi and T. Soga, Mater. Lett., 2014, 117, 199 CrossRef CAS.
- I. Y. Jeon, Y. R. Shin, G. J. Sohn, H. J. Choi, S. Y. Bae, J. Mahmood, S. M. Jung, J. M. Seo, M. J. Kim, D. W. Chang, L. Dai and J. B. Baek, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5588 CrossRef CAS PubMed.
- R. P. Rocha, A. G. Gonçalves, L. M. Pastrana-Martínez, B. C. Bordoni, O. S. G. P. Soares, J. J. M. Órfão, J. L. Faria, J. L. Figueiredo, A. M. T. Silva and M. F. R. Pereira, Catal. Today, 2015, 249, 192 CrossRef CAS.
- F. N. I. Sari and J. M. Ting, Appl. Surf. Sci., 2015, 355, 419 CrossRef CAS.
- Z. Wang, B. Li, Y. Xin, J. Liu, Y. Yao and Z. Zou, Chin. J. Catal., 2014, 35, 509 CrossRef CAS.
- K. Gopalakrishnan, A. Govindaraj and C. N. R. Rao, J. Mater. Chem. A, 2013, 1, 7563 CAS.
- Y. Lu, F. Zhang, T. Zhang, K. Leng, L. Zhang, X. Yang, Y. Ma, Y. Huang, M. Zhang and Y. Chen, Carbon, 2013, 63, 508 CrossRef CAS.
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, J. Chem. Phys. C, 2011, 115, 19761 CrossRef CAS.
- J. W. Schoppelrei, M. L. Kieke, X. Wang, M. T. Klein and T. B. Brill, J. Phys. Chem., 1996, 100, 14343 CrossRef CAS.
- M. Seredych and T. Bandosz, J. Phys. Chem. C, 2007, 111, 15596 CAS.
- L. Lai, L. Chen, D. Zhan, L. Sun, J. Liu, S. H. Lim, Ch. K. Poh, Z. Shen and J. Lin, Carbon, 2011, 49, 3250 CrossRef CAS.
- R. Arrigo, M. Hävecker, R. Schlögl and D. S. Su, Chem. Commun., 2008, 40, 4891 RSC.
- M. A. Wójtowicz, J. R. Pels and J. A. Moulijn, Fuel, 1995, 74, 507 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.