
Advances in nanomaterial based approaches for enhanced fluoride and nitrate removal from contaminated water
S. P. Suriyaraj and
R. Selvakumar*
Nanobiotechnology Laboratory, PSG Institute of Advanced Studies, P.B. No: 1609, Peelamedu, Coimbatore 641004, India. E-mail: selvabiotech@gmail.com; rsk@psgias.ac.in; Tel: +91 422 4344000 extn 4327
First published on 14th January 2016
Abstract
Fluoride and nitrate contamination in drinking water sources has been a major problem in many countries. The long term health hazards and increasing levels of fluoride and nitrate in drinking water through natural and anthropogenic sources are challenging and warrant the need for advanced technologies for abating these contaminants. Various technologies have been reported so far for fluoride and nitrate removal from drinking water. Recent advances in nanotechnology and nanomaterial synthesis have demonstrated significant impacts on fluoride and nitrate removal when compared to other conventional adsorbents and process. The present review discusses the recent advances in nanotechnology and nanomaterial synthesis, modification of process parameters and use of composite nanomaterials for enhanced removal of fluoride and nitrate from contaminated water.
1. Introduction
Water pollution is a major problem in developing countries; especially in regions where people depend on groundwater for drinking.1 Nearly forty percent of the world’s population is already facing a water shortage; the cost of water infrastructure has also increased dramatically. The quality of water in rivers and underground has deteriorated, due to pollution by waste and contaminants from cities, industry and agriculture.2 Natural water bodies have impurities from various sources. The most common contaminants are microorganisms, suspended particles, colloidal materials, pesticides and various dissolved metallic and non-metallic substances.3,4 Among these compounds, anionic pollutants like fluoride and nitrate contamination in groundwater has been recognized as one of the most serious problems worldwide.5–7 Various diseases and harmful effects caused by drinking fluoride and nitrate contaminated water have been a great worry for human society.An excessive fluoride intake leads to the loss of calcium from the tooth matrix, causing cavity formation, and ultimately leads to dental fluorosis followed by skeletal fluorosis after prolonged exposure.8,9 Fluoride has been implicated in a number of adverse outcomes related to fertility and pregnancy.10 Diseases like Alzheimer’s, bladder cancer, thyroid disorder, gastrointestinal bleeding and otosclerosis, can affect the human body following an excessive intake of fluoride.11–13 Similarly, excessive nitrate intake leads to gastric cancer, goitre, congenital birth defects, hypertension and methemoglobinemia.14,15 A prolonged intake of a high level of nitrate is linked to gastric problems due to the formation of nitrosamines. N-Nitrosamine compounds have been shown to cause cancer in test animals. Studies on people exposed to high levels of nitrate or nitrite have not provided convincing evidence of an increased risk of cancer.14–17 Other health effects following fetal exposure to elevated levels of nitrates in drinking water have included intrauterine growth retardation, an increased incidence of Sudden Infant Death Syndrome (SIDS), cardiac defects, and an increased risk of nervous system defects.18–20 Few studies have reported other health effects that are possibly associated with nitrate exposure in children, including an increased incidence of childhood diabetes, recurrent diarrhea and respiratory tract infections.14,19
Due to these clinical manifestations caused by drinking fluoride and nitrate contaminated water, the World Health Organization (WHO) has recommended 1.5 mg L−1 and 45 mg L−1 as the maximum contaminant levels (MCLs) in drinking water for fluoride and nitrate respectively.21 Hence, it is an important issue to supply water with safe F− and NO3− levels. Various methods like chemical precipitation, electrocoagulation, reverse osmosis and electrodialysis have been reported to remove fluoride and nitrate from drinking water.22–26 Despite the unique advantages of these technologies, the costs, poor regeneration, interference of other ions, customary replacement of sacrificial electrodes, consumption of electric power, membrane fouling, requirement of experienced operators and poor water recovery always remain a problem and create limited social acceptance of these technologies.23–25 Scientific evidence recommends adsorption and photocatalysis as the most efficient and cost effective methods to remove fluoride and nitrate, compared to other methods.15,27
Various types of materials like activated carbon, activated alumina, zeolites, hematite, schwertmannite, various geo-materials, bio-sorbents, industrial products, polymer resins/fibers/membranes, organics, different metal oxides, composites, and more recently a variety of nanomaterials have been studied for the removal of fluoride and nitrate from aqueous solution.23–33 Among them, selective nano based materials and composites have good potential for removal of fluoride and nitrate from aquatic environments. Nanoscience and technology have opened new avenues to many useful and powerful applications in the field of environmental safety such as water purification.34–37 Nanomaterials offer the possibility of efficient removal of various pollutants like fluorides, nitrates and various toxic ions from drinking water.15,27,28 It has been found that the unique properties of various metal and non metal nanomaterials can be used to develop high capacity and selective sorbents for metal ion removal from drinking water.38–40 Hence, this review focuses on a conceptual overview of the recent trends, principles, and applications of advanced nanomaterials for the removal of F− and NO3− from aqueous solution.
2. Advanced nanomaterials for fluoride removal
2.1. Metal oxide nanomaterials
In the past ten years, many researchers have devoted attention to develop low cost and effective adsorbent nanomaterials for the removal of fluoride from aqueous solution and contaminated water. Fluoride adsorption is determined by various factors like the effect of contact time, adsorbent dosage, adsorbate concentration, pH and temperature.29,30 Various inorganic nanomaterials play a vital role in the removal of fluoride from aqueous solution. Among them iron, alumina, titania, magnesia, silica, zirconium, cerium and calcium based metal oxide nanomaterials and composites play an important role in defluorination processes.27–30 These types of oxide nanomaterials are rich in surface functional groups, highly stable for adsorbing fluorides from aqueous solution. The increased surface area of the metal oxide nanoparticles highly favour fluoride adsorption.41–45 Their high adsorption capacity, non-toxic nature, limited solubility in water and good desorption potential makes metal oxides a material of choice.41–45 These metal oxides have been modified using various methods for preparing hybrid/core–shell nanomaterials that can increase further fluoride removal. The porous nature of the adsorbent also plays a major role in fluoride adsorption. Highly mesoporous CeO2/Al2O3 and CeO2/Mg–Fe metal oxides, having less than 50 nm porosity, were prepared by Zhang et al. using a non-thermal plasma modification method.46,47 The mesoporous metal oxide nanomaterials provided a high fluoride adsorption efficiency of 37–60 mg g−1, which might be due to the increased adsorption sites, large surface area and its mesoporous structure. The coulombic or ligand exchange type of fluoride adsorption may also be one of the important mechanisms for fluoride adsorption. Biswas et al. synthesised spherical hydrous iron(III) – chromium(III) bimetal mixed oxide spherical nanoparticles of 40–55 nm using a co-precipitation method. These hybrid bimetal nanoparticles had 16.34 mg g−1 fluoride adsorption capacity and required an optimum contact time of 30 min. The adsorption was attributed towards rapid boundary layer diffusion on to the surface of the bimetal mixed oxide nanoparticles.48 A Mn–Ce oxide adsorbent of about 4.5 nm crystals prepared by Deng et al.49 via a co-precipitation method showed high fluoride adsorption efficiency of 79.5 mg g−1 within one hour. According to the author, the surface hydroxyl group density caused by the entry of Mn species into the CeO2 lattices on the Mn–Ce adsorbent was mainly responsible for its high adsorption capacity towards fluoride. Further, the anion exchange and electrostatic interaction were also involved in the sorption of fluoride on the Mn–Ce oxide adsorbent. Similarly, the hydrous Ce(IV)–Zr(IV) oxide nanoparticles of about 60–70 nm, prepared using simple chemical precipitation method showed a fluoride adsorption capacity of ∼19.5 mg g−1.50 The mechanism proposed is similar to other previous reports for fluoride adsorption over metal oxide surfaces. CTAB mediated Mg-doped nano Fe2O3 particles with sizes ranging from 40–200 nm were synthesized using a precipitation technique by Mohapatra et al.51 and showed a fluoride adsorption efficiency of 75.2 mg g−1. Thermodynamic studies of the material revealed that the fluoride adsorption on the Mg-doped nano hematite particles was thermodynamically favourable at low temperatures and was exothermic in nature.Porous and hollow MgO microspheres with a high surface area of 130 m2 g−1 were prepared by Li et al.52 using a precursor calcination method. These microspheres were composed of MgO nanoparticles and had a high fluoride adsorption efficiency of >120 mg g−1. The reaction between MgO and water resulted in the formation of magnesium hydroxides (Mg(OH)2) which then reacted with fluoride ion to form Mg(OH)2−xFx, yielding an excellent defluoridation effect.
The adsorption of fluoride on Ti–Al binary metal oxide supported beads was studied by Thakre et al.53 by varying the initial fluoride concentration and keeping the other parameters constant (optimum dose of 4 g L−1, temperature (30 °C) and contact time of 24 h). It was observed that with an increase in the initial adsorbate concentration, the percentage removal decreased. Native TiO2, Ti–Ce, and Ti–La hybrid oxide materials for fluoride adsorption were reported by Li et al.54 The Ti–Ce and Ti–La hybrid adsorbents obtained by the hydrolysis–precipitation method had a much higher sorption capacity for fluoride than the TiO2 adsorbent prepared through hydrolysis. The adsorption capacities of fluoride on the Ti–Ce and Ti–La adsorbents with respect to fluoride were 9.6 and 15.1 mg g−1 respectively.
Recently, the adsorption of fluoride using bio-titania nanoparticles synthesized using a microbial approach was reported by our group (Suriyaraj et al.,27). A metal resistant Bacillus NARW 11 species was used for the synthesis of native TiO2 nanoparticles (Fig. 1(A) and insert). The native microbially synthesized bio-titania nanoparticles had an anatase crystalline nature Fig. 1(B). The native TiO2 nanoparticles were subjected to calcination at various temperatures and different crystalline phase structures like pure anatase Fig. 1(C), anatase mixed with rutile Fig. 1(D) and pure rutile Fig. 1(E) were achieved. Among the various phases of TiO2 nanoparticles, anatase phase showed the highest fluoride adsorption capacity (0.85 mg g−1). The nanoparticles also had good desorption potentials indicating that the adsorbents could be reused. These nanoparticles, synthesised using an eco-friendly and green approach, could be an alternative to those made using caustic chemical synthesis protocols.
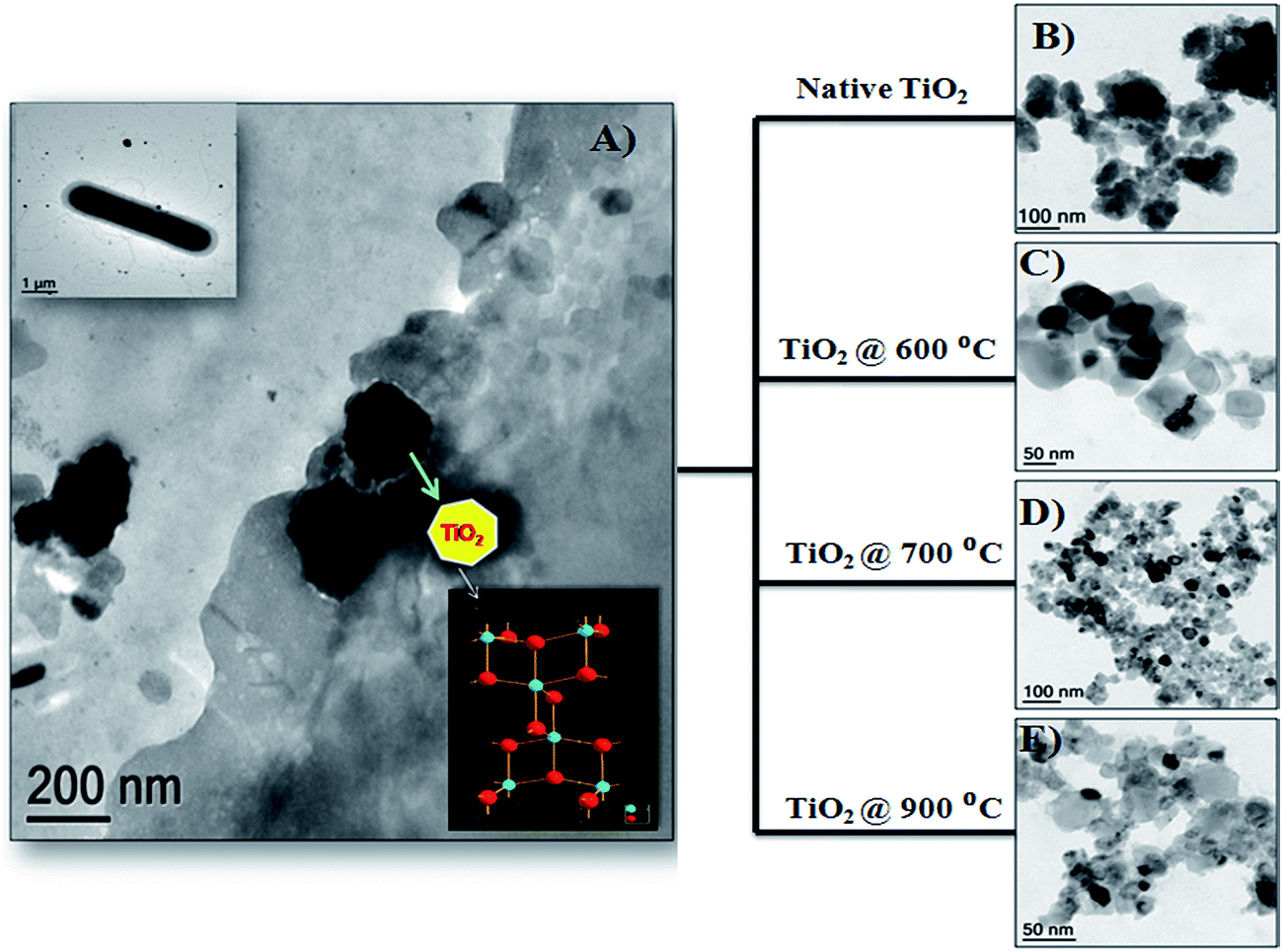 | ||
| Fig. 1 (A) Biosynthesis of TiO2 nanoparticles using a microbial approach, (A) insert shows the Bacillus NARW11 species used for the TiO2 synthesis, (B) native TiO2 without calcination, (C) anatase pure TiO2 calcined at 600 °C, (D) anatase mix rutile TiO2 calcined at 700 °C, and (E) rutile pure TiO2 calcined at 900 °C (adapted from ref. 27 with permission from Elsevier). | ||
The fundamental mechanism of fluoride adsorption using various nanomaterials is based on the electrostatic attraction or ligand-exchange reaction between fluoride and hydroxyl ions, which in turn takes place according to the pH of the medium. At an acidic pH positively charged surface sites develop, which attract the negatively charged fluoride ions by electrostatic attraction resulting in enhanced fluoride removal at acidic pH. At neutral pH, fluoride adsorption can be caused by the ligand-exchange reaction between fluoride and hydroxyl ions. In the presence of other ions like nitrate, sulphate and phosphate, etc., in water, preferential adsorption of fluoride may take place due to its smaller ionic radius (0.133 nm). Hence it can easily be accommodated in nanomaterials, especially in porous structures. Moreover, the process may be a chemical adsorption process involving valence forces through sharing or exchange of electrons between the nanomaterial and fluoride ion. In the case of mesoporous structured nanoadsorbents, the adsorption of fluoride ions mostly takes place by an intra particle diffusion mechanism.27,28,55
2.2. Magnetic nanomaterials
Some metal oxide adsorbents with magnetic properties are highly efficient for fluoride adsorption as well as the removal of adsorbent after the reaction. Even though nanocomposite adsorbents show good adsorbent potential, the removal of the adsorbent from the aqueous solution after completion of the reaction is still the main problem even now, and the nanoparticles can easily be leached into the water system.29,30 In order to overcome this problem, various metal oxides prepared with magnetic properties provide a promising way to perform solid–liquid separation of the nano-adsorbent and water. Various iron oxide based nanomaterials/composites play a significant role in this category.Sulfate-doped Fe3O4/Al2O3 prepared by a chemical co-precipitation method showed maximum fluoride adsorption of about 70.4 mg g−1 at pH 7.0 (Chai et al.56). The results revealed that the sulfate–fluoride displacement and the decreased sulfur content on the nanomaterial surface contributed to the anion exchange process and was an important mechanism for fluoride adsorption by the sulfate doped Fe3O4/Al2O3 nanoparticles. The Fe–Ti bimetallic oxide nano-adsorbents synthesized by Chen et al.57 and Zhang et al.58 using a co-precipitation method showed a better adsorption capacity which was much more superior than the pure Fe oxide or Ti oxide adsorbents. The fluoride adsorption may be due to the interaction between Fe and Ti as Fe–O–Ti bonds on the nanomaterial surface, and OH groups which provide the active sites and allow formation of the Fe–O–Ti–F bonds during the adsorption process. Zirconium(IV)–metalloporphyrin grafted Fe3O4 nanoparticles with an average size of 56 nm were synthesized by a co-precipitation method.59 The material had a removal efficiency of 92.0 ± 1.7% for fluoride for an initial adsorbate concentration of 10 mg L−1 within a contact time of 20 min. Experiments showed that the functionalization of zirconium(IV)–metalloporphyrin showed good efficiency for fluoride adsorption with high specificity and selectivity. Similarly magnesium oxide (MgO)-coated magnetite (Fe3O4) nanoparticles with magnetic properties were synthesized by Minju et al.60 using a modified sol–gel method for analyzing the fluoride scavenging potential. The adsorption capacity was found to be 10.96 mg g−1. A magnetic nanosized adsorbent made from hydrous aluminum oxide embedded with Fe3O4 nanoparticles was prepared by Zhao et al.61 to study fluoride removal from aqueous solutions. The maximum adsorption capacity was found to be 88.48 mg g−1 under neutral pH at room temperature. A distinct advantage of this defluoridation procedure is that the adsorbents can readily be isolated from the sample solutions by the application of an external magnetic field. According to the author, the material has three attractive properties: a high adsorption capacity, rapid defluoridation treatment and easy preparation, meeting the needs of practical application for treatment of large volume high-fluoride contaminated water. A polypyrrole (PPy)/Fe3O4 magnetic nanocomposite adsorbent was prepared by Bhaumik et al.62 via in situ polymerization. The fluoride adsorption capacity of the nanocomposites was found to be in the range of 17.6–22.3 mg g−1. Easy and effective magnetic based Mg–Al-LDH nanoflake impregnated magnetic alginate beads (LDH-n-MABs) were prepared by Gao et al.63 and showed an increased adsorption efficiency of about 32.4 mg g−1. The nanomaterial had many potential advantages like low leaching due to its immobilisation in the alginate beads, high magnetic sensitivity, biodegradability etc. The author explained that a complex adsorption process is responsible for the higher fluoride adsorption efficiency of the LDH-n-MABs (Fig. 2). Similarly, Fe3O4 nanoparticles in a network of Zr(IV) complexed poly(acrylamide) (Zr–PAM) prepared by an encapsulation method is one of the best fluoride ion selective magnetic sorbents.64 The material has a high adsorption efficiency of 124.5 mg g−1. The pictorial representation of the magnetic separation of nanoparticles after the completion of fluoride adsorption is shown in the Fig. 3. Hence, magnetic separation is a promising method for solid–liquid phase separation with various advantages like a high absorption capacity, high speed, accuracy, simplicity and efficient solid–liquid separation when compared to conventional separation methods.56–58
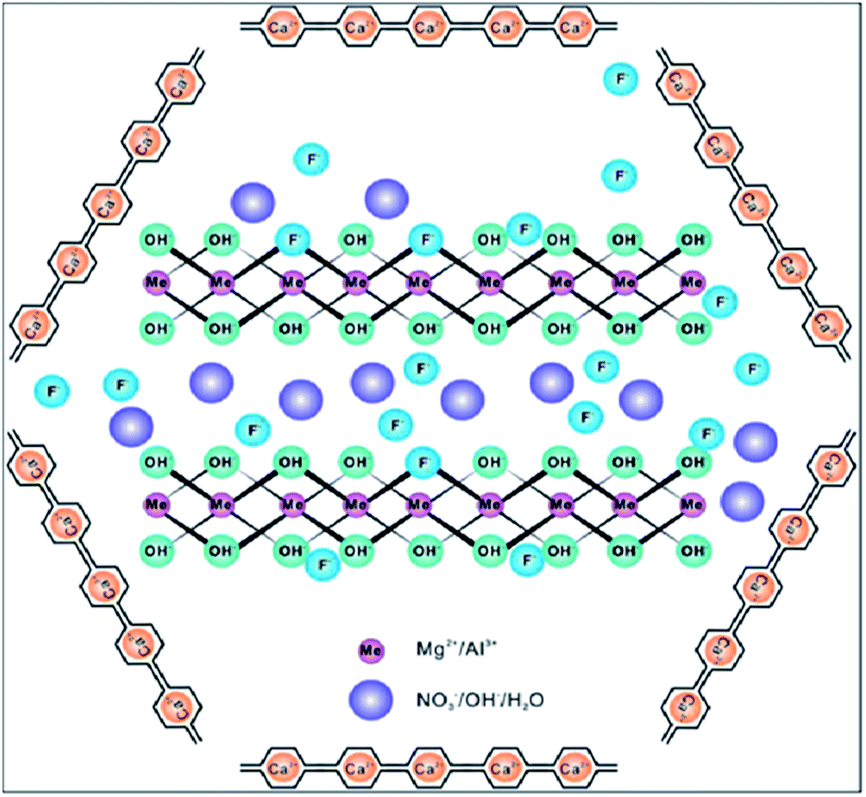 | ||
| Fig. 2 Complex adsorption process of LDH-n-MABs for fluoride removal. Reprinted from ref. 63 with permission from the Royal Society of Chemistry. | ||
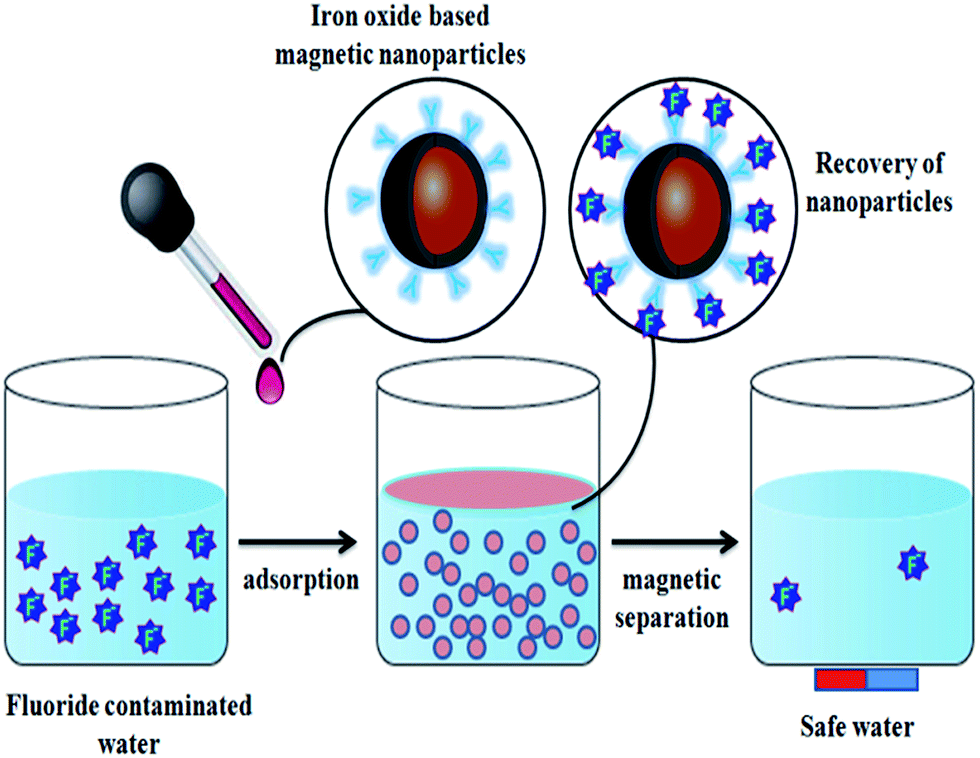 | ||
| Fig. 3 Pictorial representation of the magnetic separation of nanoparticles after the completion of fluoride adsorption. | ||
2.3. Polymer, carbon and hydroxyapatite based nanomaterials
The incorporation of nanomaterials into a polymer matrix increases the stability of the materials for fluoride adsorption. Fluoride adsorption by Al/Fe-doped nano (∼100 nm) sized porous polymeric adsorbent beads was reported by Kumar et al.65 The material was produced by a suspension polymerization method and showed significant adsorption of F− (100 mg g−1). The fluoride adsorption studies performed by Mahapatra et al.66 using electrospun alumina nanofibers showed a maximum fluoride adsorption capacity of 1.2 mg g−1. The nanoparticle impregnated hybrid nanofibers have unique and interesting features such as a high surface area to volume ratio, large porosity, good mechanical properties, good water permeability and increased elution quality due to complete solid/liquid separation, which makes them suitable for drinking water treatment applications.34,35,67 The nanoparticle impregnated nanofiber membranes can be used for fluoride removal through simple dip mode adsorption in the future. Recently, a hybrid Al2O3/bio-TiO2 nanocomposite was prepared by our group and further impregnated into a thermoplastic polyurethane (TPU) nanofibrous membrane and studied for its efficiency for fluoride removal from an aqueous solution using a dip mode adsorption method.68 The adsorption capacities (Q0) of the nanocomposite and the nanocomposite impregnated TPU nanofiber were found to be 2.73 and 1.9 mg g−1 respectively. This facile approach of designing nanocomposite impregnated nanofiber membranes can be used for F− removal from drinking water. In Fig. 4 the pictorial image gives an idea about the process of F− removal using the simple dip mode adsorption. This type of fluoride adsorption could be a quick process, safe, portable and have easy operation. Similarly the incorporation of a zirconium(IV) tungstophosphate (ZrWP) ion exchanger into the chitosan biopolymeric matrix, synthesized by Viswanathan and Meenakshi69 showed excellent defluoridation efficiency. An adsorption/complexation and ion exchange mechanism was reported as a possible mechanism for fluoride adsorption by the ZrWPCs composite (Fig. 5(a)). Positive charges like Zr4+ and W6+ on the nanocomposites’ surface attract the negatively charged fluoride ions by means of electrostatic attraction as well as surface complexation. Further, the –PO43− groups present in the ZrWPCs composite act as charge carriers and may be exchanged for F− ions by an ion exchange mechanism.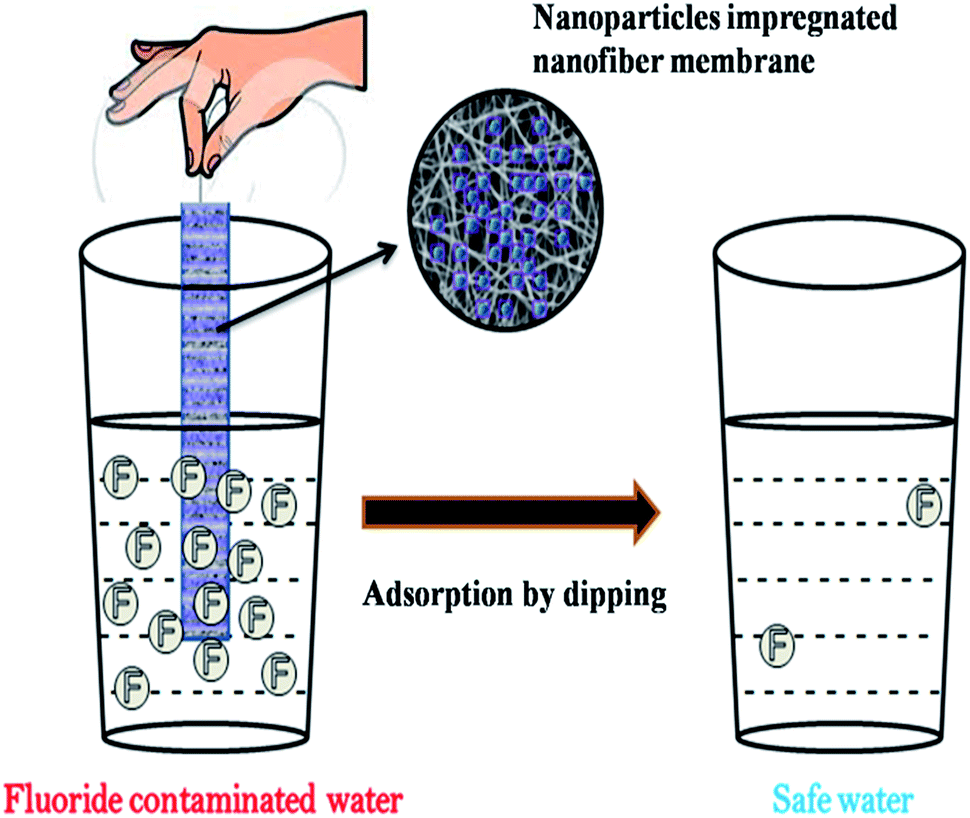 | ||
| Fig. 4 Pictorial representation of fluoride adsorption using nanoparticle impregnated nanofiber membranes by a dip mode adsorption method. Adapted from ref. 68 with permission from the Royal Society of Chemistry. | ||
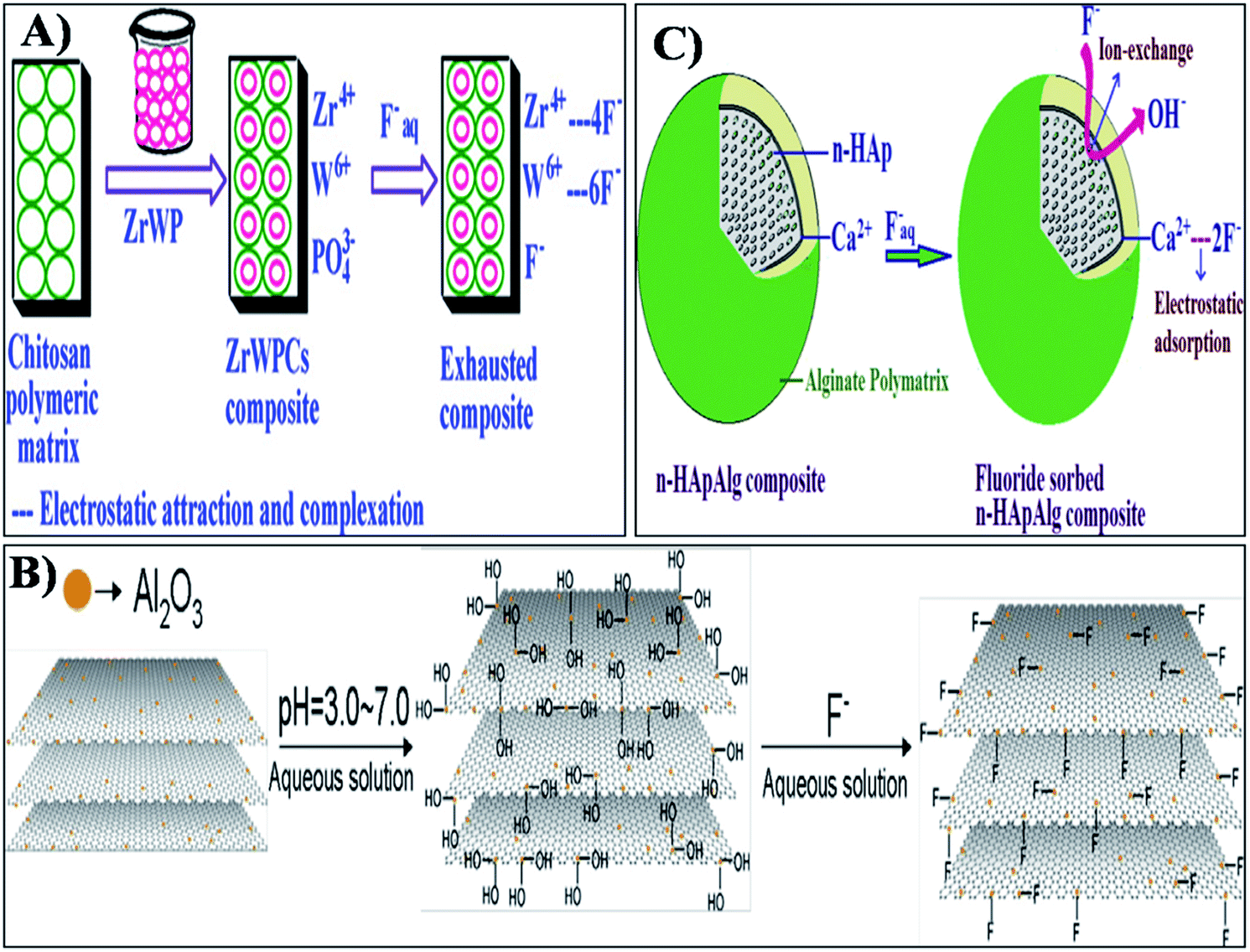 | ||
| Fig. 5 Adsorption and ion exchange mechanism of fluoride adsorption by (A) ZrWPCs composite, (B) alumina-modified expanded graphite (Al2O3/EG) nanocomposite and (C) alginate bioencapsulated nano-hydroxyapatite composite. Reprinted from ref. 69, 74 and 81 with permission from Elsevier. | ||
Novel nanoparticle impregnated carbon based adsorbents obtained by carbonization and activation of natural materials possess appreciable defluoridation efficiency depending on the specific surface area, mesoporous structure and specific surface charge of the adsorbent.23–25 Titanium and lanthanum oxides impregnated on granular activated carbon (TLAC) developed by Jing et al.70 showed a maximum fluoride adsorption capacity of 27.8 mg g−1. This nanomaterial can be used as an effective adsorbent for simultaneous removal of F−, As(V) etc. Materials like titanium and lanthanum oxide, evenly distributed on the surfaced of the TLAC, preferably form inner-sphere surface complexes with F− ions and are thus involved in F− adsorption. Gupta et al.71 developed a micro–nano hierarchal web (MiNaHiWe) consisting of activated carbon fibers (ACF) and carbon nanofibers (CNF), impregnated with Al for the removal of fluoride from wastewater. Ma et al.72 reported the fluoride adsorption properties of manganese oxides impregnated granular activated carbon (GAC-MnO2) using a redox process. The fluoride adsorption capacities of GAC-MnO2 exhibited an efficiency three times greater than uncoated GAC. The pH of the solution influenced F− removal. Zirconium impregnated activated charcoals prepared by Janardhana et al.73 showed 3–5 times higher efficiency for F− adsorption compared to plain activated charcoal. The mesoporosity and surface charge of the material influenced fluoride sorption very effectively. Similarly, an amorphous alumina-modified expanded graphite (Al2O3/EG) nanocomposite with a diameter of 10–30 nm was prepared using a facile solution method followed by thermal treatment.74 The maximum adsorption capacity was found to be 1.18 mg g−1 which is due to the generation of abundant hydroxyl ions on the surface of the adsorbent material (Fig. 5(b)). Aligned carbon nanotubes (ACNT) were used for F− adsorption studies by Li et al.75 They found that the material had an adsorption capacity of 4.5 mg L−1 with 15 ppm of fluoride at pH 7. The higher positive surface charge of the material increases the tendency of the material to adsorb negatively charged fluoride ions. Both intra-particle diffusion and pore diffusion also play important roles in the F− adsorption process. If the radius of F− is smaller than the pore size of the adsorbent, increased penetration of F− into the inner layer of the synthetic adsorbent is possible, which can make them a potential candidate for the drinking water defluoridation process.76,77
Recently a hydroxyapatite (HAp) based nanomaterial and its composites were revealed to be very good prospects for defluoridation from aqueous solution. It was found that nanosized HAp was more efficient than the bulk due to its high surface area, porosity and highly reactive surface charges. The simple wet chemical method of developing low cost nano-HAp was studied for fluoride adsorption by Sundaram et al.78 and the results revealed that the maximum fluoride adsorption was about 1845 mg F− per kg. The removal of fluoride ions by the nano-HAp was mainly due to both ion-exchange and adsorption processes.
The fluoride adsorption using nano-HAp developed by a combined ultrasonic and microwave technique showed a maximum monolayer adsorption capacity of 5.5 mg g−1.79 Nano-HAp can be modified using various dopants to increase its fluoride adsorption efficiency compared to native HAp. Zhang et al.80 studied pure nano-HAp with a hexagonal structure and a particle size of about 20 nm × 60 nm for fluoride adsorption at various reaction temperatures. In this study, the monolayer adsorption efficiency of HAp at different temperatures was found to be 19.742, 26.108, 36.914 and 40.818 mg g−1 at 298, 308, 318 and 328 K respectively. Alginate bioencapsulated nano-hydroxyapatite composite (n-HApAlg) was studied for fluoride adsorption by Pandi and Vishwanathan.81 The nanocomposite showed excellent fluoride adsorption. Both adsorption as well as ion-exchange were the main mechanisms of fluoride removal by the n-HApAlg composite as shown in Fig. 5(c). The mechanism reveals that the positively charged Ca2+ present in the nanocomposite is attracted by negatively charged fluoride ions by means of an electrostatic attraction. The OH− ions of the n-HAp lattice are replaced by the F− ions by means of an ion-exchange mechanism which further induces fluoride adsorption by the nanocomposite. As reported earlier, electrostatic interactions and hydrogen bonding are the main driving forces for fluoride uptake onto HAp. Nanomaterials like nano-hydroxyapatite/chitosan (n-HApC) composite,82 cellulose/HAp nanocomposites,83 aluminum-modified HAp84 and cationic surfactant modified HAp85 are widely used for better fluoride adsorption than native nano-HAp. At low pH conditions, the HAp nanomaterials were protonated such that they were positively charged and could attract fluoride anions by electrostatic attraction. Under a neutral pH, the intra-particle diffusion and the ligand exchange process help the fluoride removal during adsorption. Thus HAp based nanomaterials play a vital role in fluoride adsorption studies.23–25 Table 1 gives the summary of the F− adsorption capacity of various nanomaterial based adsorbents.
| S. no | Nano-adsorbents | Nanomaterial characteristics | Working pH | AC mg g−1 | Short summary | Ref. |
|---|---|---|---|---|---|---|
| a CP: crystalline phase; SA: surface area; PV: pore volume; M: morphology; PS: particle size; PD: pore diameter; SM: saturation magnetization. | ||||||
| 1 | Nano-alumina | CP: α alumina; SA: 151.7 m2 g−1; PV: 1.09 cm3 g−1 | 6.15 | 14.0 | Adsorbent: commercial grade (Sigma-Aldrich). The batch sorption of F− on nano-alumina was found to be strongly pH dependent with maximum F− removal occurring at pH 6.15. The interaction between F− and nano-alumina moieties results in the formation of aluminum–fluoro complexes. F− sorption was influenced more by the presence of PO43−, SO42− and CO32− | 43 |
| 2 | Nano-AlOOH | CP: boehmite; CS: 133 Å; M: wire-like fibre (Dia: 15–25 nm); SA: 240.38 m2 g−1 | 7 | 3.259 | In batch mode, F− removal efficiency was greater than 90% between pH 6 and 8 and decreased as pH values increased to 11. The presence of SO42− or PO43− in aqueous solution has been found to reduce the F− uptake. Desorption studies showed that F− can easily be desorbed at pH 13 | 86 |
| 3 | Nano-MgO | PS: 18 nm; SA: 92.46 m2 g−1; PV: 0.4313 cm3 g−1 | 10–11 | 14.0 | Adsorbent: commercial grade (Sigma-Aldrich). F− adsorption studies were performed by batch mode. The F− adsorption by nano-MgO was less sensitive to pH variations. At a 0.6 g L−1 dosage concentration, maximum (90%) F− removal was obtained. F− sorption was mainly influenced by the presence of hydroxide followed by sulphate, bicarbonate and chloride | 41 |
| 4 | Nano-sized goethite (α′-FeOOH) | PS: 1–10 nm; CP: α FeOOH | 6–8 | 59 | Adsorbent preparation: precipitation method. The batch adsorption studies fitted well to a pseudo-second-order kinetic model. The isothermic data showed a good fit to a Freundlich isotherm model. A contaminated ground water sample containing 10.25 mg L−1 fluoride could be purified in three stages using 8 g L−1 adsorbent dose | 87 |
| 5 | CeO2–ZrO2 nanocages | PS: 80–100 nm; M: hollow nanospheres/nanocages; CP: fcc; SA: 29.61 m2 g−1 | 4.0 | 175 | CeO2–ZrO2 nanocages were prepared by the Kirkendall process, and their F− removal performance was investigated in batch studies. The adsorption mechanism of the adsorbent for F− involves anion exchange and electrostatic interaction | 88 |
| 6 | Mn–Ce oxide nanocrystals | PS: 4.5 nm; SA: 41 m2 g−1 | 6 | 79.5 | Adsorbent preparation: co-precipitation method. The adsorption was fast within the initial 1 h. The surface hydroxyl group density on the Mn–Ce adsorbent was mainly responsible for its high sorption capacity for F−. Both anion exchange and electrostatic interaction were involved in the sorption of F− | 89 |
| 7 | Hydrous Ce(IV) + Zr(IV) mixed oxide nanoparticles (HCZMO) | CP: tetragonal; CS: 55–65 nm; PS: 60–70 nm; SA: 185.04 m2 g−1; PV: 0.12 cm3 g−1 | 5.8 | 12.4 | HCZMO was prepared by a low temperature and green method. The kinetics of F− adsorption followed a pseudo-second order kinetic model. A low activation energy (1.16 kJ mol−1) and high adsorption energy (15.05 kJ mol−1) indicated high affinity of fluoride at the HCZMO surface. >95% of desorption achieved using 1.0 M NaOH solution | 50 |
| 8 | Mesoporous CoAl2O4 nanoparticles | CP: hexagonal; SA: 379 m2 g−1; PV: 0.984 cm3 g−1; PD: 1.035 nm | 7 | 14.80 | Adsorbent preparation: micro-emulsion method. The high fluoride adsorption was mainly attributed to the adsorbent high specific surface areas and abundant pore structures | 90 |
| 9 | Fe–Ti bimetallic oxide/Fe3O4 particles core–shell nanocomposite | M: core–shell nanostructure (Dia: 10–20 nm shell thickness: 4.7 nm) | — | 57.22 | Adsorbent preparation: co-precipitation method. Batch adsorption studies indicated that the adsorption process was fast and reached equilibrium within 2 min. The nanoadsorbent was superparamagnetic with a saturation magnetization of 18.4 emu g−1. This allowed rapid separation of adsorbent from water using an external magnet | 91 |
| 10 | CeO2/Al2O3 nanoporous composites | M: irregular sheet like structure; SA: 266.1 m2 g−1 | 3–10 | 37.0 | Adsorbent preparation: co-precipitation method. During the batch adsorption process, the coexisting anions had little effect on the F− adsorption, except HPO42− and C2O42−. The F− adsorption followed pseudo second order kinetic model and Redlich–Peterson model isotherms | 46 |
| 11 | Bio TiO2 nanoparticles | CP: anatase; M: spherical; PS: 47.26 ± 13.5 nm | 7 | 0.85 | Adsorbent preparation: microbial synthesis | 27 |
| 12 | Fe–Al–Ce nanoadsorbent | M: agglomerated tiny granules of size 2–3 mm | 7 | 2.22 | Adsorption method: column mode. 300 bed volumes can be treated with the effluent F− below 1 mg L−1 for an influent F− concentration of 5.5 mg L−1, pH of 5.8, and SV of 5 h−1. The coating of Fe–Al–Ce adsorbent can produce granules that can be used in a packed bed for the removal of F− from drinking water | 92 |
| 13 | Hydrous iron(III)–chromium(III) mixed oxide (HICMO) nanoparticles | M: agglomerated spherical nanoparticles (Dia: 40–55 nm) | 6.5 | 16.34 | Adsorbent preparation: co-precipitation method. The ion-exchange mechanism was suggested for the fluoride adsorption process. The adsorption reaction was endothermic and spontaneous. The presence of bicarbonate in the solution reduced fluoride adsorption in HICMO | 48 |
| 14 | Activated nano-gibbsite | CP: χ alumina; M: spherical; SA: 399 m2 g−1 | 6 | 39 | The increased surface area resulted in high fluoride sorption capacity (4 × 10−6 mol m−2) which is equal to 39 mg g−1. No pH or ionic strength dependency was observed in the pH range 4–6.5, suggesting an inner-sphere binding mechanism for low surface loads (0.526 mM) | 93 |
| 15 | Nanosized fluorapatite (nFAP) | M: cylindrical rod like shape (Dia: 50–60 nm) | 3.0 | 7.45 | Adsorbent preparation: solution–precipitation method. Adsorption of F− by nFAP was fast and reached equilibrium in 120 min of contact time. An increase in the initial F− concentration could effectively increase the F− adsorption capacity | 94 |
| 16 | Highly ordered mesoporous alumina | CP: α alumina; CS: 7–10 nm, PS: 150–400 nm, SA: 163–338 m2 g−1, PS: 7–14 nm | 6.0 | 135 | The adsorbent had a high surface area and hydroxyl groups along with ordered mesoporous tunnel which favored diffusion and transportation of fluoride species | 95 |
| 17 | Magnesium oxide (MgO)-coated magnetite (Fe3O4) nanoparticles | M: core–shell nanostructure (Dia: 98.3 nm) | 5–7 | 10.96 | Adsorbent preparation: sol–gel method. Maximum removal of F− was estimated as 98.6% for an initial adsorbate concentration of 13.6 mg L−1 at optimal conditions: pH 6.0, adsorbent dosage of 2 g L−1 and contact time of 120 min by batch process | 60 |
| 18 | Fe3O4@Al(OH)3 magnetic nano-particles | CP: bayerite; PS: 240–340 nm; SA: 147 m2 g−1 | 6.5 | 88.48 | Adsorbent preparation: chemical co-precipitation method. Sorbents had higher surface areas and shorter diffusion route and the Al(OH)3 surface layer of Fe3O4@Al(OH)3 NPs possessed a specific affinity toward fluoride | 61 |
| 19 | CTAB mediated Mg-doped nano Fe2O3 | CP: hematite; M: spherical; PS: 40 to 200 nm | 7 | 75.2 | Adsorbent preparation: surfactant mediation–precipitation method. The F− adsorption studies were carried by varying the time, pH, temperature and amount of adsorbent and adsorbate. The contaminated water collected from the Nayagarh district of Orissa was defluoridated in a single stage batch mode | 51 |
| 20 | Sulfate-doped Fe3O4/Al2O3 core–shell nanoparticles | SA: 63.37 m2 g−1; M: nano-needle/spheres; PS: 15–20 nm; SM: 16 emu g−1 | 7 | 70.4 | Adsorbent preparation: chemical co-precipitation method. Anion exchange of sulfate by F− and formation of inner-sphere fluoride complex were the important mechanisms for fluoride removal by the sulfate-doped Fe3O4/Al2O3 nanoparticles | 56 |
| 21 | Fe–Ti bimetallic oxide nanoparticles | M: nano-needle/sphere granules; PS: 5–7 nm | — | 47.0 | Adsorbent preparation: co-precipitation method. During batch adsorption, Fe and Ti in the Fe–Ti oxide adsorbent interacted synergistically to give increased F− adsorption capacity higher than that of pure Fe oxide and Ti oxide adsorbent | 96 |
| 22 | Fe–Al-impregnated granular nanoporous ceramic | M: white and carmine colour spherical nanoporous particles (Dia: 2–3 mm); SA: 40.22 m2 g−1; PV: 0.078 cm3 g−1; PD: 5 to 60 nm | 4–9 | 3.56 | More than 96% removal of fluoride was achieved within 48 h from 10 mg L−1 initial fluoride solution at neutral pH by batch adsorption process. The F− removal efficiency was significantly decreased in the presence of carbonate and phosphate anions | 97 |
| 23 | Al- and Fe-doped micro/nanoadsorbent | M: smooth bead morphology; SA: ∼750 m2 g−1; PS: ∼200 nm | 7.4–8.0 | ∼100 | Adsorbent preparation: suspension polymerization method. The methodology adopted in this study prepared bi-metal porous adsorbents toward developing multi-functional adsorbents | 65 |
| 24 | Polypyrrole/Fe3O4 magnetic nanocomposite | M: agglomerated spherical nanoparticles (Dia: 10 nm); SA: 1206.53 m2 g−1 | 6.5 | 22.3 | The F− uptake was very rapid and depended on the initial concentration, temperature, adsorbent dose and pH. Adsorption of F− was not affected due to the presence of other anions such as chloride, nitrate, sulphate and phosphate | 62 |
| 25 | Nanomagnetite aggregated schwertmannite | M: pincushion structure (width: 2–4 nm; length: 60–90 nm); SA: 276.15 m2 g−1; PV: 0.139 cm3 g−1; PD: < 40 nm | 5.8 | 17.24 | Adsorbent preparation: chemical precipitation/wet chemical process. The permissible limit defined by the WHO for defluoridation was achieved using a 2 g L−1 adsorbent dose within 90 min contact time at pH 5.7 in a batch adsorption process | 98 |
| 26 | Zr–PAM/Fe3O4 nanocomposite | M: microporous (irregular pattern); SM: 4.5 emu g−1 | 3 | 124.5 | The adsorbent developed had the super paramagnetic properties of Fe3O4 nanoparticles and a rapid F− sorption property. Repeated sorption–regeneration cycles indicated the reusability of the sorbent for F− removal | 64 |
| 27 | Iron oxide–hydroxide nanoparticles | CP: orthorhombic; M: flower structure with several strings extended; PS: 20 nm; SA: 6.57 m2 g−1 | — | 16.7 | The adsorbent showed good F− removal. There was no significant influence of other co-anions like chloride, iodate, iodide and sulphate on the defluoridation capacity of the nanoparticles except OH−. The F− adsorbed nanoparticles was regenerated by up to 70% using sodium hydroxide or hydrochloric acid solution | 44 |
| 28 | Lanthanum-impregnated activated alumina (LAA) nanoflakes | CP: amorphous; M: thin flakes (5–20 nm thickness; SA: 31.235 m2 g−1 | 3.9 to 9.6 | 16.9 | LAA could adsorb 70.5–77.2% F− in the pH range of the actual groundwater, about four times higher than activated alumina. Meanwhile, the Al release could be substantially reduced due to LaOOH formation. In addition, LAA could be regenerated and reused | 99 |
| 29 | Mg–Al-LDH nanoflake impregnated magnetic alginate beads (LDH-n-MABs) | M: nanoflakes (thickness: 18 nm; Dia: 20 nm) | 5.0 | 32.4 | Mg–Al-LDH nanoflakes with a higher adsorption capacity for F− were immobilized into alginate beads without leaching, taking the safety issues of nanomaterials into consideration. The beads had a high magnetic sensitivity to an external magnetic field providing an easy and efficient way to separate them from aqueous solution | 63 |
| 30 | Alumina nanofibers | M: ultrafine cylindrical smooth nanofiber (Dia: 200−600 nm) | 7 | 1.2 | Adsorbent preparation: electrospinning method. Adsorption of F− ions onto the alumina nanofiber surface followed a Freundlich isotherm model indicating a heterogeneous adsorption process | 66 |
| 31 | Nanoporous Mg–Al–CO3 layered double hydroxides | — | 6.0 | 80.12 | Adsorbent preparation: co-precipitation method. Maximum removal of F− from aqueous solution was obtained in 6 h at pH 6.0 with an initial concentration of 50 mg L−1, and the retention of F− ions by the material was 98%. The influence of co-existing anions in F− aqueous solution indicated that the percentage of removal of F− increases in the order PO43− < Cl− ≈ SO42− < Br− ≪ NO3− | 100 |
| 32 | Aluminum-impregnated micro-nanohierarchal web (MiNaHiWe) of carbon fibers | M: smooth narrow range nanofiber (Dia: 30–40 nm; length: several micrometers) | 6 | 17.0 | Adsorbent preparation: chemical vapor deposition. The materials has been tested for the adsorption of F− ions over the concentration range of 1–50 ppm in water under both batch and flow conditions. The adsorbent web showed significant adsorption of F− ions. In addition, the total F− uptake was observed to be higher on the aluminum impregnated ACF/CNF web than on its parent material | 71 |
| 33 | Polyacrylonitrile coated with iron oxide nanoparticles | — | — | 250 | The influence of contact time, initial F− concentration, and adsorbent dosage were investigated by batch equilibrium studies. The rate of adsorption was rapid with equilibrium being attained after 100 min | 101 |
| 34 | Aluminium-cerium alginate mixed metal oxide nanomaterial | PS: 3.14 nm; SA: 89.23 m2 g−1; PV: 0.202 cm3 g−1; PD: 46.32 Å | — | 1.438 | The material was able to remove 98% of F− at the natural pH of water bodies (pH = 7.0), the adsorption process followed pseudo-second-order kinetics and the Freundlich isotherm | 102 |
| 35 | ZnCr layered double hydroxides | CP: hydrotalcite; SA: 12–26 m2 g−1; PV: 0.04–0.11 cm3 g−1; PD: 135–173 Å | 3–10 | 31 | Adsorbent preparation: co-precipitation method. Adsorption studies: batch and column mode. The uptake capacity of the composite materials increased with an increase in LDH content in both batch and column studies. Further, the composite materials showed better aqueous diffusivity than powdered materials | 103 |
| 36 | Fe(III)–Zr(IV)Zr–alginate (FZCA) micro/nanoparticles | — | 7 | 0.981 | The sorption of F− followed pseudo-second order kinetics. The positive value of the thermodynamic parameter ΔH0 indicated increasing randomness during the sorption process. The desorption characteristics of the hybrid material show that nearly 89% of F− could be leached out at pH 12 | 104 |
| 37 | Synthetic hydroxyapatite | M: sphere like particles (dimensions of 40–100 nm) | 5.0 | 0.489 | Adsorbent preparation: thermal decomposition. Adsorption studies: batch. Maximum adsorption of 83.2% was attained within 20 min and equilibrium was established almost after 80 min. | 105 |
| 38 | Cellulose@hydroxyapatite (HA) nanocomposites | M: irregular flakes; CS: 20–50 nm; SA: 76.257 m2 g−1 | 6.5 | 4.2 | Adsorbent preparation: in situ hybridization method. Adsorption equilibrium of F− was reached within 360 min. The coexisting anions like nitrate, sulfate and phosphate had no significant effect on F− adsorption | 106 |
| 39 | Nano-hydroxyapatite chitin-composite (n-HApCh) | — | 7 | 8.4 | Adsorbent preparation: co-precipitation method. The main advantages of the n-HApCh composite are biocompatibility, low cost, and indigenous synthesis enabling its effective utilization as a promising defluoridating agent | 82 |
| 40 | Li–Al layered double hydroxides | CP: hexagonal/monoclinic, M: rosette-shaped particles | 6–7 | 47.24 | The maximum percentage removal (97.36%) could be reached and the adsorption equilibrium could be attained within 1 h. Based on FT-IR and kinetic analysis, the “memory effect” may play an important role in the early adsorption stage, while the ion-exchange process may control the adsorption rate at the second stage adsorption | 107 |
| 41 | Mg/Fe layered double hydroxides | M: wizened nanosheets (Dia: 100 nm) | 7 | 28.65 | Adsorbent preparation: hydrothermal process. When compared with commercial activated alumina, the synthesized adsorbent was devoid of potential risks associated with alumina and had a lower leaching of metal ions. Maximum F− desorption of 97.2% was achieved | 108 |
| 42 | Hybrid Al2O3/bio-TiO2 nanocomposite | CP: orthorhombic (alumina), anatase/rutile (titania); PS: 50 ± 6 nm | 7 | 2.73 | Adsorbent preparation: microbial and chemical method adsorption method: batch process. The adsorption capacity was reported to be 2.73 mg L−1 | 68 |
| 43 | Hybrid Al2O3/bio-TiO2 nanocomposite impregnated TPU nanofiber membrane | M: smooth narrow range nanofiber (Dia: 239 + 33 nm, thickness: 21.94 ± 1.3 μm); swelling ratio: 6.02 ± 0.7%; porosity: 43.34 ± 4.3% | 7 | 1.9 | Adsorbent preparation: electrospinning/spraying, adsorption method: dip mode process. The adsorption capacity was reported to be 1.9 mg L−1 | 68 |
| 44 | MWCNTs | — | 5 | 3.50 | The performance of the MWCNTs was good at lower F− concentrations (less than 2 mg L−1) | 109 |
| 45 | ZrO2 hybrid CNTs | — | — | 60.57 | Adsorbent preparation: microwave assisted process. Fluoride uptake reached 79% of the equilibrium value within the first five minutes, indicating a high rate of adsorption. The fluoridated ZrO2/MWCNT could be regenerated easily by treating with an alkaline solution | 110 |
| 46 | CNTs supported alumina | SA: 165 m2 g−1: PV: 0.176 cm3 g−1; PD: 4.26 nm | 6.0–9.0 | 24.15 | Adsorption capacity was maximum at alumina loading of 30 wt%. All adsorption isotherms at different pHs followed the Freundlich model. Kinetic studies showed that fluoride adsorption onto Al2O3/CNTs was a second order rate reaction | 111 |
| 47 | CTAB functionalized MWCNTs | SA: 75.2 m2 g−1: PV: 0.0925 cm3 g−1; PD: 5.2 nm | 2–11 | 20.10 | A novel CTAB grafted MWCNT (CGCNT) adsorbent was synthesized by a green route using ultrasonication and using a green solvent (isoamyl alcohol). It was found that the removal of F− could be achieved over a wide range of pH from 2 to 11. This could be attributed to the positive zeta potential of the CGCNTs in both acidic and basic media. The recyclability and applicability of the adsorbent to F− spiked high alkaline ground water was also demonstrated | 112 |
| 48 | Amorphous alumina-modified expanded graphite (Al2O3/EG) nanocomposite | M: peas-like structure (Dia: 10–30 nm, length: 20–50 nm) | 3–7 | 1.18 | Adsorbent preparation: solution method followed by thermal treatment. With the optimum parameters of the batch adsorption experiment (initial F− = 5 mg L−1, pH = 4.0, temperature = 30 °C, time = 120 min and adsorbent dosage = 0.2 g) the removal rate was 94.4%. The adsorption was multi-molecular layer | 74 |
| 49 | Aligned carbon nanotubes (ACNTs) | Diameter – 20–80 nm; SA: 74 m2 g−1: PV: 0.15 cm3 g−1; PD: 3.8 nm | 7.0 | 4.5 | Adsorbent preparation: catalytic decomposition method. The kinetics experiment of ACNTs shows that the fluoride adsorption rate was fast in the first 60 min and reached equilibrium gradually in about 180 min. The highest adsorption capacity of ACNTs was 4.5 mg g−1 at pH 7 with a fluoride concentration of 15 mg L−1 | 75 |
| 50 | Aluminum sulfate@graphene hydrogel (BAS@GHG) nanocomposite | CP: amorphous; M: spherically shaped GHG onto CCG 2D thin sheets | 7.2 | 33.4 | Adsorbent preparation: homogeneous precipitation method. In the adsorbent, the presence of BAS was believed to provide high F− adsorption capacity through a ligand exchange mechanism between hydroxide ions and F− ions. Simultaneously the GHG acts as a porous matrix which can provide a large surface area and facilitate the diffusion of adsorbate | 113 |
3. Advanced nanomaterials for nitrate removal
Similar to the applications of nano-based adsorbent materials for fluoride removal, extensive research has attracted many researchers in recent years for nitrate removal from aqueous solution. When compared to other methods like biological nitrate reduction,7 adsorption and photocatalytic processes are widely used to remove NO3− very effectively from water. Various advanced nanomaterials used for the adsorption and photocatalytic degradation of nitrates and their reaction mechanisms are discussed in the following sections.3.1. Nanomaterials for nitrate removal by adsorption
Selection of a suitable adsorbent media for NO3− removal from aqueous solution is similar to that of fluoride due to their identical ionic charge. The mechanism of nitrate adsorption by various nanomaterials is based on the electrostatic attraction or ligand-exchange reaction between nitrate and hydroxyl ions depending on the pH of the medium.114,115 Recently, the feasibility of nano-alumina for NO3− removal from aqueous solutions was explored by Bhatnagar et al.116 The maximum sorption capacity of nano-alumina for NO3− removal was found to be 4.0 mg g−1. The ligand exchange reactions between the nitrate ions and surface charge of the adsorbent material took a major role in the NO3− adsorption process. Zhang et al.117 synthesized a highly porous nanocomposite material consisting of MgO nano-flakes within a biochar matrix for the removal of nitrate from water. The nitrate adsorption capacity for this material was found to be 95 mg g−1. The nano-flake structures were regular morphology and dispersed uniformly on the surface of the biochar matrix, making it a potential candidate for nitrate removal from aqueous solution.Saad et al.118 synthesized amino-functionalized mesoporous silica materials and successfully applied them to remove NO3− from water. The aminated and protonated mesoporous silica showed a high adsorption capacity for nitrate reaching 46.5 mg g−1 compared to an adsorption capacity of 2.4 mg g−1 for the unmodified mesoporous silica. A Fe3O4/ZrO2/chitosan nanocomposite having the surface area 212.9 m2 g−1 was prepared by Jiang et al.119 using a simple wet chemical method, and studied for its NO3− adsorption ability. The material exhibited a maximum NO3− adsorption of about 89.3 mg g−1 and was attributed to physical forces besides electrostatic interaction during adsorption. It has also been noted that the saturated Fe3O4/ZrO2/chitosan nanocomposite adsorbent cannot be thoroughly regenerated in strong alkali conditions. Graphene oxide (GO) coated Fe, Ni and Co nanoparticles were prepared by Motamedi et al.120 using a simple chemical reduction method which showed good NO3− removal efficiency. According to the author the NO3− adsorption efficiency of Fe-GO was higher than that of the reported unanchored Fe nanoparticles. Similarly, the Ni-GO and Co-GO showed notable nitrate removal potentials similar to that of Fe-GO. This observable fact is attributed to the greater dispersion of pure zero-valent Ni and Co over GO. It is also proved that this material is highly efficient in adsorbing NO3− due to its magnetic properties and oxidation potential. The quasi-spherical shaped Fe NPs were successfully synthesized by Wang et al.121 via a facile one-step, biosynthetic route using green tea (GT) and eucalyptus leaf (GL) extracts. The maximum nitrate adsorption efficiency was found to be 13.06 mg g−1 for GT-Fe and 9.698 mg g−1 for EL-Fe NPs. The efficiency was further compared with the native zero-valent iron nanoparticles (nZVI) and Fe3O4 nanoparticles which had 87.6% and 11.7% nitrate removal respectively. The adsorption of nitrate by Pd/Cu supported on active carbon bimetallic materials was studied by Mikami et al.122 The results revealed that the NO3− was strongly adsorbed on specific Pd–Cu bimetallic sites.
Suitable metal oxides can be introduced into CNTs to make efficient adsorbent materials for nitrate removal. The maximum adsorption efficiency of the nitric acid and liquid ammonia functionalized carbon nanotube (CNT) sheets were found to be 90.9 and 142.85 mg g−1 respectively. These results demonstrate that nitrate adsorption by the modified CNTs sheets is only due to the strong electrostatic interactions between the negative charge of NO3− and the positive charge of the CNT surface.123 Similarly the NO3− adsorption capacity of powdered activated carbon (PAC) was compared with CNTs. The NO3− adsorption capacity of CNTs (25 mmol g−1) was found to be higher than PAC (10 mmol g−1).124
In recent research, efforts have been taken to impregnate nanoparticles or composite materials on suitable matrices in-order to increase the efficiency of adsorbent–adsorbate separation after the reaction without any gradual decrease in the adsorption capacity. Alumina nanoparticles with an average diameter of 62 nm were impregnated on to a polyacrylonitrile mixed hollow fiber membrane, which showed a nitrate adsorption capacity of 15 mg g−1.125 The pore size of the membrane and the charge of the alumina nanoparticles dually contributed to the nitrate removal efficiency in aqueous solution. The removal of nitrate on ZnCl2 modified lignite granular activated carbon (LGAC) was investigated by Khan et al.126 The maximum adsorption capacity was found to be 4.4 mg g−1. The NO3− adsorption was mainly due to the active sites –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OZn2+ and –COO–Zn2+, rather than the –OH sites present on the material surface. Hence according to the author the NO3− adsorption by LGAC was due to the physical adsorption process. Hydrotalcite-like mesoporous compounds, also known as layered double hydroxides (LDH), constitute an important class of inorganic materials with desirable properties to remove NO3− from aqueous solution. The impregnation of various metals onto the LDH increases the NO3− adsorption capacity. Zn–Al–Cl and Mg–Al composite impregnated LDH has been synthesized by a co-precipitation method and tested for NO3− removal from water. The adsorption capacities were found to be 40.26 and 35 mg g−1 respectively. The percentage NO3− removal was found to decrease gradually with increasing pH and the optimum pH was found to be 6. The presence of competitive anions reduced the NO3− adsorption in the order carbonate > phosphate > chloride > sulphate. Thus the nature and content of divalent cations in LDH provide a strong influence on the NO3− adsorption process. Similarly, various metal impregnated mesoporous LDHs like Ni–Fe-LDH, Mg–Fe-LDH have been reported for the adsorption of nitrate from aqueous solution.127,128 Biosorbents like chitin and chitosan-derivative based mesoporous materials have gained wide attention with regard to nitrate adsorption. High contents of amino and hydroxyl functional groups in these biosorbents were responsible for their significant adsorption potential towards nitrate. The adsorptive mechanism is mainly due to the electrostatic interactions between the negatively charged NO3− group and the positively charged amine group. The NO3− adsorption capacity of mesoporous chitosan hydro beads prepared by Chatterjee and Woo was found to be 92.1 mg g−1.129 Similarly microalgae immobilized on chitosan nanofiber mats were recently developed by Eroglu et al.130 for the efficient removal of nitrate with its potential, simplicity and highly durable polymer capability. This type of bio-based nanomaterial support, which is water-insoluble and non-toxic, is needed for the efficient adsorption of nitrate from aqueous solution. The pH of the medium plays a major role in nitrate adsorption by most of the nanoadsorbents. At high basic pHs, most of materials do not favour adsorption due to electro static repulsion between the negatively charged adsorbent surface and the NO3− anion. At high acidic pH, the electrostatic attraction takes place between the positively charged adsorbent and the negatively charged adsorbate and makes for an effective adsorption process. In neutral pH nitrate adsorption is still possible by ion exchange and the intraparticle diffusion process. Hence the process of nitrate adsorption by nanoadsorbents mostly takes place via (1) ion exchange, (2) intraparticle diffusion and (3) electrostatic attraction between the adsorbent surface and the pollutant ion.
OZn2+ and –COO–Zn2+, rather than the –OH sites present on the material surface. Hence according to the author the NO3− adsorption by LGAC was due to the physical adsorption process. Hydrotalcite-like mesoporous compounds, also known as layered double hydroxides (LDH), constitute an important class of inorganic materials with desirable properties to remove NO3− from aqueous solution. The impregnation of various metals onto the LDH increases the NO3− adsorption capacity. Zn–Al–Cl and Mg–Al composite impregnated LDH has been synthesized by a co-precipitation method and tested for NO3− removal from water. The adsorption capacities were found to be 40.26 and 35 mg g−1 respectively. The percentage NO3− removal was found to decrease gradually with increasing pH and the optimum pH was found to be 6. The presence of competitive anions reduced the NO3− adsorption in the order carbonate > phosphate > chloride > sulphate. Thus the nature and content of divalent cations in LDH provide a strong influence on the NO3− adsorption process. Similarly, various metal impregnated mesoporous LDHs like Ni–Fe-LDH, Mg–Fe-LDH have been reported for the adsorption of nitrate from aqueous solution.127,128 Biosorbents like chitin and chitosan-derivative based mesoporous materials have gained wide attention with regard to nitrate adsorption. High contents of amino and hydroxyl functional groups in these biosorbents were responsible for their significant adsorption potential towards nitrate. The adsorptive mechanism is mainly due to the electrostatic interactions between the negatively charged NO3− group and the positively charged amine group. The NO3− adsorption capacity of mesoporous chitosan hydro beads prepared by Chatterjee and Woo was found to be 92.1 mg g−1.129 Similarly microalgae immobilized on chitosan nanofiber mats were recently developed by Eroglu et al.130 for the efficient removal of nitrate with its potential, simplicity and highly durable polymer capability. This type of bio-based nanomaterial support, which is water-insoluble and non-toxic, is needed for the efficient adsorption of nitrate from aqueous solution. The pH of the medium plays a major role in nitrate adsorption by most of the nanoadsorbents. At high basic pHs, most of materials do not favour adsorption due to electro static repulsion between the negatively charged adsorbent surface and the NO3− anion. At high acidic pH, the electrostatic attraction takes place between the positively charged adsorbent and the negatively charged adsorbate and makes for an effective adsorption process. In neutral pH nitrate adsorption is still possible by ion exchange and the intraparticle diffusion process. Hence the process of nitrate adsorption by nanoadsorbents mostly takes place via (1) ion exchange, (2) intraparticle diffusion and (3) electrostatic attraction between the adsorbent surface and the pollutant ion.
3.2. Nanomaterials for nitrate removal by photocatalysis
Photocatalytic removal of nitrates from drinking water using various nanomaterials is an important and developing area of research. A number of research studies have reported that the photocatalytic reduction of nitrate in the aqueous solution could be achieved with high efficiency using various types of photocatalysts like TiO2,131 ZnO,132 SiO2,133 CeO2,134 SnO2,135 ZrO2,136 ZnS,137 CdS138 and nZVI.139 Among the above, TiO2 nanoparticles have emerged as promising photocatalysts for water purification units especially for the removal of ions like nitrate, nitrite, arsenic, chromium etc.140,141 TiO2 nanoparticles are very versatile in nature and can act as both oxidative and reductive catalysts to degrade various pollutant ions present in water. The photocatalytic removal of NO3− using titania based nanoparticles is well reported. In brief, the illumination of the TiO2 surface with UV radiation (<385 nm) of energy greater than the band gap energy of the semiconductor TiO2 (3.2 eV) generates valence band holes and conduction band electrons (Fig. 6(a)).144 Holes and electrons react with species adsorbed on the catalyst surface. Valence band holes reacts with water (H2O/OH−) to generate hydroxyl radicals (HO˙), while electrons react with adsorbed molecular oxygen (O2) reducing it to the superoxide radical anion, which in turn reacts with protons to form peroxide radicals. Hydroxyl radicals can further oxidize pollutants ions.142–144 The above mechanism is proposed for the conversion of nitrate to nitrite and further to nitrogen/ammonia in drinking water. The NO3− ions formed during the photocatalytic reaction are further reduced to form ammonia, which in turn reacts with the valence band holes and decomposes to nitrogen gas. As we know generally, titania exhibits in different crystalline phases like anatase, rutile and brookite. The photocatalytic efficiency of titania may vary based on its crystalline phase, Fermi level position, electron mobility, concentration of hydroxyl groups, electronic and surface properties. The brookite phase of titania is rare and unstable in nature. The anatase is more highly photocatalytic than rutile due to its structural differences, electronic band structure, subsequent charge transfer, different mass densities, recombination rate and average effective mass of photo-generated electrons and holes.145–148 Therefore, it is not surprising that anatase usually shows a higher photocatalytic activity than rutile. Recent reports indicate that a mixed phase of anatase and rutile in nanocrystalline TiO2 nanoparticles shows superior photocatalytic efficiency when compared to free anatase and rutile structured materials.149–151 One of the best examples are the commercially available Degussa P25-TiO2 nanoparticles with mixed anatase and rutile phases. This is generally attributed to the formation of n–p junctions at the phase interface which improves the efficiency of charge carrier separation.152 Another requirement of the photocatalytic nitrate reduction process is the addition of simple organic compounds that act as electron hole scavengers to provide electrons to fill the electron holes in the valence band. Previous studies have reported that formic acid is one of the most efficient electron hole scavengers for nitrate reduction over TiO2.153,154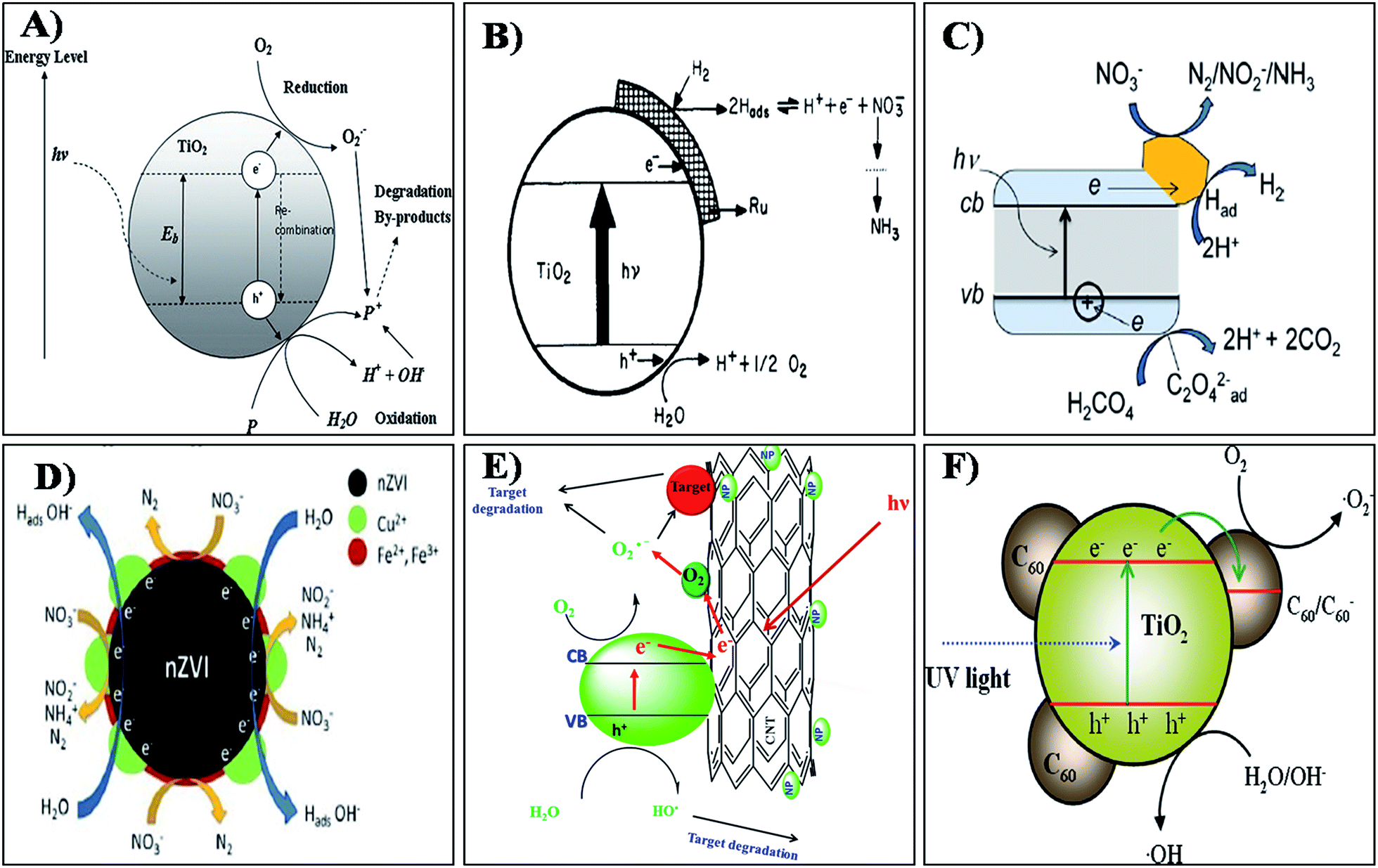 | ||
| Fig. 6 Mechanism of photocatalysis by (A) TIO2, (B) Ru/TiO2, (C) Au/TIO2, (D) Cu-nZVI, (E) metal oxide doped CNT and (F) C60/TiO2 nanocomposites. Reprinted from ref. 144, 156, 163, 175, 192 and 193 with permission from Elsevier, Applied Environmental Research, INTECH, and the Royal Society of Chemistry. | ||
One of the major challenges for the scientific and industrial communities involved in photocatalytic research is to increase the spectral sensitivity of TiO2-based photocatalysts to visible light. The TiO2 nanoparticles are only sensitive to UV radiation, because of which the photocatalytic reactions can take place only under UV light. TiO2 is further modified with various conductive metal nanoparticles to increase the efficiency of NO3− degradation in the visible region. Jin et al.155 studied the metal Cu loaded catalysts Cu/TiO2 and Cu/MgTiO3–TiO2, which exhibited higher photocatalytic activity, indicating that metal loading onto the catalyst was essential for the reduction of NO3−. The composite semiconductor catalyst induces superior photocatalytic activity in the process of NO3− ion reduction. Composite formation and crystalline phase transformation of TiO2 were the important factors in the NO3− reduction.
The ruthenium doped TiO2 catalyst developed by Ranjit et al.156 showed efficient nitrate degradation. The metal loaded on to the semiconductor catalyst can be advantageously used for hydrogenation/reduction reactions. The author exemplifies the NO3− degradation mechanism using the photocatalytic reaction of nitrate into nitrite and further into ammonia by the Ru/TiO2 catalyst. The scheme suggested for the reduction process by the catalyst materials is shown in Fig. 6(b).156 According to the author, photocatalytic reduction of nitrite and nitrate ions is dependent on many factors such as the nature of the metal loaded on the TiO2, the nature of the sacrificial agent, pH of the medium and irradiation time. However, here the author could not observe any other intermediates in his photocatalytic experiments. The photocatalytic reduction of nitrate ions was investigated using pure TiO2 and Ag–TiO2 thin films prepared using a simple sol–gel dip coating technique.157 The optical band gaps of the TiO2 and Ag–TiO2 thin films were found to be 3.27 and 2.70 eV respectively. The net efficiencies of the photocatalytic nitrate reduction of TiO2 and 0.1% Ag–TiO2 were 41.4% and 70.0%, respectively. Sa et al.158 studied the simultaneous degradation of NO3− and organic acids using Ag/TiO2. A 100% nitrate removal was achieved and the residual nitrite and ammonium levels were below the EU guideline levels. Similarly, a report by Zhang et al.159 stated that using an Ag/TiO2 catalyst resulted in a total nitrate conversion of 98% at a N2 selectivity of 100%. This ideal metal doped TiO2 gives better photocatalytic efficiency in the removal of NO3− when compared to plain TiO2.
Likewise Au/TiO2 composite materials have been extensively reported for the removal of NO3− using photocatalysis.160–163 The incorporation of Au results in a significant improvement in the photonic efficiency for titania catalysts due to the generation of Schottky barriers that inhibit the recombination of electron–hole pairs, and the process under UV activation, which is represented by the schematic shown in Fig. 6(c).163 For metals like Au, Cu, and Ag a Schottky barrier is formed and thus electrons flow into the metal and become trapped. This allows for electron storage, facilitating the multi-electron nitrate reduction reactions. Similar photocatalytic reduction of nitrates was reported by Rengaraj and Li,164 Lozovskii et al.165 and Doudrick et al.166 However, the practicability of applying doped nanocrystalline TiO2 catalysts in photocatalytic water treatment needs reconsideration because of the low catalytic activity of the doped TiO2 catalysts under visible light and the possibility of dopant leaching.
The use of nZVI based materials is one promising approach in NO3− removal studies. Various studies have been done on the removal of NO3− using nZVI.167–169 The reaction and its mechanisms between NO3− and nZVI is a true redox reaction.170 Several studies have indicated that the final products of chemical reduction of NO3− by nZVI, could be N2 or NH3 depending on the experimental conditions. Nanoscale zero valent iron supported on pillared clay (nZVI/PILC) was prepared and used for NO3− removal by Zhang et al.171 The as-prepared nZVI of 30–70 nm particle size was evenly distributed along the clay phase. It was reported that, using nZVI/PILC, almost all NO3− was converted to other nitrogen species, so the removal of nitrate by nZVI/PILC was dominantly attributed to the reduction by nZVI instead of adsorption by PILC. Similar conversion of nitrate into nitrogen gas has also been observed in various reports.172–174 Enhanced nitrate reduction by copper impregnated nanoscale zero valent iron (Cu-nZVI) was reported by Krasae et al.175 The results showed that nitrate removal over Cu-nZVI was greater than that of nZVI. By coating nZVI with novel metal catalysts such as Pt, Pd, and Cu, the performance of nZVI toward nitrate removal was found to be increased. The possible reaction mechanism for bimetallic Cu–nZVI nitrate removal is illustrated in Fig. 6(d). Hence Cu coating can enhance nitrate removal performance since Cu is reduced by nZVI to Cu0 on the nZVI surface. Cu can effectively adsorb hydrogen produced from the reaction between nZVI and water, which can then react with nitrate to produce nitrite and continuously react to form ammonia and nitrogen gas. Liou et al.176 found that Cu-nZVI shows a higher photocatalytic efficiency than Pt and Pd loading because Cu can extract oxygen from NO3− and adsorb hydrogen with both adsorbed species reacting continuously. Photocatalytic removal of NO3− was performed by Pan et al.177 using TiO2, nano zero valent iron (nZVI) and nano-TiO2–Fe0 (NTFC) composites. The experimental results confirm that NTFC can effectively remove nitrate when compared to others. The composition of NTFC at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 of TiO2 to nZVI ratio greatly influences the conversion of nitrate to N2, which can be attributed to the maintenance of high level of ferrous ions in the NTFC system due to its reducing condition. The efficiency of functional kaolinite, in supporting the Fe/Ni nanoparticles for the removal of NO3− was reported by Shi et al.178 and Cai et al.179 In this system, Fe0 is said to act as the reducing agent, Ni and Cu act as the catalysts for the hydrogen generation and kaolin serves as a support. The results revealed that, by introducing nickel, the rate of nitrate degradation increases enormously. Removal of nitrate using nano SiO2–FeOOH–Fe core–shell particles was studied by Ensie and Samad.180 The loading of nZVIs was found to play a significant role in NO3− removal: by doubling nZVI loading, removal of NO3− increases from 69% to 86%. This type of core–shell nanostructure increases their stability and also prevents agglomeration in the reaction medium. Su et al.181 studied doping of nZVI with Au, which resulted in a significant reduction in nitrite yield ratio and was found to be much better than nZVI only, or doping nZVI with Cu or Ag. Catalytic hydrogenation of NO3− using bimetallic catalysts has been noted as a promising denitrification method. The ZrO2 supported Pd–Cu bimetallic catalysts were used for the photocatalytic removal of NO3−. Materials containing a high Pd/Cu ratio exhibit a high catalytic activity.182 Palladium doped gold nanoparticles were prepared by Qian et al.183 to study the catalysis mechanisms for NO3− reduction. The results revealed that bimetallic Pd-on-Au NPs had catalytic properties toward rapid and selective reduction of NO3−.
:10 of TiO2 to nZVI ratio greatly influences the conversion of nitrate to N2, which can be attributed to the maintenance of high level of ferrous ions in the NTFC system due to its reducing condition. The efficiency of functional kaolinite, in supporting the Fe/Ni nanoparticles for the removal of NO3− was reported by Shi et al.178 and Cai et al.179 In this system, Fe0 is said to act as the reducing agent, Ni and Cu act as the catalysts for the hydrogen generation and kaolin serves as a support. The results revealed that, by introducing nickel, the rate of nitrate degradation increases enormously. Removal of nitrate using nano SiO2–FeOOH–Fe core–shell particles was studied by Ensie and Samad.180 The loading of nZVIs was found to play a significant role in NO3− removal: by doubling nZVI loading, removal of NO3− increases from 69% to 86%. This type of core–shell nanostructure increases their stability and also prevents agglomeration in the reaction medium. Su et al.181 studied doping of nZVI with Au, which resulted in a significant reduction in nitrite yield ratio and was found to be much better than nZVI only, or doping nZVI with Cu or Ag. Catalytic hydrogenation of NO3− using bimetallic catalysts has been noted as a promising denitrification method. The ZrO2 supported Pd–Cu bimetallic catalysts were used for the photocatalytic removal of NO3−. Materials containing a high Pd/Cu ratio exhibit a high catalytic activity.182 Palladium doped gold nanoparticles were prepared by Qian et al.183 to study the catalysis mechanisms for NO3− reduction. The results revealed that bimetallic Pd-on-Au NPs had catalytic properties toward rapid and selective reduction of NO3−.
Various bimetallic catalyst nanomaterials like Pd–Cu/Al2O3,184 Pd–Sn/Al2O3,185 Pd–In/Al2O3,186 Pd–Cu/TiO2187 and Rh–Cu/Al2O3188 have been reported for the efficient removal of nitrate with high selectivity and specificity. The catalytic behaviors of bimetallic catalysts can also be adjusted by using different supports. Membranes, fibers, resins and ceramics were used as supports in bimetallic catalysts to circumvent the diffusion problem. Deganello et al.189 selected pumice as the support to change the electronic properties of Pd sites to enhance nitrate reduction. Dual processes like photocatalysis and adsorption can be achieved by the hydrotalcite supported Pd–Cu catalyst due to the decreased mass transfer limitation by adsorption of nitrate in the interlayer of hydrotalcite and reduction by the Pd–Cu catalyst. Roveda et al.190 used acrylic resins as supports in Pd–Sn bimetallic catalysts to provide a buffering environment. The Pd–Cu bimetallic catalyst with ZrO2 as the support was found to be more active as compared to the SnO2 supported Pd–Cu catalyst. NaTaO3 nanoparticles were prepared by an ultrasonic method, and Pd was deposited onto the surface of NaTaO3 via photo assisted deposition.191 The different ratio of palladium composition in the nanoparticles yielded different properties. The crystalline size of NaTaO3, 0.2 Pd/NaTaO3, 0.4 Pd/NaTaO3, 0.6 Pd/NaTaO3, and 0.8 Pd NaTaO3 was found to be 15, 12, 10, 8 and 6 nm, respectively. Among the above ratios the 0.6 Pd/NaTaO3 showed better nitrate degradation efficiency when compared to the others. The author experimentally proved that the 1.2 g of 0.6Pd/NaTaO3 in 1000 mL of a 100 mg L−1 nitrate solution yielded a nitrate reduction efficiency of 100% within 50 min of irradiation by visible light. An attempt was made by our group using a co-electro spraying method to deposit TiO2 nanoparticles on to the PAN nanofiber membrane for the photocatalytic removal of nitrates from aqueous solution.15 The maximum nitrate removal was found to be 39% for the initial nitrate concentration of 10 mg L−1. However at higher concentrations, the TiO2/PAN nanofiber membrane was inefficient to remove nitrate. Hence, there is a need for an alteration technique to coat nanoparticles on to the membrane that will not block the photocatalytic process.
The TiO2 based semiconducting nanoparticles were impregnated onto the carbon nanotubes to increase the efficiency of the nitrate removal. CNTs can be used as a catalytic supporter in order to increase not only the specific surface area providing more hydroxyl radicals, but also the quantum efficiency by retarding charge carrier recombination and scavenging photo-generated electrons through the interface between TiO2–carbon nanotube and make TiO2 more sensitive to the visible light. The pictorial representation of the general photocatalytic activity of the metal oxide doped CNT is shown in Fig. 6(e).192 By considering this concept efficient nitrate degradation may also be possible even in visible light. Yu et al.193 reported that the fullerene modified TiO2(C60/TiO2) nanocomposite may also have a better catalytic efficiency. C60 molecules can be dispersed as a monolayer onto bimodal mesoporous TiO2 via covalent bonding. The C60 molecules doped onto TiO2 act as “electron acceptors” responsible for the efficient separation of photo-generated charge carriers and the enhancement of photocatalytic activity.194,195 A schematic illustration of the mechanism of the enhanced photo activity of the C60/TiO2 nanocomposite is shown in Fig. 6(f).195 These separated electrons and holes are then trapped by surface catalysts and increase the recombination time in a favourable way. Hence the development of hybrid catalyst materials using these types of functional materials can facilitate the photo degradation of nitrate molecules.
4. Conclusion
Various advanced nanomaterials used for the removal of fluoride and nitrate from aqueous solution have been summarized briefly in this review. This review gives a clear idea about the tuning of metal oxide based nanoparticles with suitable metal ions which might be helpful in the dual efficiency of adsorption and photocatalytic degradation for the removal of fluoride and nitrate. Alumina, iron oxide, LDH and HAp based hybrid nanomaterials have shown high fluoride and nitrate adsorption efficiency when compared to other nanomaterials. Metal dopants like Cu, Mg Ru Ag Au Pt, Pd influence the photocatalytic performance of the semiconducting nanomaterials like TiO2, SiO2 and CdS etc. Various conventional and non-conventional advanced nanomaterials have been assessed for the removal of fluoride and nitrate from water. Though many of them still have both merits and demerits for practical applications. Researchers have to choose a material that offers satisfactory results in the removal of fluoride or nitrate to below the permissible limit along with low cost, simplicity of design and operation.Acknowledgements
The authors are thankful to PSG Management and the PSG Institute of Advanced studies for their valuable support.Notes and references
- A. Zaporozec, Geol. J., 1981, 5, 457–471 Search PubMed.
- R. E. S. Bain, J. A. Wright, E. Christenson and J. K. Bartram, Sci. Total Environ., 2014, 490, 509–513 CrossRef CAS PubMed.
- H. H. Dieter, Drinking-Water Criteria (Safety, Quality, and Perception) Encyclopedia of Toxicology, 3rd edn, 2014, pp. 227–235 Search PubMed.
- D. A. Birkholz, S. M. Stilson and H. S. Elliott, Reference Module in Earth Systems and Environmental Sciences, 2014, 2, 212–229 Search PubMed.
- E. Kumar, A. Bhatnagar, W. Hogland, M. Marques and M. Sillanpaa, Adv. Colloid Interface Sci., 2014, 203, 11–21 CrossRef CAS PubMed.
- A. Banerjee, Geosci. Front., 2015, 6, 277–284 CrossRef CAS.
- S. Kavitha, R. Selvakumar, M. Sathishkumar, K. Swaminathan, P. Lakshmanaperumalsamy, A. Singh and S. Jain, Water Sci. Technol., 2009, 60(2), 517–524 CrossRef CAS PubMed.
- P. Denbesten and W. Li, Monogr. Oral Sci., 2011, 22, 81–96 Search PubMed.
- M. Abdollahi and F. Momen-Heravi, Fluoride. Encyclopedia of Toxicology, 3rd edn, 2014, pp. 606–610 Search PubMed.
- Y. Zhou, H. Zhang, J. He, X. Chen, Y. Ding, Y. Wang and X. Liu, Food Chem. Toxicol., 2013, 56, 297–303 CrossRef CAS PubMed.
- O. Barbier, L. A. Mendoza, L. María and D. Razo, Chem.-Biol. Interact., 2010, 188, 319–333 CrossRef CAS PubMed.
- K. Yang and X. Liang, Encyclopedia of Environmental Health, 2011, pp. 769–775 Search PubMed.
- H. Liu, Y. Gao, L. Sun, M. Li, B. Li and D. Sun, Int. J. Hyg. Environ. Health, 2014, 217, 413–420 CrossRef CAS PubMed.
- A. M. Fan, Nitrate. Encyclopedia of Toxicology, 3rd edn, 2014, pp. 523–527 Search PubMed.
- S. P. Suriyaraj, M. BenasirBegam, S. G. Deepika, P. Biji and R. Selvakumar, Water Sci. Technol.: Water Supply, 2014, 14, 554 CrossRef CAS.
- N. S. Bryan, D. D. Alexander, J. R. Coughlin, A. L. Milkowski and P. Boffetta, Food Chem. Toxicol., 2012, 50, 3646–3665 CrossRef CAS PubMed.
- N. E. Herrera, K. P. Cantor, N. Malats, D. T. Silverman, A. Tardón, R. García-Closas, C. Serra, M. Kogevinas and C. M. Villanueva, Environ. Res., 2015, 137, 299–307 CrossRef PubMed.
- M. George, L. Wiklund, M. Aastrup, J. Pousette, B. Thunholm, T. Saldeen, L. Wernroth, B. Zaren and L. Holmberg, Eur. J. Clin. Invest., 2001, 31, 1083–1094 CrossRef CAS PubMed.
- C. Kross, J. Prev. Med., 2002, 10, 3–10 Search PubMed.
- A. Cockburn, C. W. Heppner and J. L. C. M. Dorne. Encyclopedia of Food Safety, 2014, vol. 2, pp. 332–336 Search PubMed.
- WHO: World Health Organization, Guidelines for Drinking-water Quality, 4th edn, 2011, http://www.apps.who.int/iris/bitstream/10665/44584/1/9789241548151_eng.pdf Search PubMed.
- S. Jagtap, M. K. Yenkie, N. Labhsetwar and S. Rayalu, Chem. Rev., 2012, 112, 2454–2466 CrossRef CAS PubMed.
- S. Ayoob, A. K. Gupta, T. Venugopal and A. Bhat, Environ. Sci. Technol., 2008, 38, 401–470 CrossRef CAS.
- M. Mohapatra, S. Anand, B. K. Mishra, D. E. Giles and P. Singh, J. Environ. Manage., 2009, 91, 67–77 CrossRef CAS PubMed.
- A. Bhatnagar and M. Sillanpaa, Chem. Eng. J., 2011, 168, 493–504 CrossRef CAS.
- M. Bandpi, D. J. Elliott and M. A. Zazouli, J. Environ. Health Sci. Eng., 2013, 11, 35 CrossRef PubMed.
- S. P. Suriyaraj, T. Vijayaraghavan, P. Biji and R. Selvakumar, J. Environ. Chem. Eng., 2014, 2, 444–454 CrossRef CAS.
- A. Bhatnagar, E. Kumar and M. Sillanpa, Chem. Eng. J., 2011, 171, 811–840 CrossRef CAS.
- P. Loganathan, S. Vigneswaran, J. Kandasamy and R. Naidu, J. Hazard. Mater., 2013, 248, 1–19 CrossRef PubMed.
- V. Tomar and D. Kumar, Chem. Cent. J., 2013, 7, 1–15 CrossRef PubMed.
- M. G. Sujana, H. K. Pradhan and S. Anand, J. Hazard. Mater., 2008, 16, 120–125 Search PubMed.
- P. Loganathan, S. Vigneswaran and J. Kandasamy, J. Environ. Manage., 2013, 131, 363–374 CrossRef CAS PubMed.
- P. Loganathan, S. Vigneswaran and J. Kandasamy, J. Environ. Manage., 2013, 15, 363–374 CrossRef PubMed.
- A. O. Sadik, N. Du, I. Yazgan and V. Okello, Nanotechnology Applications for Clean Water, 2nd edn, 2014, pp. 95–108 Search PubMed.
- M. M. Khin, A. S. Nair, V. JagadeeshBabu, R. Murugan and S. Ramakrishna, Energy Environ. Sci., 2012, 5, 8075–8109 CAS.
- T. Pradeep and Anshup, Thin Solid Films, 2009, 517, 6441–6478 CrossRef CAS.
- M. T. M. Pendergast and E. M. V. Hoek, Energy Environ. Sci., 2011, 4, 1946–1971 CAS.
- S. Kar and P. K. Tewari, A volume in Woodhead Publishing Series in Civil and Structural Engineering, 2013, pp. 364–427 Search PubMed.
- X. Qu, P. J. J. Alvarez and Q. Li, Water Res., 2013, 47, 3931–3946 CrossRef CAS PubMed.
- T. Pradeep and M. S. Bootharaju, Water Reclam. Sustainability, 2014, 317–342 CAS.
- R. R. Devi, I. M. Umlong and P. K. Raul, J. Exp. Nanosci., 2014, 9, 512–524 CrossRef CAS.
- S. M. Maliyekkal, A. K. Sharma and L. Philip, Water Res., 2006, 40, 3497–3506 CrossRef CAS PubMed.
- E. Kumar, A. Bhatnagar, U. Kumar and M. Sillanpaa, J. Hazard. Mater., 2011, 186, 1042–1049 CrossRef CAS PubMed.
- P. K. Raul, R. R. Devi, I. M. Umlong, S. Banerjee, L. Singh and M. Purkait, J. Nanosci. Nanotechnol., 2012, 12, 3922–3930 CrossRef CAS PubMed.
- K. S. Prasad, Y. Amin and K. Selvaraj, J. Hazard. Mater., 2014, 276, 232–240 CrossRef CAS PubMed.
- T. Zhang, Q. Li, Z. Mei, H. Xiao, H. Lu and Y. Zhou, Desalin. Water Treat., 2013, 52, 16–18 CrossRef.
- T. Zhang, Q. Li, H. Xiao, Z. Mei, H. Lu and Y. Zhou, Appl. Clay Sci., 2013, 72, 117–123 CrossRef CAS.
- K. Biswas, S. Debnath and U. C. Ghosh, Sep. Sci. Technol., 2010, 45, 472–485 CrossRef CAS.
- S. Deng, H. Liu, W. Zhou, J. Huang and G. Yu, J. Hazard. Mater., 2011, 186, 1360–1366 CrossRef CAS PubMed.
- A. Ghosh, S. Chakrabarti, K. Biswas and U. C. Ghosh, Appl. Surf. Sci., 2014, 307, 665–676 CrossRef CAS.
- M. Mohapatra, T. Padhi, S. Anand and B. K. Mishra, Desalin. Water Treat., 2012, 50, 376–386 CrossRef CAS.
- L. Li, D. Xu, X. Li, W. Liu and Y. Jia, New J. Chem., 2014, 38, 5445–5452 RSC.
- D. Thakre, S. Rayalu, R. Kawade, S. Meshram, J. Subrt and N. Labhsetwar, J. Hazard. Mater., 2010, 180, 122–130 CrossRef CAS PubMed.
- Z. Li, S. Deng, X. Zhang, W. Zhou, J. Huang and G. Yu, Front. Environ. Sci. Eng. China, 2010, 4, 414–420 CrossRef CAS.
- S. Chidambaram, S. Manikandan, A. L. Ramanathan, M. V. Prasanna, C. Thivya, U. Karmegam, R. Thilagavathi and K. Rajkumar, Appl. Water Sci., 2013, 3, 741–751 CrossRef CAS.
- L. Chai, Y. Wang, N. Zhao, W. Yang and X. You, Water Res., 2013, 1, 4040–4049 CrossRef PubMed.
- L. Chen, B. Y. He, S. He, T. J. Wang, C. L. Su and Y. Jin, Powder Technol., 2012, 227, 3–8 CrossRef CAS.
- C. Zhang, L. Chen, T.-J. Wang, C.-L. Su and Y. Jin, Appl. Surf. Sci., 2014, 317, 552–559 CrossRef CAS.
- M. Poursaberi, K. Hassanisadi, K. Torkestani and M. Zare, Chem. Eng. J., 2012, 189, 117–125 CrossRef.
- N. Minju, K. V. Swaroop, K. Haribabu, V. Sivasubramanian and P. Senthil Kumar, Desalin. Water Treat., 2013, 1–10 Search PubMed.
- X. Zhao, J. Wang, F. Wu, T. Wang, Y. Cai, Y. Shi and G. Jiang, J. Hazard. Mater., 2010, 173, 102–109 CrossRef CAS PubMed.
- M. Bhaumik, T. Y. Leswifi, A. Maity, V. V. Srinivasu and M. S. Onyango, J. Hazard. Mater., 2011, 186, 150–159 CrossRef CAS PubMed.
- C. Gao, X. Yu, T. Luo, Y. Jia, B. Sun, J. Liu and X. Huang, J. Mater. Chem. A, 2014, 2, 2119–2128 CAS.
- N. Thakur, S. A. Kumar, H. Parab, A. K. Pandey, P. Bhatt, S. D. Kumar and A. V. R. Reddy, RSC Adv., 2014, 4, 10350–10357 RSC.
- V. Kumar, N. Talreja, D. Deva, N. Sankararamakrishnan, A. Sharma and N. Verma, Desalination, 2011, 282, 27–38 CrossRef CAS.
- A. Mahapatra, B. G. Mishra and G. Hota, Ind. Eng. Chem. Res., 2013, 52, 1554–1561 CrossRef CAS.
- B. Y. Lee, K. Behler, M. E. Kurtoglu, M. A. Wynosky-Dolfi, R. F. Rest and Y. Gogotsi, J. Nanopart. Res., 2010, 12, 2511–2519 CrossRef CAS.
- S. P. Suriyaraj, M. M. Pillai, A. Bhatacharya and R. Selvakumar, RSC Adv., 2015, 5, 68420–68429 RSC.
- N. Viswanathan and S. Meenakshi, J. Hazard. Mater., 2010, 176, 459–465 CrossRef CAS PubMed.
- C. Jing, J. Cui, Y. Huang and A. Li, ACS Appl. Mater. Interfaces, 2012, 4, 714–720 CAS.
- A. K. Gupta, D. Deva, A. Sharma and N. Verma, Ind. Eng. Chem. Res., 2009, 48, 9697–9707 CrossRef CAS.
- Y. Ma, S. Wang, M. Fan, W. Gong and B. Gao, J. Hazard. Mater., 2009, 168, 1140–1146 CrossRef CAS PubMed.
- C. Janardhana, G. N. Rao, R. S. Sathish, P. S. Kumar, V. A. Kumar and M. V. Madhav, Indian J. Chem. Technol., 2007, 14, 350–354 CAS.
- H. Jin, Z. Ji, J. Yuan, J. Li, M. Liu, C. Xu, J. Dong, P. Hou and S. Hou, J. Alloys Compd., 2015, 620, 361–367 CrossRef CAS.
- Y. Li, S. Wang, X. Zhang, J. Wei, C. Xu, Z. Luan and D. Wu, Mater. Res. Bull., 2003, 38, 469–476 CrossRef CAS.
- N. Chen, Z. Y. Zhang, C. P. Feng, N. Sugiura, M. Li and R. Z. Chen, J. Colloid Interface Sci., 2010a, 348, 579–584 Search PubMed.
- N. Chen, Z. Y. Zhang, C. P. Feng, N. Sugiura, M. Li, R. Z. Chen and D. R. Zhu, J. Hazard. Mater., 2010b, 183, 460–465 Search PubMed.
- S. Sundaram, N. Viswanathan and S. Meenakshi, J. Hazard. Mater., 2008, 155, 206–215 CrossRef PubMed.
- G. E. Poinern, M. K. Ghosh, Y. J. Ng, T. B. Issa, S. Anand and P. Singh, J. Hazard. Mater., 2011, 185, 29–37 CrossRef CAS PubMed.
- D. Zhang, H. Luo, L. Zheng, K. Wang, H. Li, Y. Wang and H. Feng, J. Hazard. Mater., 2012, 241, 418–426 CrossRef PubMed.
- K. Pandi and N. Viswanathan, Carbohydr. Polym., 2014, 112, 662–667 CrossRef CAS PubMed.
- S. Sundaram, N. Viswanathan and S. Meenakshi, Bioresour. Technol., 2008, 99, 8226–8230 CrossRef PubMed.
- X. Yu, S. Tong, M. Ge and J. Zuo, Carbohydr. Polym., 2013, 92, 269–275 CrossRef CAS PubMed.
- Y. Nie, C. Hu and C. Kong, J. Hazard. Mater., 2012, 233, 194–199 CrossRef PubMed.
- S. M. Prabhu and S. Meenakshi, Powder Technol., 2014, 268, 306–315 CrossRef.
- S. G. Wang, Y. Ma, Y.-J. Shi and W.-X. Gong, J. Chem. Technol. Biotechnol., 2009, 84, 1043–1050 CrossRef CAS.
- M. Mohapatra, K. Rout, S. K. Gupta, P. Singh, S. Anand and B. K. Mishra, J. Nanopart. Res., 2010, 12, 681–686 CrossRef CAS.
- J. Wang, W. Xu, L. Chen, Y. Jia, L. Wang, X. Huang and J. Liu, Chem. Eng. J., 2013, 231, 198–205 CrossRef CAS.
- S. Deng, H. Liu, W. Zhou, J. Huang and G. Yua, J. Hazard. Mater., 2011, 186, 1360–1366 CrossRef CAS PubMed.
- X. Zhao, L. Zhang, P. Xiong, M. Wenjing, N. Qian and W. Lu, Microporous Mesoporous Mater., 2015, 201, 91–98 CrossRef CAS.
- C. Zhang, L. Chen, T. Wang, C. Su and Y. Jin, Appl. Surf. Sci., 2014, 317, 552–559 CrossRef CAS.
- L. Chen, H.-X. Wu, T. Wang, Y. Jin, Y. Zhang and X. Dou, Powder Technol., 2009, 193, 59–64 CrossRef CAS.
- M. Vithanage, A. U. Rajapaksh, M. S. Bootharaju and T. Pradeep, Colloids Surf., A, 2014, 462, 124–130 CrossRef CAS.
- W. Wei, X. Wang, Y. Wang, M. Xu, J. Cui and Z. Wei, Desalin. Water Treat., 2014, 52, 6219–6229 CrossRef CAS.
- C. Yang, L. Gao, Y. Wang, X. Tian and S. Komarneni, Microporous Mesoporous Mater., 2014, 197, 156–163 CrossRef CAS.
- L. Chen, B.-Y. He, S. He, T.-J. Wang, C.-L. Su and Y. Jin, Powder Technol., 2012, 227, 3–8 CrossRef CAS.
- N. Chen, C. Feng and M. Li, Clean Technol. Environ. Policy, 2013, 16, 609–617 CrossRef.
- A. Goswami and M. K. Purkait, J. Water Process Eng., 2014, 1, 91–100 CrossRef.
- Q. Shi, Y. Huangb and C. Jing, J. Mater. Chem. A, 2013, 1, 12797–12803 CAS.
- L. Lv, J. He, M. Wei, D. G. Evans and X. Duan, J. Hazard. Mater., 2006, 133, 119–128 CrossRef CAS PubMed.
- J. Nouri, R. Nabizadeh, M. J. rad, M. Yunesian and F. Moattar, Desalin. Water Treat., 2014, 52, 4369–4375 CrossRef CAS.
- S. K. Swain, T. Patnaik and R. K. Dey, Desalin. Water Treat., 2013, 51, 4368–4378 CrossRef CAS.
- P. Koilraj and S. Kannan, Chem. Eng. J., 2013, 234, 406–415 CrossRef CAS.
- S. K. Swain, T. Patnaik, P. C. Patnaik, U. Jha and R. K. Dey, Chem. Eng. J., 2013, 215, 763–771 CrossRef.
- S. Gao, R. Sun, Z. Wei, H. Zhao, H. Li and F. Hu, J. Fluorine Chem., 2009, 130, 550–556 CrossRef CAS.
- X. Yu, S. Ton, M. Ge and J. Zuo, Carbohydr. Polym., 2013, 92, 269–275 CrossRef CAS PubMed.
- T. Zhang, Q. Li, H. Xiao, H. Lu and Y. Zhou, Ind. Eng. Chem. Res., 2012, 51, 11490–11498 CrossRef CAS.
- T. Wu, L. Mao and H. Wang, RSC Adv., 2015, 5, 23246–23254 RSC.
- M. Ansari, M. Kazemipour, M. Dehghani and M. Kazemipour, J. Fluorine Chem., 2011, 132, 516–520 CrossRef CAS.
- S. S. Ramamurthy, Y. Chen, M. K. Kalyan, G. N. Rao, J. Chelli and S. Mitra, J. Nanosci. Nanotechnol., 2011, 11, 3552–3559 CrossRef CAS PubMed.
- Y. H. Li, S. Wang, X. Zhang, J. Wei, C. Xu, Z. Luan, D. Wu and B. Wei, Environ. Technol., 2003, 24, 391–398 CrossRef CAS PubMed.
- N. Sankararamakrishnan, N. Singha and A. Gupta, RSC Adv., 2013, 3, 22421–22429 RSC.
- Y. Chen, Q. Zhang, L. Chen, H. Bai and L. Li, J. Mater. Chem. A, 2013, 1, 13101–13110 CAS.
- H. Golestanifar, A. Asadi, A. Alinezhad, B. Haybati and M. Vosoughi, Desalin. Water Treat., 2015, 1–8 Search PubMed.
- H. Demiral and G. Gündüzoğlu, Bioresour. Technol., 2010, 101, 1675–1680 CrossRef CAS PubMed.
- A. Bhatnagar, E. Kumar and M. Sillanpaa, Chem. Eng. J., 2010, 163, 317–323 CrossRef CAS.
- M. Zhang, B. Gao, Y. Yao, Y. Xue and M. Inyang, Chem. Eng. J., 2012, 210, 26–32 CrossRef CAS.
- R. Saad, S. Hamoudi and K. Belkacemi, J. Porous Mater., 2008, 15, 315–323 CrossRef CAS.
- H. Jiang, P. Chen, S. Luo, X. Tu, Q. Cao and M. Shu, Appl. Surf. Sci., 2013, 284, 942–949 CrossRef CAS.
- E. Motamedi, M. T. Atoueiand and M. Z. Kassaee, Mater. Res. Bull., 2014, 54, 34–40 CrossRef CAS.
- T. Wang, J. Lin, Z. Chen, M. Megharaj and R. Naidu, J. Cleaner Prod., 2014, 83, 413–419 CrossRef CAS.
- I. Mikami, Y. Sakamoto, Y. Yoshinaga and T. Okuhara, Appl. Catal., B, 2003, 44, 79–86 CrossRef CAS.
- M. A. Tofighy and T. Mohammadi, Chem. Eng. Res. Des., 2012, 90, 1815–1822 CrossRef CAS.
- A. Khani and M. Mirzaei, 2nd International IUPAC Conference on Green Chemistry, Russia, September 2008, pp. 14–19 Search PubMed.
- R. Mukherjee and S. De, J. Membr. Sci., 2014, 466, 281–292 CrossRef CAS.
- M. A. Khan, Y. Ahn, M. Kumar, W. Lee, B. Min, G. Kim, D. Cho, W. Park and B. Jeon, Sep. Sci. Technol., 2011, 46, 2575–2584 CrossRef CAS.
- A. Halajnia, S. Oustan, N. Najafi, A. R. Khataee and A. Lakzian, Appl. Clay Sci., 2012, 70, 28–36 CrossRef CAS.
- S. Tezuka, R. Chitrakar, A. Sonoda, K. Ooi and T. Tomida, Adsorption, 2005, 11, 751–755 CrossRef.
- S. Chatterjee and S. H. Woo, J. Hazard. Mater., 2009, 164, 1012–1018 CrossRef CAS PubMed.
- E. Eroglu, V. Agarwa, M. Bradshaw, X. Chen, S. M. Smith, C. L. Raston and K. SwaminathanIyer, Green Chem., 2012, 14, 2682–2685 RSC.
- T. Yang, K. Doudrick and P. Westerhoff, Water Res., 2013, 47, 1299–1307 CrossRef CAS PubMed.
- S. Park, H. J. Kim, J. S. Kim, K. Yoo, J. C. Lee, W. A. Anderson and J. H. Lee, J. Nanosci. Nanotechnol., 2007, 7, 4069–4072 CrossRef CAS PubMed.
- H. Kato and A. Kudo, Phys. Chem. Chem. Phys., 2002b, 4, 2833 Search PubMed.
- A. Devadas, S. Vasudevan and F. Epron, J. Hazard. Mater., 2011, 185, 1412–1417 CrossRef CAS PubMed.
- Y. N. Guo, J. H. Cheng, Y. Y. Hu and D. Li, Appl. Catal., B, 2012, 125, 21–27 CrossRef CAS.
- Z. Xu, L. Chen, Y. Shao, D. Yin and S. Zheng, Ind. Eng. Chem. Res., 2009, 48, 8356–8363 CrossRef CAS.
- O. Hamanoi and A. Kudo, Chem. Lett., 2002, 31, 838–839 CrossRef.
- T. Sato, K. I. Sato, Y. Fujishiro, T. Yoshioka and A. Okuwaki, J. Chem. Technol. Biotechnol., 1996, 67, 345–349 CrossRef CAS.
- Y. Liu, S. Li, Z. Chen, M. Megharaj and R. Naidu, Chemosphere, 2014, 108, 426–432 CrossRef CAS PubMed.
- A. O. Ibhadon and P. Fitzpatrick, Catalysis, 2013, 3, 189–218 CAS.
- B. Ayati, A. Ahmadpour, F. Bamoharram, B. Tanhaei, M. Manttari and M. Sillanpaa, Chemosphere, 2014, 107, 163–174 CrossRef PubMed.
- U. I. Gayaa and H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef.
- S. Emad, S. Elmolla and M. Chaudhuri, Desalination, 2010, 252, 46–52 CrossRef.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- J. Zhang, P. Zhou, J. Liub and J. Yu, Phys. Chem. Chem. Phys., 2014, 16, 20382 RSC.
- S. D. Mo and W. Y. Ching, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13023–13032 CrossRef CAS.
- T. Luttrell, S. Halpegamage and J. Tao, Sci. Rep., 2014, 4, 1–8 Search PubMed.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- Z. Yigit and H. Inan, Water, Air, Soil Pollut.: Focus, 2009, 9, 237–243 CrossRef CAS.
- Z. Luo, A. S. Poyraz, C. H. Kuo, R. Miao, Y. Meng, S. Y. Chen, T. Jiang, C. Wenos and S. L. Suib, Chem. Mater., 2015, 27, 6–17 CrossRef CAS.
- A. D. Paola, M. Bellardita and L. P. Brookite, Catalysis, 2013, 3, 36–73 Search PubMed.
- D. C. Hurum, A. G. Agrios and K. A. Gray, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- J. Sa, T. Berger, K. Fottinger, A. Riss, J. A. Anderson and H. Vinek, J. Catal., 2005, 234, 282–291 CrossRef CAS.
- N. Wehbe, M. Jaafar, C. Guillard, J. M. Herrmann, S. Miachon, E. Puzenat and N. Guilhaume, Appl. Catal., A, 2009, 368, 1–8 CrossRef CAS.
- R. Jin, W. Gao, J. Chen, H. Zeng, F. Zhang, Z. Liu and N. Guan, J. Photochem. Photobiol., A, 2004, 162, 585–590 CrossRef CAS.
- K. T. Ranjit, T. K. Varadarajan and B. Viswanathan, J. Photochem. Photobiol., A, 1995, 89, 67–68 CrossRef CAS.
- K. Kobwittaya and S. Sirivithayapakorn, J. Saudi Chem. Soc., 2014, 18, 291–298 CrossRef.
- J. Sa, C. A. Aguera, S. Gross and J. A. Anderson, Appl. Catal., B, 2009, 85, 192–200 CrossRef CAS.
- F. Zhang, R. Jin, J. Chen, C. Shao, W. Gao, L. Li and N. Guan, J. Catal., 2005, 232, 2424–2431 CrossRef.
- M. Shand and J. A. Anderson, Catal. Sci. Technol., 2013, 3, 879–899 CAS.
- M. C. Raphu, J. McPherson, E. van der Lingen, J. A. Anderson and M. S. Scurrell, Gold Bull., 2010, 43, 334 CrossRef.
- J. A. Anderson, Catal. Today, 2011, 175, 316–321 CrossRef CAS.
- J. A. Anderson, Catal. Today, 2012, 181, 171–176 CrossRef CAS.
- S. Rengaraj and X. Z. Li, Chemosphere, 2007, 66, 5930–5938 CrossRef PubMed.
- A. V. Lozovskii, I. V. Stolyarova, R. V. Prikhod and V. V. Goncharuk, J. Water Chem. Technol., 2009, 31, 360–366 CrossRef.
- K. Doudrick, O. Monzon, A. Mangonon, K. Hristovski and P. Westerhoff, J. Environ. Eng., 2012, 138, 852–861 CrossRef CAS.
- J. Hsu, C. Liao and Y. Wei, Sustainable Environ. Res., 2011, 21, 353–359 CAS.
- A. Su and R. W. Puls, Environ. Sci. Technol., 2004, 38, 2715–2720 CrossRef PubMed.
- S. Ziajahromi, M. Khanizadeh, M. Khiadani, A. D. Zand and M. Mehrdad, Middle East J. Sci. Res., 2013, 16, 205–209 CAS.
- C. Yang and H. L. Lee, Water Res., 2005, 39, 884–894 CrossRef PubMed.
- Y. Zhang, Y. Li, J. Li, L. Hu and X. Zheng, Chem. Eng. J., 2011, 171, 526–531 CrossRef CAS.
- S. Choe, Y. Chang, K. Hwang and J. Khim, Chemosphere, 2000, 41, 1307–1311 CrossRef CAS PubMed.
- C. Yang and H. L. Lee, Water Res., 2005, 39, 884–894 CrossRef PubMed.
- W. Wang, Z. Jin, T. Li, H. Zhang and S. Gao, Chemosphere, 2006, 65, 1396–1404 CrossRef CAS PubMed.
- N. Krasae, K. Wantala, P. Tantriratna and N. Grisdanurak, Appl. Environ. Res., 2014, 36, 15–23 Search PubMed.
- Y. H. Liou, C. J. Lin, S. C. Weng, H. H. Ou and S. L. Lo, Environ. Sci. Technol., 2009, 43, 2482–2488 CrossRef CAS PubMed.
- J. R. Pan, C. Huang, W. P. Hsieh and B. J. Wu, Sep. Purif. Technol., 2012, 84, 52–55 CrossRef CAS.
- Q. Shi, Y. Huang and C. Jing, J. Mater. Chem. A, 2013, 1, 12797–12803 CAS.
- X. Cai, Y. Gao, Q. Sun, Z. Chen, M. Megharaj and R. Naidu, Chem. Eng. J., 2014, 244, 19–26 CrossRef CAS.
- A. Ensie and S. Samad, Desalination, 2014, 347, 1–9 CrossRef.
- B. Su and R. W. Puls, Environ. Sci. Technol., 2004, 38, 2715–2720 CrossRef PubMed.
- S. Xu and D. D. Sun, Int. J. Hydrogen Energy, 2009, 34, 6096–6104 CrossRef CAS.
- H. Qian, Z. Zhao, J. C. Velazquez, L. A. Pretzer, K. N. Heck and M. S. Wong, Nanoscale, 2014, 6, 358–364 RSC.
- S. Jung, S. Bae and W. Le, Environ. Sci. Technol., 2014, 48, 9651–9658 CrossRef CAS PubMed.
- S. Ambonguilat, H. Gallard, A. Garron, F. Epron and J. P. Croue, Water Res., 2006, 40, 675–682 CrossRef CAS PubMed.
- Z. Gao, Y. Zhang, D. Li, C. J. Werth, Y. Zhang and X. Zhou, J. Hazard. Mater., 2015, 3, 425–431 CrossRef PubMed.
- M. S. Kim, S. H. Chung, C. J. Yoo, M. S. Lee, I. H. Cho and D. W. Lee, Appl. Catal., B, 2013, 142, 354–361 CrossRef.
- I. Witońska, S. Karski and J. Gołuchowska, Kinet. Catal., 2007, 48, 823–828 CrossRef.
- F. Deganello, L. F. Liotta, A. Macaluso, A. M. Venezia and G. Deganello, Appl. Catal., B, 2000, 24, 265–273 CrossRef CAS.
- A. Roveda, A. Benedetti, F. Pinna and G. Strukul, Inorg. Chim. Acta, 2003, 349, 203–208 CrossRef CAS.
- R. M. Mohameda and E. S. Baeissa, J. Ind. Eng. Chem., 2014, 20, 1367–1372 CrossRef.
- T. A. Saleh, The Role of Carbon Nanotubes in Enhancement of Photocatalysis, Synthesis and applicationsof Carbon Nanotubes and their composites, ed. Satoru Suzuki, InTech, 2013, ISBN: 978-953-51-1125-2, DOI:10.5772/51050.
- J. Yu, T. Ma, G. Liu and B. Cheng, Dalton Trans., 2011, 40, 6635–6644 RSC.
- L. Zhang, Y. Wang, T. Xu, S. Zhu and Y. Zhu, J. Mol. Catal. A: Chem., 2010, 331, 7–14 CrossRef CAS.
- Z. D. Meng, L. Zhu, S. Ye, Q. Sun, K. Ullah, K. Y. Cho and W. C. Oh, Nanoscale Res. Lett., 2013, 8, 189 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |