
Excellent lithium ion storage property of porous MnCo2O4 nanorods†
Peiyuan Zeng,
Xiaoxiao Wang,
Ming Ye,
Qiuyang Ma,
Jianwen Li,
Wanwan Wang,
Baoyou Geng* and
Zhen Fang*
Key Laboratory of Functional Molecular Solids, Ministry of Education, Center for Nano Science and Technology, College of Chemistry and Materials Science, Anhui Normal University, East Beijing Road 1#, Wuhu, 241000, P. R. China. E-mail: bygeng@mail.ahnu.edu.cn; fzfscn@mail.ahnu.edu.cn
First published on 23rd February 2016
Abstract
In this report, we present a facile method for the synthesis of porous MnCo2O4 nanorods. When the as-synthesized MnCo2O4 nanorods are applied as the anode material for lithium-ion batteries, they exhibit excellent electrochemical performance owing to their porous nature. Because of the delicate balance between structural stability and specific surface area, the porous MnCo2O4 nanorods show high discharge capacity and excellent cycling stability. The initial discharge capacity is 1845 mA h g−1 at a current density of 0.4 A g−1. Even being cycled at a current density of 30 A g−1, the discharge capacity is still about 533 mA h g−1. The effects of binders on the electrochemical performance of the electrode have also been investigated, showing that the CMC/SBR binder is more suitable than PVDF for the as-synthesized porous MnCo2O4 nanorods. The influences of the calcination temperature also have been systematically investigated, which suggest the electrochemical performance is determined by both the structural stability and surface area of the samples.
1. Introduction
Nowadays, lithium ion batteries (LIBs) are playing more and more important roles in daily life because of their relatively high energy densities and widely applications in portable devices. Though graphite has been widely used as the anode material, the low theoretical capacity (372 mA h g−1) of graphite severely limits its application in high performance LIBs. Therefore, searching for new anode materials with high capacity, high rate performance and long cycle life have been the hot spot of research in recent years. Among these anode materials, Co3O4 is considered to be a good candidate because of its high theoretical specific capacity (890 mA h g−1) and electrochemical activity.1 As a result, Co3O4 micro-/nano-structures with different morphologies such as flower-like, mesoporous microdisks, nanowires, nanoparticles, etc. had been successfully prepared. Although these Co3O4 micro-/nano-structures exhibit excellent electrochemical performances when used as anode material in LIBs, the electrochemical performances is expected to be further improved to satisfy the large-scale applications in LIBs.2–5 According to the previous reports, two formidable challenges still remain. One centers on the high cost of Co3O4, which greatly restrict its large-scale applications. A second entails the easily agglomeration and intrinsically low conductivity during the cycling process, which severely decrease the cycle life in practical use.The introduction of manganese (Mn) to the A sites of the spinel structured Co3O4 had been proved to be an effective method to improve the electrochemical performance of Co3O4. On one hand, Mn is more abundant in nature and 20 times cheaper than cobalt, which will effectively lower the cost of the anode material.6 On the other hand, the introduction of Mn will increase the intrinsic conductivity and decrease the operation voltage of Co3O4, which will be beneficial for the improvement of cycling performance. As a result, varieties of MnCo2O4 structures with different morphologies have been successfully prepared. Wang and co-workers reported the synthesis of flake-like MnCo2O4 structures through the hydrothermal method, the initial discharge capacity of which was measured to be 1460 mA h g−1 at a current density of 100 mA g−1. And a reversible discharge capacity of 952 mA h g−1 can still be retained after 100 cycles.7 Kim and co-workers also succeeded in the synthesis of MnCo2O4 hollow nanofibers via the electrospun method, which can deliver initial discharge and charge capacities of 1402 mA h g−1 and 1029 mA h g−1, respectively (current density: 100 mA g−1). Even after 50 cycles, the discharge capacity was still about 997 mA h g−1.8 Xiong and co-workers have synthesized uniform hollow MnCo2O4 submicrospheres with multilevel interiors by controlling the heating rate during the calcination process. This unique structures could supply a high contact area and structural stability, which deliver an initial discharge and charge specific capacity of 1425 and 1119 mA h g−1 at a current density of 400 mA g−1, respectively.9 Chen and co-workers reported the synthesis of hierarchical MnCo2O4 nanosheet arrays on carbon cloths, which present an initial columbic efficiency of 80%. And even being cycled at a current density of 800 μA cm−2, the integrated anodes exhibited a high areal capacity of 3.0 mA h cm−2.10 Mohamed and co-workers reported a facile hydrothermal method to synthesize a series of nanostructured cobaltates.11 These mesoporous structures could enable the flow of oxygen and provide good two-phase interface for catalysis. What's more, the unique structure also could provide additional storage space for lithium peroxide and improved the capacity. Li and co-workers synthesized the porous MnCo2O4.5 hierarchical architectures via a hydrothermal method with uniform morphology.12 The unique electrode demonstrated excellent rate capability and long term stability, which may ascribe to the unique porous and robust hierarchical structure. In this aspect, the as-prepared MnCo2O4.5 can be regarded as a promising material for high-rate supercapacitors. Despite continuous progress that have been made in this field, the large-scale synthesis of MnCo2O4 nanostructures with excellent electrochemical performance is still a big challenge, especially by a simple and economic method.
In this paper, we report a two-step method for the synthesis of porous MnCo2O4 nanorods, using oxalic acid as the precipitation reagent and sodium dodecyl sulfonate (SDS, CH3(CH2)11SO3Na) as the surfactant. The room temperature reaction will lead to the formation of metal oxalate precursor (Mn0.33Co0.67C2O4·2H2O), which will then transform to porous MnCo2O4 nanorods during the following calcination process. It should be noted that the yield of the product is in gram-scale, which make it a promising candidate for large-scale application. The sample obtained by calcining at 500 °C for 2 h (denoted as MCO) shows the best electrochemical performance, which can be ascribed to the synergistic effect of optimized surface area and structural stability. The as-prepared porous MnCo2O4 nanorods exhibit excellent electrochemical performance at high current density, especially when CMC/SBR is used as the binder. The electrodes using CMC/SBR as the binder show high initial discharge capacity of 1845 mA h g−1 and excellent capacity retention of 1620 mA h g−1 after 700 cycles at current density of 0.4 A g−1. Remarkably, the as-prepared porous MCO electrode using CMC/SBR as the binder performs excellently at a high rate of 0.4–30 A g−1. Even being cycled at 30 A g−1, the capacity is still about 533 mA h g−1. The effect of binder on the electrochemical performance of the electrode also has been investigated, which suggest that the electrode using PVDF exhibits poorer performance and the capacity faded very fast to less than 500 mA h g−1 after 70 cycles at a current density of 0.4 A g−1. To investigate the influence of calcination temperature on the electrochemical performance, the precursor was calcined at 400 °C (MCO-1) and 600 °C (MCO-2) for 2 h, respectively. The initial discharge capacities for samples MCO-1 and MCO-2 are 1527 mA h g−1 and 1522 mA h g−1, respectively. After 200 cycles, the capacities are only 851 mA h g−1 for sample MCO-1 and 555 mA h g−1 for sample MCO-2. The difference in the electrochemical performance can be ascribed to the differences in BET surface areas and structural stabilities.
2. Experimental section
2.1 Materials
Cobalt chloride hexahydrate (CoCl2·6H2O), oxalic acid (H2C2O4·2H2O), manganese(II) acetate tetrahydrate (Mn(CH3COO)2·4H2O), sodium dodecyl sulfonate (SDS, CH3(CH2)11SO3Na) were purchased from Tianjin Fengchuan Chemical Reagent Technologies Co., Ltd. (Tianjin, China). All reagents were used as received without further purification.2.2 Synthesis of porous MnCo2O4 porous structures
In a typical synthetic process, 50 mmol SDS was (CH3(CH2)11SO3Na) firstly dissolved in a mixed solution containing 500 mL ethylene glycol (EG) and 200 mL deionized water. In the next step, the metal salts (16.5 mmol Mn (CH3COO)2·4H2O, 33.5 mmol CoCl2·6H2O) and H2C2O4·2H2O (50 mmol) were added into two identical solutions with the above compositions, respectively. The two above solutions were mixed and stirred at 25 °C for another 6 h. The as-obtained yellow precipitation was collected by centrifugation, washed several times with distilled water, absolute ethanol and dried in vacuum at 60 °C for 12 h (6.4 g). The MnCo2O4 products were obtained by calcining the above precursor in air at different temperature for 2 h with a heating rate of 1 °C min−1 (2.5 g).2.3 Sample characterizations
The phase purities of the samples were characterized by X-ray diffraction (Bruker D8 ADVANCE) with irradiation from a Cu target (Kα, λ = 0.15406 nm). X-ray photoelectron spectra (XPS) spectra of the samples were recorded on an ESCALAB 250. The morphologies and sizes of the sample were observed by a field-emission scanning electron microscopy (FESEM) (Hitachi S-4800) and transmission electron microscope (TEM) (FEI TECNAI-G2). High-resolution transmission electron microscopy (HRTEM) images were obtained using a transmission electron microscopy (TEM, JEOL-2010) with an accelerating voltage of 200 kV. Thermogravimetric analysis (TGA) was measured by SDT 2960 with a heating rate of 10 °C min−1 from 20 °C to 800 °C. The Brunauer–Emmett–Teller (BET) tests were determined via a Quantachrome autosorb IQ-C nitrogen adsorption apparatus. All the as-prepared samples were degassed at 150 °C for 4 h prior to nitrogen adsorption measurements.2.4 Electrochemical measurements
The working electrodes were consisted of 50 wt% active materials, 30 wt% Super P conductive additive and CMC/SBR (weight ratio = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5%). The slurries were coated on a copper foil, and then dried in vacuum at 120 °C for 12 h. Electrochemical experiments were performed using 2032-type coin cells, which were assembled in an argon-filled glove box (Mikrouna, Super (1220/750/900)). Both moisture and oxygen concentrations were below 1.0 ppm. The electrolyte was composed of 1 M LiPF6 in a mixture of ethylene carbonate, diethyl carbonate (EC:DEC = 1:1, in volume). The galvanostatic discharge–charge characteristics were tested between the potentials of 0.01 and 3.0 V using a Neware battery tester. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were tested on a CHI660E electrochemical workstation.
:5%). The slurries were coated on a copper foil, and then dried in vacuum at 120 °C for 12 h. Electrochemical experiments were performed using 2032-type coin cells, which were assembled in an argon-filled glove box (Mikrouna, Super (1220/750/900)). Both moisture and oxygen concentrations were below 1.0 ppm. The electrolyte was composed of 1 M LiPF6 in a mixture of ethylene carbonate, diethyl carbonate (EC:DEC = 1:1, in volume). The galvanostatic discharge–charge characteristics were tested between the potentials of 0.01 and 3.0 V using a Neware battery tester. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were tested on a CHI660E electrochemical workstation.
3. Results and discussion
3.1 Sample characterizations
The room temperature reaction will not lead to the formation of MnCo2O4 directly, which has been confirmed by the XRD pattern of the precursor (ESI, Fig. S1a†). The phase of the precursor can be determined to be an orthorhombic cell with a = 11.877 Å, b = 5.419 Å, c = 15.624 Å and α = β = γ = 90°, which is similar to the structure of CoC2O4·2H2O. Compared to the structure of CoC2O4·2H2O, the as-obtained precursor can be regarded as a partially substituted CoC2O4·2H2O, in which the Co atoms are partially substituted by Mn atoms. As a result, the formula of the precursor can be tentatively described as Mn0.33Co0.67C2O4·2H2O. The composition of the precursor can be further proved by the thermogravimetric analysis (TGA) (Fig. 1a). The total weight loss is about 56%, which is consistent with the transformation from Mn0.33Co0.67C2O4·2H2O to MnCo2O4. The first weight loss process corresponds to the loss of the two water molecules from the precursor (measured value: 20%, theoretical value: 19.8%). The second weight loss corresponds to the transformation from Mn0.33Co0.67C2O4 to MnCo2O4 (measured value: 36%, theoretical value: 36.5%). Based on the above analysis, the formation process of the MnCo2O4 porous structures can be described as follows:| 0.33Mn2+ + 0.67Co2+ + C2O42− + 2H2O = Mn0.33Co0.67C2O4·2H2O | (1) |
| Mn0.33Co0.67C2O4·2H2O = Mn0.33Co0.67C2O4 + 2H2O | (2) |
| 3Mn0.33Co0.67C2O4 = MnCo2O4 + 2CO2 + 4CO | (3) |
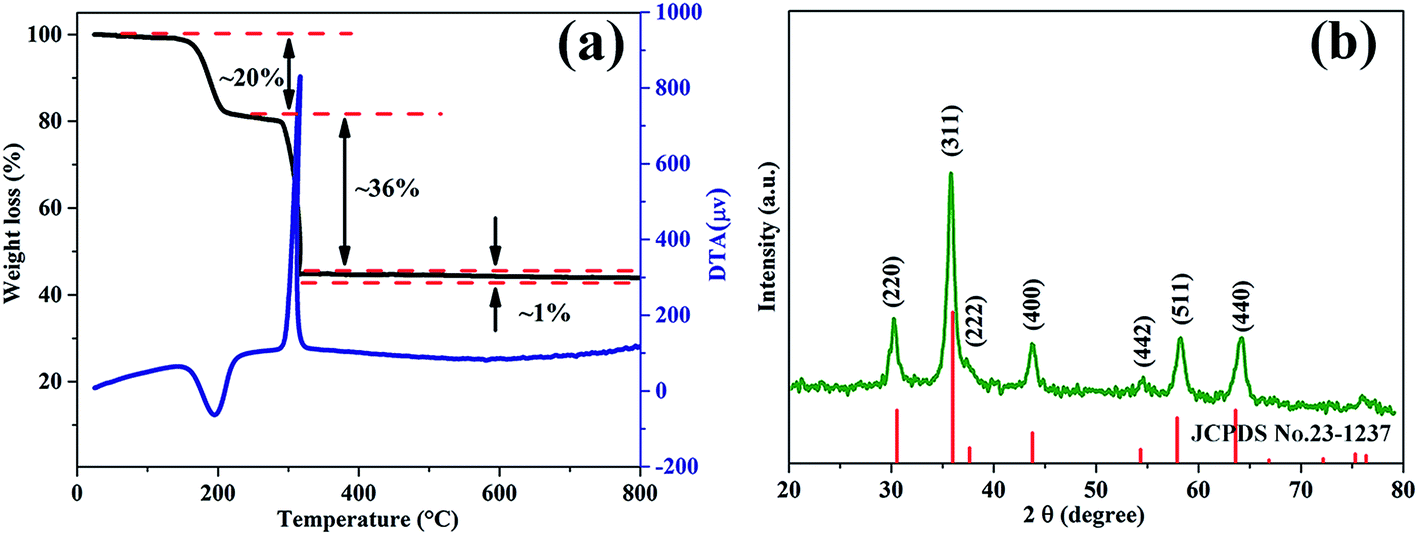 | ||
| Fig. 1 (a) TG-DTA analysis curves of the as-prepared oxalate precursor, (b) XRD pattern of the as-obtained sample obtained by calcining the precursor at 500 °C for 2 h in the air. | ||
Fig. 1b is the XRD pattern of the sample obtained by calcining the precursor at 500 °C for 2 h, on which all the diffraction peaks can be unambiguously indexed to be the cubic-phased MnCo2O4 (JCPDS no. 23-1237 space group: Fd3m, a = b = c = 8.269 Å, α = β = γ = 90°). No other impurity such as MnO2 or Co3O4 is detected, indicating high purity of the as-prepared sample. Calculated by the Scherrer equation, the average crystal size of the MnCo2O4 nanoparticles is determined to be about 11.77 nm, according to the full width at half-maximum (0.0146) of the (311) peak.
Fig. 2a and b are the SEM images of the sample obtained by calcining the precursor at 500 °C for 2 h, which clearly indicates that the whole sample is composed of MnCo2O4 nanorods with rough surfaces and porous structures. Compared to the oxalate precursor (ESI, Fig. S1b†), the overall morphology doesn't change after the calcination process. However, the sizes of these nanorods slightly decrease as compared to the precursor, which may result from the removal of CO2 and H2O during the pyrolysis process. To investigate the detailed structures of MnCo2O4 nanorods, TEM and HRTEM (High-Resolution TEM) were employed. Fig. 2c is the TEM image of a single MnCo2O4 nanorod, which clearly suggests the porous nature of the as-obtained nanorod. The as-obtained MnCo2O4 nanorod is composed of a large number of small nanoparticles which serve as the subunit for the construction of the porous structures. The average diameter of these nanoparticles is determined to be ∼10 nm, which is consistent with the result calculated by the Scherrer equation. Fig. 2d is the representative HRTEM image of the as-obtained MnCo2O4 nanorods, on which a series of lattice fringes can be clearly observed. The typical inter-planar distance, being determined to be 0.249 nm, corresponds to the (311) plane of cubic phased MnCo2O4.
 | ||
| Fig. 2 (a and b) Low and high magnification SEM images of the as-obtained MnCo2O4 nanorods, (c and d) TEM and HRTEM images of the as-obtained porous MnCo2O4 nanorods. | ||
In order to determine the elemental composition and valence states of elements, the as-prepared MnCo2O4 nanorods was further investigated by the XPS. All of the binding energies were corrected for specimen charging by referring them to the C 1s peak (284.6 eV). Fig. 3a is the overall XPS spectrum of the as-prepared MnCo2O4 nanorods, which clearly indicate the existence of Co, Mn and O. The atom ratio between Co and Mn is determined to be 2:1, which is consistent with the theoretical value. The XPS spectrum of Co 2p (Fig. 3b) can be best fitted by considering two spin–orbit doublet characteristics of Co3+ and Co2+, which clearly demonstrate the existence of Co with different valence states (Co3+ and Co2+). The two peaks locating at 780.1 (Co 2p3/2) and 795.7 eV (Co 2p1/2) can be ascribed to the Co3+, while the peaks locating at 780.9 (Co 2p3/2) and 795.5 eV (Co 2p1/2) can be ascribed to Co2+. Similarly, the XPS spectrum of Mn 2p (Fig. 3c) also suggests the existence of Mn3+ and Mn2+. The two main peaks locating at 642.7 (Mn 2p3/2) and 654.1 eV (Mn 2p1/2) belong to Mn3+, while the two peaks locating at 641.9 (Mn 2p3/2) and 652.5 eV (Mn 2p1/2) could be ascribed to Mn2+.13,14 The existence of Mn3+/Mn2+ and Co3+/Co2+ solid-state redox couples could provide a notable electrochemical activity, which would be beneficial for the improvement of the LIBs.15
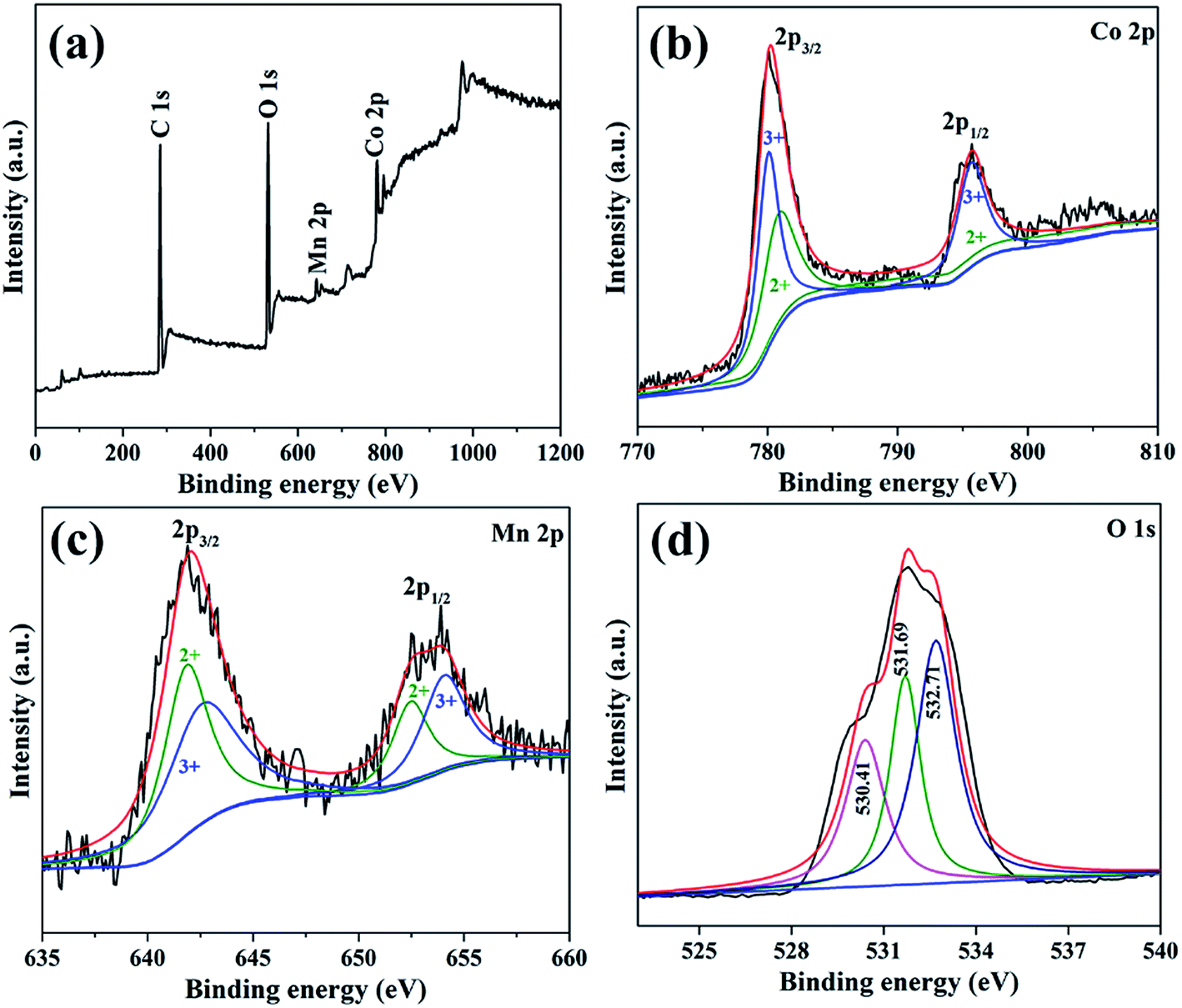 | ||
| Fig. 3 XPS spectra of (a) survey spectrum, (b) Co 2p, (c) Mn 2p and (d) O 1s for the MnCo2O4 nanorods obtained by calcining the precursor at 500 °C for 2 h in air. | ||
To further investigate the detailed porous nature of MnCo2O4 nanorods, Brunauer–Emmett–Teller (BET) gas-sorption measurements were performed. The N2 adsorption–desorption isotherms and Barrett–Joyner–Halenda (BJH) pore-size distribution are shown in ESI (Fig. S2†). The N2 adsorption–desorption isotherm of the as-obtained MnCo2O4 nanorods can be classified as type-IV with a type-H4 hysteresis loop, which clearly indicates the mesoporous nature of the product.16–18 By calculation, the BET surface area of the as-prepared MnCo2O4 nanorods is determined to be 70.0 m2 g−1. The average pore size is determined to be ∼13 nm using the BJH method, suggesting mesoporous nature of the product. The large surface area and uniform pore size distribution of the product will facilitate the insertion and extraction of Li+, which will be beneficial for the improvement of electrochemical performance.
3.2 Electrochemical performance of the as-prepared porous MnCo2O4 nanorods
The electrochemical property of the as-prepared MnCo2O4 nanorods was firstly investigated using the cyclic voltammetry (CV) method. Fig. 4a is the first three consecutive CV curves at a scanning rate of 0.1 mV s−1, with the voltage sweeping from 0.01 to 3.0 V. The first cycle of the CV curve is obviously different from the other two cycles, while the second and third curve almost overlaps. In the first cycle, there is a sharp reduction peak at ∼0.72 V in the cathodic process, which corresponds to the reduction of MnCo2O4 to Mn0 and Co0 and the irreversible formation of solid electrolyte interphase (SEI) layer. In the anodic process, two oxidation peaks can be observed at 1.52 V and 2.09 V, corresponding to the oxidation of Mn0 and Co0 to Mn2+ and Co2+, respectively.19 Because of the irreversible electrochemical reaction in the first cycle, the reduction peak shifted from 0.72 V to 0.84 V and the intensity also dramatically decreases in the following cycles. Both the shift of reduction peak and overlapped CV curves indicate the good reversibility and excellent performance of the as-prepared sample during the cycling process. The electrochemical reaction of this process can be expressed as follows:13| MnCo2O4 + 8Li+ + 8e− ↔ Mn + 2Co + 4Li2O | (4) |
| Mn + 2Co + 3Li2O ↔ MnO + 2CoO + 6Li+ + 6e− | (5) |
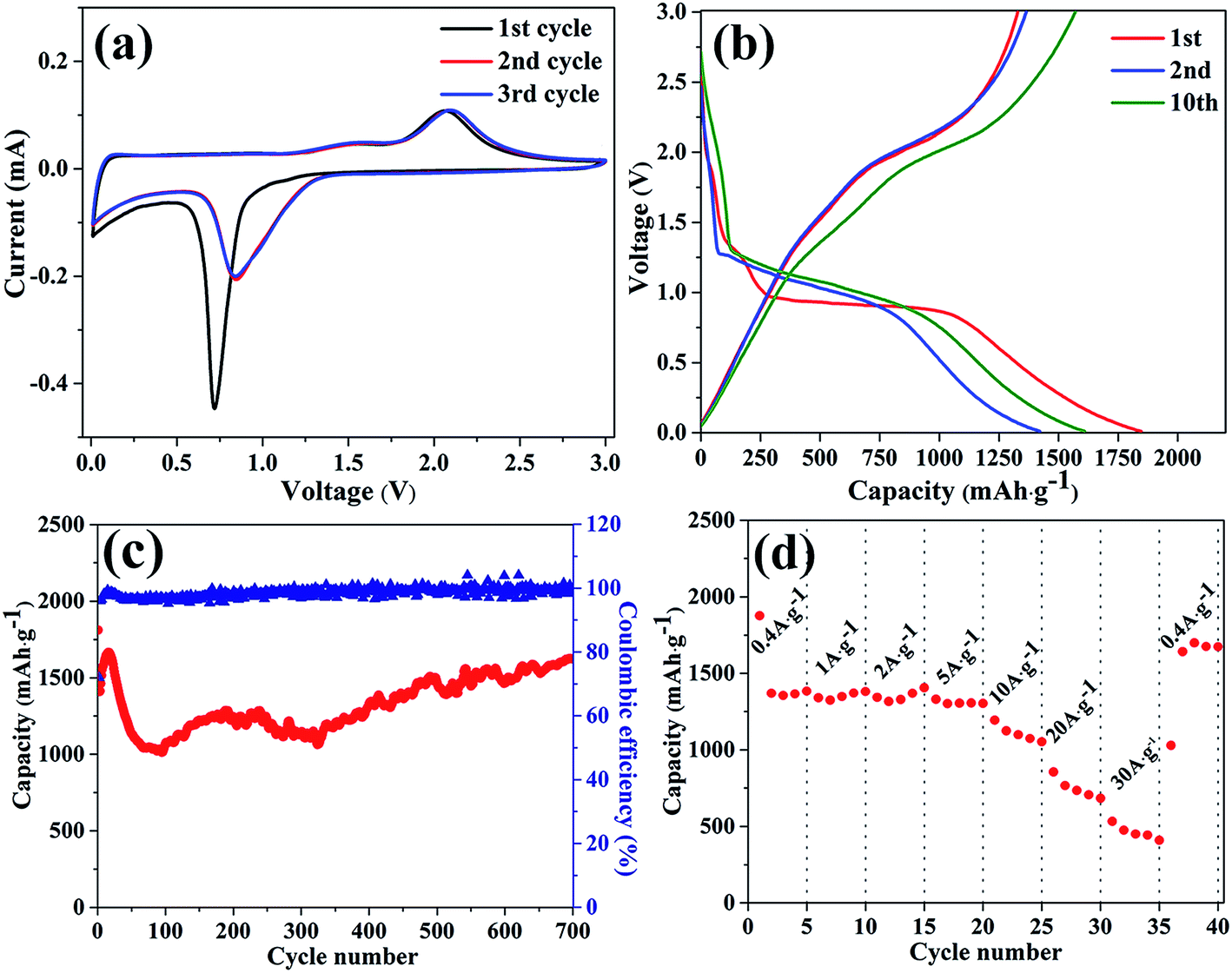 | ||
| Fig. 4 (a) The first three consecutive cycles of CVs for the porous MnCo2O4 nanorods at a scan rate of 0.1 mV s−1, (b) the 1st, 2nd and 10th galvanostatic charge–discharge profiles for the porous MnCo2O4 nanorods at a constant current density of 0.4 A g−1. (c) Cycling performance of the porous MnCo2O4 nanorods at a constant current density of 0.4 A g−1. (d) Rate performance of porous MnCo2O4 nanorods. | ||
Fig. 4b is the galvanostatic charge/discharge curves of the as-prepared MnCo2O4 nanorods, under a current density of 0.4 A g−1. The initial discharge and charge capacities for the as-prepared MnCo2O4 nanorods are 1845.8 mA h g−1 and 1328.7 mA h g−1 respectively and the corresponding columbic efficiency is 71.99%. The specific capacities are much higher than the theoretical capacity of MnCo2O4 (906 mA h g−1), and this phenomenon has also been reported for other transitional oxides nanostructure, such as iron oxide,20–22 cobalt oxide,23 manganese oxide,24 and nickel oxide.25 Though the exact reason for the excessive capacity is not clear, the deduced reason for this phenomenon can be clarified from two aspects. The first aspect relates to the decomposition of the SEI layer and organic polymeric/gel-like film. Previous reports have pointed that the decomposition of electrolyte and the formation of SEI layer will lead to the excessive discharge capacity during the electrochemical process, which is regarded as an important factor for the excessive capacity in our experiment.20 Meanwhile, at low voltage process, the decomposition of electrolyte also leads to the formation of organic polymeric/gel-like film. Unlike SEI layer, organic polymeric/gel-like film vanishes as the oxidation potential beyond 2 V and reforms between 0.02 and 1.8 V.26,27 And the reversible formation and dissolution of the organic film could enhance the specific capacity, delivering an extra capacity through a so-called “pseudo-capacitive behavior”.9 Secondly, the porous MnCo2O4 is obtained by calcination of the oxalate precursor, during which a large number of lattice defects in the typical nanostructure will form. According to previous reports,28,29 these lattice defects will provide more active sites for Li+ embedding/disembedding, which would improve the specific capacity of the active material. Both of the two processes mentioned above could provide more active sites for Li+ embedding/disembedding, leading to the excessive capacity during this process.
Fig. 4c is the cycling performance of the as-obtained MnCo2O4 nanorods at a constant current density of 0.4 A g−1. A slightly increase in capacity can be observed after the initial several cycles, which can be ascribed to the gradual activation of the electrode materials. After 16 cycles, the discharge capacity reaches a maximum value of about 1674 mA h g−1. Such phenomena have also been observed for a variety of transitional metal oxides, such as iron oxide and cobalt oxide.23,30–33 In the following cycles, the specific capacity of MnCo2O4 nanorods gradually decreases, which can be ascribed to the pulverization of the active materials.21,34 After 70 cycles, the discharge capacity is about 1042 mA h g−1. Then, the specific capacity of the electrodes become steady, which could be ascribed to the complete pulverization of the structure and the reformed connection between electrode materials and electrode collector. When further prolonging the test cycles, the specific capacity of MnCo2O4 exhibits a gradual increase. After 700 cycles, the discharge capacity increases to 1620 mA h g−1. It should be noted that this phenomenon (increase in capacity) is also observed in long time cycles when using other transitional metal oxides as anode materials.35–39 To further understand the electrochemical behavior after discharge/charge test, the phase transitions of the active material after discharge/charge process was investigated by XRD and the corresponding result is shown in Fig. S3.† The diffraction pattern can be designated to be a mixture of CoO (JPCDS no. 78-0431), Co (JPCDS no. 01-1259), MnO (JPCDS no. 65-0639) and Mn (JPCDS no. 01-1234), which is consistent with the CV result. Meanwhile, both SEM and TEM were employed to observe the morphological changes of the active material. As it is shown in Fig. S4,† the pulverization of the active material can be clearly observed, which may result from the volume changes of the active material upon the Li+ insertion/disinsertion process. On the basis of the experimental results and previous reports, this phenomenon can be explained in the following three aspects: (i) the first aspect concerns on pulverization of the active materials during the charge/discharge process. The pulverization of active materials could provide more active sites for the storage of lithium ions, which is beneficial to maintain the electrochemical performance during long cycles. (ii) During the charge/discharge process, the irreversible electrochemical reaction will lead to the formation of metallic cobalt (0) and manganese (0).34,40 The formation of metallic cobalt and manganese will increase the electrical conductivity of the electrode during the electrochemical process. Considering the fact that the paths for Li+ insertion/extraction have already formed in the previous charge/discharge process, the increase in electrical conductivity will greatly facilitates the insertion/extraction of lithium ions and the transport of electrons, resulting in the increment of capacity in the subsequent cycles. (iii) The as-formed metallic cobalt (0) could act as catalyst for the decomposition of SEI layer and the reversible formation/dissolution of the organic polymeric/gel-like film, which would lead to the reversible increase in specific capacity via the so-called “pseudo-capacitance behavior”.41,42
Besides the high specific capacity and good cycling performance, the rate performances of the as-synthesized MnCo2O4 nanorods at different current densities (0.4 A g−1 to 30 A g−1) were also investigated and the corresponding result is shown in Fig. 4d. The as-formed porous MnCo2O4 nanorods show good rate capability, with discharge capacities of 1340, 1343, 1274, 1107 and 750 mA h g−1 at current densities of 1, 2, 5, 10 and 20 A g−1. Even at a high current density of 30 A g−1, the as-formed electrodes can still deliver a capacity of 533 mA h g−1. When the current density returned to 0.4 A g−1, the discharge capacity can recover to the pervious value, indicating the integrity of the electrode. These findings demonstrate that the porous structure endows the as-synthesized MnCo2O4 nanorods with excellent electrochemical property, such as long cycling life and high rate performance, which can be attributed to the large BET surface area and proper pore size. Meanwhile, it also indicates that it is a hoping material for high-power LIBs. A comparison between the reported cobaltates and our product is summarized in Table 1. Obviously, the porous as-synthesized MnCo2O4 nanorods exhibit good electrochemical performance, both in cycling performance and discharge/charge capacity.
| Material | Current density (mA g−1) | Discharge capacity (mA h g−1) | Ref. |
|---|---|---|---|
| Porous MnCo2O4 | 1000 | 740/1000th cycle | 39 |
| MnCo2O4 hollow nanofibers | 200 | 997/50th cycle | 8 |
| Flake-like MnCo2O4 | 100 | 952/100th cycle | 7 |
| MnCo2O4 microspheres | 400 | 553/200th cycle | 19 |
| Co3O4/ZnCo2O4 hollow spheres | 230 | 934/300th cycle | 43 |
| ZnCo2O4 hollow powders | 3000 | 586/200th cycle | 44 |
| NiCo2O4 microspheres | 800 | 705/500th cycle | 45 |
| MnCo2O4 nanowire | 100 | 895.8/50th cycle | 46 |
| Core−shell ellipsoidal MnCo2O4 | 100 | 750/70th cycle | 47 |
| Porous MnCo2O4 nanorods | 400 | 1620/700th cycle | This work |
EIS were performed to provide more information about porous MnCo2O4 nanorods. Nyquist impedance plots show the impedances obtained before discharging and after 100 cycles of charging/discharging at a current density of 0.4 A g−1 (ESI, Fig. S5†). The Nyquist plots are composed of semicircle in the high-to-middle frequency regions and a sloping long line in the low frequency region. High-to-middle frequency semicircle can be ascribed to the charge transfer impedance and surface film impedance (Rsf+ct). The sloping long line in the low frequency region means the Warburg impedance (W). After 100 cycles, the semicircle diameter becomes smaller and the sloping long line in low frequency becomes more vertical, which indicated the resistance becomes smaller (102 Ω for fresh vs. 42 Ω for after 100 cycles). Both of the results coincidence with the cycling performance results in Fig. 4c, which can further confirm that the electrode materials have been active and the paths for lithium ion embedding/disembedding formatted after initial cycles.
3.3 The influence of binders
Binder is an essential part of LIBs electrodes, which performs in the role of dispersion agent to connect the materials together and adhere them to the current collector. A proper binder can effectively alleviate the volume changes during the charge/discharge process and thus prolong the cycling life of LIBs.48,49 To investigate the effects of binders on the electrochemical performance of the MnCo2O4 porous nanorods, the lithium-ions cell using PVDF as the binder have also been assembled and the electrochemical performance is investigated. All the experimental conditions remain constant except for replacing the CMC/SBR with PVDF during the assembly of the lithium-ions cells. The cycling performance of MnCo2O4 porous nanorods when using PVDF as the binder is illustrated in Fig. 5 (blue line). Compared with the situation when CMC/SBR was employed, both the two curves show similar tendency in the first 70 cycles, which indicate that the as-synthesized MnCo2O4 nanorods underwent similar electrochemical process when PVDF was employed. However, the overall discharge capacity of the electrode is much lower than the corresponding values when CMC/SBR is employed. After cycling for 75 times, the discharge capacity began to decrease. The discharge capacity dramatically decreases to about 300 mA h g−1 after recycling for 200 times, indicating that PVDF is not a proper binder for the as-synthesized MnCo2O4 nanorods.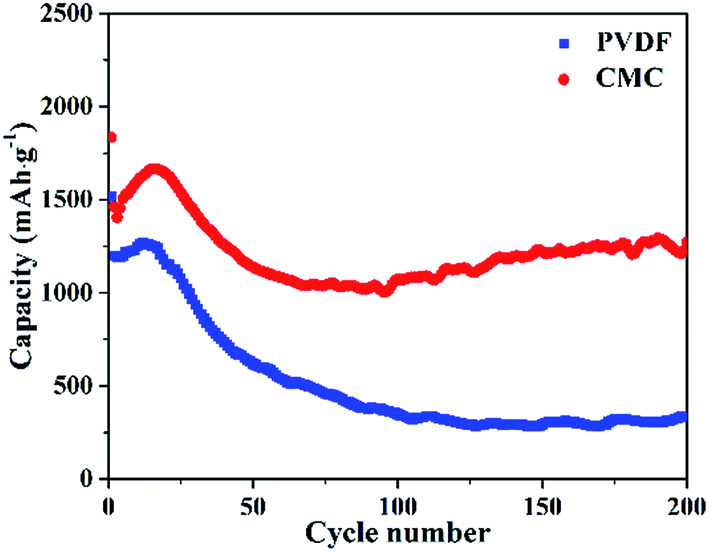 | ||
| Fig. 5 Cycling performance of the as-synthesized MnCo2O4 nanorods at a constant current density of 0.4 A g−1 using different binders: PVDF (the blue line) and CMC/SBR (the red line). | ||
Such a big difference in electrochemical performance can be ascribed to the different functional mechanism of the two binders. The electrodes use PVDF binder usually suffer from agglomerating of active materials, poor mechanical strength and separation electrode materials from the current collector because PVDF has weak hydrogen bonds with active materials and the current collector. Meanwhile, PVDF is readily swollen during the charge/discharge progress, which will cause the electrode material desquamation from the copper current collector. The mixture of CMC/SBR binder exhibits a different mechanical property and swelling behavior from PVDF. CMC dissolves in water with an almost fully stretched molecular conformation due to its rigid cellulose backbone and electrostatic repulsive interactions between ionized carboxyl groups, resulting in a tightened electrode architecture.50,51 Nevertheless, CMC is extremely stiff and brittle which would lead to the electrode materials separate from the copper foil or form cracks when used alone as binder.52,53 As an elastomer, SBR has higher flexibility, stronger binding force and better heat resistance. The addition of SBR into CMC can weaken the brittle and stiff. And the mixture binder has stronger hydrogen bonding with the active material and the current collector than PVDF binder, resulting in a firm electrode architecture.54 Recent years, the CMC/SBR binder has shown enhanced performance for a number of electrode materials, especially those suffering from huge volume variation during discharge/charge cycles such as transitional metal oxides,55,56 germanium based materials,57,58 and silicon based materials.59,60 In our experiment, the fast fading of the electrode may result from the weak bonding between the active material and the current collector. During this process, the electrode materials slide off from current collector in the prolonged cycles, resulting in the fast decrease in specific capacity.
To further disclose the electrochemical process when PVDF is employed, the SEM and TEM observation of the active material is carried out after the discharge/charge process. According to the previous reports, the intercalation/deintercalation of Li+ will cause severe volume changes and the intrinsic structure will be destroyed when using transition metal oxides as the active materials. As it is shown in Fig. S4,† the nanorods underwent pulverization and turn into nanoparticles after the discharge/charge cycles, this process will expose more active sites. For this reason, the capacity of the material using PVDF as the binder keeps increasing during the initial 70 cycles. In the following cycles, the active material gradually separate from the current collector and lead to the continuously decrease in capacity.
3.4 The influence of calcination temperature
The previous researches have suggested that the calcination temperature will exert tremendous influence on the electrochemical lithium storage behavior of the metal oxides.61 In order to confirm the effect of calcination temperature on the electrochemical properties of the final products, the as-prepared rod-like precursor were also calcined at 400 °C and 600 °C. For comparison purpose, the samples obtained by calcining the precursor at 500 °C, 400 °C and 600 °C are denoted as MCO, MCO-1 and MCO-2, respectively. The XRD patterns of samples MCO, MCO-1 and MCO-2 are shown in Fig. 6, which clearly demonstrate that all the three samples can be unambiguously designated to be the cubic phased MnCo2O4. No additional peaks can be detected, indicating the high purities of the samples. As the calcination temperature elevated, the intensities of the diffraction peaks increase, indicating the increase in crystallinity of the final samples.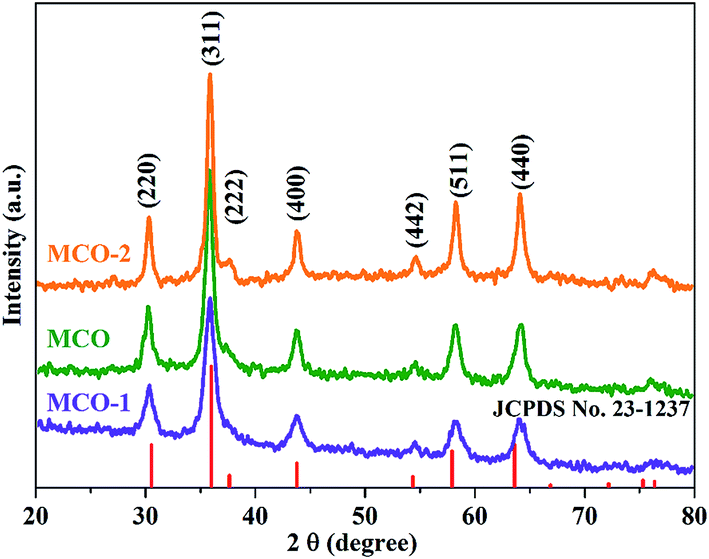 | ||
| Fig. 6 XRD patterns of the sample obtained by calcining the precursor at 400 °C, 500 °C and 600 °C. | ||
In our study, the calcination temperature is proved to have little influence on the morphologies of the final products. According the SEM and TEM image of samples MCO-1 and MCO-2 (ESI, Fig. S6†), the porous natures of samples MCO-1 and MCO-2 can still be retained after the calcination process. Both samples MCO-1 and MCO-2 are mainly composed of porous nanorods, which is similar to sample MCO. The typical lattice fringes taken from sample MCO-1 and MCO-2 are determined to be 0.291 nm and 0.247 nm, correspond to the lattice distances of (220) and (311) planes respectively. The composition and chemical states of samples MCO-1 and MCO-2 are also investigated by XPS, which clearly indicate the existence of the Co2+/Co3+, Mn2+/Mn3+ solid redox couples (ESI, Fig. S7†). For comparison purpose, the oxidation state ratios and the average cation charges of Co and Mn of MCO, MCO-1 and MCO-2 were calculated based on the peak areas in XPS spectra (ESI, Table S1†). The average cation charge for samples MCO, MCO-1 and MCO-2 are determined to be 2.46, 2.50 and 2.51 respectively, indicating that the chemical environment of Co and Mn atoms are nearly the same in the three samples.
Though the porous MnCo2O4 structures can be obtained under the three calcination temperature, the detailed microstructures are totally different according to the N2 adsorption–desorption analysis (ESI, Fig. S8†). The isotherm curves of MCO-1 and MCO-2 can be classified as type-IV with a type-H4 hysteresis loop, indicating the mesoporous natures of samples MCO-1 and MCO-2. The BET surface areas for samples MCO-1 and MCO-2 are determined to be 101.4 m2 g−1 and 36.6 m2 g−1, respectively. The average pore sizes of the two samples are determined to be 7.66 nm (MCO-1) and 30–45 nm (MCO-2) using the BJH method. It should be noted that the BET surface areas of the three samples decrease when the calcination temperature increase, which would be of great influence on the electrochemical performance of the final products.
Generally speaking, increasing the annealing temperature can enhance the materials' crystallinity, decrease the surface area and change the inner structure of the materials. To investigate the influence of calcination temperature on the electrochemical properties of the final products, the cycling performance of samples MCO-1 and MCO-2 are investigated. Fig. 7a are the cycling curves for samples MCO, MCO-1 and MCO-2 at a constant current density of 0.4 A g−1, which clearly indicate that the calcination temperature do have a great influence on the electrochemical performance. Among the three samples, sample MCO possess the most excellent electrochemical performance. The discharge curves of the three samples are similar in shapes, which indicate the occurrence of similar electrochemical process during the discharging process. Both sample MCO-1 and MCO-2 display poorer electrochemical performance than sample MCO, exhibiting fast fading or low discharge capacity during the discharge process. The discharge capacities of samples MCO-1 and MCO-2 after 200 cycles are 851 and 555 mA h g−1 respectively, which is much lower than the corresponding value of sample MCO. As it is shown in Fig. 7b, the calcination temperature is also believed to have great influence on the rate performances of the final products. Among the three samples, sample MCO displays the best rate performance. As for the other two samples, sample MCO-2 possesses better rate performance than MCO-1 at higher current density (from 5–30 A g−1). To explain the different electrochemical behaviors among the three sample, the phase transition as well as the morphological transition of samples MCO-1 and MCO-2 were investigated by XRD, TEM and SEM. And the corresponding result is shown in Fig. S9.† According to the XRD pattern, CoO (JPCDS no. 78-0431), Co (JPCDS no. 01-1259), MnO (JPCDS no. 65-0639) and Mn (JPCDS no. 01-1234) can be detected, which is similar to the situation when sample MCO is employed as the active material. Meanwhile, SEM and TEM observation indicate that both of the two samples will underwent pulverization during the discharge/charge process (ESI, Fig. S10†). Both of the two samples (MCO-1 and MCO-2) can maintain the rod-like morphology before the discharge/charge process. After cycling process, the materials turned into particles via the same process as sample MCO does. These experimental facts clearly indicate that all the three samples underwent the similar electrochemical changes during discharge/charge process. Thus, the electrochemical changes will not be the main reason for the observed difference in electrochemical performances of the three samples.
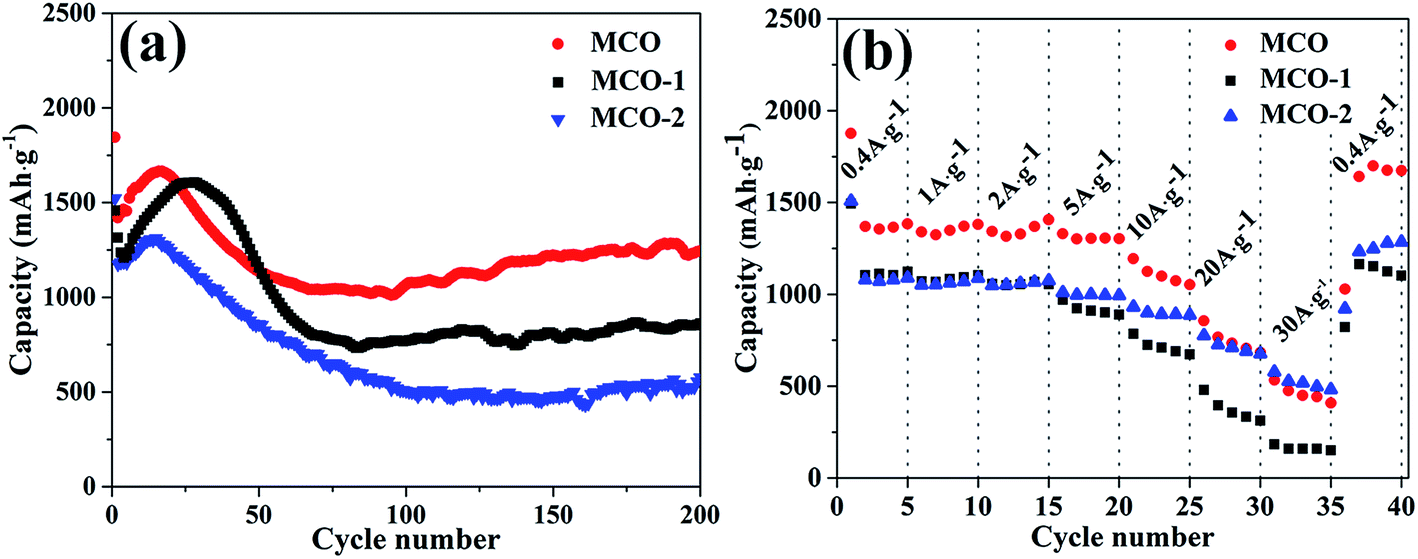 | ||
| Fig. 7 (a) Cycling performance of samples MCO, MCO-1 and MCO-2 at a constant current density of 0.4 A g−1. (b) Rate performance of samples MCO, MCO-1 and MCO-2 at different current densities. | ||
When considering the electrochemical performances of these samples, the structural stability as well as the surface area should also be taken into consideration. The enhancement in structural stability can alleviate the pulverization of the active material, while the enlargement in surface areas will facilitate the transport of lithium ions. All of these two aspect will exert positive influence on the electrochemical performance of the LIBs. However, these two aspects usually vary in the opposite direction. To be specific, the structural stability usually improves with the cost of reducing specific surface area. Thus, the electrochemical performance of the material will be determined by the two factors mentioned above. In our study, although sample MCO-1 possesses the highest surface area, the poor structural stability greatly restricts its cycling stability during the charge/discharge process. Although sample MCO-2 possesses the highest crystallinity, the small surface area greatly restricts the transport of lithium ion and the reversible electrochemical process, leading to the low capacity during the discharging process. Because of the high crystallinity and structural stability of sample MCO-2, this sample can effectively buffer higher current during discharge/charge progress, leading to the stable cycling performance under high current density than sample MCO-1.62
To further confirm the difference of cycling results mentioned above, CV tests were performed for MCO, MCO-1 and MCO-2 at different scan rates after 15 cycles. As shown in Fig. 8, one can see the peak current rise gradually as the scan rate elevated. According to the equation proposed by Randles–Sevcik:63
| Ip = (2.69 × 105)n3/2AD1/2υ1/2C |
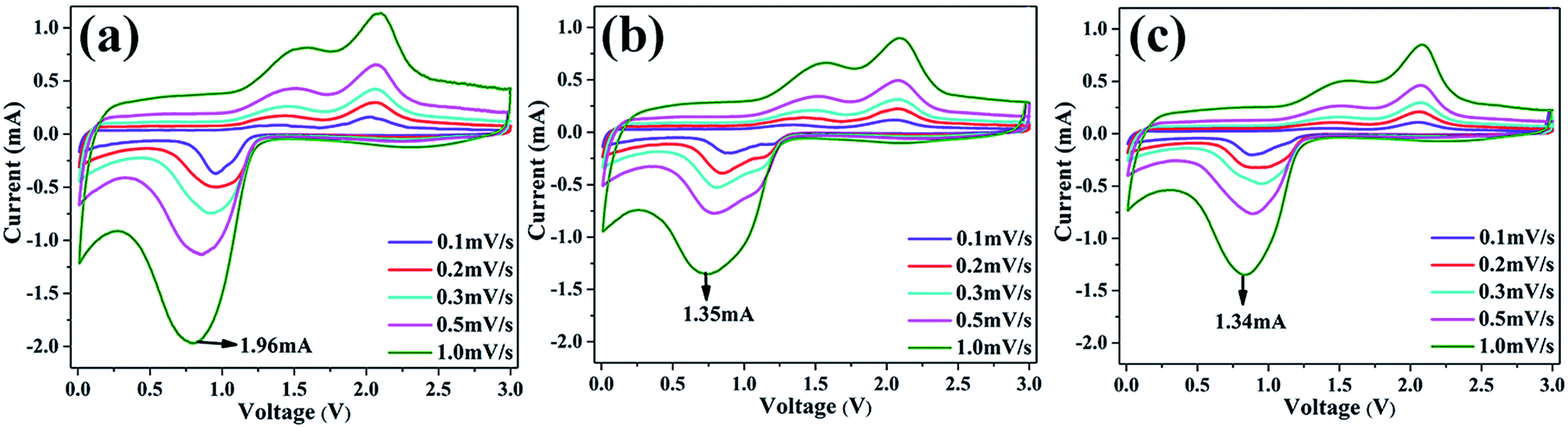 | ||
| Fig. 8 CV curves at different scan rates after 15 cycles for MCO (a), MCO-1 (b) and MCO-2 (c), respectively. | ||
4. Conclusions
In summary, we present a facile method for the synthesis of porous MnCo2O4 nanorods by calcination of the oxalate precursor with formula of Mn0.33Co0.67C2O4·2H2O. The as-prepared MnCo2O4 nanorods exhibit high specific capacity, excellent rate performances and stable cycling performance when used as the anode materials in LIBs. The as-obtained porous MnCo2O4 (MCO) nanorods could deliver an initial discharge capacity of 1845 mA h g−1 under current density of 0.4 A g−1. Even being cycled at high current density of 30 A g−1, the capacity is still about 533 mA h g−1. The effect of binder is also investigated, which suggest that CMC/SBR is more proper than PVDF when used as the binder for the as-synthesized MnCo2O4 nanorods. The electrodes use PVDF binder not only exhibit a lower initial discharge capacity (1217 mA h g−1 at 0.4 A g−1), but also suffer from a rapid capacity decay in 70 cycles. The influence of calcination temperature is also systematically investigated, which suggest the electrochemical performance is determined by both the structural stability and surface area of the samples.Acknowledgements
The present work financially supports from National Natural Science of China (NSFC 21171007) and the Programs for Science and Technology Development of Anhui Province (1501021019).Notes and references
- J. C. Liu, Y. J. Xu, X. J. Ma, J. K. Feng, Y. T. Qian and S. L. Xiong, Nano Energy, 2014, 7, 52–62 CrossRef CAS.
- J. J. Zhang, T. Huang and A. S. Yu, J. Power Sources, 2015, 273, 894–903 CrossRef CAS.
- G. H. An and H. J. Ahn, J. Power Sources, 2014, 272, 828–836 CrossRef CAS.
- Y. H. Jin, L. Wang, Y. M. Shang, J. Gao, J. J. Li and X. M. He, Electrochim. Acta, 2015, 151, 109–117 CrossRef CAS.
- Y. A. Xu, X. F. Wang, C. H. An, Y. J. Wang, L. F. Jiao and H. Yuan, J. Power Sources, 2014, 272, 703–710 CrossRef CAS.
- L. Yu, L. Zhang, H. B. Wu, G. Zhang and X. W. Lou, Energy Environ. Sci., 2013, 6, 2664–2671 Search PubMed.
- A. K. Mondal, D. Su, S. Chen, A. Ung, H. S. Kim and G. Wang, Chem.–Eur. J., 2015, 21, 1526–1532 CrossRef CAS PubMed.
- S. M. Hwang, S. Y. Kim, J. G. Kim, K. J. Kim, J. W. Lee, M. S. Park, Y. J. Kim, M. Shahabuddin, Y. Yamauchi and J. H. Kim, Nanoscale, 2015, 7, 8351–8355 RSC.
- J. Li, J. Wang, X. Liang, Z. Zhang, H. Liu, Y. Qian and S. Xiong, ACS Appl. Mater. Interfaces, 2014, 6, 24–30 Search PubMed.
- X. Hou, X. Wang, B. Liu, Q. Wang, T. Luo, D. Chen and G. Shen, Nanoscale, 2014, 6, 8858–8864 RSC.
- S. G. Mohamed, Y. Q. Tsai, C. J. Chen, Y. T. Tsai, T. F. Hung, W. S. Chang and R. S. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 12038–12046 Search PubMed.
- W. Li, K. Xu, G. Song, X. Zhou, R. Zou, J. Yang, Z. Chen and J. Hu, CrystEngComm, 2014, 16, 2335–2339 RSC.
- J. Li, S. Xiong, X. Li and Y. Qian, Nanoscale, 2013, 5, 2045–2054 RSC.
- J. G. Kim, Y. Kim, Y. Noh and W. B. Kim, ChemSusChem, 2015, 8, 1752–1760 CrossRef CAS PubMed.
- T. Y. Ma, Y. Zheng, S. Dai, M. Jaroniec and S. Z. Qiao, J. Mater. Chem. A, 2014, 2, 8676–8682 RSC.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- A. Vinu, D. P. Sawant, K. Ariga, M. Hartmann and S. B. Halligudi, Microporous Mesoporous Mater., 2005, 80, 195–203 CrossRef CAS.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169–3183 CrossRef CAS.
- C. Fu, G. Li, D. Luo, X. Huang, J. Zheng and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 2439–2449 Search PubMed.
- K. Cao, L. Jiao, H. Liu, Y. Liu, Y. Wang, Z. Guo and H. Yuan, Adv. Energy Mater., 2015, 5, 1401421 CrossRef.
- M. Chen, J. Liu, D. Chao, J. Wang, J. Yin, J. Lin, H. Jin Fan and Z. Xiang Shen, Nano Energy, 2014, 9, 364–372 CrossRef CAS.
- X. Zhu, X. Song, X. Ma and G. Ning, ACS Appl. Mater. Interfaces, 2014, 6, 7189–7197 Search PubMed.
- H. S. Jadhav, A. K. Rai, J. Y. Lee, J. Kim and C. J. Park, Electrochim. Acta, 2014, 146, 270–277 CrossRef CAS.
- J. Yue, X. Gu, L. Chen, N. Wang, X. Jiang, H. Xu, J. Yang and Y. Qian, J. Mater. Chem. A, 2014, 2, 17421–17426 RSC.
- W. Liu, C. Lu, X. Wang, K. Liang and B. K. Tay, J. Mater. Chem. A, 2015, 3, 624–633 RSC.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dolle, L. Dupont and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A627–A634 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A1212–A1217 CrossRef CAS.
- C. Hu, L. Lv, J. Xue, M. Ye, L. Wang and L. Qu, Chem. Mater., 2015, 27, 5253–5260 CrossRef CAS.
- X. H. Huang, J. P. Tu, B. Zhang, C. Q. Zhang, Y. Li, Y. F. Yuan and H. M. Wu, J. Power Sources, 2006, 161, 541–544 CrossRef CAS.
- Y. Liu, J. Xu, X. Qin, H. Xin, X. Yuan, J. Zhang, D. Li and C. Song, J. Mater. Chem. A, 2015, 3, 9682–9688 RSC.
- Z. Wang, D. Luan, S. Madhavi, Y. Hu and X. W. Lou, Energy Environ. Sci., 2012, 5, 5252–5256 Search PubMed.
- W. J. Yu, P. X. Hou, F. Li and C. Liu, J. Mater. Chem., 2012, 22, 13756–13763 RSC.
- J. G. Kim, Y. Kim, Y. Noh and W. B. Kim, RSC Adv., 2014, 4, 27714–27721 RSC.
- J. Luo, J. Liu, Z. Zeng, C. F. Ng, L. Ma, H. Zhang, J. Lin, Z. Shen and H. J. Fan, Nano Lett., 2013, 13, 6136–6143 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- C. Ban, Z. Wu, D. T. Gillaspie, L. Chen, Y. Yan, J. L. Blackburn and A. C. Dillon, Adv. Energy Mater., 2010, 22, E145–E149 CrossRef CAS PubMed.
- J. C. Guo, Q. Liu, C. S. Wang and M. R. Zachariah, Adv. Funct. Mater., 2012, 22, 803–811 CrossRef CAS.
- Y. Shi, B. Guo, S. A. Corr, Q. Shi, Y. S. Hu, K. R. Heier, L. Chen, R. Seshadri and G. D. Stucky, Nano Lett., 2009, 9, 4215–4220 CrossRef CAS PubMed.
- G. Li, L. Xu, Y. Zhai and Y. Hou, J. Mater. Chem. A, 2015, 3, 14298–14306 RSC.
- X. Huang, J. Chen, Z. Lu, H. Yu, Q. Yan and H. H. Hng, Sci. Rep., 2013, 3, 2317 CrossRef PubMed.
- L. Zhang, P. Hu, X. Zhao, R. Tian, R. Zou and D. Xia, J. Mater. Chem., 2011, 21, 18279 RSC.
- S. Grugeon, S. Laruelle, L. Dupont and J. M. Tarascon, Solid State Sci., 2003, 5, 895–904 CrossRef CAS.
- Q. Wang, B. Yu, X. Li, L. Xing and X. Xue, J. Mater. Chem. A, 2016, 4, 425–433 RSC.
- S. H. Choi and Y. C. Kang, ChemSusChem, 2013, 6, 2111–2116 CrossRef CAS PubMed.
- J. Li, S. Xiong, Y. Liu, Z. Ju and Y. Qian, ACS Appl. Mater. Interfaces, 2013, 5, 981–988 Search PubMed.
- L. Li, Y. Q. Zhang, X. Y. Liu, S. J. Shi, X. Y. Zhao, H. Zhang, X. Ge, G. F. Cai, C. D. Gu, X. L. Wang and J. P. Tu, Electrochim. Acta, 2014, 116, 467–474 CrossRef CAS.
- G. Huang, S. Xu, Z. Xu, H. Sun and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 21325–21334 Search PubMed.
- J. P. Yen, C. C. Chang, Y. R. Lin, S. T. Shen and J. L. Hong, J. Electrochem. Soc., 2013, 160, A1811–A1818 CrossRef CAS.
- Y. S. Park, E. S. Oh and S. M. Lee, J. Power Sources, 2014, 248, 1191–1196 CrossRef CAS.
- B. Lestriez, S. Bahri, I. Sandu, L. Roue and D. Guyomard, Electrochem. Commun., 2007, 9, 2801–2806 CrossRef CAS.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- R. Y. Zhang, X. Yang, D. Zhang, H. L. Qiu, Q. Fu, H. Na, Z. D. Guo, F. Du, G. Chen and Y. J. Wei, J. Power Sources, 2015, 285, 227–234 CrossRef CAS.
- J. Chong, S. D. Xun, H. H. Zheng, X. Y. Song, G. Liu, P. Ridgway, J. Q. Wang and V. S. Battaglia, J. Power Sources, 2011, 196, 7707–7714 CrossRef CAS.
- B.-R. Lee and E.-S. Oh, J. Phys. Chem. C, 2013, 117, 4404–4409 CrossRef CAS.
- Z. Wang, S. Madhavi and X. W. Lou, J. Phys. Chem. C, 2012, 116, 12508–12513 CrossRef CAS.
- C. Zhong, J. Z. Wang, S. L. Chou, K. Konstantinov, M. Rahman and H. K. Liu, J. Appl. Electrochem., 2010, 40, 1415–1419 CrossRef CAS.
- T. T. Qiang, J. X. Fang, Y. X. Song, Q. Y. Ma, M. Ye, Z. Fang and B. Y. Geng, RSC Adv., 2015, 5, 17070–17075 RSC.
- J. W. Liang, X. N. Li, Z. G. Hou, T. W. Zhang, Y. C. Zhu, X. D. Yan and Y. T. Qian, Chem. Mater., 2015, 27, 4156–4164 CrossRef CAS.
- H. S. Kim, K. Y. Chung and B. W. Cho, J. Power Sources, 2009, 189, 108–113 CrossRef CAS.
- P. J. Zuo, W. G. Yang, X. Q. Cheng and G. P. Yin, Ionics, 2011, 17, 87–90 CrossRef CAS.
- L. Zhang, H. B. Wu and X. W. Lou, J. Am. Chem. Soc., 2013, 135, 10664–10672 CrossRef CAS PubMed.
- N. Yan, L. Hu, Y. Li, Y. Wang, H. Zhong, X. Hu, X. Kong and Q. Chen, J. Phys. Chem. C, 2012, 116, 7227–7235 CrossRef CAS.
- C. Comminges, R. Barhdadi, M. Laurent and M. Troupel, J. Chem. Eng. Data, 2006, 51, 680–685 CrossRef CAS.
- S. R. Das, S. B. Majumder and R. S. Katiyar, J. Power Sources, 2005, 139, 261–268 CrossRef CAS.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS PubMed.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26176g |
| This journal is © The Royal Society of Chemistry 2016 |