
Assembly of substituted phenanthridines via a cascade palladium-catalyzed coupling reaction, deprotection and intramolecular cyclization†
Jun Geab,
Xiaojian Wang*ab,
Tianqi Liua,
Zeyu Shib,
Qiong Xiaob and
Dali Yin*ab
aState Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing 100050, P. R. China. E-mail: wangxiaojian@imm.ac.cn; yindali@imm.ac.cn; Fax: +86 10 63165248; Tel: +86 10 63165248
bDepartment of Medicinal Chemistry, Beijing Key Laboratory of Active Substances, Discovery and Drugability Evaluation, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing 100050, P. R. China
First published on 12th February 2016
Abstract
The discovery and development of a general method for the one-pot synthesis of substituted phenanthridines is presented. In the presence of Pd(PPh3)4, accessible precursors undergo a Suzuki cross-coupling reaction with 2-(Boc-amino)benzeneboronic acid pinacol ester and then spontaneously undergo deprotection and intramolecular condensation to form the corresponding phenanthridines in one step. This reaction has a wide range of substrates with various functional groups, and the corresponding products have been obtained in good yields.
Introduction
Substituted phenanthridines are an important class of heterocyclic compounds in materials science and in medicinal chemistry due to their significant biological activities.1–3 Molecules containing the phenanthridine core are the subject of considerable interest as potent antitumor agents,4 anti-tuberculosis agents,5,6 and antibacterial agents.7 Structures, shown in Fig. 1, are representative examples of natural products and biologically relevant compounds containing phenanthridine core structures.8–17 Because of these applications, the synthesis of the substituted phenanthridine motif has spurred vigorous research for the development of new methodologies.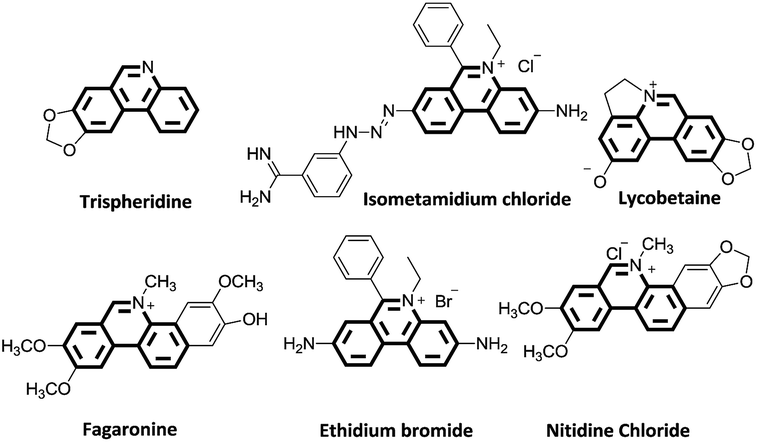 | ||
| Fig. 1 Examples of natural products and biologically relevant compounds. | ||
Traditionally, phenanthridines are synthesized by the Pictet–Hubert reaction, which usually performed in the presence of P4O10, POCl3, or PCl5 at elevated temperatures.18–21 The limitation of functional group tolerance was observed in the Pictet–Hubert reaction due to the harsh conditions. Besides the traditional methods, phenanthridines have recently been successfully prepared by transition metal catalyzed approaches,22–30 radical promoted cyclization,31–41 cycloaddition,42–45 and other methods.46–48 The general synthetic approaches leading to phenanthridines are summarized in Scheme 1. Although a number of methods are available for the synthesis of these phenanthridines, many are limited because of the lack of generality, limited functional group tolerance, and lengthy synthetic time. Thus, a simple, efficient, and general method to synthesize phenanthridines would be still attractive.
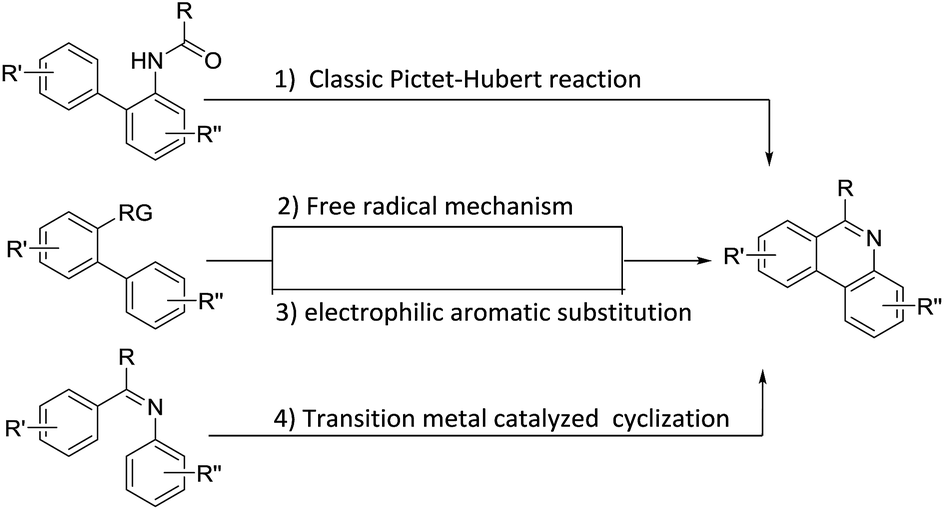 | ||
| Scheme 1 Synthetic approaches leading to phenanthridines. | ||
Among synthesis methodology research towards phenanthridines, James and his coworker first attempted to use 2′-bromoacetophenone and (2-Boc-aminophenyl)boronic acid to construct the phenanthridines scaffold using a stepwise strategy by a Suzuki cross-coupling reaction, followed by acid-catalyzed N-Boc removal, intramolecular cyclization and basification.49 Subsequently, Marko published the synthesis method of a lycobetaine–tortuosine analogue, which also included the metal catalyzed coupling and acid hydrolysis steps.50 Obviously, amide or ester substituted materials were not tolerated during the second acid work up step. Additionally, Graham also mentioned a Suzuki coupling/ring closure to prepare the norallonitidine and nornitidine. However, the reactions were claimed to be sluggish and the products were achieved with low yields.51 Recently, Dhara also reported a method to synthesize substituted phenanthridines using substituted aromatic ortho-bromoaldehydes and ortho-aminobenzeneboronic acid as starting substrates.52 In our previous studies, we showed a new approach for constructing 1-substituted-4-methylisoquinolines via a cascade Pd-catalyzed Heck reaction, intramolecular cyclization and isomerization.53 In order to extend our study about synthetic methodology of heterocyclic compound,54–57 we applied this strategy to the construction of phenanthridines (Scheme 2).
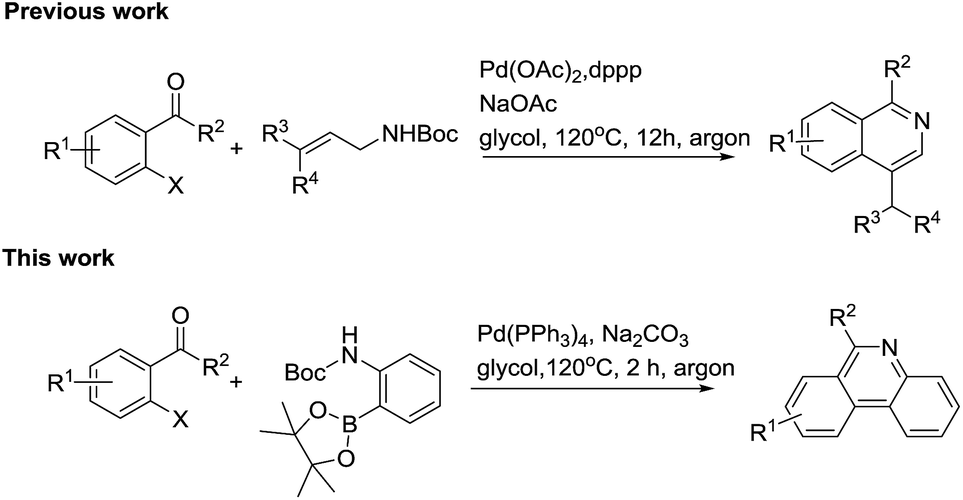 | ||
| Scheme 2 Strategies for the construction substituted phenanthridines. | ||
Results and discussion
Initially, 2-iodoacetophenone (1a) and 2-(Boc-amino)benzeneboronic acid pinacol ester (2) were chose as the model substrates to explore the possible formation of 6-methyl phenanthridine (3a) and the results were summarized in Table 1. The reaction proceeded in glycol at 120 °C for 2 hours with Pd(OAc)2 as the catalyst, 1,3-bis(diphenylphosphine)propane (dppp) as the ligand, Cs2CO3 as the base. To our delight, 3a was obtained in 44% yield (Table 1, entry 1), indicating that it was feasible to construct the phenanthridine scaffold based on our hypothesis. However, changing the base Cs2CO3 to NaOAc or organic base Et3N failed to show any improvement (Table 1, entries 2–3). When ligand dppp was replaced with 1,1′-bis(diphenylphosphino)ferrocene (dppf), the result was similar to dppp with a yield of 37% (Table 1, entry 4). We then screened catalysts such as Pd(PPh3)2Cl2, Pd2(dba)3, and Pd(PPh3)4 (Table 1, entries 5–7).
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Catalyst | Ligand | Base | Solvent | Yieldb |
| a All reactions were conducted using 1a (1 mmol, 1.0 equiv.), 2 (1.2 mmol, 1.2 equiv.), base (2 mmol, 2.0 equiv.), Pd catalyst (3 mmol%), solvent (3.0 mL), stirred at 120 °C for 2 h under argon.b Isolated yields.c The reaction was conducted at 85 °C for 4 h under argon. | ||||||
| 1 | I | Pd(OAc)2 | dppp | Cs2CO3 | Glycol | 44% |
| 2 | I | Pd(OAc)2 | dppp | NaOAc | Glycol | 0 |
| 3 | I | Pd(OAc)2 | dppp | Et3N | Glycol | 10% |
| 4 | I | Pd(OAc)2 | dppf | Cs2CO3 | Glycol | 37% |
| 5 | I | Pd(PPh3)2Cl2 | Cs2CO3 | Glycol | 14% | |
| 6 | I | Pd2(dba)3 | Cs2CO3 | Glycol | 17% | |
| 7 | I | Pd(PPh3)4 | Cs2CO3 | Glycol | 73% | |
| 8 | I | Pd(PPh3)4 | Na2CO3 | Glycol | 80% | |
| 9 | I | Pd(PPh3)4 | K2CO3 | Glycol | 76% | |
| 10 | I | Pd(PPh3)4 | K3PO4 | Glycol | 67% | |
| 11 | OTf | Pd(PPh3)4 | Na2CO3 | Glycol | 75% | |
| 12 | Br | Pd(PPh3)4 | Na2CO3 | Glycol | 71% | |
| 13c | I | Pd(PPh3)4 | Na2CO3 | EtOH | 6% | |
| 14 | I | Pd(PPh3)4 | Na2CO3 | DMF | 20% | |
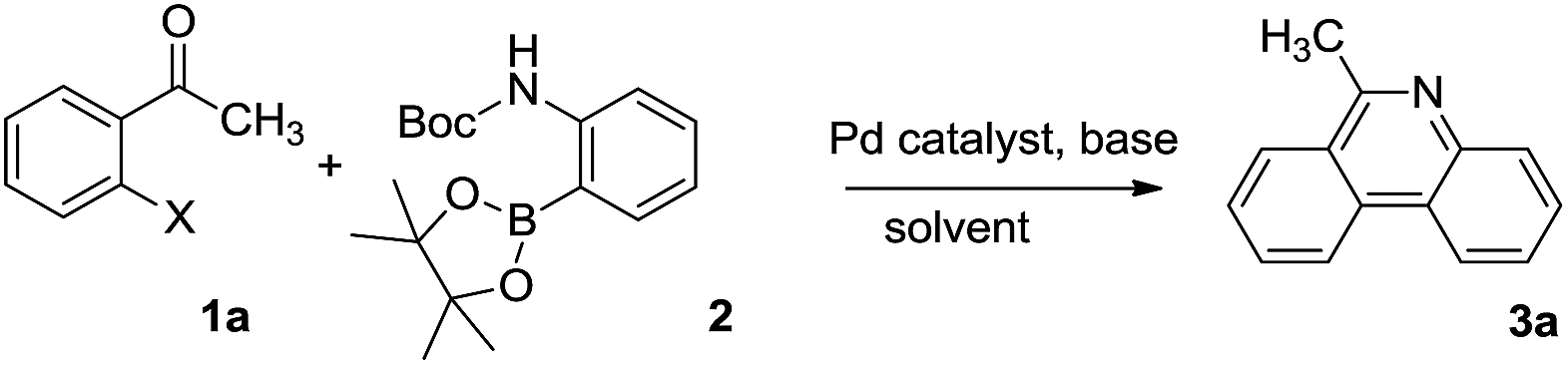
The results showed that Pd(PPh3)4 displayed the highest catalytic activity toward the formation of 3a in 80% yield. Keeping the Pd(PPh3)4 as the catalyst, other bases such as Na2CO3, K2CO3 and K3PO4 were examined subsequently (Table 1, entries 8–10). The results showed that carbonates were better than K3PO4, but there was little difference among Na2CO3, K2CO3 and Cs2CO3. We also investigated the effect of reactive group X in substrates. It was found that Br and OTf gave a similar performance as I (Table 1, entries 11 and 12). Varying the solvents proved glycol to be optimal (Table 1, entries 8, 13 and 14). Consequently, we decided to use entry 8 as the optimal condition to investigate the application of this transformation.
After the optimized reaction condition was established, we then attempted to apply it to synthesize a series of 6-alkylphenanthridines. As shown in Table 2, when R1 were electron-donating groups such as alkyl and alkoxyl as well as electron-withdrawing groups like fluoro, trifluoromethyl, substituted 6-methylphenanthridines were successfully obtained in good yields (Table 2, 3b–3f). We were pleased to find that amide group, sensitive to acid condition, was observed to tolerate this condition (Table 2, 3g). A chloro group substituent of acetophenone had a negative effect on this reaction (Table 2, 3h). This observation, combined with the fact that some aryl chlorides failed to be compatible well with palladium-catalyzed coupling reaction.58,59
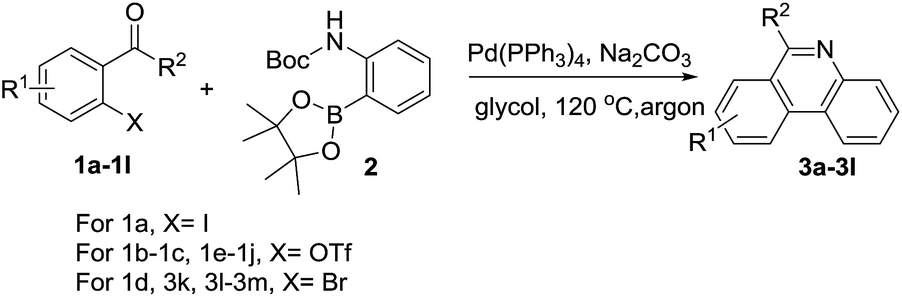
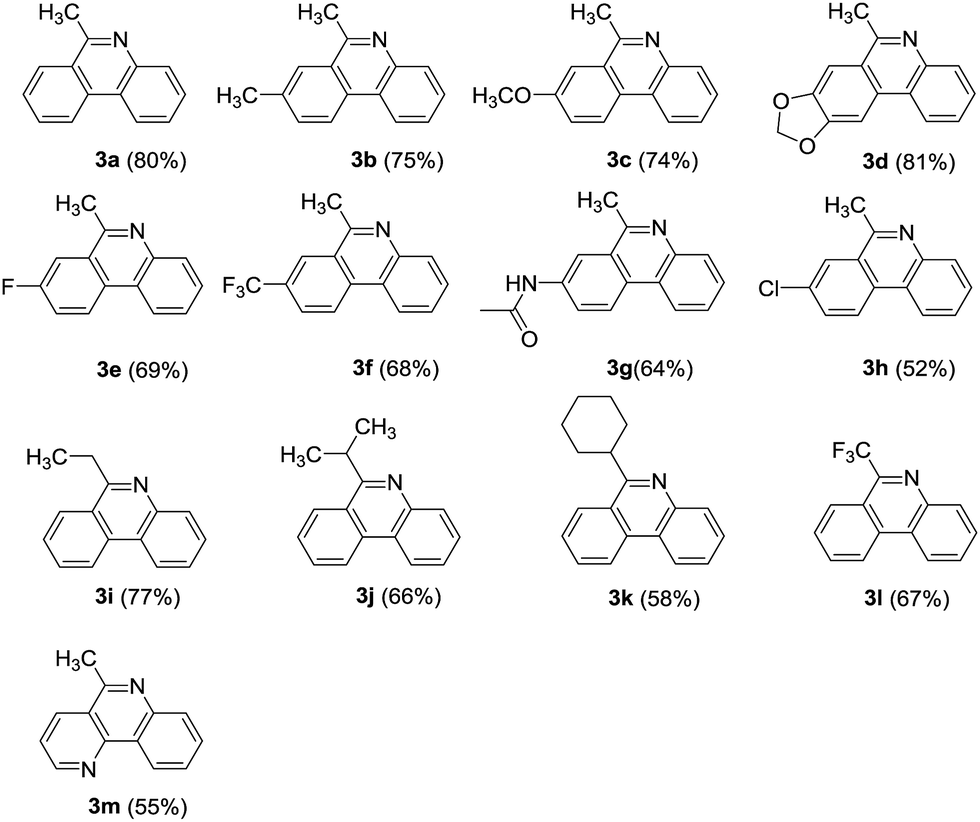
On the other hand, the R2 substitution effect of phenones was then investigated in reactions with 2. The ethyl, isopropyl and cyclohexyl substituted phenones reacted efficiently, affording corresponding phenanthridine derivatives in yields ranging from 58 to 77% (Table 2, 3i–3k). Interestingly, when R2 was trifluoromethyl group, the corresponding product was obtained in 67% yield (Table 2, 3l). Additionally, pyridine fused phenanthridine was also constructed in the current reaction system and obtained with 55% yield (Table 2, 3m).
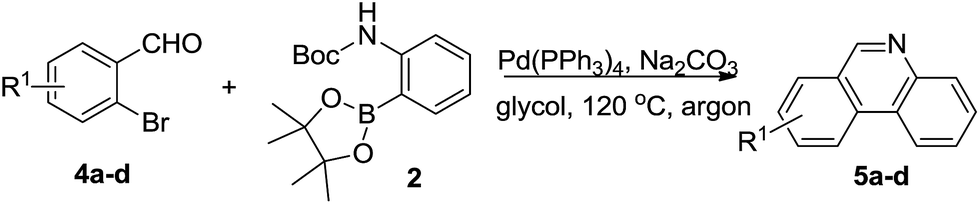
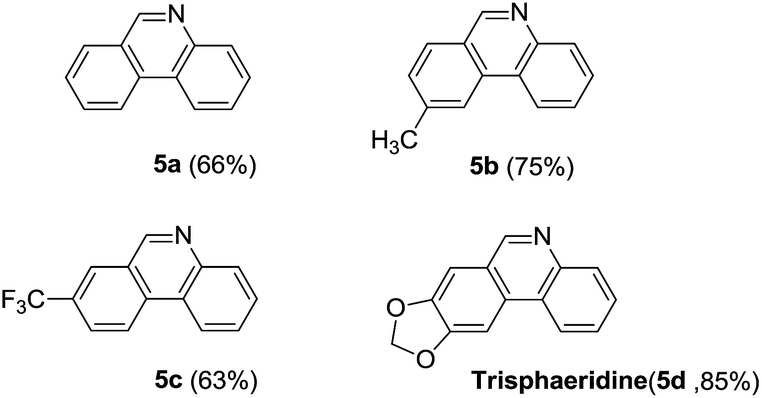
Next, we turned our attention to explore whether substituted bromobenzaldehyde could work well in this reaction system. It was found that substrate bearing an electron-donating group smoothly afforded the corresponding phenanthridines in good yield (Table 3, 5b). Moreover, trifluoromethyl group substituted phenanthridine was obtained in 63% yield (Table 3, 5c). Trisphaeridine, a natural product isolated from plants of Amaryllidaceae, was then synthesized through this method with 85% yield (Table 3, 5d).8
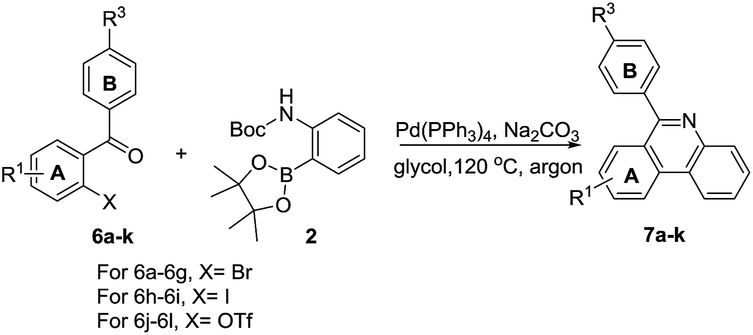
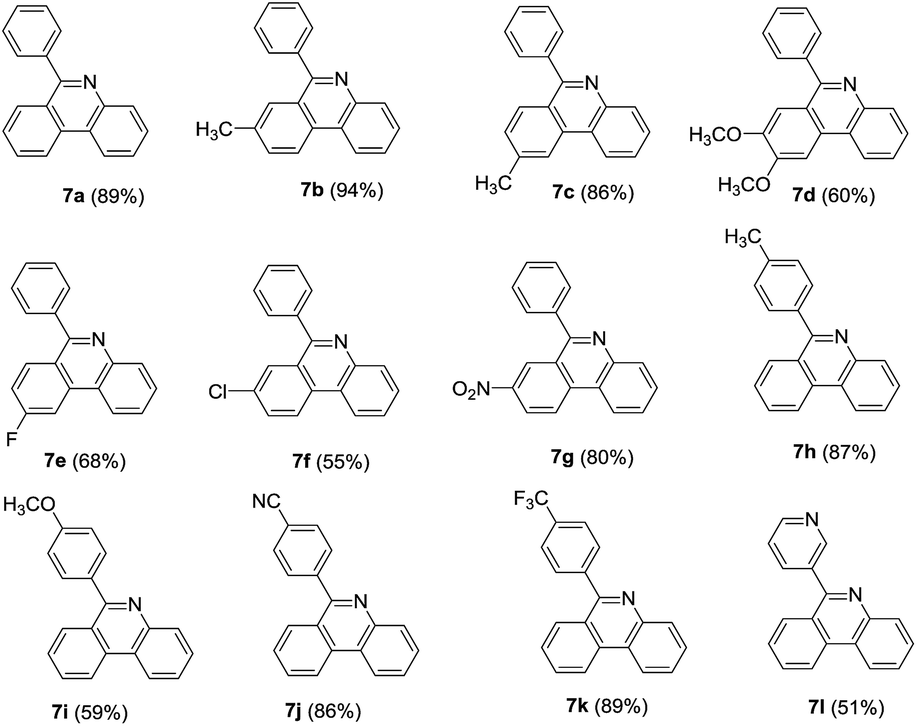
In light of the above results, 2-OTf/halide substituted benzophenones were then subjected to the same conditions to test the scope of the reaction. o-Bromobenzophenones reacted smoothly with 2 affording corresponding phenylphenanthridines in good to excellent yields. In general, electron-donating and electron-withdrawing groups were found to be compatible on both the A and B phenyl rings. For A phenyl ring, after two monosubstituted-6-phenyl-phenanthridines were obtained (Table 4, 7b–7c), the 8,9-dimethoxy-6-phenyl-phenanthridine was also obtained in 60% yield (Table 4, 7d). With respect to halide-substituted substrate, the chloro-substituted substrate afforded the product in a lower yield than the corresponding fluoro-substituted substrate (Table 4, 7e–7f), which was consistent with our previous finding. Moreover, 3-nitro-6-phenylphenanthridine was also successfully obtained in 80% yield (Table 4, 7g). When the substituted groups at the para-position of B phenyl ring were methyl, nitrile and trifluoromethyl groups, the corresponding substituted phenanthridines were obtained in good yields (Table 4, 7h, 7j–7k). However, when the R3 group of B phenyl ring was methoxy group, the yield was only 59% (Table 4, 7i). Noticeably, changing the B phenyl ring to pyridine ring, the corresponding product was obtained in 51% yield (Table 4, 7l).
On the basis of above observations, a plausible mechanism for the reaction is proposed in Scheme 3. First, the substrate 1a and 2 undergo a typical Suzuki coupling reaction to afford intermediate 8a. Then the deprotection of N-Boc group occurs by the promotion of glycol to afford intermediate 8b. Finally, product 3a is obtained by the intramolecular condensation reaction between amino and carbonyl group.
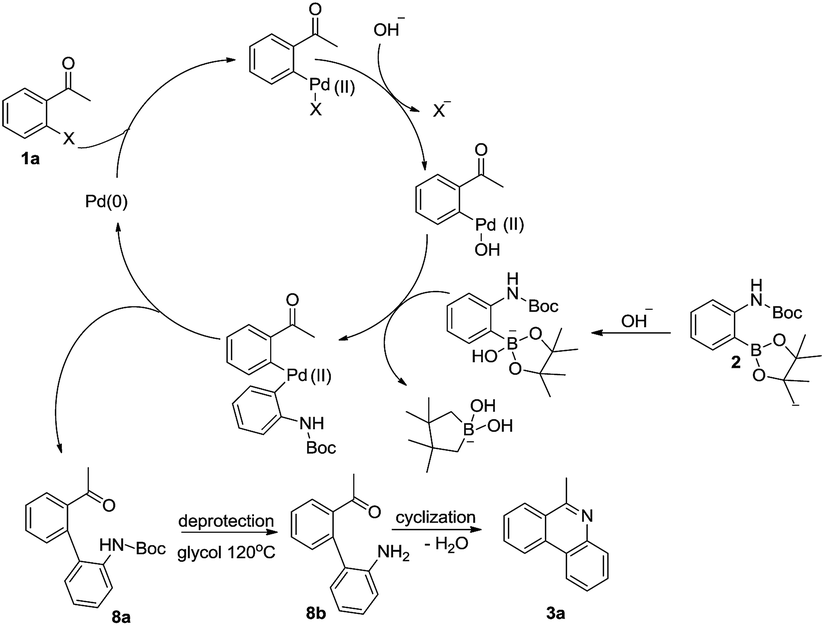 | ||
| Scheme 3 A plausible reaction mechanism. | ||
Conclusions
In summary, we have reported a practical method for one-pot synthesis of substituted phenanthridines, using 2-acylphenyl triflates/halide and 2-(Boc-amino)benzeneboronic acid pinacol ester as the starting materials. A series of substituted phenanthridines with various functional groups are obtained by this method. Our method has several advantages as follows: (a) easily accessible precursors; (b) short reaction time; (c) good functional groups compatibility. The present method therefore would serve as a powerful tool to construct heterocyclic scaffolds that are beneficial for medicinal chemistry and material science.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 81473096).Notes and references
- P. H. Bernardo, K.-F. Wan, T. Sivaraman, J. Xu, F. K. Moore, A. W. Hung, H. Y. Mok, V. C. Yu and C. L. Chai, J. Med. Chem., 2008, 51, 6699–6710 CrossRef CAS PubMed.
- E. Dubost, N. Dumas, C. Fossey, R. Magnelli, S. Butt-Gueulle, C. Ballandonne, D. H. Caignard, F. Dulin, J. Sopkova de-Oliveira Santos and P. Millet, J. Med. Chem., 2012, 55, 9693–9707 CrossRef CAS PubMed.
- T. Nakanishi and M. Suzuki, J. Nat. Prod., 1998, 61, 1263–1267 CrossRef CAS PubMed.
- D. Lamoral-Theys, A. Andolfi, G. Van Goietsenoven, A. Cimmino, B. Le Calvé, N. Wauthoz, V. Mégalizzi, T. Gras, C. Bruyère and J. Dubois, J. Med. Chem., 2009, 52, 6244–6256 CrossRef CAS PubMed.
- D. Cappoen, P. Claes, J. Jacobs, R. Anthonissen, V. Mathys, L. Verschaeve, K. Huygen and N. D. Kimpe, J. Med. Chem., 2014, 57, 2895–2907 CrossRef CAS PubMed.
- T. Ishikawa, Med. Res. Rev., 2001, 21, 61–72 CrossRef CAS PubMed.
- K. K. Schrader, F. Avolio, A. Andolfi, A. Cimmino and A. Evidente, J. Agric. Food Chem., 2013, 61, 1179–1183 CrossRef CAS PubMed.
- F. Viladomat, M. Selles, C. Cordina and J. Bastida, Planta Med., 1997, 63, 583 CrossRef CAS PubMed.
- M. Cushman and L. Cheng, J. Org. Chem., 1978, 43, 286–288 CrossRef CAS.
- I. Cesari, P. Grisoli, M. Paolillo, C. Milanese, G. Massolini and G. Brusotti, J. Pharm. Biomed. Anal., 2015, 105, 115–120 CrossRef CAS PubMed.
- K.-H. Merz, T. Muller, S. Vanderheiden, G. Eisenbrand, D. Marko and S. Bräse, Synlett, 2006, 20, 3461–3463 Search PubMed.
- S. A. Baechler, M. Fehr, M. Habermeyer, A. Hofmann, K.-H. Merz, H.-H. Fiebig, D. Marko and G. Eisenbrand, Bioorg. Med. Chem., 2013, 21, 814–823 CrossRef CAS PubMed.
- W. M. Messmer, M. Tin-wa, H. H. S. Fong, C. Bevelle, N. R. Farnsworth, D. J. Abraham and J. Trojánek, J. Pharm. Sci., 1972, 61, 1858–1859 CrossRef CAS PubMed.
- M. Rivaud, A. Mendoza, M. Sauvain, A. Valentin and V. Jullian, Bioorg. Med. Chem., 2012, 20, 4856–4861 CrossRef CAS PubMed.
- P. P. Slonimski, G. Perrodin and J. H. Croft, Biochem. Biophys. Res. Commun., 1968, 30, 232–239 CrossRef CAS PubMed.
- J. Huang, Z. Wang, J.-K. Kim, X. Su and Z. Li, Anal. Chem., 2015, 87, 12254–12261 CrossRef CAS PubMed.
- A. Melaku and B. Birasa, Egypt. J. Biomed. Sci., 2013, 5, 82–89 Search PubMed.
- A. Pictet and A. Hubert, Ber. Dtsch. Chem. Ges., 1896, 29, 1182–1189 CrossRef CAS.
- G. T. Morgan and L. P. Walls, J. Chem. Soc., 1931, 2447–2456 RSC.
- J. J. Li and E. J. Corey, Name Reactions in Heterocyclic Chemistry, John Wiley & Sons, Inc., 2005, ch. 9, pp. 375–494 Search PubMed.
- R. K. Chinnagolla and M. Jeganmohan, Chem. Commun., 2014, 50, 2442–2444 RSC.
- D. A. Candito and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 6713–6716 CrossRef CAS PubMed.
- T. Gerfaud, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 572–577 CrossRef CAS PubMed.
- G. Maestri, M. H. Larraufie, E. Derat, C. Ollivier, L. Fensterbank, E. Lacote and M. Malacria, Org. Lett., 2010, 12, 5692–5695 CrossRef CAS PubMed.
- J. Peng, T. Chen, C. Chen and B. Li, J. Org. Chem., 2011, 76, 9507–9513 CrossRef CAS PubMed.
- R. D. Grigg, R. Van Hoveln and J. M. Schomaker, J. Am. Chem. Soc., 2012, 134, 16131–16134 CrossRef CAS PubMed.
- R. Pearson, S. Zhang, G. He, N. Edwards and G. Chen, Beilstein J. Org. Chem., 2013, 9, 891–899 CrossRef CAS PubMed.
- X. Bao, W. Yao, Q. Zhu and Y. Xu, Eur. J. Org. Chem., 2014, 33, 7443–7450 CrossRef.
- H. Jiang, H. Gao, B. Liu and W. Wu, RSC Adv., 2014, 4, 17222–17225 RSC.
- B. Wang, Y. Dai, W. Tong and H. Gong, Org. Biomol. Chem., 2015, 13, 11418–11421 CAS.
- R. Alonso, P. J. Campos, B. Garcia and M. A. Rodriguez, Org. Lett., 2006, 8, 3521–3523 CrossRef CAS PubMed.
- M. E. Buden, V. B. Dorn, M. Gamba, A. B. Pierini and R. A. Rossi, J. Org. Chem., 2010, 75, 2206–2218 CrossRef CAS PubMed.
- A. M. Linsenmeier, C. M. Williams and S. Brase, J. Org. Chem., 2011, 76, 9127–9132 CrossRef CAS PubMed.
- Z. Li, F. Fan, J. Yang and Z. Q. Liu, Org. Lett., 2014, 16, 3396–3399 CrossRef CAS PubMed.
- L. Wang, W. Sha, Q. Dai, X. Feng, W. Wu, H. Peng, B. Chen and J. Cheng, Org. Lett., 2014, 16, 2088–2091 CrossRef CAS PubMed.
- Z. Xia, J. Huang, Y. He, J. Zhao, J. Lei and Q. Zhu, Org. Lett., 2014, 16, 2546–2549 CrossRef CAS PubMed.
- Z. Xu, C. Yan and Z. Q. Liu, Org. Lett., 2014, 16, 5670–5673 CrossRef CAS PubMed.
- T. H. Zhu, S. Y. Wang, Y. Q. Tao, T. Q. Wei and S. J. Ji, Org. Lett., 2014, 16, 1260–1263 CrossRef CAS PubMed.
- Z. Zhang, X. Tang and W. R. Dolbier Jr, Org. Lett., 2015, 17, 4401–4403 CrossRef CAS PubMed.
- H. Jiang, X. An, K. Tong, T. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2015, 54, 4055–4059 CrossRef CAS PubMed.
- B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 RSC.
- B. K. Mehta, K. Yanagisawa, M. Shiro and H. Kotsuki, Org. Lett., 2003, 5, 1605–1608 CrossRef CAS PubMed.
- W. G. Shou, Y. Y. Yang and Y. G. Wang, J. Org. Chem., 2006, 71, 9241–9243 CrossRef CAS PubMed.
- L. Sripada, J. A. Teske and A. Deiters, Org. Biomol. Chem., 2008, 6, 263–265 CAS.
- R. Yanada, K. Hashimoto, R. Tokizane, Y. Miwa, H. Minami, K. Yanada, M. Ishikura and Y. Takemoto, J. Org. Chem., 2008, 73, 5135–5138 CrossRef CAS PubMed.
- C. Tang, Y. Yuan and N. Jiao, Org. Lett., 2015, 17, 2206–2209 CrossRef CAS PubMed.
- J. Li, H. Wang, J. Sun, Y. Yang and L. Liu, Org. Biomol. Chem., 2014, 12, 7904–7908 CAS.
- J. Tummatorn, S. Krajangsri, K. Norseeda, C. Thongsornkleeb and S. Ruchirawat, Org. Biomol. Chem., 2014, 12, 5077–5081 CAS.
- J. J. S. Lamba and J. M. Tour, J. Am. Chem. Soc., 1994, 116, 11723–11736 CrossRef CAS.
- S. Bräse, D. Marko, K.-H. Merz, T. Muller, S. Vanderheiden and G. Eisenbrand, Synlett, 2006, 20, 3461–3463 Search PubMed.
- G. R. Geen, I. S. Mann, M. V. Mullane and A. McKillop, Tetrahedron, 1998, 54, 9875–9894 CrossRef CAS.
- S. Dhara, M. Ghosh and J. K. Ray, Synlett, 2013, 24, 2263–2265 CrossRef CAS.
- Y. Tian, J. Qi, C. Sun, D. Yin, X. Wang and Q. Xiao, Org. Biomol. Chem., 2013, 11, 7262–7266 CAS.
- Y. Tian, X. Wang, Q. Xiao, C. Sun and D. Yin, J. Org. Chem., 2014, 79, 9678–9685 CrossRef CAS PubMed.
- T. Liu, X. Wang and D. Yin, RSC Adv., 2015, 5, 75794–75805 RSC.
- J. Qi, C. Sun, Y. Tian, X. Wang, G. Li, Q. Xiao and D. Yin, Org. Lett., 2014, 16, 190–192 CrossRef CAS PubMed.
- Q.-Y. Zhang, X.-J. Wang, Y.-L. Tian, J.-G. Qi, C. Li and D.-L. Yin, Chin. Chem. Lett., 2013, 24, 825–828 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- J. J. Li, Name Reactions for Homologations, John Wiley & Sons, Inc., 2009, ch. 1, pp. 163–185 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra. See DOI: 10.1039/c6ra00249h |
| This journal is © The Royal Society of Chemistry 2016 |