
Synthesis and catalytic performance of ruthenium complexes ligated with rigid o-(diphenylphosphino)aniline for chemoselective hydrogenation of dimethyl oxalate†
Xiaolong Fang,
Chunyan Zhang,
Jin Chen,
Hongping Zhu* and
Youzhu Yuan*
State Key Laboratory of Physical Chemistry of Solid Surfaces, National Engineering Laboratory for Green Chemical Productions of Alcohols-Ethers-Esters, iChEM, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, China. E-mail: yzyuan@xmu.edu.cn; hpzhu@xmu.edu.cn
First published on 4th May 2016
Abstract
A series of new ruthenium complexes with rigid ligand o-(diphenylphosphino)aniline, including [(PPh3)(o-PPh2C6H4NH2)RuCl2]2 (1), (o-PPh2C6H4NH2)2RuCl2 (2), [(o-PPh2C6H4NH2)2(o-PPh2C6H4NH)Ru]+Cl− (3), Ph3P(η2-H2)Ru(μ-H)(μ-o-PPh2C6H4NH)2RuH(PPh3) (4), (o-PPh2C6H4NH2)(o-PPh2C6H4NH)RuCl(CO) (5), (o-PPh2C6H4NH2)(o-PPh2C6H4NH)RuH(CO) (6), and [(o-PPh2C6H4NH)2Ru(CO)]2 (7) were synthesized and employed as catalysts for chemoselective hydrogenation of esters. Among them, complexes 1, 2, and 5 exhibited excellent performance in hydrogenation of dimethyl oxalate to methyl glycolate, in comparison with the ruthenium complexes with a flexible aminophosphine ligand, such as (Ph2P(CH2)2NH2)2RuCl2, (Ph2P(CH2)3NH2)2RuCl2, and (o-Ph2PC6H4CH2NH2)2RuCl2, under identical conditions. Complexes 1 and 2 also displayed good activities in the hydrogenation of other aliphatic and cyclic esters. The catalytic mechanism of hydrogenation was discussed according to the results of NMR spectroscopic studies and control experiments.
Introduction
Selective hydrogenation of esters to alcohols is an important transformation from both conceptual and practical perspectives.1 For example, the chemoselective hydrogenation of dimethyl oxalate (DMO) to methyl glycolate (MG) and further to ethylene glycol (EG) is a key step in the process so-called “coal to EG”.2 Compared with stoichiometric reactions with hydride reagents (e.g., LiAlH4 or NaBH4), catalytic hydrogenation with dihydrogen (H2) inhibits the production of metal salt waste and is more economical and environmentally benign.1,3 Therefore, catalytic hydrogenation with H2 is widely accepted and adopted. Industrially, this process is commonly accomplished by using heterogeneous supported metal catalysts at relatively high reaction temperatures and H2 pressures.1b,4 In contrast, molecularly-defined organometallic complexes are usually considered to be more active at lower reaction temperatures and H2 pressures, which might be beneficial for getting higher selectivity to the desired products.3,5Grey et al.6 and Matteoli et al.7 used ruthenium-hydride anions and ruthenium-cluster complexes coordinated with phosphine ligands as homogeneous catalysts, respectively, for ester hydrogenation. However, these complexes exhibited low activities and required drastic reaction conditions (180 °C and 200 bar H2) to achieve complete conversion. Subsequently, Elsevier et al.8 developed an in situ catalyst system of Ru(acac)3/MeC(CH2PPh2)3/Zn (acac = acetylacetonate); this catalyst was used for the DMO hydrogenation at 100 °C and 70 bar H2 for 16 h and generated 94% EG. Milstein et al.9 reported the first example of homogeneous non-activated aromatic and aliphatic ester hydrogenation by using pincer-type ruthenium complex a (Scheme 1) under 5.3 bar of H2 at 115 °C. Saudan et al.10 demonstrated the outstanding performance of ruthenium complexes b and c (Scheme 1) for ester and lactone reduction, which contain bidentate N,P-chelate and tetradentate P,N,N′,P′-chelate ligands, respectively. Since then, scientists worldwide have paid much attention on homogeneous ester hydrogenation and developed several other efficient catalysts.5,11,12 As we know, the ligands used in these catalysts are relatively flexible and the complexes with soft structures are preferred for the hydrogenation of esters.11a,13
 | ||
| Scheme 1 Selected homogeneous catalysts. | ||
o-(Diphenylphosphino)aniline14 is an N,P-chelate ligand containing an NH2 group. After ligating with ruthenium, the complexes thus formed contain a rigid five-member chelate ring.15 They have been applied to the conversion of nitroarene into secondary amines and tertiary amines by using primary alcohols as sources of hydrogen and N-alkylation groups.15a,b
However, the application of these complexes in the hydrogenation of esters is rarely reported. In this work, we have synthesized and characterized a series of new o-(diphenylphosphino)anilinoruthenium complexes. The catalytic results manifested that the rigid o-(diphenylphosphino)anilinoruthenium complexes with proper structure configuration could show high performance for the hydrogenation of esters.
Results and discussion
Synthesis of ruthenium(II) complexes
Complexes [(PPh3)(o-PPh2C6H4NH2)RuCl2]2 (1, 87% isolated yield) and (o-PPh2C6H4NH2)2RuCl2 (2, 92% isolated yield) were prepared by ligand substitution reactions of RuCl2(PPh3)3 and o-PPh2C6H4NH2 in toluene at 100 °C, with the molar ratios of these two complexes setting to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2, respectively. The reaction of (PPh3)3RuHCl with 3 equiv. of o-PPh2C6H4NH2 afforded complex [(o-PPh2C6H4NH2)2(o-PPh2C6H4NH)Ru]+Cl− (3, 77% isolated yield) under identical conditions. Similarly, complex (o-PPh2C6H4NH2)(o-PPh2C6H4NH)RuCl(CO) (5, 86% isolated yield) was prepared from the reaction of (PPh3)3RuHCl(CO) and 2 equiv. of o-PPh2C6H4NH2 (Scheme 2).
:1 and 1:2, respectively. The reaction of (PPh3)3RuHCl with 3 equiv. of o-PPh2C6H4NH2 afforded complex [(o-PPh2C6H4NH2)2(o-PPh2C6H4NH)Ru]+Cl− (3, 77% isolated yield) under identical conditions. Similarly, complex (o-PPh2C6H4NH2)(o-PPh2C6H4NH)RuCl(CO) (5, 86% isolated yield) was prepared from the reaction of (PPh3)3RuHCl(CO) and 2 equiv. of o-PPh2C6H4NH2 (Scheme 2).
 | ||
| Scheme 2 Synthesis of complexes 1–3 and 5. | ||
The reactions of 1, 2 and 5 with hydride reagent K[HBsBu3] could readily afford corresponding ruthenium hydride complexes. Treatment of 1 with K[HBsBu3] in THF from −75 °C to room temperature yielded complex Ph3P(η2-H2)Ru(μ-H)(μ-o-PPh2C6H4NH)2RuH(PPh3) (4) with 85% isolated yield (Fig. 1). The formation of 4 might proceed through the dihydride intermediate (o-PPh2C6H4NH2)Ru(H)2(PPh3) (4a), similar to the transformation of dihydride complexes (PPh3)2(cydn)Ru(H)2 (cydn = (R,R)-cyclohexyldiamine) and (R-binap)(tmen)Ru(H)2 (tmen = NH2CMe2CMe2NH2).16,17 The reaction of 2 with K[HBsBu3] under similar condition produced a mixture of complexes, but isolation of the pure complex was unsuccessful. Finally, the reaction of 5 with K[HBsBu3] successfully produced complex (o-PPh2C6H4NH2)(o-PPh2C6H4NH)RuH(CO) (6) (Fig. 2). Notably, heat treatment at 70 °C converted 6 into [(o-PPh2C6H4NH)2Ru(CO)]2 (7) by eliminating one H2 molecule (Fig. 2).
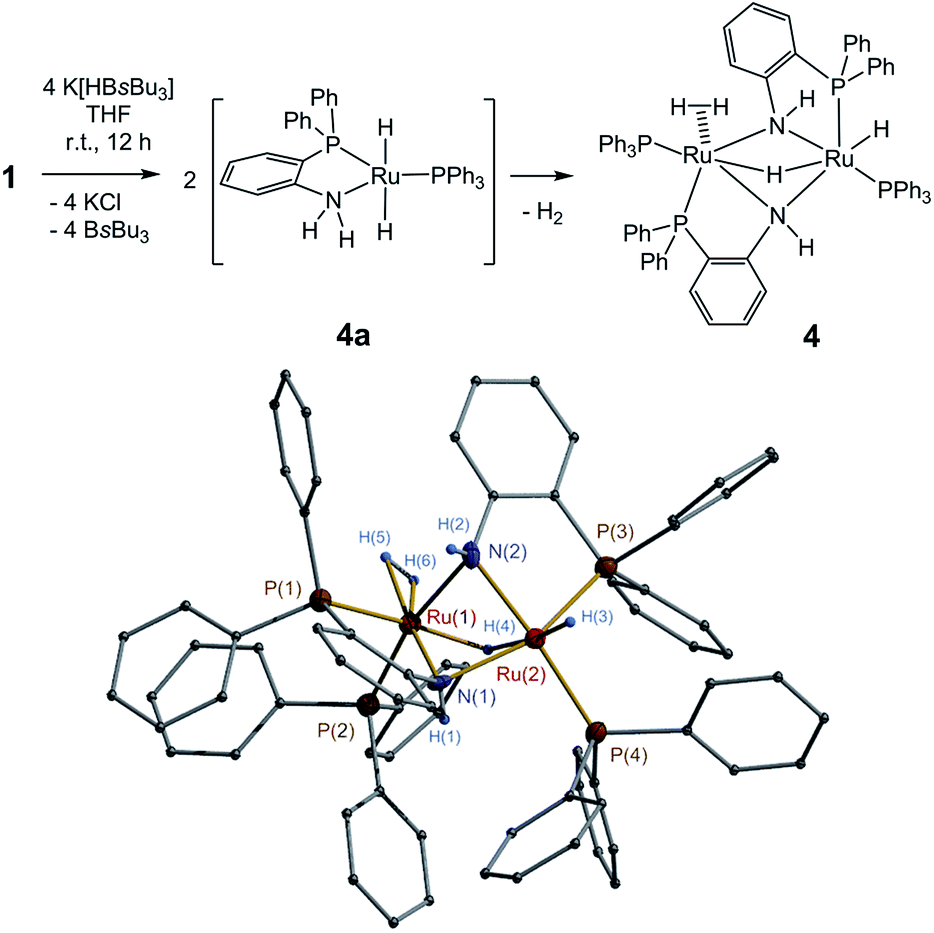 | ||
| Fig. 1 Synthesis and X-ray structure of complex 4. | ||
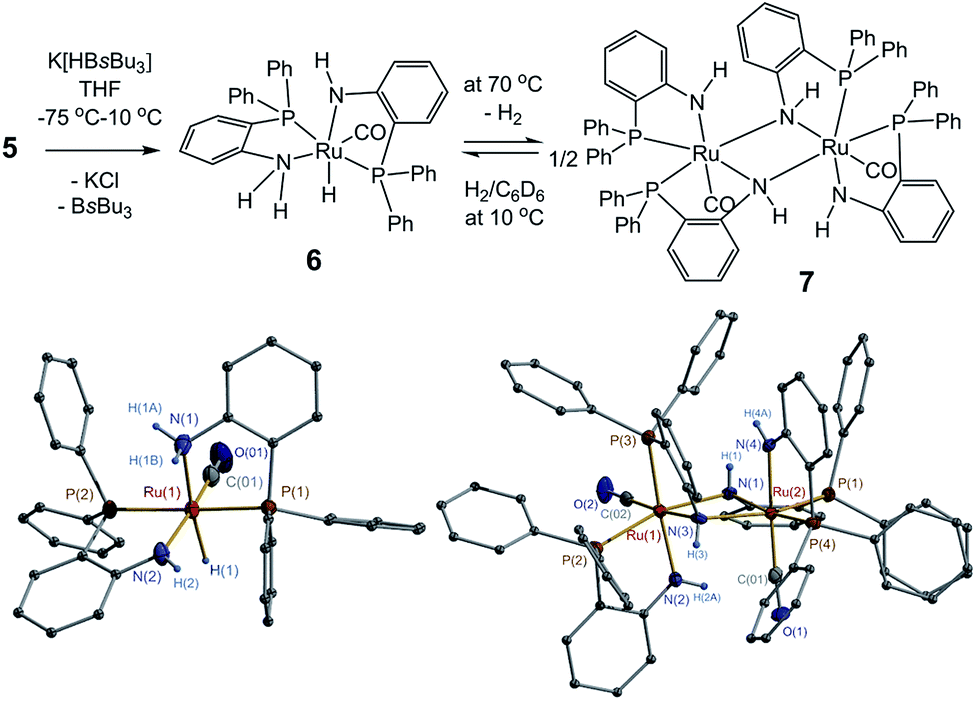 | ||
| Fig. 2 Synthesis and X-ray structure of complexes 6–7. | ||
Characterization of ruthenium(II) complexes
The complexes 1–7 were characterized by NMR and IR spectroscopy and CHN elemental analysis, of which 1–2 and 4–7 were further studied by X-ray crystallography. X-ray structure analysis confirmed that 1 was a dimer in solid state (Fig. S1 in ESI†). In solution, 1 can dissociate into (PPh3)(o-PPh2C6H4NH2)RuCl2 (1a),15d as indicated by the variable-temperature (25 to −75 °C) 31P{1H} NMR studies (Fig. S7 in ESI†). The mononuclear complex 2 possesses a structure configuration comparable with those of Noyori's N,P-chelate18 and P,N,N′,P′-chelate19 ruthenium complexes (Fig. S2 in ESI†).The complex 4 exhibited an asymmetric dinuclear structure (Fig. 1). The terminal Ru–H bond length [1.65(6) Å] was close to that of Ru–η2-H2 [1.68(7) Å (av)] but shorter than that of Ru–μ-H [1.77(5) Å (av)]. In the 1H NMR spectrum, resonances at δ −12.27, −8.38, and −8.22 ppm are assigned to the protons of Ru–μ-H, Ru–H, and Ru–η2-H2, respectively. These resonance values are comparable with those of previously reported related moieties.16c,20 The IR bands for the bonds of these moieties were found at 2113, 1956, and 1901 cm−1.
The complexes 5 and 6 both had two N,P-chelates by o-PPh2C6H4NH2 and o-PPh2C6H4NH at ruthenium (Fig. S4 in ESI† and 2). The Ru–H bond length in 6 was 1.67(2) Å, close to the corresponding value in 4. The complex 5 was insoluble in organic solvent and was only subjected to solid-state 31P NMR and IR spectral analyses. The complex 6 was soluble in aromatic hydrocarbons and was characterized by solution NMR (1H and 31P{1H}) spectroscopy together with solid-state IR spectrometry. The 1H NMR spectrum of 6 exhibited a proton resonance at δ −12.14 ppm, assignable to the Ru–H, which gave an IR vibration of Ru–H bond at 2190 cm−1.
Compound 7 was a dimer with two o-PPh2C6H4NH ligands served as a μ-κ1(N):η2(N,P)-type bridge (Fig. 2). However, complex 7 lacked symmetry probably because of the equatorial/axial location difference among the four o-PPh2C6H4NH ligands around each ruthenium center. The solution NMR spectra displayed two groups of data for the CO carbon resonances and four groups of data for the 1H, 13C{1H}, and 31P{1H} resonances of the four o-PPh2C6H4NH ligands.
Kinetic studies on transformation between 6 and 7
The thermal conversion of 6 into 7 under elimination of one H2 molecule prompted us to investigate the possibility of reversing the reaction of 7 and H2 to produce the original complex 6. This process is also considered in catalytic H2 hydrogenation reactions.16a,b,21 At a reaction temperature of 10 °C, exposure of the C6D6 solution of 7 to H2 for 56 h expectedly led to the complete formation of 6, as traced by 1H and 31P{1H} NMR spectra (Fig. S8 and S9 in ESI†). Therefore, while the complex 6 eliminated one H2 molecule through the Ru(H) ← NH2 unit to form 7, the complex 7 underwent H2 addition through the as-formed Ru–NH unit to be transformed into 6, exhibiting heterolytic splitting of H2. H2 elimination/addition switched between 6 and 7 was realized by monitoring the reaction temperature. To understand this reaction process in detail, we performed a reaction using 7 with D2 under the same condition as that of 7 with H2. Fig. 3 combines the 1H NMR data of this reaction with that of the reaction of 7 and H2 to 6. By comparison, after reaction completion, RuH proton resonance remained present with NH and NH2 resonances, although under low integral intensity. This finding revealed that in the final formation of 6, the reaction may have occurred through an alternative switch between 7 and 6 under D2/DH addition/elimination despite carrying out the reaction at 10 °C (Scheme S1 in ESI†).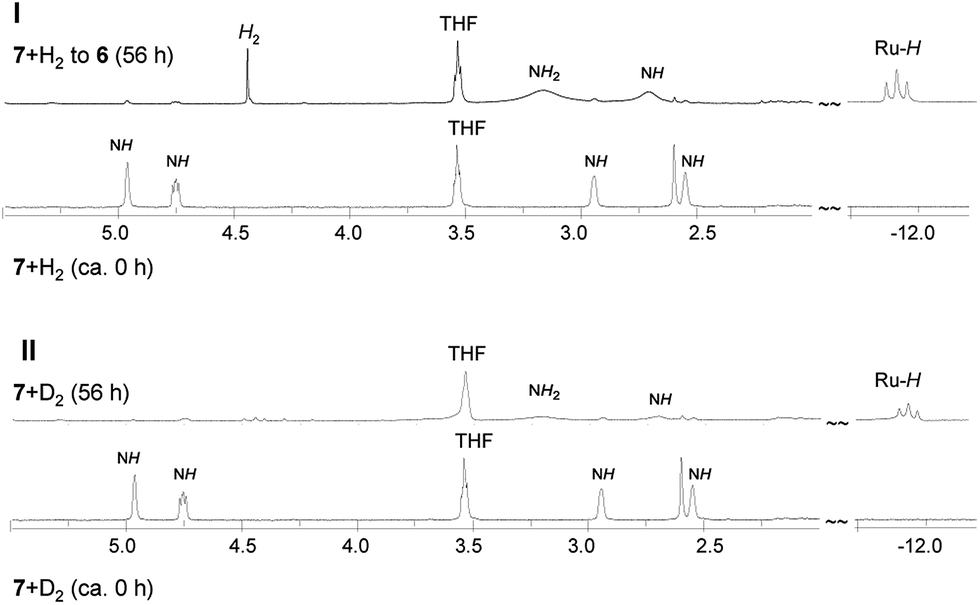 | ||
| Fig. 3 Comparison of 1H NMR spectra for reactions of 7 with H2 (I) and D2 (II) recorded in C6D6 at 10 °C (δNH/NH2, 5.5 to 2.0 ppm; δRuH, −11.0 to −12.7 ppm). | ||
In the presumed 4a-mediated production of 4, 4a also indeed formed the Ru(H) ← NH2 unit through the metathesis reaction of 1 and K[HBsBu3] and was able to eliminate one H2 molecule in a similar manner to that of 6. Nonetheless, a reaction of 1 with K[HBsBu3] in D2 atmosphere was carried out. By comparing the 1H NMR data of the obtained complex and 4 (Fig. S10 in ESI†), a partially D/H-exchange occurred.
Catalytic hydrogenation of DMO
The complexes 1–5 and 7 were firstly applied in DMO hydrogenation. Under the conditions at 100 °C and 50 bar H2 in THF solvent, both 1 and 2 could catalyze the reaction to give MG in excellent yield (entries 1 and 2, Table 1). Moreover, 5 yielded a quantitative conversion and a 99% yield after 3 h (entry 6). The performance of these rigid complexes were better than those of the flexible ruthenium complexes b, (Ph2P(CH2)3NH2)2RuCl2 (d), or (o-Ph2PC6H4CH2NH2)2RuCl2 (e) (46%, 16%, and 49% yield of MG within 4 h, entries 9–11), which have configuration similar to 2 and are good catalysts for the hydrogenation of other esters, ketones and amines.10,18a,22 Under the same conditions, 3 showed no activity (entry 3). The results indicate the superiority of rigid o-(diphenylphosphino)anilinoruthenium complexes with proper structure for the chemoselective hydrogenation of DMO to MG.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | NaOMe/Rub | Time (h) | Conv.c (%) | Yield of MGc (%) |
| a DMO (7.57 mmol) in THF (10 mL) was hydrogenated by the catalyst at 100 °C; the molar ratio of DMO to ruthenium was 200.b Molar ratio.c Determined by GC. | |||||
| 1 | 1 | 10 | 1 | 97 | 97 |
| 2 | 2 | 10 | 1 | 97 | 97 |
| 3 | 3 | 10 | 1 | 0 | 0 |
| 4 | 4 | 0 | 20 | 97 | 92 |
| 5 | 4 | 10 | 20 | 100 | 96 |
| 6 | 5 | 10 | 3 | 100 | 99 |
| 7 | 7 | 0 | 3 | 86 | 86 |
| 8 | 7 | 10 | 3 | 99 | 99 |
| 9 | b | 10 | 4 | 46 | 46 |
| 10 | d | 10 | 4 | 17 | 16 |
| 11 | e | 10 | 4 | 50 | 49 |
Without using NaOMe, the hydrogenation with complex 4 led to 97% conversion and 92% yield in a relatively longer time of 20 h (entry 4). When combined with NaOMe, improved result was obtained (100% conversion and 96% yield in 20 h, entry 5). Similar performance was also obtained using complex 7 (entries 7–8).
We then studied the effect of reaction conditions on the performance of complex 2 for the DMO hydrogenation. As listed in Table 2, 2 could convert DMO into MG easily at either low H2 pressure (entries 12–13) or mild temperature (entries 14–21). Especially, 86% yield of MG was afforded at room temperature after 24 h (entry 21). The amount of NaOMe used significantly influenced DMO transformation in a volcano-type trend (entries 16–20), and 40 equiv. of NaOMe over 2 gave the optimal result (81% MG yield at 40 °C in 1 h). NaOMe not only promoted the Cl− to H− metathesis in the presence of H2,23 but also accelerated the deprotonation of the NH or NH2 group. The latter function has been studied in detail by Bergens et al.24 for the hydrogenation of amide and imide carbonyls. Finally, 2 was investigated to catalyze the hydrogenation of DMO in 2000 molar equiv. relative to 2. An excellent result with 98% MG yield was obtained within 16 h (entry 22), indicating the high performance of 2 for this chemoselective hydrogenation.
| Entry | Catalyst | NaOMe/Rub | DMO/Rub | T (°C) | P (bar) | Time (h) | Conv.c (%) | Yield of MGc (%) |
|---|---|---|---|---|---|---|---|---|
| a DMO (7.57 mmol) in THF (10 mL) was hydrogenated by the catalyst.b Molar ratio.c Determined by GC. | ||||||||
| 12 | 2 | 10 | 200 | 100 | 20 | 1 | 74 | 74 |
| 13 | 2 | 10 | 200 | 100 | 20 | 3 | 98 | 98 |
| 14 | 2 | 10 | 200 | 60 | 50 | 1 | 81 | 81 |
| 15 | 2 | 10 | 200 | 60 | 50 | 2 | 98 | 97 |
| 16 | 2 | 5 | 200 | 40 | 50 | 1 | 31 | 31 |
| 17 | 2 | 10 | 200 | 40 | 50 | 1 | 35 | 35 |
| 18 | 2 | 15 | 200 | 40 | 50 | 1 | 44 | 44 |
| 19 | 2 | 40 | 200 | 40 | 50 | 1 | 81 | 81 |
| 20 | 2 | 80 | 200 | 40 | 50 | 1 | 65 | 65 |
| 21 | 2 | 10 | 200 | 25 | 50 | 24 | 86 | 86 |
| 22 | 2 | 10 | 2000 | 100 | 50 | 16 | 98 | 98 |
Catalytic hydrogenation of DMO to EG by complexes 1 and 2
The results in Tables 1 and 2 indicated that DMO could be converted into MG as the major product by using the o-(diphenylphosphino)anilinoruthenium complexes under the conditions of 100 °C and 50 bar H2. Under these conditions, negligible activity was observed for converting MG (200 equiv.) into EG by using 2 and 10 equiv. of NaOMe. With increased temperature to 120 °C, MG started to be transformed into EG, but only 25% conversion was achieved within 8 h. At this temperature, the NaOMe amount was further increased from 10 equiv. to 20 equiv., and improved conversion by 92% was obtained (entry 40 in Table 4, vide infra). The influence of NaOMe amount on the catalytic activity was found for the DMO conversion into MG. Taking the results into account, we performed the reaction for EG production by DMO hydrogenation. However, the reaction produced only 4% yield of EG but 94% yield of MG by 2/NaOMe/DMO (1/20/200) within 16 h (entry 26 in Table 3). At this stage, when we reduced the DMO/Ru ratio to 100/1 and prolonged the reaction time to 36 h, conversion into EG at a yield of 97% was achieved (entry 29). In comparison, the 1/NaOMe/DMO (1/40/200) system required a higher temperature of 140 °C to gain 94% yield of EG within 36 h (entry 25).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | DMO/Rub | Time (h) | Conv.c (%) | Yield of MGc (%) | Yield of EGc (%) |
| a DMO (3.49 mmol) in THF (10 mL) was hydrogenated by the catalyst at 120 °C; the molar ratio of NaOMe to ruthenium was 20.b Molar ratio.c Determined by GC.d At 140 °C. | ||||||
| 23 | 1 | 100 | 36 | 100 | 31 | 61 |
| 24 | 1 | 100 | 72 | 100 | 25 | 68 |
| 25 | 1 | 100 | 36d | 100 | 0 | 94 |
| 26 | 2 | 200 | 16 | 100 | 94 | 4 |
| 27 | 2 | 100 | 16 | 100 | 51 | 47 |
| 28 | 2 | 100 | 24 | 100 | 20 | 75 |
| 29 | 2 | 100 | 36 | 100 | 0 | 97 |
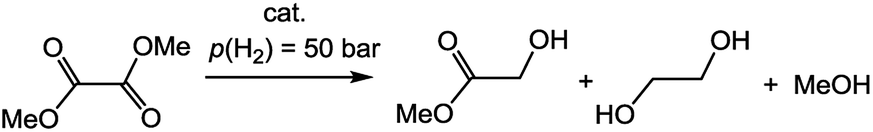
Compared with DMO, MG was a less activated ester because of the loss of one ester substituent.8,13 Mostly due to this electronic characteristic, the reaction conditions were more severe for the conversion of MG into EG or DMO into EG than those for converting DMO into MG. Increasing the reaction temperature could be an effective approach for the former conversion, as shown in previous reports.8b Meanwhile, using more NaOMe greatly promoted the deprotonation of the NH or NH2 group24 and then increased the catalytic reactivity toward either DMO or MG. Finally, MeOH was produced as one of the products during hydrogenation of DMO. The amount of MeOH that accumulated in this closed reaction system may greatly influence its kinetic conversion. Apparently, this influence was greater for conversion into EG than that into MG because the former underwent an additional hydrogenation process. Reducing the amount of DMO relative to the catalyst is benefit for conversion into EG.
Catalytic hydrogenation of other esters by complexes 1 and 2
We used 1 and 2 for the hydrogenation of other esters. In general, the catalytic activities for these reactions by 2 were better than those by 1 (Table 4). Moreover, 2 showed slightly better activity for quantitative conversion of methyl lactate into 1,2-propanediol (entry 41) than that of MG into EG (entry 40). This result suggested the electronic effect of the Me group attached onto the substituent of MG on the reduction of the adjacent ester group. In methyl pyruvate hydrogenation, methyl lactate was obtained under mild conditions (entries 32 and 42). Increase the temperature and H2 pressure, both the ketone and ester groups of methyl pyruvate were hydrogenated (entries 33 and 43). The result indicate that the complexes 1 and 2 can be also useful for the hydrogenating the other carbonyl derivatives. Excellent performances were achieved for transforming cyclic esters to diols (entries 44–47). However, low activities were found for phenyl group-containing methyl phenylacetate and methyl benzoate. The phenyl group, especially in the latter, probably exerted a significant steric influence in consideration of the rigid five-member chelate ring on the catalyst structure. Similar results of steric influence have been observed in reactions involving sterically hindered substrates.11a,b,n
|
||||
|---|---|---|---|---|
| Entry | Ester | Catalyst | Conv.b (%) | Yieldb (%) |
| a Ester (7.57 mmol) in THF (10 mL) was hydrogenated under 50 bar H2 and 120 °C for 8 h; the molar ratio of NaOMe to ruthenium was 20 and that of ester to ruthenium was 200.b Determined by GC.c Performed at 40 °C and 10 bar H2 for 1 h; the molar ratio of ester to ruthenium was 1000.d Methyl lactate.e 1,2-Propanediol. | ||||
| 30 | I | 1 | 62 | 61 |
| 31 | II | 1 | 82 | 80 |
| 32c | III | 1 | 100 | 100d |
| 33 | III | 1 | 82 | 82e |
| 34 | IV | 1 | 96 | 92 |
| 35 | V | 1 | 80 | 80 |
| 36 | VI | 1 | 93 | 84 |
| 37 | VII | 1 | 100 | 95 |
| 38 | VIII | 1 | 39 | 35 |
| 39 | IX | 1 | 17 | 9 |
| 40 | I | 2 | 92 | 92 |
| 41 | II | 2 | 100 | 100 |
| 42c | III | 2 | 100 | 100d |
| 43 | III | 2 | 100 | 98e |
| 44 | IV | 2 | 97 | 88 |
| 45 | V | 2 | 100 | 99 |
| 46 | VI | 2 | 92 | 91 |
| 47 | VII | 2 | 100 | 95 |
| 48 | VIII | 2 | 68 | 63 |
| 49 | IX | 2 | 15 | 12 |

Proposed hydrogenation mechanism
As mentioned above, the complexes 1, 2, and 5 were active in the DMO hydrogenation with the aid of NaOMe. In contrast, the complexes 4 and 7 were catalytically active without NaOMe. NaOMe is the Cl− to H− metathesis reagent during the reaction,23 so the ruthenium hydride complex 4a (or 6) might be the active state of 1 (or 5). The inactivity of 3 was probably due to no available space for ruthenium to generate the active hydride group. This result suggested the importance of Ru–H group in the catalytic reaction.The reversible transformation of 6 and 7 clearly indicates a cooperative function exists between metal and o-PPh2C6H4NH2 ligand. In order to clarify the specific function of NH2 group in o-PPh2C6H4NH2 during the catalytic cycle, we chose the non-NH2-group-containing ligand o-PPh2C6H4NMe2 and prepared complexes (PPh3)(o-PPh2C6H4NMe2)RuCl2 (8)25 and (o-PPh2C6H4NMe2)2RuCl2 (9). Both 8 and 9 were examined for the reaction, but no catalytic activities were found under similar conditions. The complete shutdown of the catalysis activity after NMe2 group substitution either in 1 or 2 indicates that the NH2 moiety is indispensible in the catalytic reaction.10,11b
Based on these observations, we proposed a bifunctional mechanism for the hydrogenation of DMO to MG (Scheme 3). At first, the active species like 4a (or 6) transfers the H−/H+ equivalents into the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the ester group through an outer-sphere interaction. Then, a hemiacetal forms and arranges into MeOC(O)CHO by eliminating one MeOH molecule. The as-produced complex 4 (or 7) recovers to 4a (or 6) by H2 addition through the as-produced Ru–NH unit. Finally, MeOC(O)CHO undergoes a similar cycle and is hydrogenated to MG.
O bond of the ester group through an outer-sphere interaction. Then, a hemiacetal forms and arranges into MeOC(O)CHO by eliminating one MeOH molecule. The as-produced complex 4 (or 7) recovers to 4a (or 6) by H2 addition through the as-produced Ru–NH unit. Finally, MeOC(O)CHO undergoes a similar cycle and is hydrogenated to MG.
 | ||
| Scheme 3 Proposed mechanism for the reduction of DMO to MG. | ||
Conclusions
New well-defined ruthenium complexes 1–7 coordinated with rigid o-(diphenylphosphino)aniline ligand were synthesized and structurally characterized. The results indicated that the complexes 1 and 5 reacted with K[HBsBu3] to produce the ruthenium hydride complexes 4 and 6, respectively. The complex 6 further underwent H2 elimination to produce 7 and 7 split H2 to form back to 6. The D2 experiment confirmed the reversible transformation between 6 and 7. The catalytic tests proved the high efficiency of 1, 2 and 5 in the hydrogenation of DMO to MG, affording improved activities than those of the flexible ruthenium complexes under the same conditions. The complexes 1 and 2 also displayed satisfactory activities in the selective hydrogenations of other aliphatic and cyclic esters. All these results demonstrated that the rigid o-(diphenylphosphino)anilinoruthenium complexes are a class of suitable catalysts for the ester hydrogenation. Mechanistic studies revealed a metal–NH ligand bifunctional mechanism for ester hydrogenation. The inactivity of 3 and 8 (or 9) convinced that both the Ru–H group and NH2 group are indispensable in the catalytic reaction of ester hydrogenation with homogeneous ruthenium complexes.Acknowledgements
This work was supported by the National Basic Research Program of China (2011CBA00508), the National Nature Science Foundation of China (21403178, 21473145, 21473142, and 2013B019), and the Science Foundation of Ministry of Education of China (20110121130002) and the Program for Innovative Research Team in Chinese Universities (IRT_14R31).Notes and references
- (a) H. Adkins and K. Folkers, J. Am. Chem. Soc., 1931, 53, 1095–1097 CrossRef CAS; (b) T. Turek, D. Trimm and N. Cant, Catal. Rev. – Sci. Eng., 1994, 36, 645–683 CrossRef CAS; (c) J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- (a) L. F. Chen, P. J. Guo, M. H. Qiao, S. R. Yan, H. X. Li, W. Shen, H. L. Xu and K. N. Fan, J. Catal., 2008, 257, 172–180 CrossRef CAS; (b) Z. He, H. Q. Lin, P. He and Y. Z. Yuan, J. Catal., 2011, 277, 54–63 CrossRef CAS.
- (a) P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS; (b) B. G. Zhao, Z. B. Han and K. L. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744–4788 CrossRef CAS PubMed; (c) S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS; (d) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- Y. Pouilloux, F. Autin and J. Barrault, Catal. Today, 2000, 63, 87–100 CrossRef CAS.
- W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2012, 16, 166–171 CrossRef CAS.
- (a) R. A. Grey, G. P. Pez and A. Wallo, J. Am. Chem. Soc., 1981, 103, 7536–7542 CrossRef CAS; (b) R. A. Grey, G. P. Pez, A. Wallo and J. Corsi, J. Chem. Soc., Chem. Commun., 1980, 783–784 RSC.
- (a) U. Matteoli, M. Bianchi, G. Menchi, P. Frediani and F. Piacenti, J. Mol. Catal., 1985, 29, 269–270 CrossRef CAS; (b) U. Matteoli, G. Menchi, M. Bianchi and F. Piacenti, J. Organomet. Chem., 1986, 299, 233–238 CrossRef CAS.
- (a) H. T. Teunissen and C. J. Elsevier, Chem. Commun., 1997, 667–668 RSC; (b) M. C. van Engelen, H. T. Teunissen, J. G. de Vries and C. J. Elsevier, J. Mol. Catal. A: Chem., 2003, 206, 185–192 CrossRef CAS.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed.
- L. A. Saudan, C. M. Saudan, C. Debieux and P. Wyss, Angew. Chem., Int. Ed., 2007, 46, 7473–7476 CrossRef CAS PubMed.
- (a) W. Kuriyama, Y. Ino, O. Ogata, N. Sayo and T. Saito, Adv. Synth. Catal., 2010, 352, 92–96 CrossRef CAS; (b) Z. B. Han, L. C. Rong, J. Wu, L. Zhang, Z. Wang and K. L. Ding, Angew. Chem., Int. Ed., 2012, 51, 13041–13045 CrossRef CAS PubMed; (c) D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2012, 51, 2772–2775 CrossRef CAS PubMed; (d) S. Chakraborty, H. G. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. R. Guan, J. Am. Chem. Soc., 2014, 136, 7869–7872 CrossRef CAS PubMed; (e) S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS; (f) S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed; (g) S. Takebayashi and S. H. Bergens, Organometallics, 2009, 28, 2349–2351 CrossRef CAS; (h) M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 4240–4242 CrossRef CAS PubMed; (i) D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2013, 52, 2538–2542 CrossRef CAS PubMed; (j) X. F. Tan, Y. Wang, Y. H. Liu, F. Y. Wang, L. Y. Shi, K. H. Lee, Z. Y. Lin, H. Lv and X. M. Zhang, Org. Lett., 2015, 17, 454–457 CrossRef CAS PubMed; (k) G. Q. Zhang, B. L. Scott and S. K. Hanson, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS PubMed; (l) G. Q. Zhang, K. V. Vasudevan, B. L. Scott and S. K. Hanson, J. Am. Chem. Soc., 2013, 135, 8668–8681 CrossRef CAS PubMed; (m) D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12357–12360 CrossRef CAS PubMed; (n) C. Ziebart, R. Jackstell and M. Beller, ChemCatChem, 2013, 5, 3228–3231 CrossRef CAS.
- (a) E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS PubMed; (b) Y. S. Sun, C. Koehler, R. Tan, V. T. Annibale and D. T. Song, Chem. Commun., 2011, 47, 8349–8351 RSC; (c) E. Fogler, E. Balaraman, Y. Ben-David, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2011, 30, 3826–3833 CrossRef CAS; (d) E. Balaraman, E. Fogler and D. Milstein, Chem. Commun., 2012, 48, 1111–1113 RSC.
- B. Boardman, M. J. Hanton, H. van Rensburg and R. P. Tooze, Chem. Commun., 2006, 2289–2291 RSC.
- O. Herd, A. Heßler, M. Hingst, M. Tepper and O. Stelzer, J. Organomet. Chem., 1996, 522, 69–76 CrossRef CAS.
- (a) C. C. Lee, H. C. Huang, S. T. Liu, Y. H. Liu, S. M. Peng and J. T. Chen, Polyhedron, 2013, 52, 1024–1029 CrossRef CAS; (b) C. C. Lee and S. T. Liu, Chem. Commun., 2011, 47, 6981–6983 RSC; (c) A. Bacchi, M. Balordi, R. Cammi, L. Elviri, C. Pelizzi, F. Picchioni, V. Verdolino, K. Goubitz, R. Peschar and P. Pelagatti, Eur. J. Inorg. Chem., 2008, 2008, 4462–4473 CrossRef; (d) C. C. Lee, Y. H. Liu, S. M. Peng, P. T. Chou, J. T. Chen and S. T. Liu, Polyhedron, 2012, 35, 23–30 CrossRef CAS.
- (a) K. Abdur-Rashid, S. E. Clapham, A. Hadzovic, J. N. Harvey, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2002, 124, 15104–15118 CrossRef CAS PubMed; (b) K. Abdur-Rashid, M. Faatz, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2001, 123, 7473–7474 CrossRef CAS PubMed; (c) J. H. Choi, N. E. Schloerer, J. Berger and M. H. G. Prechtl, Dalton Trans., 2014, 43, 290–299 RSC.
- K. Abdur-Rashid, A. J. Lough and R. H. Morris, Organometallics, 2000, 19, 2655–2657 CrossRef CAS.
- (a) K. Abdur-Rashid, R. W. Guo, A. J. Lough, R. H. Morris and D. T. Song, Adv. Synth. Catal., 2005, 347, 571–579 CrossRef CAS; (b) R. Morris, A. Habtemariam, Z. J. Guo, S. Parsons and P. J. Sadler, Inorg. Chim. Acta, 2002, 339, 551–559 CrossRef CAS.
- (a) J. X. Gao, T. Ikariya and R. Noyori, Organometallics, 1996, 15, 1087–1089 CrossRef CAS; (b) J. X. Gao, H. Zhang, X. D. Yi, P. P. Xu, C. L. Tang, H. L. Wan, K. R. Tsai and T. Ikariya, Chirality, 2000, 12, 383–388 CrossRef CAS PubMed; (c) V. Rautenstrauch, H. C. Xuân, R. Churlaud, K. Abdur-Rashid and R. H. Morris, Chem. –Eur. J., 2003, 9, 4954–4967 CrossRef CAS PubMed.
- (a) M. M. Bhadbhade, L. D. Field, R. Gilbert-Wilson, R. W. Guest and P. Jensen, Inorg. Chem., 2011, 50, 6220–6228 CrossRef CAS PubMed; (b) E. S. F. Ma, D. C. Mudalige, B. O. Patrick and B. R. James, Dalton Trans., 2013, 42, 7614–7621 RSC; (c) K. Namura, M. Ohashi and H. Suzuki, Organometallics, 2012, 31, 5979–5982 CrossRef CAS; (d) K. A. Smart, M. Grellier, L. Vendier, S. A. Mason, S. C. Capelli, A. Albinati and S. Sabo-Etienne, Inorg. Chem., 2013, 52, 2654–2661 CrossRef CAS PubMed.
- (a) R. Abbel, K. Abdur-Rashid, M. Faatz, A. Hadzovic, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2005, 127, 1870–1882 CrossRef CAS PubMed; (b) C. A. Sandoval, T. Ohkuma, K. Muñiz and R. Noyori, J. Am. Chem. Soc., 2003, 125, 13490–13503 CrossRef CAS PubMed.
- W. L. Jia, X. H. Chen, R. W. Guo, C. Sui-Seng, D. Amoroso, A. J. Lough and K. Abdur-Rashid, Dalton Trans., 2009, 8301–8307 RSC.
- (a) R. J. Hamilton and S. H. Bergens, J. Am. Chem. Soc., 2006, 128, 13700–13701 CrossRef CAS PubMed; (b) R. Hartmann and P. Chen, Angew. Chem., Int. Ed., 2001, 40, 3581–3585 CrossRef CAS.
- J. M. John, S. Takebayashi, N. Dabral, M. Miskolzie and S. H. Bergens, J. Am. Chem. Soc., 2013, 135, 8578–8584 CrossRef CAS PubMed.
- D. C. Mudalige, S. J. Rettig, B. R. James and W. R. Cullen, J. Chem. Soc., Chem. Commun., 1993, 830–832 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental results, NMR spectra of complexes 1–7 and 9 and CIF data of compounds 1–2 and 4–7. CCDC 1443566–1443570 and 1468780. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra00320f |
| This journal is © The Royal Society of Chemistry 2016 |