
The environmental impact of unsaturated fluoroesters: atmospheric chemistry towards OH radicals and Cl atoms, radiative behavior and cumulative ozone creation†
Ana Rodríguez*a,
Iván Bravob,
Diana Rodrígueza,
Mercedes Tajueloa,
Yolanda Diaz-de-Merac and
Alfonso Arandac
aFaculty of Environmental Sciences and Biochemistry, University of Castilla La Mancha, Avenida Carlos III, s/n, 45071 Toledo, Spain. E-mail: anamaria.rodriguez@uclm.es
bFaculty of Pharmacy, University of Castilla La Mancha, Edificio Polivalente, s/n, 02071 Albacete, Spain
cFaculty of Chemical Sciences, University of Castilla La Mancha, Avenida Camilo José Cela 10, 13071 Ciudad Real, Spain
First published on 17th February 2016
Abstract
Smog chamber/GC techniques were used to investigate the atmospheric degradation of two hydrofluoroesters (allyl trifluoroacetate (CF3C(O)OCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) and vinyl trifluoroacetate (CF3C(O)OCHCH2)) by oxidation with OH radicals and Cl atoms at 298 K and an atmospheric pressure of N2 or air. The measured rate coefficients were (in units of cm3 per molecule per s): kallyl trifluoroacetate+OH = (9.27 ± 3.81) × 10−12; kvinyl trifluoroacetate+OH = (8.07 ± 1.92) × 10−12; kallyl trifluoroacetate+Cl = (1.75 ± 0.21) × 10−10 and kvinyl trifluoroacetate+Cl = (2.08 ± 0.16) × 10−10. In the OH-initiated oxidation of allyl trifluoroacetate, the identified product can arise from OH addition to both carbons in the double bond and the later decomposition of the alkoxy radical formed. However, in the reactions of both fluoroesters with Cl atoms, the main product detected arises from Cl addition to the terminal carbon atom and the subsequent reaction of the chloroalkoxy radical formed with O2. Infrared spectra of the studied esters were collected and their contribution to global warming was assessed by calculating their radiative efficiencies. Combining these results with the kinetic data we found that their global warming potentials are negligible. Finally, the photochemical ozone creation potentials were calculated, obtaining values lower than those of non-fluorinated unsaturated hydrocarbons.
CH2) and vinyl trifluoroacetate (CF3C(O)OCHCH2)) by oxidation with OH radicals and Cl atoms at 298 K and an atmospheric pressure of N2 or air. The measured rate coefficients were (in units of cm3 per molecule per s): kallyl trifluoroacetate+OH = (9.27 ± 3.81) × 10−12; kvinyl trifluoroacetate+OH = (8.07 ± 1.92) × 10−12; kallyl trifluoroacetate+Cl = (1.75 ± 0.21) × 10−10 and kvinyl trifluoroacetate+Cl = (2.08 ± 0.16) × 10−10. In the OH-initiated oxidation of allyl trifluoroacetate, the identified product can arise from OH addition to both carbons in the double bond and the later decomposition of the alkoxy radical formed. However, in the reactions of both fluoroesters with Cl atoms, the main product detected arises from Cl addition to the terminal carbon atom and the subsequent reaction of the chloroalkoxy radical formed with O2. Infrared spectra of the studied esters were collected and their contribution to global warming was assessed by calculating their radiative efficiencies. Combining these results with the kinetic data we found that their global warming potentials are negligible. Finally, the photochemical ozone creation potentials were calculated, obtaining values lower than those of non-fluorinated unsaturated hydrocarbons.
1. Introduction
Hydrofluoroethers (HFEs) have been designed and are widely recommended as a third generation replacement for chlorofluorocarbons (CFCs) and their derivatives, hydrochlorofluorocarbons (HCFCs) and hydrofluorocarbons (HFCs), in industrial applications such as cleaning solvents, fire suppression agents or heat transfer fluids;1,2 or in pharmaceutical applications as, for example, anesthetic agents.3 The insertion of an ether oxygen atom activates the molecule towards tropospheric oxidant attack, which leads to shorter atmospheric lifetimes than those for any HCFCs or HFCs. The primary removal of HFEs in the troposphere will mainly be initiated by reaction with OH radicals forming the corresponding hydrofluorinated esters (FESs).4,5 Moreover, recent studies have shown that saturated and unsaturated FESs can be used as potential solvents or co-solvents for Li-ion batteries6–8 or as precursors for reactive polymers in various kinds of functional materials,9,10 which could result in release of these compounds into the atmosphere.The main fate of FESs in the troposphere is again OH radical-initiated photo-oxidation, without forgetting dissolution in clouds and seawater,11,12 and reaction with Cl atoms in coastal environments and contaminated urban areas, where high Cl levels may originate from industrial and urban emissions.13–16 The degradation of FESs may contribute to the environmental burden of products like trifluoroacetic acid (TFA) (or derivatives), which may affect agricultural and aquatic systems.17 Moreover, most FESs contribute more actively to global warming than the parent HFEs, even when they both have the same number of C–F bonds in their molecular structure. This fact has been observed in work carried out in our laboratory,18 where the radiative properties in terms of radiative efficiencies (REs) and global warming potentials (GWPs) of several FESs were investigated and compared to their parent HFEs. This behavior is probably due to the presence of an additional strong band, corresponding to C–O–C(O)R stretching, in the region of the spectrum where the radiative forcing function is large. Thus, these secondary products may significantly increase the total GWP of the emitted HFE, due to their cumulative effect.19
In order to evaluate the environmental compatibility of HFEs, it is necessary to obtain a full understanding of the atmospheric chemistry of FESs. Although in recent years experimental and theoretical studies on the decomposition kinetics of saturated FESs have been performed,13–16,20–23 the database for saturated fluorinated esters is very limited compared to the numerous studies on hydrogenated esters, and non-existent for unsaturated fluorinated esters. Thus, the degradation mechanisms of unsaturated FESs, the distribution of products, their GWPs calculated using REs and lifetimes, and photochemical ozone creation potentials, are necessary to know the role of HFEs and FESs in the atmosphere.
To the best of our knowledge, this work provides the first kinetic and mechanistic study for the reaction of OH radicals and Cl atoms with two unsaturated fluorinated esters: allyl trifluoroacetate (ATFA) and vinyl trifluoroacetate (VTFA). The photochemical interactions of oxygenate organic compounds with nitrogen oxides (NOx) in the troposphere lead to the formation of secondary pollutants such as ozone,4 which is a serious problem in many urban areas. In this sense, one of the objectives of this work will be to determine the reaction mechanisms of the title FESs in different scenarios of NOx: in the absence of NOx so as to simulate remote or rural areas; and with large amounts of NOx to simulate the chemistry of polluted areas. Moreover, the ‘climate friendship’ of these FESs will be examined through the calculation of lifetimes and GWPs, and integration with possible effects due to secondary species originated.
2. Experimental
The kinetic study and determination of products was performed using a 200 L Teflon bag housed in an isothermal cabinet with six fluorescent lamps (Philips TUV G13, 36 W, λ = 254 nm) mounted on the walls. Monitoring of reactants and products was carried out by capillary gas chromatography (GC) coupled with flame ionization detection (FID) or mass spectrometry (MS). Infrared spectra of the studied FESs were measured using a 56 L quartz-glass reactor coupled to Fourier-transform infrared spectrophotometer (FTIR). The experimental systems and the procedures used in this work are briefly described below. Further details can be found elsewhere.19,242.1. Kinetic study
All experiments were performed at 298 ± 1 K in ∼760 Torr total pressure of N2/O2 diluent. Cl and OH radicals were generated by the photolysis of Cl3CCOCl and H2O2, respectively:| Cl3CCOCl + hν → Cl3CCO + Cl |
| H2O2 + hν → 2OH |
In smog chamber experiments, unwanted loss of reactants and products via photolysis, dark chemistry and heterogeneous reactions have to be considered. For this reason, Cl2 was not used as the source of Cl atoms since Cl2 reacts with the substrates in the dark. However, the losses with the walls and by photolysis were negligible.
The relative rate method is a well-established technique for measuring the reactivity of Cl and OH radicals with organic compounds. Kinetic data were derived by monitoring the loss of the substrate relative to the different reference compounds:
| Cl/OH + FES → products kFES |
| Cl/OH + reference → products kR |
The decays of the reactant and the reference compounds were then plotted using the expression:
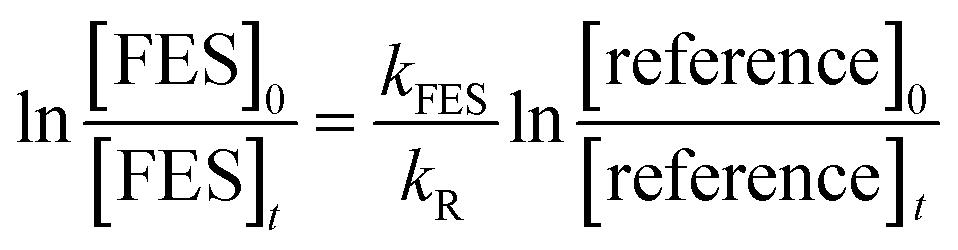 | (1) |
The decay of reactants and reference compounds was monitored by gas chromatography with flame ionization detection, GC-FID (Shimadzu 2010), using a capillary column (size: 30 m × 0.32 mm × 1 m, Meta.X5 Teknokroma) maintained isothermally at 100 °C. Gas samples from the bag were injected into a split/splitless injector using a port gas sampling valve (Valco Instruments Co. Inc). Once started photolysis, the injection step was repeated 10 to 15 times in each kinetic experiment, and this set of experiments was performed four to six times on different dates, changing the reactant concentrations. The concentration ranges of the reactants were as follows: (in molecules per cm3): FESs (0.3–5.1) × 1015, reference compounds (1.4–9.9) × 1014, H2O2 (4.0–7.3) × 1015 and Cl3CCOCl (2.5–7.1) × 1015. For each mixture of organic compounds, a number of injections of the unreacted mixture, usually 10 or more, were carried out in order to obtain an estimate of the precision associated with the measurements, to be used in the error analysis. The standard deviations (2σ) of these replicate injections were typically 1% for FESs and the reference compounds. The reproducibility of the results and the absence of a decreasing profile versus time show that the wall losses of the reactants were not significant.
2.2. Product study
Reactants and formed products were monitored by gas chromatography with mass detection, GC-MS (Shimadzu QP2010). The temperature program used in the chromatograph was the following: 40 °C (hold 4 min), 15 °C min−1 to 130 °C and hold for 6 min. All experiments were carried out in the absence and presence of NOx, with initial concentrations as follows: (in molecules per cm3) FESs (0.2–6.3) × 1015, H2O2 (3.8–6.8) × 1015, Cl3CCOCl (1.9–7.0) × 1015 and NO (2.2–6.5) × 1015. Irradiation was performed in steps of 60–300 s, followed by turning off the lamps and sampling of the reaction mixture. The total photolysis time was 60–150 min in order to have a conversion of reactants between 60 and 90%. The identification of the products was made by analysis of the mass spectrum and by comparison with a library of spectra and, where possible, was also performed by comparing with the retention time of a commercial sample of the detected product.2.3. Infrared spectra
Infrared spectra of ATFA and VTFA were measured at 298 ± 1 K using a 56 L quartz-glass reactor coupled to a Bruker VERTEX-80V-FTIR spectrophotometer equipped with a liquid nitrogen-cooled mercury cadmium telluride (MCT) detector. The reactor was equipped with a White-type multiple-reflection mirror system (Saturn Series Multi-Pass cells) with a base length of 1.37 m and a total optical path length of 197 m. The spectrometer was operated at a spectral resolution of 0.03 cm−1 with interferograms being obtained from 64 co-added scans in the range 600–2000 cm−1.The experimental procedure was similar to that described in previous studies.19,25 No dependence on total pressure was observed for esters used in the studied range. Mixtures of the title compounds in air were mixed in this way and allowed to stand overnight to allow good mixing. A pumping system consisting of a rotary pump (Varian DS 402) was used to evacuate the reactor after every experiment. Peak absorbance was plotted as a function of each compound concentration to ensure that saturation was not a problem in the measurements (see ESI, S1 and S2†). Points at higher concentrations showing non-linear behavior were ignored. Plots of absorbance vs. compound concentration showed good linearity, and zero intercepts.
The absorption cross section at temperature T, wavenumber (cm−1) and at the experimental resolution was determined through the relationship:
σ(![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) , T) = ln(I0/I)/cl , T) = ln(I0/I)/cl
| (2) |
, T) is the absorption cross section in cm2 per molecule at temperature T and wavenumber ; I0 is the intensity of radiation reaching the detector when the cell is empty; I is the intensity of radiation reaching the detector when the cell contains a sample; c is the concentration of sample (molecules per cm3); and l is the pathlength (cm). Then, integrated absorption cross sections, S(T) in cm2 per molecule per cm were determined according to the expression:
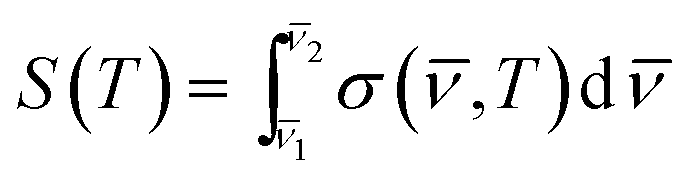 | (3) |
2.4. Chemicals
The chemicals used (with their purities) were: ATFA and VTFA (98%, Aldrich); cyclohexane and heptane (99.9%, Aldrich); octane (>99%, Acros Organics); trichloroacetyl chloride (99%, Aldrich); hydrogen peroxide (>60%, Fisher Chemical) and chloroacetaldehyde (∼50% wt in H2O, Aldrich). Synthetic air and N2 (99.999%, Air Liquide) were employed as bath gases for the experiments and He (99.998%, Air Liquide) was used as GC carrier gas.3. Results and discussion
3.1. Kinetic study
The rate coefficient measurements for the reactions of OH radicals and Cl atoms with ATFA and VTFA were each performed relative to three reference compounds, cyclohexane, n-heptane and n-octane, whose rate coefficients are: kOH+cyclohexane = (6.97 ± 1.39) × 10−12;26 kOH+heptane = (6.76 ± 2.7) × 10−12;26 kOH+octane = (8.11 ± 1.62) × 10−12;26 kCl+cyclohexane = (2.91 ± 0.31) × 10−10;27 kCl+octane = (3.22 ± 0.36) × 10−10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 and kCl+heptane = (3.50 ± 0.13) × 10−10.28 All the k values are in units of cm3 per molecule per s. Representative plots of the kinetic data for these reactions are displayed in Fig. 1. The rate coefficient ratios (kFES/kR) are listed in Table 1 together with the absolute rate coefficients at 298 ± 1 K and atmospheric pressure. The errors quoted are a combination of twice the standard deviation arising from the least-squares fit of the straight lines and the corresponding error to the reference rate coefficients. The experiments were performed in N2 and synthetic air, to test for potential systematic errors due to secondary reactions and to obtain a better approach to atmospheric conditions. The obtained results were independent of the bath gas used, which suggests that there is no interference from secondary reactions with O2. The uncertainty in rate coefficients with OH radicals are bigger than those of Cl atoms. This fact is due to the smog chamber experiments and the analysis precision of the detection system used are more compatible with the rate coefficients for Cl atoms than for OH radicals.14
27 and kCl+heptane = (3.50 ± 0.13) × 10−10.28 All the k values are in units of cm3 per molecule per s. Representative plots of the kinetic data for these reactions are displayed in Fig. 1. The rate coefficient ratios (kFES/kR) are listed in Table 1 together with the absolute rate coefficients at 298 ± 1 K and atmospheric pressure. The errors quoted are a combination of twice the standard deviation arising from the least-squares fit of the straight lines and the corresponding error to the reference rate coefficients. The experiments were performed in N2 and synthetic air, to test for potential systematic errors due to secondary reactions and to obtain a better approach to atmospheric conditions. The obtained results were independent of the bath gas used, which suggests that there is no interference from secondary reactions with O2. The uncertainty in rate coefficients with OH radicals are bigger than those of Cl atoms. This fact is due to the smog chamber experiments and the analysis precision of the detection system used are more compatible with the rate coefficients for Cl atoms than for OH radicals.14
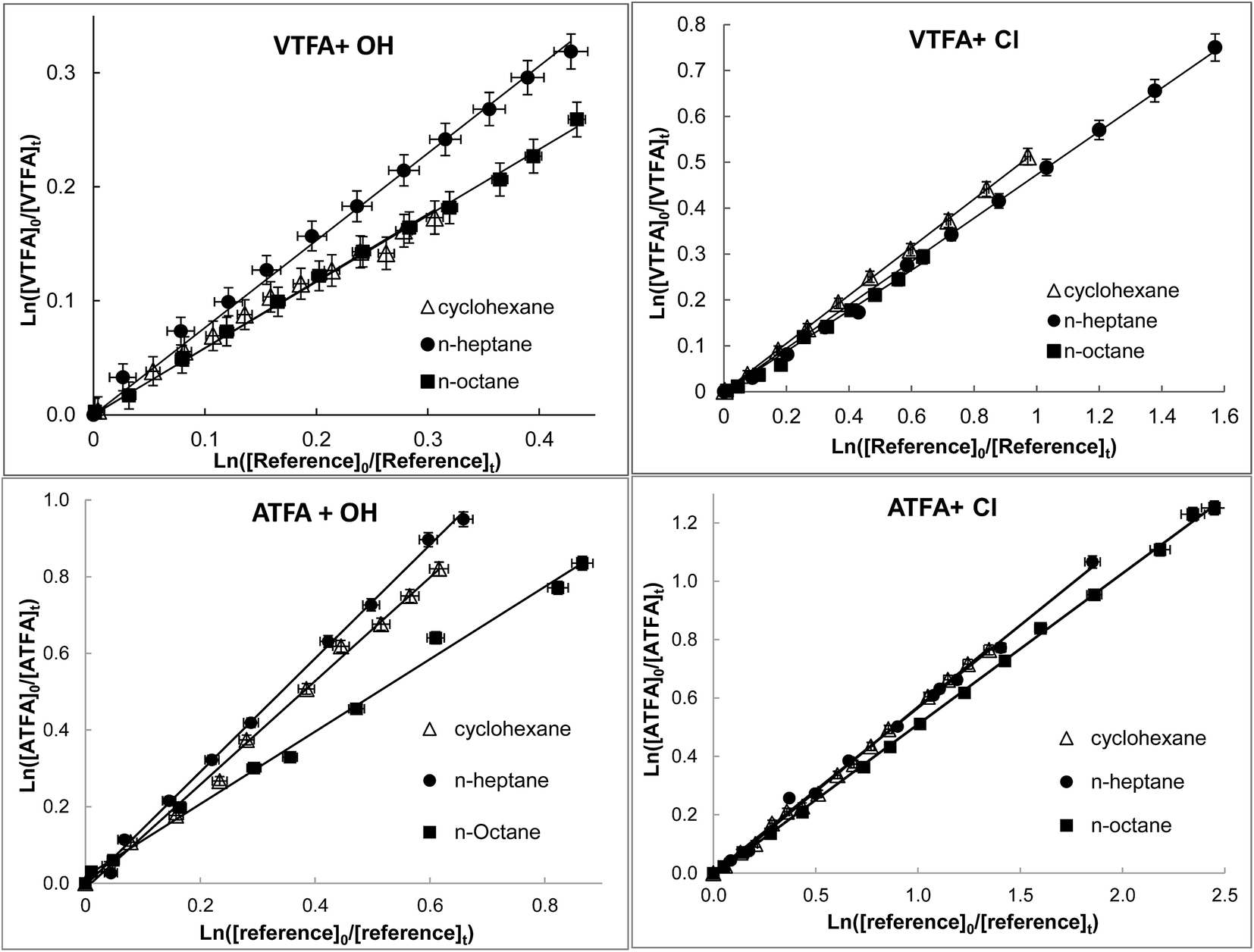 | ||
| Fig. 1 Examples of the relative loss of title FESs vs. reference compounds in the presence of OH radicals or Cl atoms at 298 ± 1 K and atmospheric pressure of air. | ||
| Reference compound | Allyl trifluoroacetate | Vinyl trifluoroacetate | ||
|---|---|---|---|---|
| kFES/kR | kFES (cm3 per molecule per s) | kFES/kR | kFES (cm3 per molecule per s) | |
| OH | ||||
| Cyclohexane | 1.26 ± 0.13 | (8.76 ± 1.82) × 10−12 | 1.22 ± 0.02 | (8.52 ± 0.90) × 10−12 |
| n-Heptane | 1.48 ± 0.12 | (9.57 ± 2.05) × 10−12 | 1.11 ± 0.02 | (7.52 ± 0.98) × 10−12 |
| n-Octane | 1.09 ± 0.10 | (8.84 ± 1.82) × 10−12 | 1.01 ± 0.03 | (8.19 ± 1.01) × 10−12 |
| Average (9.27 ± 1.91) × 10−12 | Average (8.07 ± 0.96) × 10−12 | |||
|
||||
| Cl | ||||
| Cyclohexane | 0.57 ± 0.03 | (1.67 ± 0.21) × 10−10 | 0.66 ± 0.01 | (1.93 ± 0.17) × 10−10 |
| n-Heptane | 0.54 ± 0.02 | (1.90 ± 0.18) × 10−10 | 0.62 ± 0.01 | (2.16 ± 0.13) × 10−10 |
| n-Octane | 0.51 ± 0.02 | (1.65 ± 0.23) × 10−10 | 0.70 ± 0.03 | (2.25 ± 0.19) × 10−10 |
| Average (1.75 ± 0.21) × 10−10 | Average (2.08 ± 0.16) × 10−10 | |||
This work reports the first study of the degradation of ATFA and VTFA with OH radicals and Cl atoms and, therefore, no direct comparison with the literature can be made. However, the rate coefficients from this work can be compared to the rate coefficients of compounds with similar structures, establishing structure–reactivity relationships that can be applied to other similar compounds not previously studied. In this regard, Table 2 shows the rate coefficients of the reactions of OH radicals and Cl atoms with the FESs studied in this work together with rate coefficients of other hydrogenated esters and fluorinated compound analogues (saturated esters and alkenes) from the literature.
| Compounds | kOH (cm3 per molecule per s) | kCl (cm3 per molecule per s) |
|---|---|---|
| a This work.b (Blanco et al., 2009a).30c (Blanco et al., 2009b).32d (Sulbaek Andersen et al., 2005).31e (Blanco et al., 2007).20f (Blanco et al., 2008).13g (Picquet-Varrault et al., 2002).33h (Papagni et al., 2001).34i (Rodríguez et al., 2007).35 | ||
| CF3C(O)OCHCH2 (VTFA) |
(8.07 ± 0.96) × 10−12a | (2.08 ± 0.16) × 10−10a |
| CH3C(O)OCHCH2 |
(2.48 ± 0.61) × 10−11b | (2.68 ± 0.91) × 10−10c |
| CF3CHCH2 |
(1.36 ± 0.25) × 10−12d | (9.07 ± 1.08) × 10−11d |
| CF3C(O)OCH2CH3 | (2.64 ± 0.59) × 10−13e | (1.78 ± 0.57) × 10−12f |
| CF3C(O)OCH2CHCH2 (ATFA) |
(9.27 ± 1.91) × 10−12a | (1.75 ± 0.21) × 10−10a |
| CH3C(O)OCH2CHCH2 |
(3.06 ± 0.31) × 10−11g | (1.30 ± 0.45) × 10−10c |
| OHCH2CHCH2 |
(5.45 ± 0.35) × 10−11h | (1.72 ± 0.19) × 10−10i |
In the OH-induced reactions, the presence of fluorinated substituents caused a deactivation effect on the rate of the reactions. This can be explained based on a mechanism of addition of the oxidant to the double bond (the main pathway in the oxidation of unsaturated non-fluorinated esters29), where the –CF3 group reduces the partial negative charge, and thus the electrophilic addition of the OH radicals is inhibited compared to the hydrogenated ester. In this same way, the rate coefficient of the ATFA is bigger than that of the VTFA, since the ATFA accounts with a –CH2 group more than the VTFA between the double bond and the fluorinated substituents and the deactivation effect would be minor.
In the case of reactions with Cl atoms, the rate coefficients of each couple of esters (hydrogenated and fluorinated) were similar within experimental uncertainties. Therefore, the effect of fluorinated substituents on the reactivity of the double bond for Cl atoms is less important than the effect observed for OH addition. For VTFA it is possible to compare its rate coefficient with that of the analogous alkene, 3,3,3-trifluoropropene, the rate coefficient of the FES is greater both with OH radicals and Cl atoms. Thus, the presence of a –OC(O)R group attached to the double bond apparently exerts an activating effect on the bond for electrophilic addition reactions. Thus, the lone pair of electrons on the oxygen atom would lead to an increase of the electronic density on the π system.29 Moreover, the reactivity of VTFA for OH radicals and Cl atoms is higher than that of its corresponding saturated ester, ethyl trifluoroacetate.13,19 This would imply a change of mechanism in addition to the CC double bond in the unsaturated esters and H-atom abstraction for the analogous saturated ester.14,29
In the case of ATFA, the rate coefficients of the corresponding alkene with OH radicals and Cl atoms have not been studied, so comparison was not possible. However, it is also worth noting that in reactions with Cl atoms, compounds with the –CH2CHCH2 group in their structure, for example allyl alcohol or the hydrogenated ester mentioned before, have similar rate coefficients within the error limits. This fact suggests that the reactivity of the allyl group in Cl-initiated oxidation is little affected by the substituents and the chemical structure of the unsaturated compound. This does not occur in the OH additions, so the Cl atoms are, in general, less discriminating than the OH radicals in their reactions with organic compounds.18,29,31
3.2. Products and reaction mechanism of VTFA
In the OH-initiated reaction, a product was observed in the presence and absence of NOx with a retention time very similar to that of VTFA. The compounds could not be separated and, therefore, identification was not possible. Moreover, when the conversion of reactant was higher, a new product arose from secondary reactions. Comparing its spectrum with the GC-MS library, it was identified as TFA.In the reactions with Cl atoms, in presence and absence of NOx, the main product detected by the GC-MS instrument has an electron impact (EI) mass spectrum as follows: m/z = 28 (loss of CO), m/z = 35 and 37 (loss of Cl35 and Cl37, respectively), m/z = 49 and 51 (loss of CH2Cl35 and CH2Cl37, respectively), m/z = 69 (loss of CF3), and m/z = 97 (loss of CF3CO) (see ESI material, S3†). This fragmentation pattern could be consistent with the chlorinated compound with the formula CF3C(O)OC(O)CH2Cl. This product is not commercially available and its yield could not be estimated. Chloroacetaldehyde, identified by comparison with the retention time of a commercial sample, and another unidentified product were observed with an intensity of their signals much lower than that of CF3C(O)OC(O)CH2Cl. Moreover, as the reaction proceeded, a new product arose from secondary reactions, TFA, which was identified by comparison with the library of spectra.
Fig. 2 shows the atmospheric primary reaction mechanism proposed for the oxidation of VTFA with Cl atoms. The atmospheric degradation of unsaturated esters is expected to proceed predominantly through the addition of the oxidant to one of the carbon atoms of the double bond. The alkyl radical formed in this reaction adds O2 to form the corresponding alkyl peroxy radical. Depending on the experimental conditions, the absence or presence of NOx, the peroxy radicals may react with NO, HO2, other peroxy radicals or with themselves, giving rise to the corresponding alkoxy radical. This radical can decompose by C–C bond scission or react with O2, leading to different reaction products.29 In the present work, the main product appears from Cl addition to the terminal carbon atom of the double bond (addition 2). Moreover, it is interesting that the major fate of the alkoxy radical formed in the oxidation process is the reaction with O2 (2a) instead of the scission of the C–C bond (2b and 2c). This behavior has also been observed in the reactions of Cl atoms with fluorinated ethers,36,37 in which the increase of substituents that withdraw electron density, such as –CF3 groups or even in our case the Cl atom attached to the terminal carbon atom, leads to an increase in the relative importance of the reaction with O2.
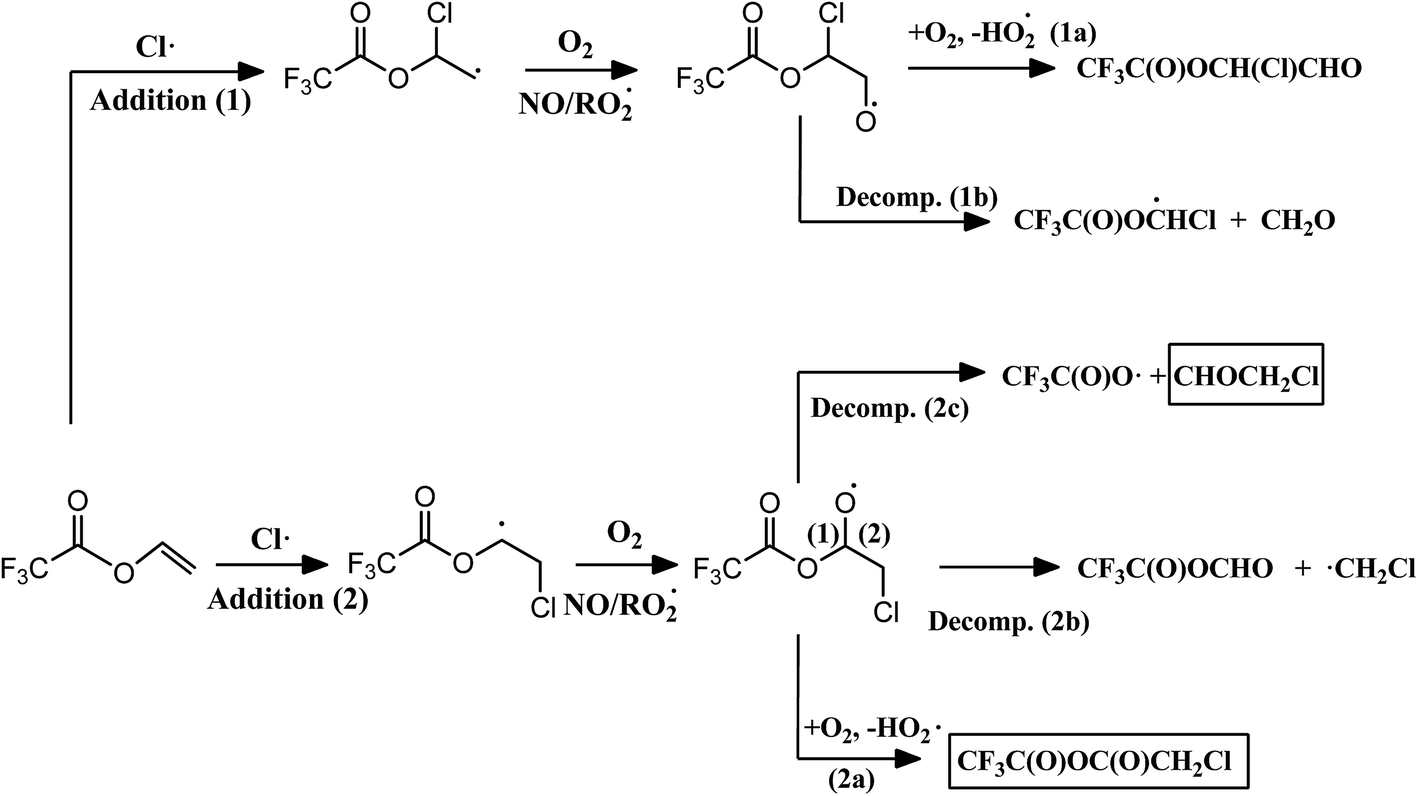 | ||
| Fig. 2 Proposed mechanism for the reaction of VTFA with Cl atoms, where the identified products are shown in a solid line box. | ||
In the case of the reaction of the analogous hydrogenated ester (vinyl acetate) with Cl atoms, different channels were observed depending on the experimental conditions.38 In presence of NOx, the reaction proceeds by decomposition and an α-ester rearrangement/decomposition of the chloroalkoxy radical formed; however, in experiments without NOx approximately 50% of the reaction proceeds by the mentioned pathways and the other 50% by reaction of the chloroalkoxy radical with O2. This fact may be due to the reactions of peroxy radicals with NO are very exothermic, the alkoxy radicals formed in presence of NOx will have higher internal excitation energy and the decomposition channels would be favored.29,39 In VTFA, no differences were observed in both conditions, with and without NOx, which could be due to the presence of electronegative substituents, such as –CF3 group, increases the reactivity of the alkoxy radical toward O2 (2a), as mentioned above, annulling the effect of NO on the decomposition pathway.
3.3. Products and reaction mechanism of ATFA
In the reaction of ATFA with OH radicals, in presence and absence of NOx, only one product was detected by the GC-MS instrument and was tentatively assigned as trifluoroacetoxyetanal (CF3C(O)OCH2CHO) based on the fragmentation pattern from the EI mass spectrum. This mass spectrum has major ion signals at m/z = 29 (loss of CHO typical of aldehydes), m/z = 42 (CH2CO), m/z = 50 (loss of CF2), m/z = 59 (loss of OCH2CHO) and m/z = 69 (loss of CF3) (see ESI material, S4†), which are consistent with this compound.In the case of the Cl-initiated reactions, four products were detected both in the presence and absence of NOx, and a fifth additional product was found exclusively upon addition of NOx. The main product, with and without NOx, was tentatively assigned as CF3C(O)OCH2C(O)CH2Cl according to its EI mass spectrum. The major ion signals were at m/z = 42 (loss of CH2CO), m/z = 49 and 51 (loss of CH2Cl35 and CH2Cl37, respectively), m/z = 69 (loss of CF3), m/z = 77 and 79 (loss of COCH2Cl35 and COCH2Cl37, respectively), m/z = 127 (loss of CF3C(O)OCH2) and m/z = 155 (loss of CF3C(O)OCH2C(O)) (see ESI material, S5†). Unfortunately, this compound is not commercially available for comparative purposes in order to verify its formation. Moreover, the product observed in the reaction with OH radicals, the fluorinated aldehyde (CF3C(O)OCH2CHO), was also observed in the Cl reactions with and without NOx, but its integrated signal in the experiments using the MS detector was significantly smaller than the signal for CF3C(O)OCH2C(O)CH2Cl (with relative integrated values in the order of 1:20). This result shows that the fluorinated aldehyde must be a minor product within the global mechanism. The other detected products could not be identified from their EI mass spectra; nevertheless, it is important to notice that the product only observed in presence of NOx has a major ion signal at m/z = 46 (loss of the –NO2 group) (see ESI material, S6†), suggesting the formation of an alkyl nitrate.
Fig. 3 shows the primary reaction mechanism proposed for the oxidation of ATFA with OH radicals and Cl atoms. In the OH-initiated oxidation, the product obtained (CF3C(O)OCH2CHO) can arise from OH addition to both carbons in the double bond, and the later decomposition of the alkoxy radical formed (1b and 2c). The same mechanism was obtained in the reaction of OH radicals with the analogous hydrogenated ester (allyl acetate).29 Unfortunately, in this work, the yield of the fluorinated aldehyde could not be calculated since this product is not commercial and we have not found a compound of similar structure suitable for our experiments. However, it should be noted that, in the GC-MS instrument, the integrated signal of this compound is higher in the presence of NOx. This may be due to the fact mentioned above, the reactions of peroxy radicals with NO are very exothermic and the decomposition pathway would be favored.29,39
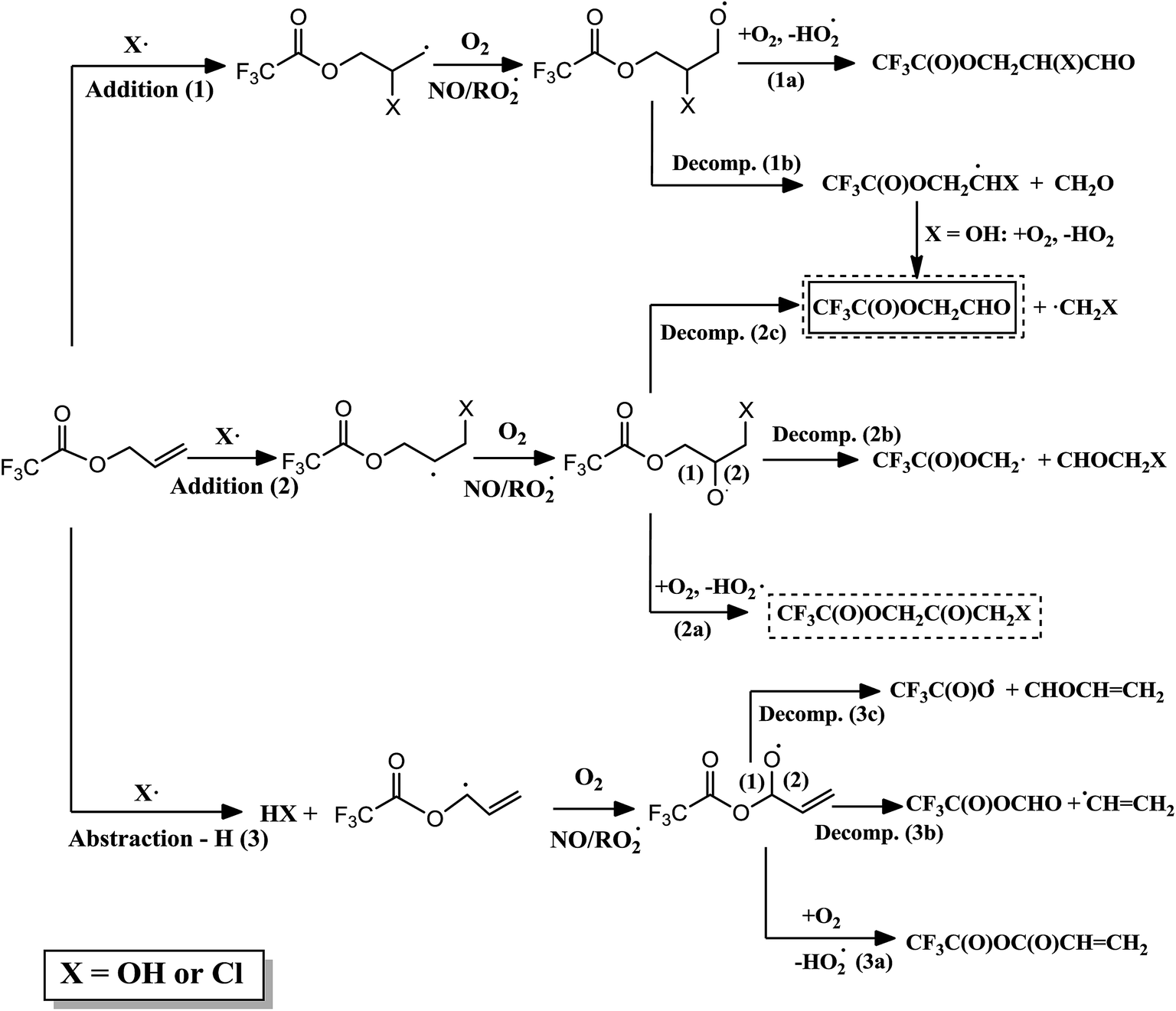 | ||
| Fig. 3 Proposed mechanism for the reaction of ATFA with OH or Cl radicals (X), where the detected product in the reaction with OH is shown in a solid line box and the detected products in the reaction with Cl are shown in dashed boxes. | ||
In contrast, in the reactions of ATFA with Cl radicals, the main product detected arises from Cl addition to the terminal carbon atom (2), and the later reaction of the chloroalkoxy radical formed with O2 (2a). Again, this fact shows the effect of the electronegative substituents on the relative importance of the reaction of alkoxy radical with O2. Moreover, the decomposition product is also present (2c) and, again in presence of NOx, the intensity of its signal is slightly higher. Comparing with the corresponding hydrogenated ester, allyl acetate, the results are similar to those obtained in the comparison between VTFA and vinyl acetate.38 The absence of electronegative substituents in the hydrogenated ester increases the yield of the decomposition pathway, especially, in NOx-rich environments. Moreover, it should be noted that the degradation of allyl acetate with Cl atoms leads to the formation of a peroxynitrate reasonably stable at room temperature and atmospheric pressure.38 A product with a major ion signal at m/z = 46 (corresponding to the loss of the –NO2 group) has been observed in the reaction ATFA with Cl atoms, which could indicate the presence of a peroxinitrate also in this fluorinated ester. Finally, it should be noted that three products have not been identified; therefore, the existence of other channels of reaction, as the H-abstraction channel (3), cannot be discarded.
4. Atmospheric implications
4.1. Atmospheric lifetimes with OH and Cl
The rate coefficients determined in this study for the reactions of OH radicals and Cl atoms with the studied FESs can be used to estimate their atmospheric lifetimes. Kinetic data are not available for the reaction of the studied FESs with NO3 radicals or O3 molecules, while photolysis losses are expected to be negligible in the actinic region of the electromagnetic spectrum,40,41 and this way it has been observed at our wavelength of photolysis. The atmospheric lifetimes can be calculated using the following expression: kx = 1/(kx[X]), where kx is the rate coefficient for the reaction of the oxidant X with the FESs obtained in this work and [X] is the typical atmospheric concentration of the oxidant (global average concentrations of 1 × 106 molecules per cm342 and 1 × 103 molecules per cm343,44 for OH and Cl, respectively).
As shown in Table 3, the reaction with OH radicals is a major atmospheric loss process for both FESs during the day, whereas the reaction with Cl atoms is of much less importance. However, the reactivity with Cl atoms is, in general, higher than that corresponding to the OH radicals. Therefore, in areas where atomic Cl concentrations are high, oxidation initiated by Cl atoms would be competitive with that initiated by OH radicals. In this sense, the production rate of the Cl atoms exceeds the production of OH for 2 or 3 h after sunrise due to the high concentration of ClNO2, precursor of the Cl atoms through its photolysis.45–48 Thus, Cl concentrations of ∼106 molecules per cm3 have been calculated from the atmospheric concentrations and photolysis rates of ClNO2 in early morning air masses.45 Under such conditions, the lifetime of VTFA and ATFA with respect to this oxidant would be ∼1.3 and 1.6 h, respectively, and therefore the reaction of Cl atoms with the studied FESs could be an important process for their atmospheric removal.
It should be noted that the short lifetimes indicate that these esters will be degraded close to their emission sources. Since the characteristic mixing time in the troposphere is around 0.5 years, VTFA and ATFA are far from being homogeneously distributed and may be considered very short-lived species (VSLS), and their lifetime may depend on when and where these compounds are emitted along with the atmosphere conditions.49
As a consequence, the fate of the products arising from OH- and Cl-initiated oxidation of the studied esters will be important since the atmospheric oxidation of these products can also contribute to ozone and photo-oxidant formation in the troposphere. Moreover, as has been shown in this work, the oxidation of FESs leads to the secondary formation of TFA, which is highly soluble and may be rapidly taken up into cloud droplets, contributing to the acidity of precipitation.14
4.2. Brief overview of the radiative behavior
It is worth noting that FESs such as VTFA and ATFA are potential greenhouse gases since they absorb infrared radiation strongly within the atmospheric window 700–1400 cm−1, due to the presence of C–F bonds in the molecule, as can be observed in Fig. 4. Besides this, radiative properties can be enhanced compared to other greenhouse gases such as HFEs, due to the presence of an additional strong band, corresponding to C–O–C(O)R stretching, in the region of the spectrum where the radiative forcing function is large.19 The integrated cross sections obtained for VTFA and ATFA are collected in Table 3. To our knowledge, here we present the first experimental study providing data for the infrared spectra for these two species.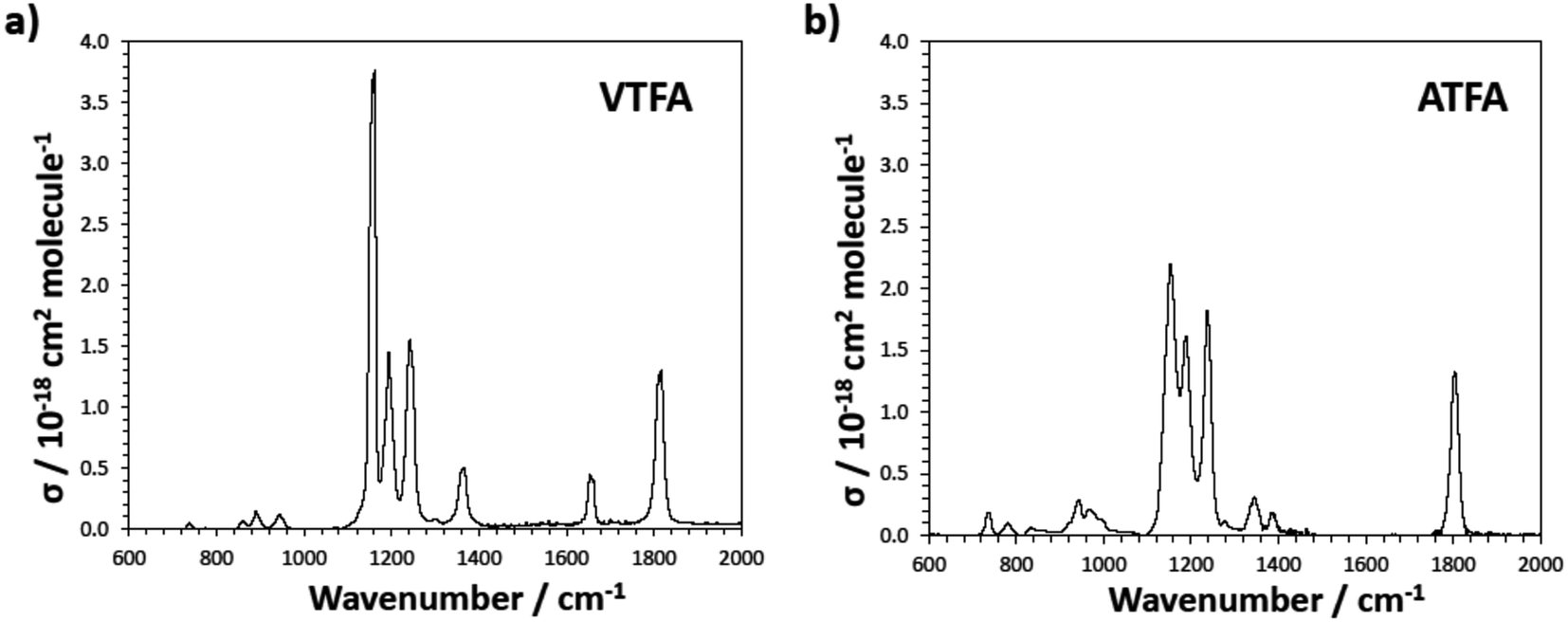 | ||
| Fig. 4 Infrared spectra of: (a) VTFA and (b) ATFA. Spectra have been smoothed to ca. 2 cm−1 resolution using a sliding average method. | ||
In order to evaluate the climate impact of emission of VTFA and ATFA into the atmosphere, the forcing REs were calculated using the methodology described by Pinnock et al.51 in combination with the updated 1 cm−1 resolution radiative forcing function recently updated by Hodnebrog et al.50 Table 3 summarizes the REs calculated assuming a perfect mixing. However, the lifetime obtained here for both compounds is evidence that these materials are relatively short lived and are unlikely to be well mixed in the troposphere. In this sense, atmospheric lifetime plays an important role in the determination of the RE. Since it depends on the atmospheric compound location, a unique RE cannot be defined for very short-lived compounds without a detailed knowledge of the spatial and temporal emission patterns. To take this into account, we used the lifetime correction proposed by Hodnebrog et al.50 for lifetimes within 10−4 to 104 years, where the “well-mixed” RE value is multiplied by a factor 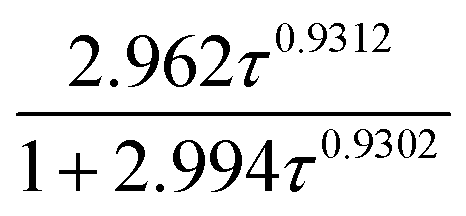 , and τ is the atmospheric lifetime in years; “well-mixed” and lifetime-corrected REs are collected in Table 3. To our knowledge, we report the first RE literature data for VTFA and ATFA.
, and τ is the atmospheric lifetime in years; “well-mixed” and lifetime-corrected REs are collected in Table 3. To our knowledge, we report the first RE literature data for VTFA and ATFA.
These RE values found for VTFA and ATFA (0.25 and 0.33 W m−2 ppb−1, respectively) are significantly large and comparable with other common greenhouse gases widely used in industry as HFEs or HFCs. For instance CFC-11, HFC-125 or HFE-134 present RE values of 0.25, 0.23 and 0.45 W m−2 ppb−1, respectively.52 However, the lifetime-corrected REs obtained here are nearly negligible since they present very short lifetimes and their contribution to warming is expected to be low.
The intergovernmental panel on climate change (IPCC)52 generally recommends the use of the GWP parameter to assess the contribution to warming of a species compared to carbon dioxide, and regularly reports 20, 100 and 500 year GWPs along with RE values for a large numbers of gases. Table 3 collects the 20, 100 and 500 year GWPs obtained here for VTFA and ATFA. Note that these values were calculated using the lifetime-corrected REs to get more realistic results. As can be appreciated from these low GWP values, the contribution to warming of both compounds may be negligible.
4.3. Estimated photochemical ozone creation potentials (εPOCP)
εPOCP estimates the total additional ozone increase in the multi-day modeling, as a by-product of the atmospheric oxidation of volatile organic compounds, relative to ethene, on the same mass basis. Jenkin et al.53 and Derwent et al.54 have developed a method for such a calculation that uses fundamental molecular properties of the considered compound and its reactivity towards OH radicals. The equation used was:| εPOCP = α1 × γS × γβR(1 − α2 × nc) | (4) |
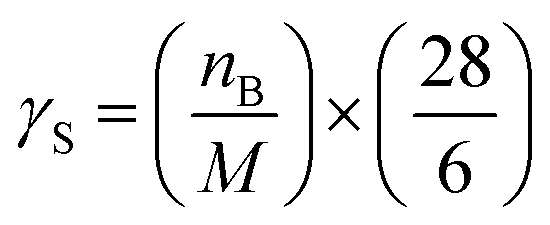 | (5) |
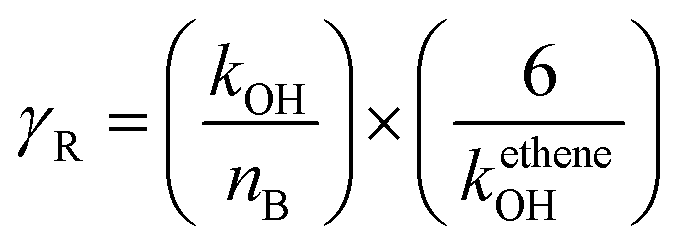 | (6) |
For reactions with kOH outside the range (4–40) × 10−12 cm3 per molecule per s, the suggested β, α1, and α2 values are 0.25, 104 and 0.03, respectively.53 Using the OH rate coefficients calculated in this work for the studied FESs, the estimated photochemical ozone creation potentials (εPOCP) were 22.2 and 18.0 for ATFA and VTFA, respectively. These values are in the same order as some hydrofluoroolefins, in which the rise of F atoms decreases εPOCP and is significantly lower than non-fluorinated unsaturated hydrocarbons.55 However, it is worth noting that Cl-initiated oxidation may be of equal importance to OH-initiated oxidation, especially in regions with high emissions such as coastal regions and polluted areas with rather high Cl atom-mixing ratios. In such cases, the Cl chemistry should be also considered, and this may lead to higher εPOCP values.
5. Conclusion
We have used the smog chamber/GC techniques to investigate the atmospheric degradation of VTFA and ATFA by oxidation with OH radicals and Cl atoms at 298 K and atmospheric pressure. Atmospheric lifetimes, infrared spectra, radiative efficiencies, the photochemical ozone creation potentials, along with the reaction mechanisms, were elucidated to assess the environmental impact of these FESs. In the OH-initiated oxidation of allyl trifluoroacetate, the identified product can arise from OH addition to both carbons in the double bond and the later decomposition of the alkoxy radical formed. However, in the reactions of both fluoroesters with Cl atoms, the main product detected arises from Cl addition to the terminal carbon atom and the subsequent reaction of the chloroalkoxy radical formed with O2, being the decomposition pathway minority. Thus, in the reaction of fluorinated esters with Cl atoms, the presence of the –CF3 group increases the reactivity of the alkoxy radical toward O2, even annulling the majority contribution of the decomposition pathway observed in hydrogenated esters in presence of NOx.29,38,39 However, the intensity of the minority pathway, the decomposition, would increase slightly in NOx-rich environments.Although the studied compounds are fluorinated esters, they have a short atmospheric lifetime that leads to low GWP and are therefore expected to have a minor impact on global warming and climate change. In addition, εPOCP is relatively low compared to non-fluorinated unsaturated hydrocarbons, so the studied FESs have no significant local effects in contribution to ozone formation, except in areas with increased levels of Cl atoms where their chemistry could increase the local ozone formation. Therefore, from an atmospheric point of view, the studied fluorinated esters show suitable characteristics as replacements for CFCs, even though we must also take into account the formation of fluorinated acetic acid as an oxidation product, which is a highly soluble compound that could be rapidly incorporated into cloud droplets, contributing to the acidity of local precipitation.
Acknowledgements
This study was supported by the Spanish Ministerio de Ciencia e Innovación (project CGL2011-4799) and the University of Castilla La Mancha (project GI20152950).References
- W. T. Tsai, J. Hazard. Mater., 2005, 119, 69–78 CrossRef CAS PubMed.
- Clean Air Act, EPA, U.S. Environmental Protection Agency, http://www.epa.gov/ozone/snap/index.html.
- Anesthetic Gases: Guidelines for Workplace Exposures, OSHA, U.S. Occupational Safety & Health Administration, http://www.osha.gov/dts/osta/anestheticgases/index.html.
- J. G. Calvert, A. Mellouli, J. J. Orlando, M. J. Pilling and T. J. Wallington, The Mechanism of Atmospheric Oxidation of the Oxygenate, Oxford University Press, New York, 2011 Search PubMed.
- L. Chen, T. Uchimaru, S. Kutsuna and A. Sekiya, Thromb. Res., 2011, 514, 207–213 CAS.
- W. Lu, K. Xie, Y. Pan, Z. Chen and C. Zheng, J. Fluorine Chem., 2013, 156, 136–143 CrossRef CAS.
- W. Lu, K. Xie, Z. Chen, Y. Pan and C. Zheng, J. Fluorine Chem., 2014, 161, 110–119 CrossRef CAS.
- L. Zhao, S. Okada and J. Yamaki, J. Power Sources, 2013, 244, 369–374 CrossRef CAS.
- K. Takagi, I. Tomita and T. Endo, Macromolecules, 1997, 30, 7386–7390 CrossRef CAS.
- E. Girard, X. Liu, J. D. Marty and M. Destarac, Polym. Chem., 2014, 5, 1013–1022 RSC.
- S. Kutsuna, L. Chen, K. Ohno, K. Tokuhashi and A. Sekiya, Atmos. Environ., 2004, 38, 725–732 CrossRef CAS.
- S. Kutsuna, L. Chen, T. Abe, J. Mizukado, T. Uchimaru, K. Tokuhashi and A. Sekiya, Atmos. Environ., 2005, 39, 5884–5892 CrossRef CAS.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Chem. Phys. Lett., 2008, 453, 18–23 CrossRef CAS.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Environ. Sci. Technol., 2010, 44, 2354–2359 CrossRef CAS PubMed.
- M. B. Blanco, C. Rivela and M. A. Teruel, Chem. Phys. Lett., 2013, 578, 33–37 CrossRef CAS.
- C. M. Tovar and M. A. Terual, Atmos. Environ., 2014, 94, 489–495 CrossRef CAS.
- A. Jordan and H. Frank, Environ. Sci. Technol., 1999, 33, 522–527 CrossRef CAS.
- I. Bravo, Y. Díaz-de-Mera, A. Aranda, E. Moreno, D. R. Nutt and G. Marston, Phys. Chem. Chem. Phys., 2011, 13, 17185–17193 RSC.
- I. Bravo, A. Rodríguez, D. Rodríguez, Y. Diaz-de-Mera, A. Notario and A. Aranda, ChemPhysChem, 2013, 14, 3834–3842 CrossRef CAS PubMed.
- M. B. Blanco and M. A. Teruel, Atmos. Environ., 2007, 41(34), 7330–7338 CrossRef CAS.
- B. K. Mishra, H. J. Singh and L. Tiwari, J. Mol. Model., 2014, 20, 2475 CrossRef PubMed.
- B. K. Mishra, J. Fluorine Chem., 2015, 172, 74–79 CrossRef CAS.
- N. K. Gour, R. C. Deka, H. J. Singh and B. K. Mishra, J. Fluorine Chem., 2014, 160, 64–71 CrossRef CAS.
- A. Rodríguez, D. Rodríguez, A. Garzon, A. Soto, A. Aranda and A. Notario, Phys. Chem. Chem. Phys., 2010, 12, 12245–12258 RSC.
- Y. Diaz-de-Mera, A. Aranda, A. Notario, A. Rodríguez, D. Rodríguez and I. Bravo, Phys. Chem. Chem. Phys., 2015, 17, 22991–22998 RSC.
- R. Atkinson, Atmos. Chem. Phys., 2003, 3, 2233–2307 CrossRef CAS.
- Z. Li and A. Pirasteh, Int. J. Chem. Kinet., 2006, 38, 386–398 CrossRef CAS.
- R. S. Anderson, L. Huang, R. Iannone and J. Rudolph, J. Phys. Chem. A, 2007, 111, 495–504 CrossRef CAS PubMed.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Environ. Sci. Technol., 2012, 46, 8817–8825 CrossRef CAS PubMed.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, J. Phys. Chem. A, 2009a, 113, 5958–5965 Search PubMed.
- M. P. Sulbaek Andersen, O. J. Nielsen, A. Toft, T. Nakayama, Y. Matsumi, R. L. Waterland, R. C. Buck, M. D. Hurley and T. J. Wallington, J. Photochem. Photobiol., A, 2005, 176, 124–128 CrossRef.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Environ. Sci. Pollut. Res., 2009b, 16, 641–648 Search PubMed.
- B. Picquet-Varrault, J. F. Doussin, R. Durand-Jolibois, O. Pirali and O. Carlier, Environ. Sci. Technol., 2002, 36, 4081–4086 CrossRef CAS PubMed.
- C. Papagni, J. Arey and R. Atkinson, Int. J. Chem. Kinet., 2001, 33, 142–147 CrossRef CAS.
- A. Rodríguez, D. Rodríguez, A. Soto, A. Notario, A. Aranda, Y. Diaz-de-Mera and I. Bravo, Atmos. Environ., 2007, 41, 4693–4702 CrossRef.
- M. Goto, Y. Inoue, M. Kawasaki, A. G. Guschin, L. T. Molina, M. J. Molina, T. J. Wallington and M. D. Hurley, Environ. Sci. Technol., 2002, 36, 2395–2402 CrossRef CAS PubMed.
- A. Rodríguez, D. Rodríguez, A. Moraleda, I. Bravo, E. Moreno and A. Notario, Atmos. Environ., 2014, 96, 145–153 CrossRef.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, RSC Adv., 2015, 5, 48154–48163 RSC.
- R. G. Gibilisco, J. G. Uranga, A. N. Santiago and M. A. Teruel, J. Phys. Chem. A, 2015, 119, 8775–8783 CrossRef CAS PubMed.
- N. Oyaro, S. R. Sellevag and C. J. Nielsen, Environ. Sci. Technol., 2004, 38, 5567–5576 CrossRef CAS PubMed.
- M. A. Teruel, S. I. Lane, A. Mellouki, G. Solignac and G. Le Bras, Atmos. Environ., 2006, 40, 3764–3772 CrossRef CAS.
- W. J. Bloss, M. J. Evans, J. D. Lee, R. Sommariva, D. E. Heard and M. J. Pilling, Faraday Discuss., 2005, 130, 425–436 RSC.
- A. A. P. Pszenny, W. C. Keene, D. J. Jacob, S. Fan, J. R. Maben, M. P. Zetwo, M. Springer-Young and J. N. Galloway, Geophys. Res. Lett., 1993, 20, 699–702 CrossRef CAS.
- O. W. Wingenter, M. K. Kubo, N. J. Blake, T. W. Smith, D. R. Blake and F. S. Rowland, J. Geophys. Res., 1996, 101, 4331–4340 CrossRef CAS.
- H. D. Osthoff, J. M. Roberts, A. R. Ravishankara, E. J. Williams, B. M. Lerner, R. Sommariva, T. S. Bates, D. Coffman, P. K. Quinn, J. E. Dibb, H. Stark, J. B. Burkholder, R. K. Talukdar, J. Meagher, F. C. Fehsenfeld and S. Brown, Nat. Geosci., 2008, 1(5), 324–328 CrossRef CAS.
- J. A. Thornton, J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dubé, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander and S. S. Brown, Nature, 2010, 464(7286), 271–274 CrossRef CAS PubMed.
- L. H. Mielke, A. Furgeson and H. D. Osthoff, Environ. Sci. Technol., 2011, 45(20), 8889–8896 CrossRef CAS PubMed.
- G. J. Phillips, M. J. Tang, J. Thieser, B. Brickwedde, G. Schuster, B. Bohn, J. Lelieveld and J. N. Crowley, Geophys. Res. Lett., 2012, 39, L10811 CrossRef.
- Scientific Assessment of Ozone Depletion: 2010, Global Ozone Research and Monitoring Project, Report No. 50, World Meteorological Organization, Geneva, Switzerland, 2011 Search PubMed.
- Ø. Hodnebrog, M. Etminan, J. S. Fuglestvedt, G. Marston, G. Myhre, C. J. Nielsen, K. P. Shine and T. J. Wallington, Rev. Geophys., 2013, 51, 300–378 CrossRef.
- S. Pinnock, M. D. Hurley, K. P. Shine, T. J. Wallington and T. J. Smyth, J. Geophys. Res., 1995, 100, 23227–23238 CrossRef CAS.
- P. M. D. Forster, V. Ramaswamy, P. Artaxo, T. Berntsen, R. Betts, D. W. Fahey, J. Haywood, J. Lean, D. C. Lowe, G. Myhre, J. Nganga, R. Prinn, G. Raga, M. Schulz, R. Van Dorland and S. Solomon, Fourth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 2007 Search PubMed.
- M. E. Jenkin, Photochemical Ozone and PAN Creation Potentials: Rationalisation and Methods of Estimation, Report AEAT-4182/20150/003, AEA Technology plc., National Environmental Technology Centre, Culham, Oxfordshire OX14 3DB, UK, 1998 Search PubMed.
- R. G. Derwent, M. E. Jenkin, S. M. Saunders and M. J. Pilling, Atmos. Environ., 1998, 32, 2429–2441 CrossRef CAS.
- T. J. Wallington, M. P. S. Andersen and O. J. Nielsen, Atmos. Environ., 2010, 44, 1478–1481 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00630b |
| This journal is © The Royal Society of Chemistry 2016 |