
An efficient catalytic synthesis and characterization of new styryl-ferrocenes and their trans-π-conjugated organosilicon materials†
M. Majchrzak*a,
S. Kosteraa,
M. Grzelaka,
B. Marciniec*ab and
M. Kubickia
aFaculty of Chemistry, Adam Mickiewicz University in Poznan, Umultowska 89b, 61-614 Poznan, Poland. E-mail: mariusz.majchrzak@amu.edu.pl; marcinb@amu.edu.pl
bCenter for Advanced Technology, Adam Mickiewicz University in Poznan, Umultowska 89c, 61-614 Poznan, Poland
First published on 11th April 2016
Abstract
A selective and efficient catalytic method was developed for the synthesis of new styryl-conjugated silyl-ferrocene materials. The use of an effective palladium catalytic system [Pd(η2-dba)(P(o-tolyl)3)2] (3) with a K3PO4 base solution allowed us to obtain new, reactive ferrocene olefins which can be used in various applications. In addition, using the stereo-control silylative coupling reaction we received unique new organosilicon trans-vinylene products with a built-in metallocene.
Introduction
Polymers and copolymers containing metal fragments have been the subject of research recently because they show the great potential which results from the unique characteristics of transition metals and the processibility of polymeric materials at the same time.1–5 In general, the metal moiety can be placed in the polymer backbone by covalently bonding directly to the main chain as well as by coordinating to ligands within the backbone. It can be pendant or attached to the side chain of the polymer, too.In the early 1960s the synthesis of the polymers containing ferrocene was described by Korshak and Nesmeyanov. The process occurred via the reaction of ferrocene with tert-butyl hydroperoxide.6,7 Since then a lot of synthetic methods have been designed to allow the formation of metal-containing polymers such as living, ionic, and controlled radical polymerization,8–10 polycondensation,11–14 electropolymerization,15 and ring opening polymerization.16,17 So far only a few examples of aryl-ferrocene derivatives have been reported.18,19 Zhang and co-workers effectively applied a 1,1′-di(4-vinylphenyl-ethane)ferrocene for acyclic diene metathesis polymerization (ADMET) to get a regular, linear polymers.20
The Suzuki–Miyaura coupling reaction is an exceptionally useful method in organic synthesis of compounds containing carbon–carbon bond, e.g. biaryl compounds.21–24 The formation of C–C bond is of vital importance in preparative organic processes from molecular model chemistry to macromolecular and nanomaterial science. Together with the silylative coupling (SC) reaction they are very effective tools in the synthesis of new materials. The research on SC reaction has been carried out in our group for the last twenty years. The silylative coupling is the reaction of olefins with vinyl-substituted organosilicon compounds which takes place in the presence of complexes containing initially or generating in situ metal–hydrogen and metal–silicon bonds.25,26 The process occurs via cleavage of the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–Si bond in the vinylsilane and the C–H bond in the olefin and is catalysed by transition metal complexes [TM–H] or silyl [TM–Si] ligands (where TM = Ru, Rh, Ir, Co).27,28 The silylative coupling reaction is an exceptional, effective and highly regio- and stereoselective way to functionalize molecular and macromolecular compounds that contain one or a few vinyl groups connected to the silicon atom. Undoubtedly, this method is a convenient and powerful tool to synthesize unsaturated, highly π-conjugated compounds.29 We previously described the catalytic transformation of vinylsilyl-ferrocene derivatives via silylative coupling. Each time this transformation led to well-defined cyclic structures.30
C–Si bond in the vinylsilane and the C–H bond in the olefin and is catalysed by transition metal complexes [TM–H] or silyl [TM–Si] ligands (where TM = Ru, Rh, Ir, Co).27,28 The silylative coupling reaction is an exceptional, effective and highly regio- and stereoselective way to functionalize molecular and macromolecular compounds that contain one or a few vinyl groups connected to the silicon atom. Undoubtedly, this method is a convenient and powerful tool to synthesize unsaturated, highly π-conjugated compounds.29 We previously described the catalytic transformation of vinylsilyl-ferrocene derivatives via silylative coupling. Each time this transformation led to well-defined cyclic structures.30
Herein, we present an efficient, stereoselective methods for the preparation of new π-conjugated ferrocenyl-organosilicon compounds. The aforementioned ferrocene derivatives were synthesized via suitable selected catalytic transformations such as Suzuki–Miyaura (SM) coupling and silylative coupling (SC) catalysed by well-defined TM complexes. We also present our preliminary studies on the application of this protocol to the synthesis of new polymeric material.
Experimental
Instruments and measurement
Materials and methods
The chemicals were obtained from the following sources: toluene, diethyl ether, dichloromethane (DCM), pentane and hexane, were purchased from Fluka, CDCl3 from Dr Glaser A. G. Basel, 1,1′-dibromoferrocene, sodium carbonate (Na2CO3), potassium carbonate (K2CO3), magnesium sulphate (MgSO4), tripotassium phosphate (K3PO4), calcium hydride (CaH2), sodium hydride (NaH), 1,2-dichloromethane, 4-bromostyrene, ferroceneboronic acid, 4-vinylphenylboronic acid, 3-vinylphenylboronic acid, ethanol, Celite 545, bis(dibenzylideneacetone)palladium(0)–[Pd(dba)2], tris(o-tolyl)phosphine from Aldrich, vinyltrimethylsilane from ABCR and bromoferrocene from Strem. Toluene was distilled from sodium and hexane, ethanol from calcium hydride under argon. The above mentioned solvents were stored over molecular sieves type 4 Å. All liquid substrates were also dried and degassed by bulb-to-bulb distillation. All the syntheses of the catalysts, molecular compounds and catalytic tests were carried out under an inert argon or air atmosphere. Palladium complexes [Pd(η2-dba)(PPh3)2] (1),31 [Pd(η2-dba)(PCy3)2] (2),31 [Pd(η2-dba)(P(o-tolyl)3)2] (3)32 and ruthenium species [RuH(CO)Cl(PCy3)2] (4)33 were prepared according to the literature procedure. The silyl derivatives of compound 4-vinyldimethylsilylbiphenyl (5) and 4,4′-bis(vinyldimethylsilyl)biphenyl (6) were synthesized according to the procedure.34Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Rf = 0.4–0.5 in relation to biphenyl) and GCMS to complete.:1, Rf = 0.3–0.5 in relation to biphenyl) and GCMS to complete.
:1, Rf = 0.4–0.5 in relation to biphenyl) and GCMS to complete.:1, Rf = 0.3–0.5 in relation to biphenyl) and GCMS to complete.1-(3-Vinylphenyl)ferrocene (7). [Pd(η2-dba)(P(o-tolyl)3)2] (3) (8.4 mg, 0.088 mmol, 1 mol%), 3-vinylphenylboronic acid or 4-vinylphenylboronic acid (0.90 mmol), bromoferrocene (235 mg, 0.88 mmol), ethanol (1.75 mL), toluene (1.75 mL) and aqueous 2 M K3PO4 (2.60 mL) were placed in a two-neck glass reactor equipped with a magnetic stirring bar and reflux condenser under an argon atmosphere. The mixture was stirred at 90 °C and the progress of the reaction was monitored by GCMS. Upon the consumption of ferrocene halide, the mixture was cooled to room temperature. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The organic phases were combined, dried over MgSO4 (6 h) and concentrated. The product was isolated by column chromatography (SiO2, Celit®, sand) with hexane and then hexane/dichloromethane (2
:1) mixture as eluents. The final product 9 was obtained with 76% yield (194 mg) as an orange solid.Analytic data: 1H NMR (300 MHz, CDCl3): δ 3.98 (s, 5H, C5H4), 4.24 (s, 2H, C5H4), 4.59 (s, 2H, C5H4), 5.20 (d, 1H, JHH = 10.8 Hz, –CHCH2), 5.71 (d, 1H, JHH = 16.8 Hz, –CHCH2), 6.67 (dd, 1H, JHH = 10.8 Hz, –CHCH2), 7.19 (m, 2H, C6H4), 7.32 (m, 1H, C6H4), 7.41 (s, 1H, C6H4). 13C NMR (75 MHz, CDCl3): δ 66.6, 68.9, 69.6, 85.3, 113.8, 123.8, 125.8, 128.4, 136.9, 137.4, 139.5. MS (EI) (m/z (relat. int.%)): 288.0 (M+˙) (100), 121 (5). HRMS (m/z) calcd for C18H16Fe: 288.06014; found 288.05956. Elemental analyses calcd for C18H16Fe: C 75.02, H 5.60; found C 75.01, H 5.59.
1-(4-Vinylphenyl)ferrocene (8). 1-(4-Vinylphenyl)ferrocene (8) was obtained according to the above-mentioned preparation procedure of 7 with 82% yield (209 mg) as an orange-yellow solid.
Analytic data: 1H NMR (300 MHz, CDCl3): δ 4.04 (s, 5H, C5H4), 4.33 (s, 2H, C5H4), 4.65 (s, 2H, C5H4), 5.25 (d, 1H, JHH = 10.8 Hz, –CHCH2), 5.76 (d, 1H, JHH = 16.8 Hz, –CHCH2), 6.71 (dd, 1H, JHH = 10.8 Hz, –CHCH2), 7.35 (d, 2H, C6H4), 7.45 (d, 2H, C6H4). 13C NMR (75 MHz, CDCl3): δ 66.4, 69.0, 69.6, 84.9, 112.8, 126.1, 126.2, 135.1, 136.6, 138.9. MS (EI) (m/z (relat. int.%)): 288.1 (M+˙) (100). HRMS (m/z) calcd for C18H16Fe: 288.06014; found 288.05998. Elemental analyses calcd for C18H16Fe: C 75.02, H 5.60; found C 75.05, H 5.61.
1,1′-Bis(3-vinylphenyl)ferrocene (9). [Pd(η2-dba)(P(o-tolyl)3)2] (3) (16.7 mg, 0.0174 mmol, 1 mol%), 4-vinylphenylboronic acid or 3-vinylphenylboronic acid (1.9 mmol), 1,1′-dibromoferrocene (299 mg, 0.870 mmol), ethanol (1.75 mL), toluene (1.75 mL) and aqueous 2 M K3PO4 (2.60 mL) were placed in a two-neck glass reactor equipped with a magnetic stirring bar and reflux condenser under an argon atmosphere. The mixture was stirred at 90 °C and the progress of the reaction was monitored by GCMS. Upon the consumption of ferrocene dihalide, the mixture was cooled to room temperature. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). The organic phases were combined, dried over MgSO4 (6 h) and concentrated. The product was isolated by column chromatography (SiO2, Celit®, sand) with hexane and then hexane/dichloromethane (1
:1) mixture as eluents. The final product 9 was obtained with 91% yield (307 mg) as an orange solid.Analytic data: 1H NMR (300 MHz, CDCl3): δ 4.30 (s, 4H, C5H4), 4.53 (s, 4H, C5H4), 5.24 (d, 2H, JHH = 17.6 Hz, –CHCH2), 5.71 (d, 2H, JHH = 11.6 Hz, –CHCH2), 6.65 (dd, 2H, JHH = 10.8 Hz, 17.5 Hz, –CHCH2), 7.15–7.25 (m, 8H, C6H4). 13C NMR (75 MHz, CDCl3): δ 68.1, 70.4, 86.0, 113.7, 123.5, 124.1, 125.5, 128.4, 136.8, 137.4, 138.2. MS (EI) (m/z (relat. int.%)): 390.7 (M+˙) (100). HRMS (m/z) calcd for C26H22Fe: 390.10709; found 390.10654. Elemental analyses calcd for C26H22Fe: C 80.01, H 5.68; found C 80.16, H 5.70.
1,1′-Bis(4-vinylphenyl)ferrocene (10). 1,1′-Bis(4-vinylphenyl)ferrocene (10) was obtained according to the above-mentioned preparation procedure of 9 with 90% yield (305 mg) as an orange-yellow solid.
Analytic data: 1H NMR (300 MHz, CDCl3): δ 4.25 (s, 4H, C5H4), 4.50 (s, 4H, C5H4), 5.27 (d, 2H, JHH = 10.8 Hz, –CHCH2), 5.76 (d, 2H, JHH = 16.8 Hz, –CHCH2), 6.71 (dd, 2H, JHH = 10.8 Hz, 17.4 Hz, –CHCH2), 7.26 (s, 8H, C6H4). 13C NMR (75 MHz, CDCl3): δ 67.8, 70.5, 85.7, 112.7, 125.9, 126.1, 135.1, 136.6, 137.6. MS (EI) (m/z (relat. int.%)): 390.3 (M+˙) (100), 388.6 (5), 167 (5). HRMS (m/z) calcd for C26H22Fe: 390.10709; found 390.10714. Elemental analyses calcd for C26H22Fe: C 80.01, H 5.68; found C 80.11, H 5.70.
1,1′-Bis-{((E,E)-2-phenyl-3-ethenyl)trimethylsilyl}ferrocene (11). In a typical experiment, the ruthenium catalyst [RuH(CO)Cl(PCy3)2] (4) (1 mol% per styryl group) was dissolved in toluene (0.75 mL, 0.75 M) and placed in a glass reactor under argon. Then 1,1′-bis(vinylphenyl)ferrocene (9 or 10) (200 mg, 0.512 × 10−1 mmol) and trimethylvinylsilane (308 mg, 3.07 mmol) were added in the molar ratio 1
:6. Then a glass reactor was heated to 80 °C and maintained at that temperature for 18 h. The progress of the reaction was monitored by GC and GCMS. The final product was separated from the residues of the catalyst via SiO2 column with hexane as an eluent. The product was obtained with 98% yield (268 mg) as an orange solid.Analytic data: 1H NMR (300 MHz, CDCl3): δ 0.16 (s, 18H, –Si–CH3), 4.29 (s, 4H, C5H4), 4.52 (s, 4H, C5H4), 6.44 (d, 2H, JHH = 19.2 Hz, –CHCH–SiMe3), 6.86 (d, 2H, JHH = 18.8 Hz, –CHCH–SiMe3), 7.19–7.28 (m, 8H, C6H4). 13C NMR (75 MHz, CDCl3): δ −1.2 (–Si–CH3), 68.1, 70.4, 86.0, 123.6, 123.9, 125.6, 128.4, 136.9, 137.2, 138.2, 143.2. 29Si NMR (60 MHz, CDCl3): δ −6.30. MS (EI) (m/z (relat. int.%)): 536.5 (17), 535.8 (43), 535.0 (M+˙) (100). HRMS (m/z) calcd for C32H38FeSi2: 534.18615; found 534.18605. Elemental analyses calcd for C32H38FeSi2: C 71.89, H 7.16; found C 71.81, H 7.13.
1,1′-Bis-{((E)-2-phenyl-4-ethenyl)trimethylsilyl}ferrocene (12). 1,1′-Bis-{((E)-2-phenyl-4-ethenyl)trimethylsilyl}ferrocene (12) was obtained according to the above-mentioned preparation procedure of 11 with 99% yield (271 mg) as an orange solid.
Analytic data: 1H NMR (300 MHz, CDCl3): δ 0.19 (s, 18H, –Si–CH3), 4.32 (s, 4H, C5H4), 4.55 (s, 4H, C5H4), 6.45 (d, 2H, JHH = 19.2 Hz, –CHCH–SiMe3), 6.85 (d, 2H, JHH = 18.8 Hz, –CHCH–SiMe3), 7.21 (d, 4H, C6H4), 7.28 (d, 4H, C6H4). 13C NMR (75 MHz, CDCl3): δ −1.16 (–Si–CH3), 67.6, 71.5, 87.1, 127.5, 127.6, 128.5, 136.2, 138.2, 143.2. 29Si NMR (60 MHz, CDCl3): δ −6.29. MS (EI) (m/z (relat. int.%)): 535.4 (M+˙) (100). HRMS (m/z) calcd for C32H38FeSi2: 534.18615; found 534.18599. Elemental analyses calcd for C32H38FeSi2: C 71.89, H 7.16; found C 71.83, H 7.14.
1,1′-Bis-{((E)-2-phenyl-3-ethenyl)dimethylbiphenylsilyl}ferrocene (13) and 1,1′-bis-{((E)-2-phenyl-4-ethenyl)dimethyl biphenylsilyl}ferrocene (14). In a typical experiment, the ruthenium catalyst [RuH(CO)Cl(PCy3)2] (4) (1 mol%) was dissolved in toluene (0.75 M) and placed in a glass reactor under argon. Then 1,1′-bis(vinylphenyl)ferrocene (9 or 10) (200 mg, 0.512 × 10−1 mmol) and 4-(vinyldimethylsilil)biphenyl (5) (305 mg, 0.128 mmol) were added in the molar ratio 1
:2.5. Then the glass reactor was heated to 90 °C and maintained at that temperature for 24 h. The progress of the reaction was monitored by GC and GCMS. The final product was separated via SiO2 column with hexane as an eluent. The final product was obtained with 81% yield (337 mg) as an orange solid.Analytic data: 1H NMR (300 MHz, CDCl3): δ 0.49 (s, 12H, –Si–CH3), 4.40 (s, 4H, C5H4), 4.66 (s, 4H, C5H4), 6.53 (d, 2H, JHH = 19.2 Hz, –CHCH–), 6.87 (d, 2H, JHH = 19.2 Hz, –CHCH–SiMe2–), 7.10–7.25 (m, 8H, C6H4), 7.35 (t, 4H, m-C6H4–C6H5), 7.44 (t, 2H, –C6H4–p-C6H5), 7.62 (m, 10H, –C6H4–C6H5), 7.67 (d, 4H, o-C6H4–C6H5). 13C NMR (75 MHz, CDCl3): δ −2.4 (–Si–CH3), 68.1, 70.4, 124.2, 124.4, 126.1, 126.5, 126.7, 127.1, 127.4, 128.2, 128.7, 134.4, 137.0, 137.3, 141.0, 141.0, 141.8, 145.5. 29Si NMR (60 MHz, CDCl3): δ −11.15. HRMS (m/z) calcd for C54H50FeSi2: 810.28005; found 810.28001. Elemental analyses calcd for C54H50FeSi2: C 79.97, H 6.21; found C 79.59, H 6.19.
1,1′-Bis-{((E)-2-phenyl-4-ethenyl)dimethylbiphenylsilyl}ferrocene (14). 1,1′-Bis-{((E)-2-phenyl-4-ethenyl)dimethylbiphenylsilyl}ferrocene (14) was obtained according to the above-mentioned preparation procedure of 13 with 87% yield (362 mg) as an orange-yellow solid.
Analytic data: 1H NMR (300 MHz, CDCl3): δ 0.45 (s, 12H, –Si–CH3), 4.40 (s, 4H, C5H4), 4.66 (s, 4H, C5H4), 6.42 (d, 2H, JHH = 20.4 Hz, –CHCH–); 6.93 (d, 2H, JHH = 18.9 Hz, –CHCH–Si–), 7.19–7.23 (m, 8H, C6H4), 7.35 (t, 4H, m-C6H4–C6H5), 7.44 (t, 2H, –C6H4–p-C6H5), 7.62 (m, 10H, –C6H4–C6H5). 13C NMR (75 MHz, CDCl3): δ −2.3 (–Si–CH3), 68.1, 70.3, 124.2, 126.1, 126.5, 127.4, 127.4, 128.2, 128.8, 134.4, 137.0, 137.3, 141.1, 141.3, 141.7, 145.5. 29Si NMR (60 MHz, CDCl3): δ −11.16. HRMS (m/z) calcd for C54H50FeSi2: 810.28005; found 810.28009. Elemental analyses calcd for C54H50FeSi2: C 79.97, H 6.21; found C 79.64, H 6.20.
Poly[(1,1′-di(3-phenylene)ferrocenylene)-(E)-vinylene-(4,4′-bis(dimethylsilylene)biphenylene)-(E)-vinylene]s (P1). In a typical experiment, the ruthenium catalyst [RuH(CO)Cl(PCy3)2] 4 (1 mol%) was dissolved in toluene and placed in a glass reactor under argon. Then 1,1′-bis(3-vinylphenyl)ferrocene (9) and 4,4′-bis(vinyldimethylsilyl)biphenyl (6) were added in the molar ratio 1
:1. After that the glass reactor was heated to 90 °C and maintained at that temperature for 24 h. The progress of the reaction was monitored by 1H NMR. The final product was separated via precipitation step with DCM/hexane as an eluent system. The products were obtained with 69% (P1) and 71% (P2) yields (as yellow solids).
Poly[(1,1′-di(3-phenylene)ferrocenylene)-(E)-vinylene-(4,4′-bis(dimethylsilylene)biphenylene)-(E)-vinylene]s (P1). Analytic data: 1H NMR (300 MHz, CDCl3): δ 0.49 (s, 12H, –Si–CH3), 4.27 (s, 4H, C5H4), 4.50 (s, 4H, C5H4), 5.21 (d, 1H, JHH = 10.8 Hz, –CH
CH2, traces), 5.21 (d, 1H, JHH = 10.8 Hz, –CHCH2, traces), 6.56 (d, 1H, JHH = 18.8 Hz, –CHCH–), 6.87 (d, 1H, JHH = 18.8 Hz, –CHCH–Si); 7.28–7.12 (m, 4H, C6H4); 7.62–7.67 (m, 4H, C12H8). 13C NMR (75 MHz, CDCl3): δ −2.3 (–Si–CH3), 68.1, 70.2, 85.8, 123.9, 124.2, 125.9, 126.6, 126.6, 128.4, 134.4, 137.6, 137.9, 138.3, 141.7, 145.5. 29Si NMR (60 MHz, CDCl3): δ −10.35. Elemental analyses calcd for (C42H40FeSi2)n: C 76.81, H 6.14; found C 76.34, H 6.10. GPC data: Mn = 3600 g mol−1, Mw = 5652 g mol−1, PDI (Mw/Mn) = 1.57, n = 9.
Poly[(1,1′-di(4-phenylene)ferrocenylene)-(E)-vinylene-(4,4′-bis(dimethylsilylene)biphenylene)-(E)-vinylene]s (P2). Poly[(1,1′-di(4-phenylene)ferrocenylene)-(E)-vinylene-(4,4′-bis(dimethylsilylene)biphenylene)-(E)-vinylene]s P2 was obtained according to the above-mentioned preparation procedure of P1.
Analytic data: 1H NMR (300 MHz, CDCl3): δ 0.47 (s, 12H, –Si–CH3), 4.41 (s, 4H, C5H4), 4.66 (s, 4H, C5H4), 5.2 (d, 1H, JHH = 10.8 Hz, –CHCH2, traces), 5.67 (d, 1H, JHH = 10.8 Hz, –CHCH2, traces), 6.51 (d, 1H, JHH = 17.4 Hz, –CHCH–Si), 6.96 (d, 1H, JHH = 18.9 Hz, –CHCH–Si), 7.16 (C6H4), 7.31 (C6H4), 7.61–7.66 (m, C12H8). 13C NMR (75 MHz, CDCl3): δ −2.6 (–Si–CH3), 68.3, 70.9, 124.0, 124.2, 125.9, 126.6, 128.4, 134.4, 137.6, 137.9, 138.3, 141.7, 145.5. 29Si NMR (60 MHz, CDCl3): δ −10.36. Elemental analyses calc. for (C42H40FeSi2)n: C 76.81, H 6.14; found C 76.27, H 6.09. GPC data: Mn = 3370 g mol−1, Mw = 5024 g mol−1, PDI (Mw/Mn) = 1.49, n = 8.
X-Ray crystal data
800 reflections collected, 6409 independent (Rint = 0.134), 4912 with I > 2σ(I). Final R[I > 2σ(I)] = 5.99%, wR2[I > 2σ(I)] = 8.01%, R[all data] = 8.57%, wR2[all data] = 8.83%, S = 0.91, Δρmax/Δρmin = 0.58/−0.46 e Å−3.Results and discussion
The main aim of the catalytic research was to develop efficient methods for the synthesis of the new materials consisting of π-conjugated ferrocenyl units. In order to do this, two fully controlled reactions which allowed to get new stereoselective organic-ferrocene derivatives were used. Olefins were prepared in an efficient Suzuki–Miyaura coupling reaction catalysed by well-defined palladium species. A series of catalytic tests was performed to optimize the reaction conditions for the selective formation of new 1-substituted ferrocene olefins.Two substrates systems were used: first – the reaction of bromoferrocene (BrFc) with vinylboronic acid and the second one – ferroceneboronic acid ((OH)2BFc) with 4-bromostyrene in the presence of palladium(0) catalyst, i.e. [Pd(dba)(PPh3)2]31 (1), [Pd(dba)(PCy3)2]31 (2) [Pd(dba)(P(o-tolyl)3)2]32 (3), according to Scheme 1.
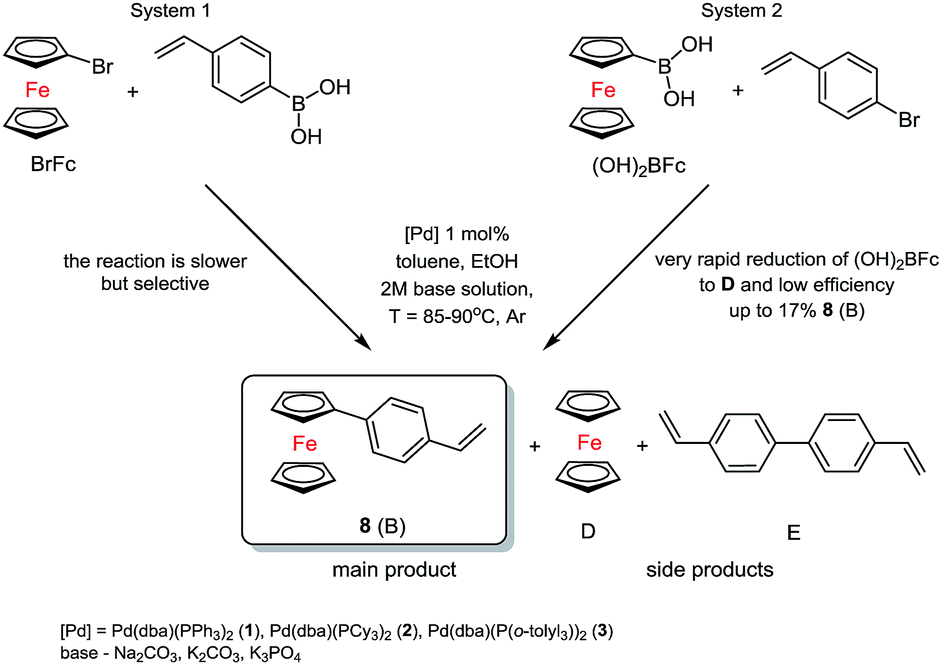 | ||
| Scheme 1 Synthesis of new 1-styrylferrocene via Suzuki coupling reaction. | ||
While examining the first system, we worked out the catalytic conditions to obtain controlled main product 8. At the same time, while testing the second system, we observed decomposition of (OH)2BFc to ferrocene and the progress of the reaction was not satisfying. In addition, we noticed an important factor, which was the aqueous solution of the base. In case of weaker bases such as sodium carbonate or potassium carbonate no formation of the product was observed in the system. The most appropriate was salt of potassium orthophosphate(V) (K3PO4). In this way it is possible to get nearly 92% conversion of BrFc to styrylferrocene (8).
In the first system we identified the by-products in small amounts as well (D = 4–6%, E = 8–10%). The best and the most selective catalyst proved to be 3. In the next step, which was based on previous research, we synthesized two substituted 1,1′-ferrocenes. As we expected a mixture of products, we conducted a series of preliminary tests again. Three complexes of palladium(0) were used. The typical catalytic system for tests consisted of toluene solution (0.5 M) of 1,1′-dibromoferrocene (Br2Fc), ethanol, 2 M solution of base, 4-vinylphenylboronic acid and suitable palladium(0) catalysts, see Scheme 2.
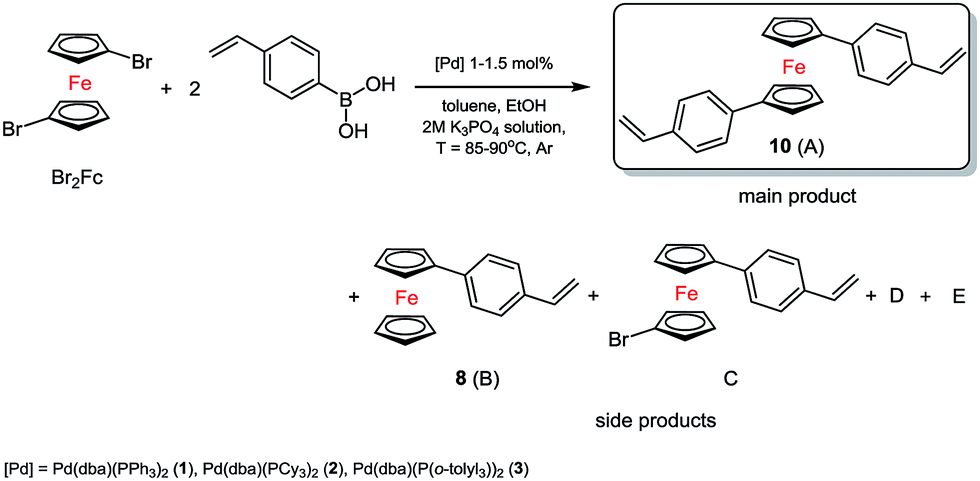 | ||
| Scheme 2 Synthesis of new styrylferrocenes via Suzuki coupling reaction. | ||
In many cases we also identified trace amount of side compounds (8) B, D, E (sometimes a lot, see Table 1) 1-bromoferrocene (BrFc) and 1,1′-dibromo ferrocene (as an unreacted substrate, Br2Fc) see Fig. S5, ESI†, based on MS spectra data. In the presence of the complexes 1 and 2 the debromination process followed quite fast.
| Cat. | Conversion of Br2Fcb (%) | A (%) | B (%) | C (%) | D (%) |
|---|---|---|---|---|---|
| a Reaction conditions: t = 24 h; T = 85–90 °C; open system under Ar were examined.b Determined by GC and GCMS methods.c 1 mol%.d 1.5 mol%.e 30 h.f 36 h. | |||||
| 1 | 58c | 0 | 6 | 47 | 5 |
| 79d | 27 | 18 | 26 | 8 | |
| 2 | 96c | 63 | 29 | 0 | 4 |
| 98d | 58 | 36 | 0 | 4 | |
| 3 | 77c | 53 | 0 | 24 | 0 |
| 73f | Trace | 3 | Trace | ||
| 98d | 93d | 0 | 4 | Trace | |
| 99d,e | 97e | Trace | 0 | Trace | |
As a result, some quantity of product 8 (B) was created and it was quite difficult to separate it from the main compound A, see Table 1.
In order to slow down the process, we reduced the amount of the catalyst 2 (0.5 mol%) and lowered the temperature to 70 °C. In this way the selectivity of the coupling reaction was controlled. However, the B product was formed as well. The most selective catalyst was 3, too. Probably, the activity and selectivity of this catalyst is caused by the type of the phosphine and its steric hindrance.
To our best knowledge, the use of well-defined palladium catalysts consisting of tris(o-tolyl) phosphine and ‘dba’ ligands in Suzuki–Miyaura coupling reactions is not known. This form of the catalyst is generated ‘in situ’ and participates in the polymerization reaction35 and the organometallic changes.36 Moreover, the catalytic mixture of Pd2(dba)3 and tris(o-tolyl) phosphine is regularly used in the common coupling reactions37 to generate catalytic species.
Therefore, we used biphasic solvents system of toluene (0.5 M solution concentration for halide)/ethanol (1.75 mL)/bases (2.6 mL, 2 M solution of K3PO4) and 3 (1.5 mol%) as the most selective and efficient palladium catalyst for the synthesis of new olefins, see Scheme 3.
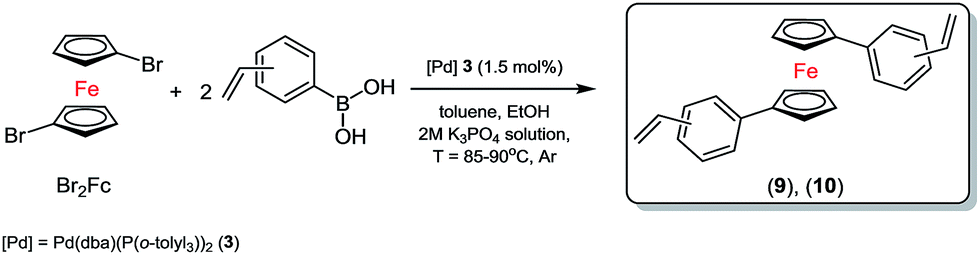 | ||
| Scheme 3 Synthesis of distyryl-ferrocene derivatives in the presence of 3. | ||
Typically, the crude mixture was extracted with CH2Cl2/water system solvents and a small amount of sodium chloride was used to disperse the slurry. Then, the mixture was left over magnesium sulfate for five-six hours. 1H NMR spectrum of 9 (see Fig. 1) below shows a new, clean ferrocene olefin which was isolated with simple and convenient methods of flash filtration (glass filter/silica gel/Celit®) and next by column chromatography (silica gel/sand).
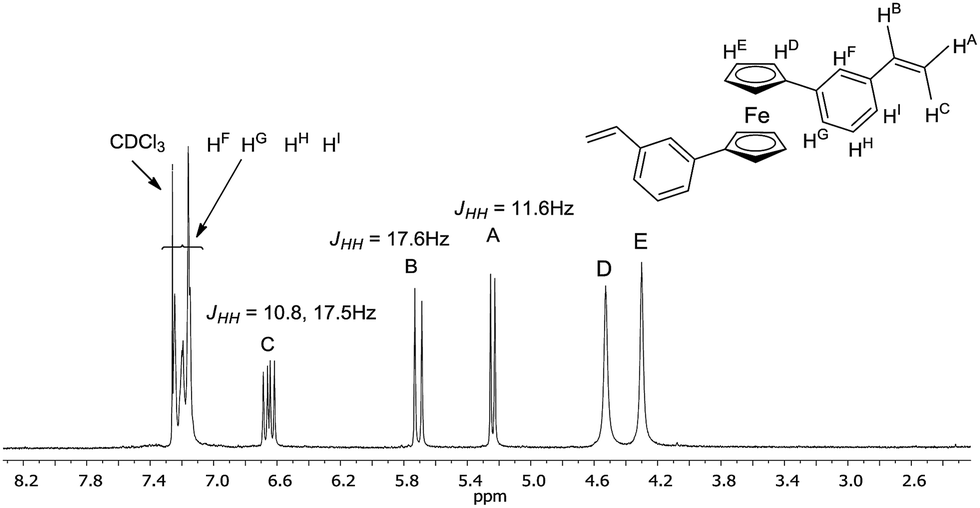 | ||
| Fig. 1 1H NMR fragment spectrum of 9 in CDCl3 at 25 °C. | ||
Because of the potential distribution (polymerization process via vinyl group), these olefins should be kept at low temperatures (2–5 °C). Compounds 9 and 10 were isolated and fully characterized spectroscopically (1H NMR spectrum show us clearly of high purity of 9). The solid state structure of 9 was determined by X-ray crystallography (Fig. 3). As far as we know this is the first crystallographic characterization of styryl ferrocene derivative.
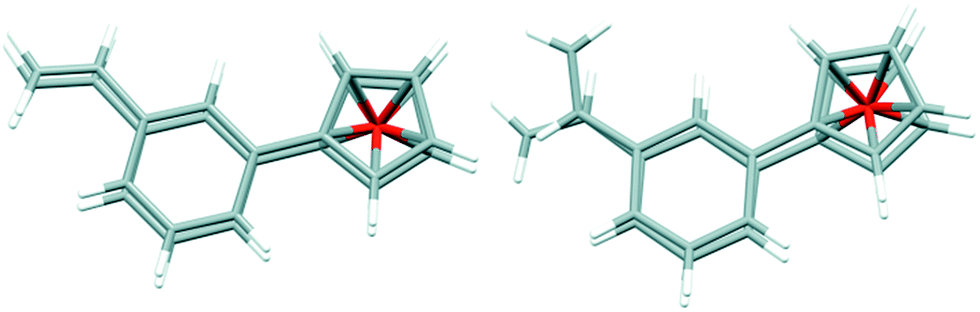 | ||
| Fig. 2 Comparison of two symmetry-independent molecules in the crystal structure of 9. | ||
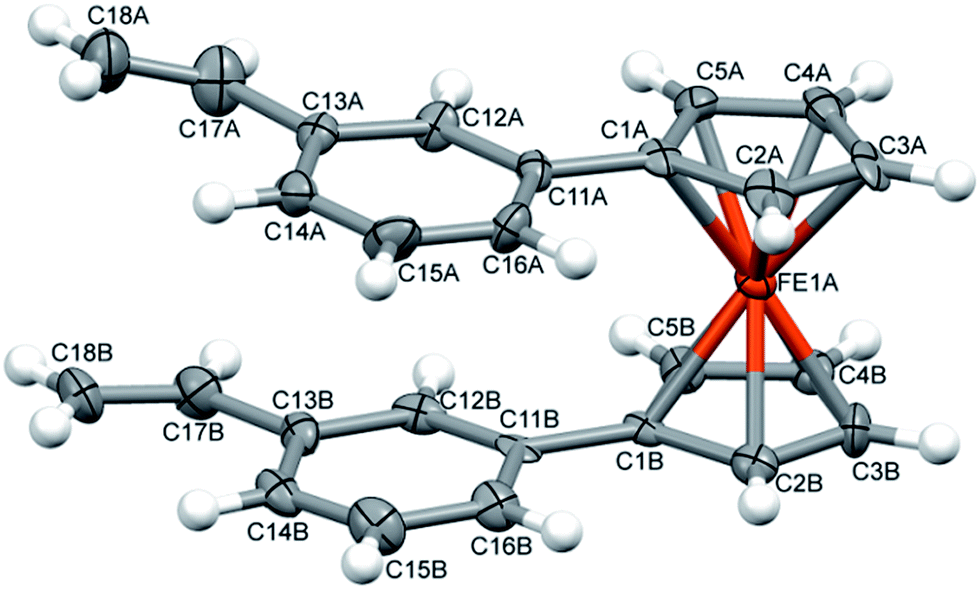 | ||
| Fig. 3 A perspective view of one of the symmetry-independent molecules of 9 with labelling scheme. The ellipsoids are drawn at the 50% probability level, hydrogen atoms are shown as spheres with arbitrary radii. | ||
X-Ray discussion
Interestingly, in the crystal structure there are two symmetry-independent molecules in the asymmetric part of the unit cell, and these molecules have different conformation (Fig. 2). In one of them, the vinyl groups are located almost exactly one over another (the improper torsion angle CC⋯CC is 1°), while in the other these groups are rotated, and the torsion angle is 54.1°. This might suggest – as the rotation barrier is obviously high and there is no possibility of interconversion in the crystal – that in the solution this compound exists as the mixture of two conformers.
The geometrical parameters of both molecules are typical, phenyl rings are almost parallel one to another (dihedral angles are 6.0 (4)° and 3.2 (4)°), and as the mean distance between the rings is ca. 3.5 Å there is some π⋯π interaction between the rings.
The arrangement of vinyl groups on the phenyl ring in no way interferes with the binding of activated carbon–hydrogen bond.
Therefore, we used a selected new distyryl-olefins in a stereo-selective synthesis of several new conjugated silicon-ferrocenyl–organic compounds.
In the next step, we used the olefin obtained for silylative coupling (SC) reaction. Ruthenium(II) hydride complexes are known to be active in SC of vinylsilanes with vinylarenes. This catalytic process of 1,1′-distyrylferrocene derivative with vinylsilane was conducted following the original procedure. The SC reaction of 9 or 10 with trimethylvinylsilane (6 equivalents) was accomplished following the well-defined procedure: ruthenium catalyst [RuH(CO)Cl(PCy3)2] (4) (2 mol% per olefin molecule), toluene (0.75 M), 80 °C for 18 h (up to 100% conversion of divinylarylene derivative), under argon atmosphere, see Scheme 4.
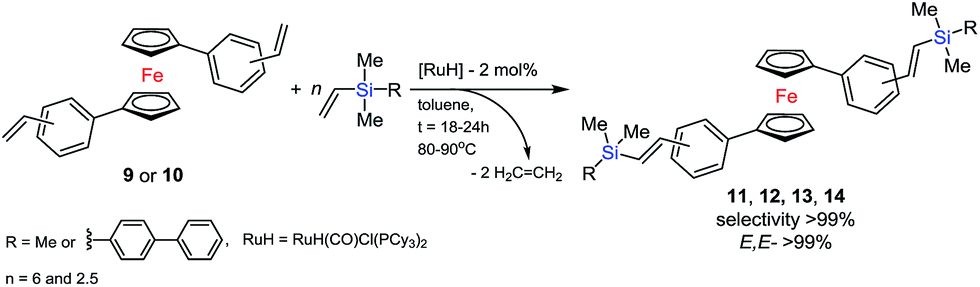 | ||
| Scheme 4 Silylative coupling of 9 and 10 with vinylsilanes. | ||
The ‘Schlenk’ closed system was used to give selectively only one isomer of the new E,E-bis((trimethylsilyl)vinylene)ferrocenes (11, 12) in very good isolated yields 98% and 99%. On the basis of MS spectral analysis we identified further by-product, i.e. the E-bis(trimethylsilyl)ethene. A typical compound is formed by reacting an excess of vinylsilane (yield 15–19%), but this one is easily separated from the main product. We observed lower reactivity of the vinyl group at 4-dimethylvinylsilylbiphenyl (5). Therefore we applied an open system at 90 °C with prolonged time to 24 hours. Such reaction conditions allowed to synthesize and isolate new ferrocene organicsilicon compounds in a good yield 81% (13), 87% (14).
The optimized conditions for the above reaction allowed us to use the most effective catalytic system for selectively controlled synthesis of new polymeric materials. A typical reaction system consisted of 0.2–0.125 M olefin solution in toluene, 4,4′-bis(dimethylvinylsilyl)biphenyl (6) and the ruthenium complex (4). The system was placed in an oil bath at 80–90 °C for 24–36 h. The molar ratio of the reaction mixture was [cat.]:[olefin]:[vinylsilane] = 1 × 10−2:1:1, according to Scheme 5.
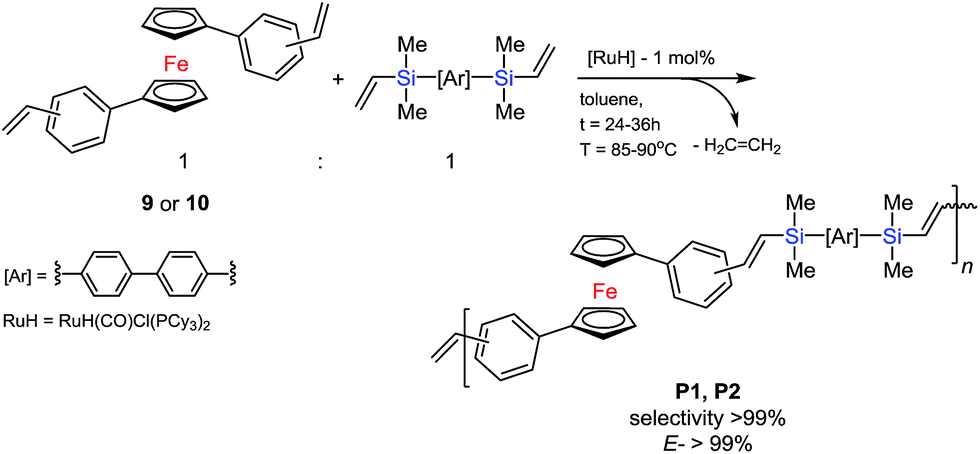 | ||
| Scheme 5 Polycondensation reaction coupling of 9 or 10 with 6 in the present 4. | ||
The low concentration of the reagents prevented the olefin from thermal-radical polymerizing. Detailed analysis of the proton spectrum confirmed the formation of an oligomer made of trans-vinylene fragments only in mers‘-A-trans-B-trans-A-trans-B-’ (coupling constants for the protons of the –Me2Si–HCCH–Ci![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) unit: JHH = 17.4 Hz for HB and 18.9 Hz for HC) in the polycondensation reaction, see Fig. 4. Polymers P1 and P2 were analysed using 1H NMR spectroscopy which, in all cases, confirmed the complete consumption of vinyl groups, with no observable resonances in the alkene region (δ = 5.0–6.4 ppm) except the terminal vinyl group of final materials.
unit: JHH = 17.4 Hz for HB and 18.9 Hz for HC) in the polycondensation reaction, see Fig. 4. Polymers P1 and P2 were analysed using 1H NMR spectroscopy which, in all cases, confirmed the complete consumption of vinyl groups, with no observable resonances in the alkene region (δ = 5.0–6.4 ppm) except the terminal vinyl group of final materials.
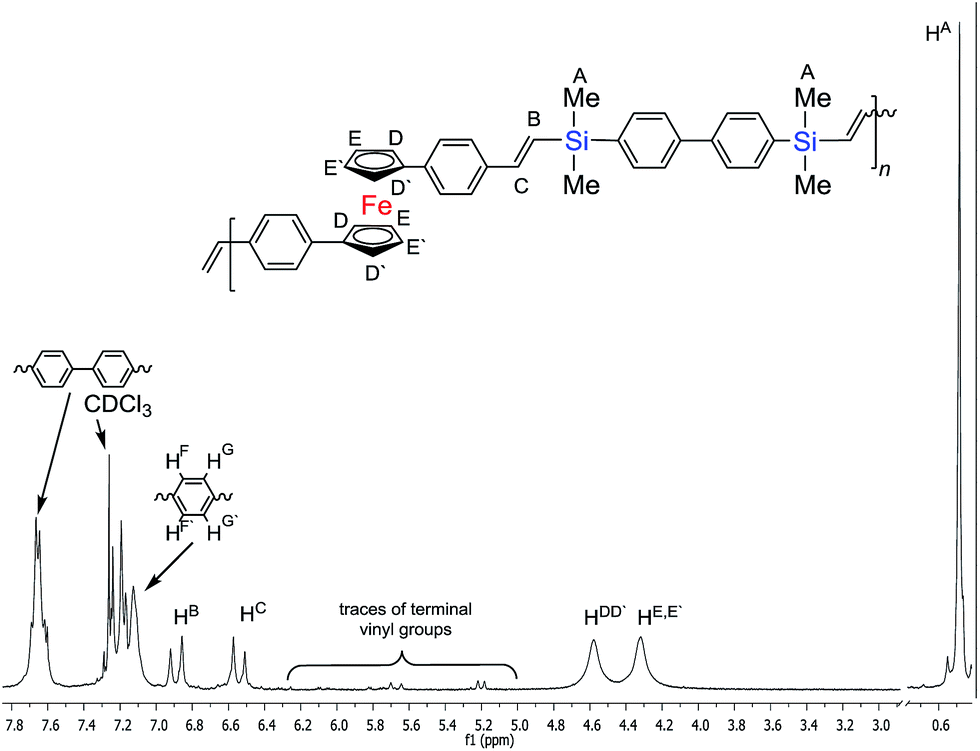 | ||
| Fig. 4 1H NMR spectrum of (P2) in CDCl3 at 25 °C. | ||
Gel permeation chromatography (GPC) in THF showed quite good molecular weight values of the polymer consisting of ferrocenylene–arylene–vinylene–silylene fragments (for P1:Mn = 3600 g mol−1, Mw = 5652 g mol−1; P2:Mn = 3370 g mol−1, Mw = 5024 g mol−1). In addition, the polymers had narrow size distributions for P1 PDI = 1.57 and for P2 PDI = 1.49 (see Fig. P1 and P2 at ESI†).
The polymers were purified by successive precipitations into hexane and vacuum-dried. Both polymeric materials showed good solubility in common organic solvents such as THF, CHCl3 and CH2Cl2. All styrylferrocenes and aryl-ferrocene silane derivatives were isolated and spectroscopically characterized. We synthesized several novel functionalized ferrocenes. All organic and silyl products were obtained in an excellent yield and selectivity. Both reaction systems are characterized by trans-stereoselectivity and selection of appropriate conditions for functionalization depending on the type compounds used.
Conclusions
To sum up, this is the first report on a fully controlled and selective method for the synthesis of new metallocene π-conjugated molecular 7–14 and macromolecular distyrylferrocenyl organosilicon compounds P1, P2. The reactions proceed with the high yields and are regio-stereo-selective giving E- and E,E-products. This methodology can be extended to the synthesis of new class E-regular controlled silyl–organic–metallocene and hybrid organic–metallocene–inorganic polymeric materials. All of the compounds were produced in good and high yields. Further results on the use of these compounds for the synthesis of high-molecular weight π-conjugated organic and hybrid compounds will be reported in due time. Now we focus on studying optoelectronic, electrochemical and thermal properties of new materials.Acknowledgements
We gratefully acknowledge financial support from the National Science Centre (Poland) (Project UMO-2011/02/A/ST5/00472).Notes and references
- I. Manners, Science, 2001, 294, 1664–1666 CrossRef CAS PubMed.
- R. D. Archer, Inorganic and Organometallic Polymers, Wiley-VCH, Weinheim, 2001 Search PubMed.
- T. Ogoshi, H. Itoh, K.-M. Kim and Y. Chujo, Macromolecules, 2002, 35, 334–338 CrossRef CAS.
- A. S. Abd-El-Aziz, Macromol. Rapid Commun., 2002, 23, 995–1031 CrossRef CAS.
- Y. Morisaki, H. Chen and Y. Chujo, J. Organomet. Chem., 2004, 689, 2684–2689 CrossRef CAS.
- V. V. Korshak, S. L. Sosin and V. P. Alekseeva, Dokl. Akad. Nauk SSSR, 1960, 132, 360–365 CAS.
- A. N. Nesmeyanov, V. V. Korshak, N. S. Voevodskii, S. L. Kochetkova, S. L. Sosin and R. B. Materikova, et al., Dokl. Akad. Nauk SSSR, 1961, 137, 1370–1375 CAS.
- R. B. Grubbs, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4323–4336 CrossRef CAS.
- Y. Qin, C. Cui and F. Jäkle, Macromolecules, 2008, 41, 2972–2974 CrossRef CAS.
- L. Ren, C. G. Hardy and C. Tang, J. Am. Chem. Soc., 2010, 132, 8874–8875 CrossRef CAS PubMed.
- J. B. Heilmann, M. Schiebitz, Y. Qin, A. Sundararaman, F. Jäkle and T. Kretz, et al., Angew. Chem., Int. Ed., 2006, 45, 920–925 CrossRef CAS PubMed.
- W. K. Chan, Coord. Chem. Rev., 2007, 251, 2104–2118 CrossRef CAS.
- W.-Y. Wong and P. D. Harvey, Macromol. Rapid Commun., 2010, 31, 671–713 CrossRef CAS PubMed.
- H. Fukumoto, K. Yamane, Y. Kase and T. Yamamoto, Macromolecules, 2010, 43, 10366–10375 CrossRef CAS.
- B. J. Holliday and T. M. Swager, Chem. Commun., 2005, 23–36 RSC.
- D. E. Herbert, U. F. J. Mayer and I. Manners, Angew. Chem., Int. Ed., 2007, 46, 5060–5081 CrossRef CAS PubMed.
- V. Bellas and M. Rehahn, Angew. Chem., Int. Ed., 2007, 46, 5082–5104 CrossRef CAS PubMed.
- R. Knapp and M. Rehahn, J. Organomet. Chem., 1993, 452, 235–240 CrossRef CAS.
- C. Imrie, P. Engelbrecht, C. Loubser, C. W. McCleland, V. O. Nyamori, R. Bogardi, D. C. Levendis, N. Tolom, J. Van Rooyen and N. J. Williams, J. Organomet. Chem., 2002, 645, 65–81 CrossRef CAS.
- H. Zhang, F. Liu, J. Cao, L. Ling and R.-F. Sun, Chin. J. Polym. Sci., 2016, 34(2), 242–252 CrossRef CAS.
- B. M. Trost and T. R. Verhoeven, in Comprehensive Organometallic Chemistry, ed. G. Wilkinson, F. G. Stone and E. W. Abel, Pergamon Press, Oxford, 1982, vol. 8, pp. 799–938 Search PubMed.
- R. F. Heck, Palladium Reagents in Organic Synthesis, Academic Press, New York, 1985 Search PubMed.
- J. Hassan and M. Sevignon, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS PubMed.
- S. Kotha, K. Lahin and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS.
- B. Marciniec and C. Pietraszuk, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley, Weinheim, 2003, ch. 2.13 Search PubMed.
- B. Marciniec and C. Pietraszuk, Curr. Org. Chem., 2003, 7, 691–735 CrossRef CAS.
- B. Marciniec, Coord. Chem. Rev., 2005, 249, 2374–2390 CrossRef CAS.
- B. Marciniec, Acc. Chem. Res., 2007, 40, 943–952 CrossRef CAS PubMed.
- M. Majchrzak, M. Hybsz, S. Kostera, M. Kubicki and B. Marciniec, Tetrahedron Lett., 2014, 55, 3055–3058 CrossRef CAS.
- M. Majchrzak, B. Marciniec, M. Kubicki and A. Pawelczyk, Organometallics, 2005, 24, 3731–3736 CrossRef CAS.
- M. Majchrzak, S. Kostera, M. Kubicki and I. Kownacki, Dalton Trans., 2013, 42, 15535–15539 RSC.
- B. A. Harding, P. R. Melvin Jr, W. Dougherty, S. Kassel and F. E. Goodson, Organometallics, 2013, 32, 3570–3573 CrossRef CAS.
- C. S. Yi, D. W. Lee and Y. Chen, Organometallics, 1999, 18, 2043–2045 CrossRef CAS.
- M. Majchrzak, B. Marciniec and Y. Itami, Adv. Synth. Catal., 2005, 347, 1285–1294 CrossRef CAS.
- J. Murage, J. W. Eddy, J. R. Zimbalist, T. B. McIntyre, Z. R. Wagner and F. E. Goodson, Macromolecules, 2008, 41, 7330–7338 CrossRef CAS.
- F. Paul, J. Patt and J. F. Hartwig, Organometallics, 1995, 14, 3030–3039 CrossRef CAS.
- B. Zimmerman, W. I. Dzik, T. Himmler and L. K. Goossen, J. Org. Chem., 2011, 76, 8107–8112 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedure of 3; GPC graphs; MS mass and NMR spectra, X-ray structure determination. CCDC 974168. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra00859c |
| This journal is © The Royal Society of Chemistry 2016 |