
Ethylene brassylate-co-δ-hexalactone biobased polymers for application in the medical field: synthesis, characterization and cell culture studies†
Jorge Fernández*a,
Aitor Larrañagaa,
Agustin Etxeberriab and
Jose-Ramon Sarasuaa
aDepartment of Mining-Metallurgy Engineering and Materials Science, POLYMAT, University of the Basque Country (UPV/EHU), School of Engineering, Alameda de Urquijo s/n, 48013 Bilbao, Spain. E-mail: jorge.fernandez@ehu.es; Tel: +34 946 017318
bDepartment of Polymer Science and Technology, POLYMAT, University of the Basque Country (UPV/EHU), M. de Lardizabal 3, 20018 Donostia-San Sebastian, Spain
First published on 17th February 2016
Abstract
Copolymers based on ethylene brassylate (EB) and δ-hexalactone (δ-HL), two monomers obtained from renewable sources, were synthesized with triphenyl bismuth by ring-opening polymerization. The poly(EB-co-δ-HL), with EB molar contents ranging from 49 to 90%, presented a slight deviation from the random distribution of sequences (R > 0.71) while WAXS measurements proved that only the EB blocks were able to crystallize (with crystallinity degrees of ∼23–39%). The low Tgs (<−27 °C), their reduced crystallization capability and melting temperatures from 50 to 65 °C make these thermoplastic elastomers (of good thermal stability) very easy to process. Likewise, their thermal properties led to mechanical behaviour with improved flexibility in comparison to poly(ε-caprolactone) or poly(ω-pentadecalactone) (elastic modulus between 57–274 MPa with high elongation at break values) at both 21 °C and 37 °C. On the other hand, the incorporation of δ-HL in addition to increase the amorphous character, brought a more disordered chain microstructure distribution, resulting in higher degradation rates in the range of 0.0028–0.0041 per day. Finally, metabolic activity and cell morphology studies using human dermal fibroblasts (HDFs) demonstrated that these materials are not cytotoxic and provide a valid substrate for cells to attach and proliferate, meaning that they are suitable for use in the biomedical field.
1. Introduction
For the formation of reliable medical devices absolute confidence in the material is essential, and its properties must include stability, durability and predictability and at the same time, meet the requirements of a specific application. Since the 1980s, most research work to obtain suitable materials for biomedical uses has been focused on aliphatic polyesters. However, there are still a large number of cyclic esters that have received hardly any attention. This paper presents the properties of statistical copolymers based on ethylene brassylate macrolactone and δ-hexalactone, which were synthesized for the first time by our group.Ethylene brassylate is a 17 member ring lactone with two ester groups, it is commercially available in large quantities and is of particular interest owing to its lower cost in comparison to other lactones and macrolactones, such as lactide, ε-caprolactone or ω-pentadecalactone. This compound can be obtained from tridecanoic acid which is itself synthesized from 10-undecanoic acid,1 an unsaturated fatty acid derived from castor oil, a renewable source. Ethylene brassylate is a colorless to very pale yellow viscous liquid with a sweet musk-like odor,2 for this reason it is widely used in many fragrances (cosmetics, fine perfumes, shampoos, toilet soaps and other toiletries), and non-cosmetic products such as household cleaners and detergents.3 The volume of ethylene brassylate used worldwide in 2008 was over than 1000 metric tonnes (International Fragrance Association).
The ring-opening polymerization of ethylene brassylate was first reported by Kobayashi in 1999 (ref. 4) but low molecular weights were obtained using lipase enzymes. More recently, Mecerreyes et al.5 employed acidic and basic organic compounds as catalysts. TBD-guanidine super base (1,5,7-triazabicyclo[4-4-0]dec-5-ene) was the most efficient catalyst in the process of synthesis, producing the fastest polymerization rates. The poly(ethylene brassylate) homopolymers, with molecular weights ranging between 2 and 14 kg mol−1, presented similar properties to poly(ε-caprolactone)6–8 but with a slightly higher melting temperature (Tm = 69 °C), a glass transition temperature at −33 °C and good thermal stability.5 In more recent works, ethylene brassylate has been also copolymerized with other monomers such as ethylene glycol9 or ε-caprolactone.10 The poly(ethylene brassylate-co-ε-caprolactone)s synthesized by the Mecerreyes' group were highly crystalline and showed a range of melting temperatures between 39 and 69 °C, depending on the comonomer content. However, the large melting enthalpy values (∼85 J g−1) and the close similarity of unit-cell lateral dimensions of ethylene brassylate and ε-caprolactone suggest a cocrystallization of both units, similar to other isomorphic copolymers containing ε-caprolactone.11–16
With the aim of preventing the isomorphic cocrystallization phenomenon, ethylene brassylate was copolymerized in this study with δ-hexalactone (δ-caprolactone),17–19 a six-membered lactone with identical structure to δ-valerolactone but possessing a methyl pendant group. This little known lactone, found in heated milk fat,20 was successfully copolymerized in a previous work by our group with ω-pentadecalactone.21 The incorporation of δ-HL allowed the reduction of the crystalline fraction of the poly(ω-pentadecalactone) homopolymer owing to the racemic stereochemistry of the methyl side chain of the δ-HL unit. As a result, copolymers with a lower level of stiffness and upgraded biodegradability were achieved.
The poly(ethylene brassylate-co-δ-hexalactone)s in this paper were synthesized using triphenyl bismuth (Ph3Bi)22 as a catalyst and characterized using proton and carbon nuclear magnetic resonance spectroscopy (1H and 13C NMR), gel permeation chromatography (GPC) measurements and thermogravimetric analysis (TGA). Their crystallization and melting behavior was studied by means of differential scanning calorimetry (DSC) and Wide Angle X-ray diffraction (WAXRD). In addition, films of EB-co-δ-HL copolymers with EB molar contents ranging from 49 to 90% were prepared for mechanical testing at room temperature (21 °C) and at human body temperature (37 °C), the typical working temperatures of biomaterials. Finally, an in vitro hydrolytic degradation study was also carried out at 37 °C for a period up to 26 weeks in phosphate buffer solution (PBS). Thus, any changes in water absorption, weight loss, macroscopic morphology, crystallinity, phase structure, molecular weight and mechanical properties of the copolymers were monitored.
Of the metal catalysts available, bismuth salts or complexes, such as Ph3Bi, are notable for their low toxicity.23 In this context, several studies24–27 have demonstrated that bismuth compounds belong to the group of least toxic heavy metal compounds, while Bi3+ performed even better than Zn2+, although small amounts of zinc are needed by the human metabolism. However, cell viability studies with some of the materials of this work were deemed necessary in order to rule out the possible cytotoxicity of these Bi-containing polymers and confirm their safe use in medical and tissue engineering applications. Therefore, human dermal fibroblasts (HDFs) were cultured on samples obtained from films of the poly(EB-co-δ-HL) and the metabolic activity and morphology were investigated by means of AlamarBlue® assay and using an inverted fluorescent microscope.
2. Materials and methods
2.1. Materials
δ-Hexalactone monomer (assay > 98%) was provided by Sigma Aldrich (W316709). Ethylene brassylate monomer (assay > 95%) was also supplied by Sigma Aldrich (W354309). The triphenyl bismuth (Ph3Bi) catalyst was obtained from Gelest. Phosphate buffer solution (PBS) (pH 7.2) was obtained from Fluka Analytical (Sigma Aldrich).Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), Hank's balanced salt solution (HBSS) and penicillin-streptomycin (PS) solution were purchased from Sigma-Aldrich (Ireland). AlamarBlue® and rhodamine phalloidin were obtained from ThermoFisher Scientific (Ireland); Hoechst staining solution was from Sigma-Aldrich (Ireland). Human dermal fibroblasts (HDFs) were obtained from Sigma-Aldrich (Ireland).
2.2. Synthesis procedure
Statistical copolymers from ethylene brassylate and δ-hexalactone (see Fig. 1 for structure) at differing compositions were synthesized in bulk by one pot-one-step ring-opening polymerizations (ROP). For the correct evaluation of the EB-co-δ-HL copolymer behavior, a poly(ethylene brassylate) (PEB) homopolymer was also synthesized. The reactions were conducted in a flask immersed in a controlled temperature oil bath. In each polymerization, predetermined amounts of the different comonomers at the chosen mass feed ratio were simultaneously added and melted into the flask. The flask was purged for 30 minutes with a nitrogen stream under the surface of the melt. Then the catalyst (Ph3Bi) was added (at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 comonomers/catalyst molar ratio) and the magnetic stirrer maintained at 100 rpm. No initiator compound was added to the reaction mix so the catalyst was activated by the ROH provided by the monomers (H2O and impurities), with the exception of the PEB homopolymer and the copolymers with higher contents of EB. In those cases 1-hexanol was added in order to control the molecular weight. The ethylene brassylate-co-δ-hexalactone polymerizations were carried out at 130 °C over 3 or 6 days.
:1 comonomers/catalyst molar ratio) and the magnetic stirrer maintained at 100 rpm. No initiator compound was added to the reaction mix so the catalyst was activated by the ROH provided by the monomers (H2O and impurities), with the exception of the PEB homopolymer and the copolymers with higher contents of EB. In those cases 1-hexanol was added in order to control the molecular weight. The ethylene brassylate-co-δ-hexalactone polymerizations were carried out at 130 °C over 3 or 6 days.
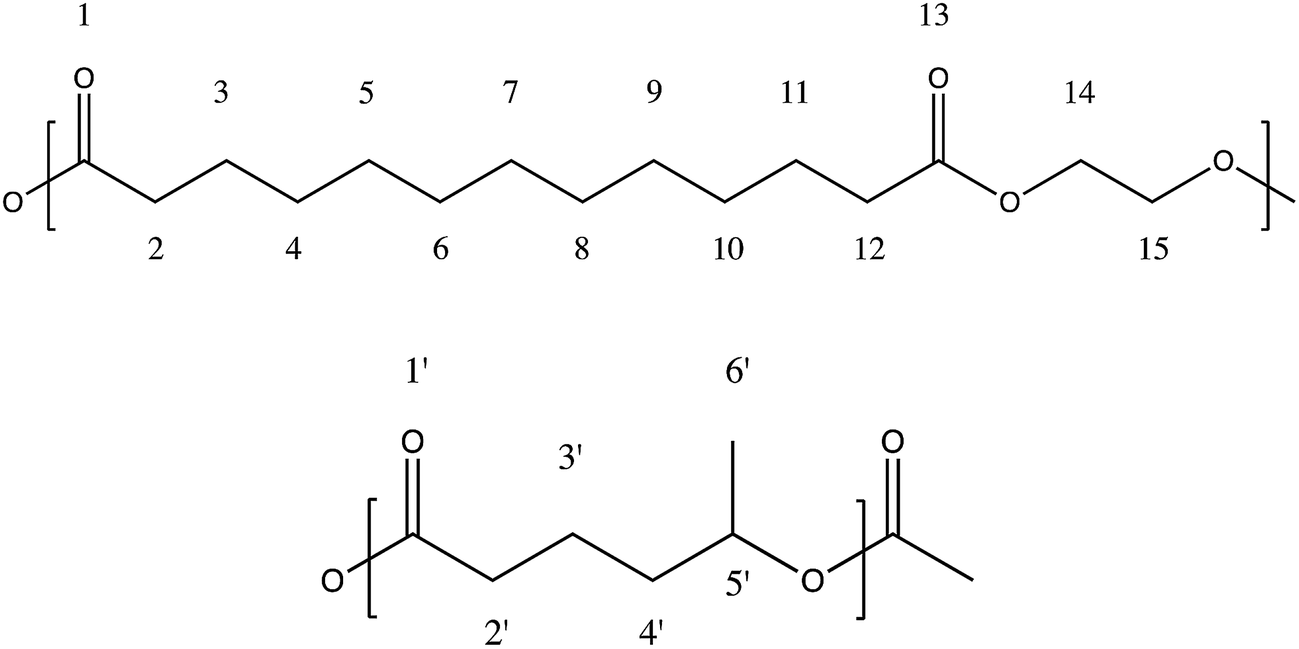 | ||
| Fig. 1 Scheme of the units of ethylene brassylate (above) and δ-hexalactone (below). | ||
After the corresponding period of reaction time the product was dissolved in chloroform and precipitated, pouring the polymer solution into an excess of methanol in order to remove the catalyst impurities and those monomers that had not reacted. Finally the product was dried at room temperature and then subjected to a heat treatment at 140 °C for 1 hour to ensure the complete elimination of any remaining solvent. Then, the polymer sample was weighed obtaining the yield of the synthesis process.
The final products were highly soluble in chloroform, dichloromethane, tetrahydrofuran, benzene, toluene, cyclohexanone and 2-nitropropane; slightly soluble in acetone, ethyl acetate, dimethylformamide, dimethyl sulfoxide and acetonitrile; and, like other polyesters, insoluble in alcohols, petroleum ether, diethyl ether and water.
2.3. Methods
Proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were recorded in a Bruker Avance DPX 300 at 300.16 MHz and at 75.5 MHz of resonance frequency respectively, using 5 mm O.D. sample tubes. All spectra were obtained at room temperature from solutions of 0.7 mL of deuterated chloroform (CDCl3). Experimental conditions were as follows: (a) for 1H NMR: 10 mg sample; 3 s acquisition time; 1 s delay time; 8.5 μs pulse; spectral width 5000 Hz and 32 scans; (b) for 13C NMR: 40 mg, inverse gated decoupled sequence; 3 s acquisition time; 4 s delay time; 5.5 μs pulse; spectral width 18800 Hz and more than 10000 scans. The assignment of the different signals was made employing the tables of structural determination from Prestch et al.28 The copolymer composition data, average sequence lengths and randomness character from Table 1 were calculated by averaging the values of molar contents and the EB-HL dyad relative molar fractions that were obtained by means of 1H and 13C NMR spectroscopy. Eqn (1)–(3)29 were employed to obtain the number-average sequence lengths (li), the Bernoullian random number-average sequence lengths (li) and the randomness character (R):
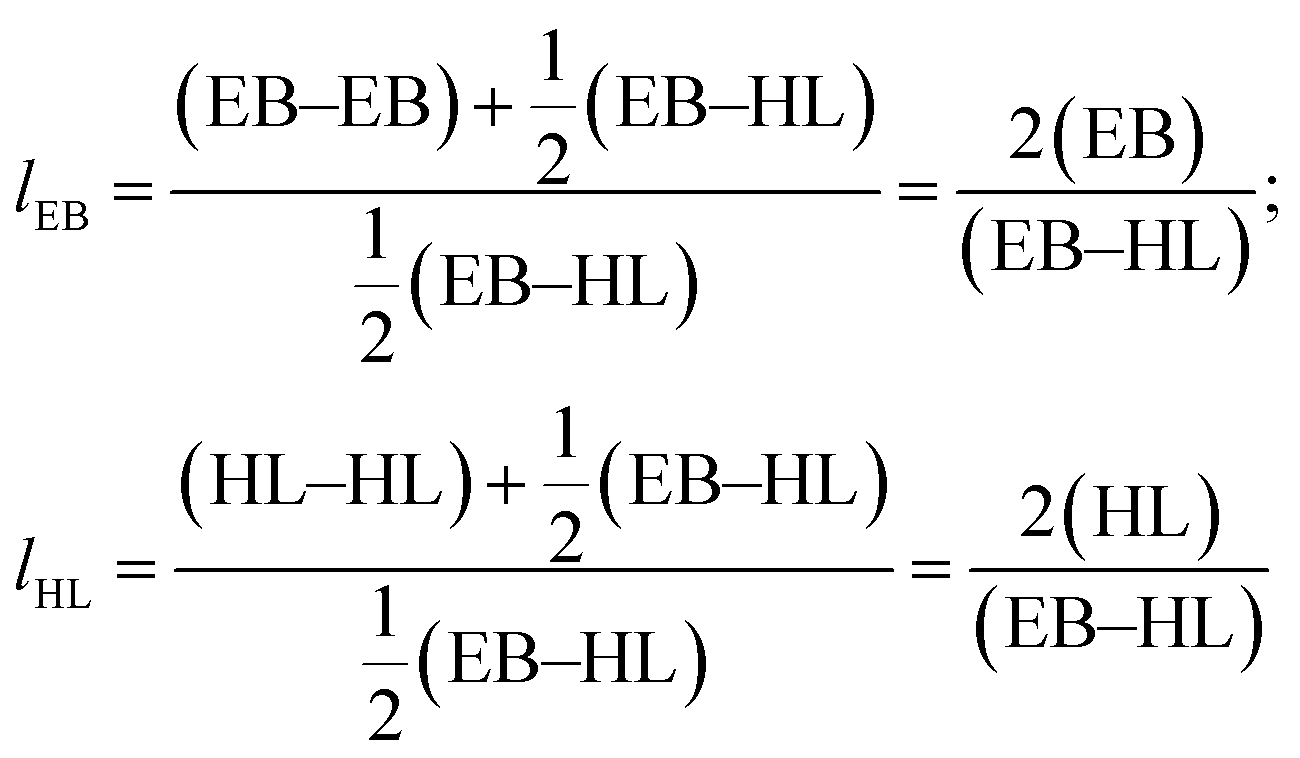 | (1) |
 | (2) |
 | (3) |
| Sample | Feed molar composition | Compositiona | Yield (%) | Conversion (%) | Mw, kg mol−1 | D | Microstructural magnitudesb | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % EB | % HL | % EB | % HL | EB | HL | lEB | lHL | R | ||||
| a Calculated averaging the copolymer molar compositions obtained by 1H and 13C NMR.b lEB and lHL are the EB and HL number average sequence lengths of the EB-co-δ-HL copolymers obtained from the average dyad relative molar fraction (EB–HL) in 13C NMR. These values are compared to the Bernoullian random number-average sequence lengths obtaining the randomness character value (R) of the different copolymers. | ||||||||||||
| PEB | 100 | 0 | 100 | 0 | 75.0 | 75.0 | — | 270.9 | 2.16 | — | — | — |
| EB–HL 90 | 79.2 | 20.8 | 89.8 | 10.2 | 86.9 | 92.1 | 39.8 | 182.4 | 1.86 | 11.69 | 1.32 | 0.84 |
| EB–HL 83 | 70.5 | 29.5 | 82.6 | 17.4 | 85.2 | 92.0 | 46.4 | 289.8 | 2.29 | 7.50 | 1.58 | 0.77 |
| EB-HL 83 after 3 days | 70.5 | 29.5 | 87.5 | 12.5 | 79.3 | 88.0 | 30.1 | 285.3 | 2.07 | 10.61 | 1.52 | 0.75 |
| EB–HL 79 | 62.8 | 27.2 | 78.8 | 21.2 | 78.9 | 88.6 | 40.2 | 228.2 | 1.96 | 6.58 | 1.77 | 0.72 |
| EB–HL 70 | 55.9 | 44.1 | 70.3 | 29.7 | 80.0 | 90.5 | 48.4 | 228.4 | 2.21 | 4.70 | 1.99 | 0.72 |
| EB-HL 70 after 3 days | 55.9 | 44.1 | 77.2 | 28.2 | 74.6 | 86.2 | 39.9 | 152.6 | 1.86 | 6.99 | 2.07 | 0.63 |
| EB–HL 63 | 47.3 | 52.7 | 63.3 | 36.7 | 76.6 | 90.5 | 47.1 | 156.4 | 1.89 | 3.82 | 2.21 | 0.71 |
| EB–HL 49 | 34.0 | 66.0 | 49.0 | 51.0 | 71.3 | 90.1 | 48.4 | 148.1 | 2.00 | 2.57 | 2.68 | 0.76 |
200–300 μm films were prepared by pressure melting at 175 °C followed by water quenching. The films were then stored for 24 hours in a fridge (at 0–5 °C), the typical storage temperature for biopolymers. From these films repetitive square samples for the in vitro degradation study (1 × 1 cm2) and repetitive samples for mechanical characterization (10 × 1 cm2) were obtained. The specimens for mechanical testing at 37 °C were stored for another 24 hours at 37 °C before tests were conducted at the same temperature. In addition, DSC scans were made at 20 °C min−1 for each polymer sample before the mechanical testing, in order to monitor the thermal properties of the specimens.
Static contact angle measurements (see the example of Fig. S1 from ESI†) were recorded on the films using a VCA 2500 XE system (AST, Billerica, MA, USA). At least five nanopure water drops (5 μL) were deposited on different parts of each sample.
The mechanical properties were determined by tensile tests with an Instron 5565 testing machine at a crosshead displacement rate of 10 mm min−1. These tests were performed at room temperature (21 ± 2 °C) and at human body temperature (37 °C) following ISO 527-3/1995. The specimens had the following dimensions: overall length = 100 mm, distance between marks = 50 mm, width = 10 mm; and were cut from 200–300 μm thick films. The mechanical properties reported (secant modulus at 2%, yield strength, ultimate tensile strength and elongation at break) correspond to average values of at least 5 determinations. Mechanical testing at 37 °C was conducted in an Instron controlled temperature chamber. The tests were stopped at 300% of strain due to the size limitations of the temperature chamber.
For the in vitro degradation study, square samples (25–35 mg (W0) (n = 3)) of the different copolymers were placed in Falcon tubes containing phosphate buffer solution (PBS) (pH = 7.2) maintaining a surface area to volume ratio equal to 0.1 cm−1. The samples were stored in an oven at 37 °C. Three samples of each polymer were removed at different times from the PBS and weighed wet (Ww) immediately after wiping the surface with filter paper to absorb any surface water. These samples were air-dried overnight at 37 °C and then weighed again to obtain the dry weight (Wd). Water absorption (% WA) and remaining weight (% RW) were calculated according to eqn (4) and (5). On day 70 and at the end of the degradation study (182 days) the mechanical properties of the poly(ethylene brassylate-co-δ-hexalactone) were also measured at 37 °C.
 | (4) |
 | (5) |
In order to compare the degradation rate of the studied copolymers, the exponential relationship between molecular weight and degradation time for biodegradable polyesters degrading under bulk degradation was used:30–33
|
lnMw = lnMw0 − KMwt
| (6) |
|
t1/2 = 1/KMw × ln2
| (7) |
The molecular weights of the polymers were determined by GPC using a Waters 1515 GPC device equipped with two Styragel columns (102 to 104 Å). Chloroform was used as eluent at a flow rate of 1 mL min−1 and polystyrene standards (Shodex Standards, SM-105) were used to obtain a primary calibration curve. The samples were prepared at a concentration of 10 mg in 2 mL. The reported values are likely to be higher than the actual molecular weights owing to the differences in the hydrodynamic volume of the copolymers and the polystyrene.
The thermal properties of the polymers were studied on a DSC 2920 (TA Instruments). Samples of 6–9 mg were melted at 130 °C and then quenched to 5 °C at different cooling rates (5, 10, 20 and 30 °C min−1) to study the crystallization process during the cooling. Finally, a scan was also made at 20 °C min−1 from −85 to 130 °C to determine the glass transition temperature (Tg), the melting temperature (Tm) and the heat of fusion or melting enthalpy per gram (ΔHm) of the samples. During the in vitro degradation study, the DSC samples were heated from 21 °C to 130 °C at 20 °C min−1, immediately after the drying at 37 °C of the polymer samples removed from the degradation medium. After this first scan, the samples were quenched in the DSC and a second scan was made from −85 °C to 130 °C at 20 °C min−1.
Wide angle X-ray diffraction (WAXRD) data were collected on a Bruker D8 Advance diffractometer operating at 30 kV and 20 mA. This device is equipped with a Cu tube (λ = 1.5418 Å), a Vantec-1 PSD detector and an Anton Parr HTK2000 high-temperature furnace. The powder patterns were recorded in 2θ steps of 0.033° in the 10 ≤ 2θ ≤ 38 range, counting for 0.2 s per step, from 30 to 120 °C every 2 °C using a heating rate of 0.16 °C s−1. The degree of crystallinity (χc) was determined as the ratio of the crystalline peak areas to the total area under the scattering curve, while the average crystal size was obtained employing the Scherrer equation34 with a shape factor “k” of 0.90. The latter data were calculated from the two most intense peaks in the diffraction pattern: at 2θ = 21.7° and at 2θ = 24.1°. On the other hand, the estimated melting temperatures (Tms) are the temperatures until the crystalline phase became stable.
Thermal degradation was studied under nitrogen by means of thermogravimetric analysis (TGA) into a TGA model Q50-0545 (TA Instruments). Samples of 10–15 mg were heated from room temperature to 500 °C at a heating rate (β) of 5 °C min−1, with the heat flow, sample temperature, residual sample weight and its time derivative being continuously recorded. In this temperature range, the polymers degraded completely.
2.4. Cell culture studies
000 cells per cm2 on a 48 well tissue culture plate and incubated in 0.5 mL of DMEM with 10% FBS and 1% PS (37 °C, 5% CO2). As a control, the cells were also seeded on a tissue culture plastic. The culture medium was replaced on the third day after seeding.With regard to the staining, HDFs were seeded on sterilized samples (circles 9 mm in diameter) at a density of 11000 cells per cm2 on a 24 well tissue culture plate and incubated in 1 mL of DMEM with 10% FBS and 1% PS (37 °C, 5% CO2). As a control, the cells were also seeded on 13 mm sterile glass coverslips onto cell culture well. The culture medium was replaced on the third day after seeding.
The morphology of HDFs seeded on the copolymers was analyzed via rhodamine phalloidin and Hoechst staining. After 1, 3 and 7 days, cells were washed with HBSS and subsequently fixed with 4% paraformaldehyde for 15 minutes at room temperature. Then, the cells were washed twice with HBSS and permeabilized with 0.5% Triton X-100 in PBS for 10 minutes. After washing twice with PBS, cells were incubated with 1% bovine serum albumin solution in PBS for 30 minutes. Afterwards, the cells were stained with rhodamine phalloidin solution for 15 minutes and thoroughly washed with PBS-T (0.1% Tween 20). Finally, the nuclei of HDFs were stained with Hoechst staining solution for 3 minutes and subsequently washed three times with PBS. Samples were observed with an inverted fluorescent microscope (Olympus IX81).
3. Results and discussion
3.1. Synthesis characterization
Table 1 summarizes the characterization data of several poly(EB-co-δ-HL) of differing compositions synthesized at 130 °C with a monomers/catalyst molar ratio (M/C) of 100. As can be seen, the yields of reaction of the PEB homopolymer (polymerized for 3 days) along with those of the two copolymers also synthesized for 3 days were under 80%. In order to ensure a higher yield and a final composition closer to the feed composition, the remaining reactions were conducted over 6 days.Regarding the molecular weights of the copolymers, these are within the 148 to 290 kg mol−1 range, while their dispersity values vary between 1.86 to 2.29. The weight average molecular weights (Mw) increase proportionally with the EB molar content, with the exception of PEB and the copolymer with 90% of EB. In these two cases, 0.05 to 0.1 g of 1-hexanol was added to limit the molecular weight because polymers with Mw (measured by GPC) above 400 kg mol−1 were obtained when 1-hexanol was not added.
The EB molar content in this set of copolymers ranges from 49% (EB–HL 49) to ∼90% (EB–HL 90) with average sequence lengths of EB (lEB) between 2.57 to 11.69 and lHL from 1.32 to 2.68. As can be seen in Table 1, higher reaction yields and molecular weights were obtained when the feed was richer in EB. Since the conversion of EB was higher than that of δ-HL in all the precipitated products, this suggests that the reactivity of EB is higher than that of δ-HL. Hence, under these conditions of synthesis, the consumption of EB was faster than δ-HL owing to the low reactivity of the latter. As a result of the different reactivity of the comonomers, the synthesis reactions lead to gradient copolymers with a slightly deviated from random distribution of sequences (R → 1). The randomness character (R) value was, in all cases, higher than 0.71 with the exception of one of the copolymers synthesized for 3 days.
3.2. NMR characterization
The calculus of the molar composition of the ethylene brassylate-co-δ-hexalactone copolymers in this study was made by averaging the results obtained from 1H and 13C NMR spectroscopy. On the contrary, the data of average sequence lengths and randomness character were estimated on the basis of the EB–HL dyad relative molar fractions that were acquired only from the 13C NMR spectra. Fig. 2 and 3 present the 1H and 13C NMR spectrum of the EB-co-δ-HL copolymer with 49% of ethylene brassylate (EB–HL 49).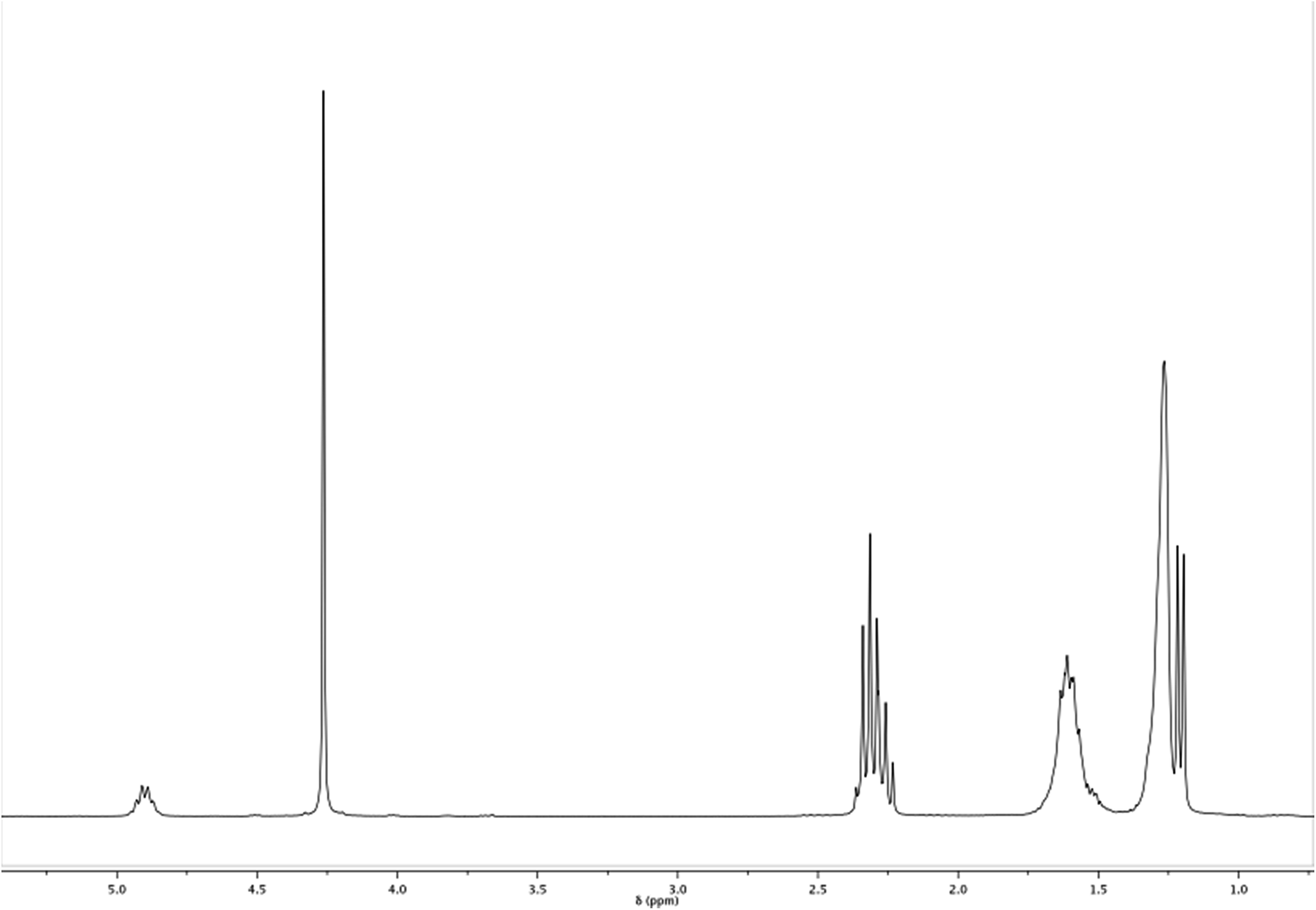 | ||
| Fig. 2 1H NMR spectrum of EB–HL 49. | ||
 | ||
| Fig. 3 13C NMR spectrum of EB–HL 49 with some regions of interest enlarged. | ||
In the 1H NMR spectrum, the analysis of the molar composition was conducted in two ways. By comparing the signals of the δ-HL methine at around 4.92 ppm (carbon 5′) with regard to the EB methylenes bonded to the ester group (carbons 14 and 15), which appear centered at 4.28 ppm, or using the signal of the δ-HL methylene (carbon 2′), which is overlapped with the hydrogens of the methylenes 2 and 12 of the ethylene brassylate at approximately 2.33 ppm. The rest of the peaks (see scheme in Fig. 1) are assigned to the other hydrogens of the copolymer in Table 2, although some signals appear overlapped.
| Number of C | H EB | H δ-HL |
|---|---|---|
| δ (ppm) | δ (ppm) | |
| 1 | — | — |
| 2 | 2.33 | 2.28, 2.36 |
| 3 | 1.63 | 1.60 |
| 4 | 1.30 | 1.60 |
| 5 | 4.92 | |
| 6 | 1.22 | |
| 7 | 1.63 | |
| 8 | 2.33 | |
| 9 | — | |
| 10 | 4.28 | |
| 11 | 4.28 |
The different signals appearing in the 13C NMR spectrum between 19 and 175 ppm of chemical shift (δ) are assigned in Table 3 to the different carbons numbered in Fig. 1. Hence, the molar composition of these copolymers can be easily determined by comparison of the areas under the peaks due to the δ-HL and EB carbons. In the case of the δ-hexalactone, the signals employed were those of the carbonyl carbon at 172.85 ppm (carbon 1′), the methine centered at 70.15 ppm (carbon 5′), the methylene 3′at 20.70 ppm and the methyl side chain (carbon 6′). Regarding the ethylene brassylate, an average relative value was calculated using the signals of the methylenes bonded to the ester group (carbons 14 and 15) which appear at 61.95 ppm, and those from the methylenes 2 and 12 at 34.04 ppm, 3 and 11 at 24.90 ppm and the seven methylenes from carbons 4 to 10, which peaks are overlapped at 29.04–29.16 ppm.
| Number of C | C EB | C δ-HL |
|---|---|---|
| δ (ppm) | δ (ppm) | |
| 1 | 173.37 ![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif) ![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif) –HL –HL |
172.80 HL–HL |
| 173.47 EB–EB | 172.92 ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) –EB –EB |
|
| 2 | 34.04 | 34.58, 34.59 |
| 3 | 24.80 EB–EB | 20.64 –EB |
| 24.97 –HL |
20.78 HL–HL | |
| 4 | 29.04–29.16 | 35.15 |
| 5 | 69.96 EB–HL–EB | |
| 70.05 HL–HL–EB | ||
| 70.21 EB–HL–HL | ||
| 70.29 HL–HL–HL | ||
| 6 | 19.6 | |
| 7 | Equal to C3 | |
| 8 | Equal to C2 | |
| 9 | Equal to C1 | |
| 10 | 61.91 EB–EB | |
| 62.07 –HL |
||
| 11 | Equal to C14 |
As can be observed in the enlargement of Fig. 3, some of the carbons show sequence sensitivity. The different dyads and triads found in the 13C NMR spectrum are also marked in Table 3 (underlining is used to emphasize that the analysed nuclei belong to that unit). For the estimation of the chain microstructure parameters the average dyad relative molar fraction (EB–HL) was determined. This variable is the sum of the (–HL) and (EB–) average dyad relative molar fractions. The first value was obtained from the –HL dyads at 173.37, 62.07 and 24.97 ppm while the (EB–) molar fraction was estimated based on the EB– dyads at 172.97 and 20.78 ppm and the value provided by the triads. From the peaks of the EB–HL–EB, HL–HL–EB, EB–HL–HL and HL–HL–HL triads, which appear from left to right at around 70.15 ppm, (EB–) average dyad relative molar fractions can also be obtained using the eqn (8) in which (HL) is the previously calculated δ-hexalactone average molar fraction.
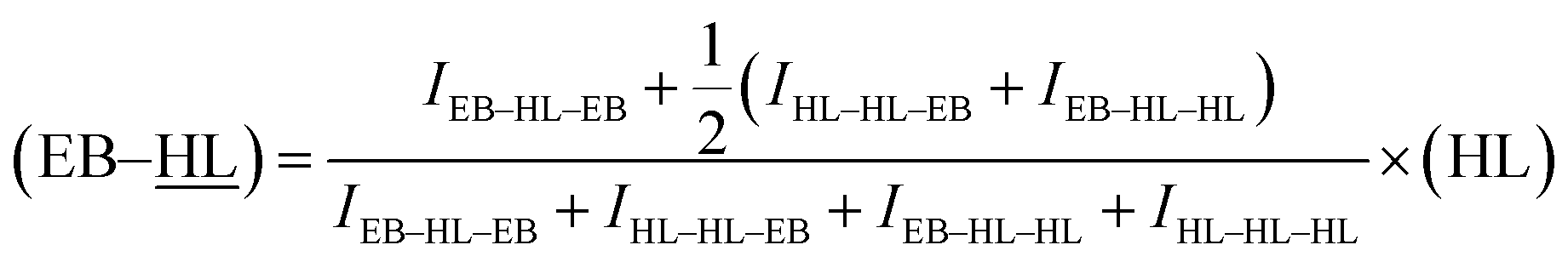 | (8) |
3.3. Physical properties
In this section the crystallization and melting behavior of the copolymers from Table 1 were studied. Firstly, several cooling treatments at different rates (5, 10, 20 and 30 °C min−1) were conducted in the DSC. The crystallization temperatures (Tc) and the enthalpy of crystallization (ΔHc) of each material at a cooling rate of 5 °C min−1 are shown in Table 4, while the rest of the data are summarized in the ESI (Table S1†). As can be seen in this Table, the differences between the respective ΔHc from the same polymer were small (the slight exception being EB–HL 49). The EB chains crystallized from the melt very fast and a cooling rate of 30 °C min−1 was sufficient for crystallization of the polymers while slower cooling rates only result in small increases in ΔHc. Likewise, the crystallization peaks of each polymer appeared at a higher temperature as the cooling rate decreased. This is due to the fact that at lower cooling rates the crystal nuclei have more time to develop.| Sample | lEB | Contact angle (°) | WAXS | Cooling at 5 °C min−1 | Heating at 20 °C min−1 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tma (°C) | χc (%) | c.s (nm) | ΔHc (J g−1) | Tc (°C) | ΔHm (J g−1) | Tmb (°C) | Tg (°C) | ΔCp (J g−1 °C−1) | |||
| a Obtained by WAXS. This is the crystalline phase-stable temperature.b Obtained from a DSC at 20 °C min−1 from −85 to 130 °C. The melting point of the peak that appears at the highest temperature was chosen because this is related to the most perfect crystals.c Not measured. The film was too sticky due to its low melting temperature. | |||||||||||
| PEB | — | 68 ± 6 | 67.0 | 47.0 | 19 | 91.1 | 52.1 | 96.4 | 70.6 | −27.0 | 0.34 |
| EB–HL 90 | 11.69 | 62 ± 3 | 64.0 | 34.6 | 30 | 89.3 | 47.9 | 92.4 | 62.7 | −31.1 | 0.31 |
| EB–HL 83 | 7.70 | 70 ± 3 | 58.0 | 38.9 | 20 | 76.0 | 41.2 | 74.4 | 65.0 | −28.3 | 0.31 |
| EB–HL 79 | 6.58 | 62 ± 5 | 56.0 | 28.1 | 32 | 74.3 | 36.4 | 64.7 | 61.4 | −31.9 | 0.36 |
| EB–HL 70 | 4.70 | 56 ± 5 | 49.0 | 29.5 | 24 | 63.4 | 28.3 | 59.6 | 57.5 | −33.4 | 0.34 |
| EB–HL 63 | 3.82 | 58 ± 3 | 47.0 | 22.6 | 41 | 58.8 | 25.5 | 55.5 | 49.6 | −36.7 | 0.35 |
| EB–HL 49 | 2.57 | c | 35.0 | 25.7 | 10 | 40.6 | 16.4 | 40.8 | 31.0 | −41.2 | 0.36 |
The DSC heating curves obtained after the cooling treatments were virtually identical for each polymer, all of them having practically the same melting temperatures and enthalpies (Tm and ΔHm) after the cooling process. Fig. 4 shows the DSC curves of scans made from −85 to 130 °C at 20 °C min−1 while the data obtained from them (the glass transition temperature (Tg) with its associated heat capacity (ΔCp) and the melting enthalpies and temperatures) are summarized in Table 4 along with the Tm, the crystal fraction (χc) and the average crystal size (c.s.) from Wide Angle X-ray Scattering (WAXS).
 | ||
| Fig. 4 DSC heating curves at 20 °C min−1 from −85 to 130 °C of the different EB-co-δ-HL copolymers. | ||
Fig. 5 shows the diffraction profiles of some poly(EB-co-δ-HL) together with the diffractogram of the reference homopolymer (PEB). As can be seen, at higher contents of δ-HL in the copolymers the reflection intensity decreases. However, the poly(EB-co-δ-HL) presented exactly the same signals as PEB at 2θ of 21.7, 24.1, 30.2 and 36.4° (the small peak at 27.3° may be due to the bismuth compounds because it also appeared at high temperatures when the polymers were fully amorphous). Hence, it was demonstrated that the EB-co-δ-HL copolymers and the PEB present the same crystal structure and confirms that only the EB units were able to crystallize.
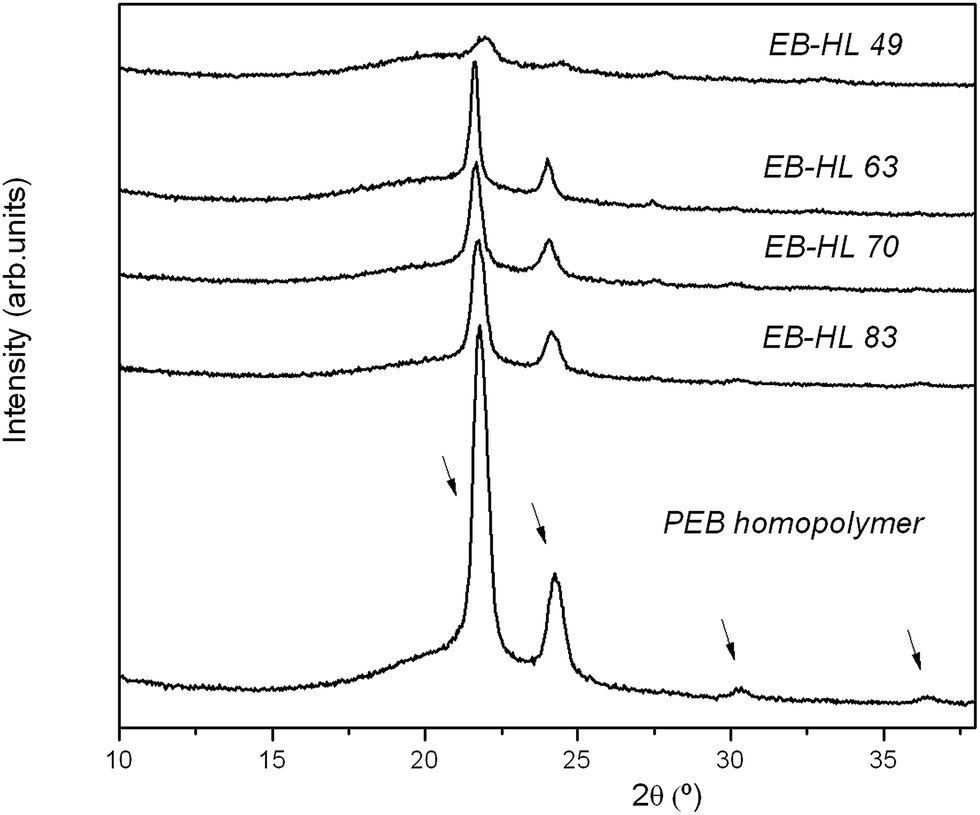 | ||
| Fig. 5 WAXS profiles of PEB and the EB-co-δ-HL copolymers in the 10 ≤ 2θ ≤ 38 range. | ||
As can be observed in Fig. 4, the PEB homopolymer and the EB rich copolymers showed narrower melting peaks and a higher crystallization capability in comparison to other copolymers such as EB–HL 49, the poly(EB-co-δ-HL) that presented the lowest melting peak. As the δ-hexalactone content increased, the melting peak split into two or more peaks while in the case of EB–HL 83 and EB–HL 79, a recrystallization peak also appeared between two of the melting peaks. During the heating, some imperfect crystals melt, then others form and finally the new crystallites and the more perfect crystal population melt. With regard to their glass transition behavior, the Tgs shifted to lower values when the δ-HL content increased and the ΔCp values stayed almost constant as the amorphous phase increased. The measured values were between −31 and −41 °C, temperatures lower than those of the PEB homopolymer (Tg at −27 °C).
The EB-co-δ-HL copolymers, with EB content between 49 to 90% and average sequence lengths of EB in the range of 2.57–11.69, presented ΔHms values between ∼41 to 92 J g−1 and Tms in the range of 31 to 62 °C (the melting point of the peak that appears at the highest temperature was chosen because this is associated with the most perfect crystals), values consistent with the Tms obtained by WAXS (from 35 to 64 °C). As expected at higher δ-HL contents, the degree of crystallinity of the copolymers fell, down to between 22.6 to 38.9%. On the other hand, PEB homopolymer showed a melting peak of 96.4 J g−1 at ∼71 °C with an associated crystalline fraction of 47.0%, a higher value than that of poly(ε-caprolactone) but lower than that of poly(ω-pentadecalactone) (with a χc of 53.8%, a ΔHm of 136.1 J g−1 and a Tm at 104 °C).35–37 The calculated average crystal sizes of the copolymers were in the 20 to 41 nm range, all values higher than the estimate for the homopolymer which had an average crystal size of 19 nm. The crystal size distribution is very wide and this is why the DSC melting peaks were found to be so broad. It might be possible that there are large differences between the thickest and the thinnest crystallites and for this reason discordant values were obtained (such as the c.s. of EB–HL 49).
With regard to the water contact angle measurements (see Table 4) it was observed that at higher δ-HL molar contents the surface hydrophilicity of the films decreased slightly. The EB-co-δ-HL copolymers had contact angles from 56 to 70° with EB–HL 70 and EB–HL 63 showing the lowest values.
3.4. Thermogravimetric analysis
Fig. 6 shows the curves of percent weight loss and first derivative of weight loss against temperature (TG and DTG curves) of the PEB homopolymer and the EB-co-δ-HL copolymers. As can be seen, the copolymers began thermal degradation at around 225 °C and were completely decomposed at 475 °C. Three well-defined peaks can be distinguished in the DTG curves. The first stage of depolymerization at around 250 °C was attributed to the decomposition of δ-HL-rich sequences, which is accelerated by the residual metal compounds from the catalyst.38 This peak did not appear in the curve of the PEB and were more pronounced in the case of EB–HL 49, the copolymer with the highest δ-HL content. The second peak, more intense than the first, appeared at 350 °C and covers the degradation of the bismuth free blocks of δ-HL and the decomposition of some metal accelerated sequences of EB. As the EB content rose, the intensity of this peak decreased, and at the same time it shifted to slightly higher temperatures. These first two peaks of thermal degradation, in which all the δ-HL decomposed, were also reported in the pyrolysis of poly(ω-pentadecalactone-co-δ-hexalactone) at the same temperatures21 but were not found in the curve of the PEB homopolymer. On the other hand, the thermal degradation of most of the ethylene brassylate was delayed to ∼425 °C, the most significant stage of depolymerization. In the case of the PEB homopolymer, a single peak appeared in the DTG curve at a temperature similar to that of the poly(ω-pentadecalactone), this only has one ester group in each structural unit instead of the two of the ethylene brassylate. | ||
| Fig. 6 Thermogravimetric (TG) and differential thermogravimetric (DTG) curves of PEB and the EB-co-δ-HL copolymers. | ||
3.5. Hydrolytic degradation study
A hydrolytic degradation study was conducted over 182 days with films of the ethylene brassylate-co-δ-hexalactone copolymers. DSC scans were made from 21 to 130 °C: at room temperature (before submerging the samples in PBS), after storage for 24 hours at 37 °C and at different degradation times (after drying). The data relating to melting enthalpies and temperatures at the start, on day 70 (when mechanical tests were performed) and at the end of the study are gathered in Table 5.| Sample | 21 °C | 37 °C | On day 70 of degradation at 37 °C | On day 182 of degradation at 37 °C | ||||
|---|---|---|---|---|---|---|---|---|
| ΔH (J g−1) | Tm (°C) | ΔH (J g−1) | Tm (°C) | ΔH (J g−1) | Tm (°C) | ΔH (J g−1) | Tm (°C) | |
| EB–HL 90 | 86.4 | 70.5 | 97.6 | 65.5 | 99.4 | 75.7 | 100.1 | 75.5 |
| EB–HL 83 | 72.8 | 66.4 | 83.7 | 66.5 | 86.0 | 67.4 | 93.9 | 71.4 |
| EB–HL 79 | 65.1 | 61.7 | 82.7 | 61.3 | 85.8 | 67.4 | 90.7 | 68.3 |
| EB–HL 70 | 50.5 | 56.3 | 64.5 | 57.9 | 74.2 | 61.8 | 74.6 | 63.0 |
| EB–HL 63 | 48.7 | 55.4 | 50.5 | 55.9 | 60.1 | 58.2 | 61.4 | 61.2 |
| EB–HL 49 | 9.9 | 36.3 | 10.6 | 49.3 | 20.0 | 52.0 | 18.9 | 50.3 |
It can be noted from the DSC results that there are some differences between the melting behaviors of the poly(EB-co-δ-HL) films at 21 °C and after storage at 37 °C for 24 hours. The copolymers presented higher melting peaks after being stored for 24 hours at 37 °C while their Tm values shifted to higher temperatures, especially in the case of EB–HL 49 (from 36.3 °C to 49.3 °C). These rearrangements in the crystalline phase occurring at 37 °C were the result of the appearance of new crystallites that melt at higher temperatures, and also the disappearance of other crystalline structures with melting points between 21 °C to 37 °C or below room temperature. Hence, it was also found that the ΔHm of EB–HL 49, which had a value of 40 J g−1 after conducting a scan from −85 to 130 °C (see Table 4), crystallized only ∼10 J g−1 in the heating scans from 21 °C. The crystallization temperature of this copolymer was the lowest and, as has be seen earlier, when it was cooled from 130 °C at a rate of 5 °C min−1 it showed a Tc of approximately 16 °C.
In addition, it can be observed that on days 70 and 182 of degradation (there are no large differences in the melting behaviour within this timeframe), the samples were slightly more crystalline with higher ΔHm values than at the start of the study. The more important changes occurred in the first week submerged in PBS and then the melting peak became almost stable. However, the Tm values shifted progressively to higher temperatures (2–7 °C higher), a tendency that is observed for all the materials owing to rearrangements of the crystalline phase. These changes in the thermal properties are more obvious in the case of the copolymers with lower EB contents. Therefore, the final ΔHm for EB–HL 49 reached a value of around 20 J g−1, twice the value recorded at the start of the study.
The weight of the samples remained almost constant during the entire study (see Fig. S2 from ESI† which shows the remaining weight (RW) and water absorption (WA) curves). The molecular weight has to be reduced substantially (Mw < 25 kg mol−1) to permit mass loss through solubilization of the oligomers and in this work no copolymer reached a low enough molecular weight. Moreover, water uptake was negligible and did not exceed a value of 3% in any case. Only EB–HL 49 might be less resistant to water absorption owing to its more amorphous structure.
Fig. 7 shows the progress of lnMw against degradation time of the poly(ethylene brassylate-co-δ-hexalactone). As the degradation progressed, the weight average molecular weight (Mw) of the samples fell while the associated dispersity rose slightly. Therefore, the Mw of EB–HL 90, EB–HL 83, EB–HL 79, EB–HL 70, EB–HL 63 and EB–HL 49 decreased to final values of 108, 134, 126, 112, 87 and 77 kg mol−1. In the latter case, this material displayed a macroscopic morphological deterioration after day 126 but as a result of the crystallization process became easier to handle from day 7 onward Table 6 presents the values recorded for the degradation rate (KMw) and half degradation times (t1/2) calculated from the slope of the fitting curve. The Mw experimental data adapts well to the fitting curve (R2 > 0.95) over the 182 days. However, it seems that the drop in lnMw is more pronounced in the first 70 days of the study, it then falls more slowly. Due to this fact, the regression line did not fit the data more perfectly. EB–HL 90, with the largest average sequence length of ethylene brassylate, exhibited the lowest degradation rate (0.0028 per day), 3 times higher than that of the PCL homopolymer (0.0010 per day),8 while the rest of poly(EB-δ-HL) presented KMw in the range of 0.0032–0.0041 per day with corresponding t1/2 of 169 to 217 days. The incorporation of δ-HL had little effect on the degradation rate and these minor variations in KMw may be due to the differences in initial molecular weight and chain microstructure.
 | ||
| Fig. 7 lnMw against degradation time of the EB-δ-HL copolymers. | ||
| Sample | KMw (per day) | Half-molecular weight degradation time t1/2 (days) |
|---|---|---|
| EB–HL 90 | 0.0028 | 248 |
| EB–HL 83 | 0.0041 | 169 |
| EB–HL 79 | 0.0033 | 210 |
| EB–HL 70 | 0.0038 | 182 |
| EB–HL 63 | 0.0032 | 217 |
| EB–HL 49 | 0.0035 | 198 |
3.6. Mechanical properties
Table 7 summarizes the mechanical testing results at 21° and at 37 °C (at the start and on day 182 of degradation) for the poly(ω-PDL-co-δ-HL) films, whose DSC data, before performing the mechanical tests, can be seen in Table 5. As can be seen in the table and in Fig. 8, which displays the typical stress–strain curves of the polymers studied at 21 °C, the EB-co-δ-HL copolymers showed a marked improvement in flexibility (with lower secant modulus and increased strain recovery rates) compared to PEB homopolymer, without losing any ductility (deformation at break values > 918%). Nevertheless, it proved impossible to measure the mechanical properties of EB–HL 49 because its film was too soft and sticky due to its low melting temperature (Tm at 30–35 °C). The PEB presented a secant modulus value of 308 MPa, ultimate strength of ∼26 MPa and an elongation at break of around 937%, results that are in accordance with those of other homopolyesters such as poly(ω-pentadecalactone) and poly(ε-caprolactone) (with secant modulus from 300 to 400 MPa, ultimate tensile strength of 28–32 MPa and deformation at break values > 750%).8,37 On the other hand, the ethylene brassylate-co-δ-hexalactone copolymers, with EB molar content from 63 to 90%, exhibited secant modulus in the range of 57.4–273.5 MPa at 21 °C, tensile strengths at break of 7.5–16.5 MPa and strain recoveries from 37.9 to 52.8%. All the polymers presented yield points which were broader and less defined as the δ-HL content increased.| Sample | Testing temperaturea | Secant modulus at 2% (MPa) | Yield strength (yield point) (MPa) | Tensile strength at 300%b (MPa) | Ultimate tensile strengthc (MPa) | Elongation at break (%) | Strain recovery after breakd (%) |
|---|---|---|---|---|---|---|---|
| a At 37 °C, the tests were stopped at 300% of strain due to the size limitations of the temperature chamber.b Some specimens broke before 300% of strain.c The tensile strength was determined as ultimate stress value (σu).d Measured 24 hours after break. | |||||||
| PEB | 21 °C | 307.6 ± 14.3 | 11.1 ± 0.5 (8.0%) | 10.5 ± 0.3 | 25.9 ± 7.0 | 937 | 34.1 ± 2.6 |
| 37 °C | 250.8 ± 8.5 | 7.3 ± 0.5 (4.4%) | 8.4 ± 0.8 | — | >300 | ||
| EB–HL 90 | 21 °C | 273.5 ± 11.4 | 8.9 ± 0.6 (6.4%) | 8.5 ± 0.5 | 16.5 ± 2.4 | 954 | 37.9 ± 1.6 |
| 37 °C | 262.3 ± 9.6 | 7.8 ± 0.5 (5.1%) | 7.7 ± 0.3 | — | >300 | ||
| On day 182 (37 °C) | 303.8 ± 10.4 | 8.8 ± 1.1 (4.1%) | — | 6.0 ± 1.0 | 98 | ||
| EB–HL 83 | 21 °C | 187.3 ± 13.8 | 5.6 ± 0.9 (5.3%) | 7.2 ± 0.1 | 16.3 ± 1.7 | 924 | 38.8 ± 3.0 |
| 37 °C | 194.8 ± 4.4 | 6.3 ± 0.2 (7.2%) | 6.4 ± 0.3 | — | >300 | ||
| On day 182 (37 °C) | 230.5 ± 1.8 | 6.5 ± 0.7 (4.4%) | 6.8 ± 1.2 | — | >300 | ||
| EB–HL 79 | 21 °C | 137.7 ± 7.9 | 5.3 ± 0.3 (17.3%) | 6.3 ± 0.1 | 16.2 ± 2.7 | 1018 | 39.0 ± 1.6 |
| 37 °C | 155.0 ± 6.1 | 4.6 ± 0.5 (5.3%) | 5.8 ± 0.4 | — | >300 | ||
| On day 182 (37 °C) | 199.6 ± 3.4 | 5.9 ± 0.4 (5.1%) | — | 5.5 ± 0.1 | 233 | ||
| EB–HL 70 | 21 °C | 92.0 ± 7.8 | 3.9 ± 0.3 (18.9%) | 5.0 ± 0.1 | 11.6 ± 1.5 | 1020 | 45.6 ± 1.2 |
| 37 °C | 94.9 ± 7.1 | 3.2 ± 0.5 (7.8%) | 4.1 ± 0.1 | — | >300 | ||
| On day 182 (37 °C) | 140.8 ± 16.1 | 4.4 ± 0.2 (5.4%) | — | 3.6 ± 0.5 | 144 | ||
| EB–HL 63 | 21 °C | 57.4 ± 5.5 | 2.9 ± 0.4 (27.7%) | 4.0 ± 0.1 | 7.5 ± 1.3 | 918 | 52.8 ± 3.1 |
| 37 °C | 48.3 ± 5.0 | 1.9 ± 0.2 (11.4%) | 2.8 ± 0.2 | — | >300 | ||
| On day 182 (37 °C) | 88.1 ± 4.2 | 2.6 ± 0.2 (5.1%) | — | 1.8 ± 0.1 | 6.3 | ||
 | ||
| Fig. 8 Tensile stress–strain curves of PEB and the EB-co-δ-HL copolymers at 21 °C. | ||
Fig. 9 shows the tensile stress–strain curves up to 25% of strain of the tensile tests conducted at 37 °C with non-degraded (curves in bold type) and degraded specimens (on days 70 and 182). The mechanical behavior of these polymers at 37 °C were, in general, more elastomeric than at 21 °C and presented yield strengths and tensile strengths at 300% which were in all cases lower than the values measured at room temperature. The best example is EB–HL 63, whose secant modulus shifted from 57.4 MPa to 48.3 MPa, its tensile strength at 300% suffered a drop from 4.0 MPa to 2.8 MPa and its yield point became narrower with a yield strength that decreased from 2.9 MPa to 1.9 MPa. Nevertheless, it should be also stated that, as a result of the different melting behaviors at both temperatures, the secant modulus of these copolymers did not fall in all cases. Thus, the corresponding secant modulus of EB–HL 83, EB–HL 79 and EB–HL 70 was higher at 37 °C than at 21 °C. Mechanical performance is greatly influenced by the crystalline phase and these were the three copolymers that showed the most significant increase in melting enthalpy at 37 °C (see Table 5). However, despite the changes in their properties mentioned above, it can be said that all the EB-co-δ-HL copolymers displayed a good mechanical stability between 21 and 37 °C and their mechanical behavior is adequate at both temperatures.
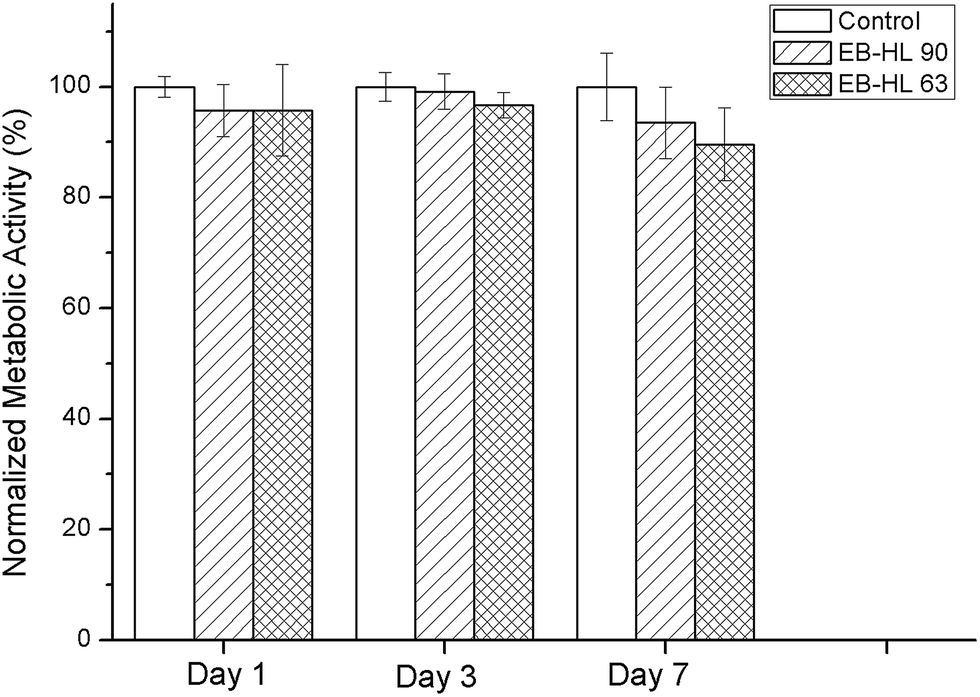 | ||
| Fig. 9 Metabolic activity of HDFs seeded on EB–HL 90 and EB–HL 63 with respect to the control on days 1, 3 and 7. | ||
As a consequence of the hydrolytic degradation process, the stress related properties were found to be higher than at the start of the study. As can be seen in Fig. S3 from ESI,† the yield strength of the samples submerged for 70 or 182 days rose compared to non-degraded films at 37 °C. Likewise, the elastic modulus at the end of the study reached values around 40–50 MPa higher than when measured at 37 °C on day 0. However, owing to the changes in molecular weight and the mechanical deterioration experienced in an aqueous medium, the EB-co-δ-HL copolymers (with the exception of EB–HL 83 which had on day 182 the highest Mw) lost deformation capability and their specimens broke earlier, before reaching 300% of strain. EB–HL 90 and EB–HL 63 were the most severely affected copolymers and exhibited elongation at break values of only 98% and 6.3%.
3.7. Cell culture studies
In vitro compatibility studies were performed to determine the possible toxicity of the ethylene brassylate-co-δ-hexalactone copolymers, as they contain a large amount of bismuth. To achieve this, circular samples of 9 mm in diameter, obtained from 150–200 μm films, were employed. Fig. 9 shows the metabolic activity of cells seeded in EB–HL 90 and EB–HL 63 samples compared to cells seeded in tissue culture plastic (control). No significant differences were observed in the metabolic activity of HDFs with respect to the control, indicating normal metabolic activity of cells seeded on the synthesized polymers.To observe the cytoskeleton organization and analyze the spreading and proliferation of HDFs seeded on EB–HL 90 and EB–HL 63, cells were stained with rhodamine phalloidin (red) and Hoechst (blue), see Fig. 10. On day 1, individual HDFs were observed in all the samples. From these micrographs it can be observed that HDFs were able to maintain their typical fibroblastic morphology38,39 when they were seeded on EB–HL 90 and EB–HL 63 and no differences were observed with respect to those cells seeded on glass coverslips (control). A higher number of cells were observed in all the samples on day 3, proving that there was adequate cell spreading and proliferation. This was also confirmed by nuclei quantification from these images. The symbols “*” and “#” from the graph in Fig. 10 indicate significant differences (p < 0.05) with respect to day 1 and day 3, respectively. Therefore, it can be stated that significant differences were observed between the number of nuclei on day 3 and on day 1 in all cases. On day 7, the surface of the samples was completely covered by cells forming a monolayer of cells. The number of nuclei also increased with respect to day 3, a good sign of the proliferation of the seeded cells on the studied materials.
 | ||
| Fig. 10 Fluorescent microscopy images of HDFs seeded on glass coverslips (control), EB-HL 90 and EB-HL 63 films on days 1, 3 and 7. F-actin was stained with rhodamine phalloidin (red) and nuclei with Hoechst (blue). | ||
4. Conclusions
In this work several statistical copolymers based on ethylene brassylate macrolactone and δ-hexalactone were synthesized with triphenyl bismuth as catalyst. The ring-opening polymerizations were carried out for 6 days at 130 °C although after 3 days of reaction copolymers of high molecular weight were already obtained. Owing to the racemic stereochemistry of the methyl side chain of the δ-HL unit, only the ethylene brassylate sequences were able to crystallize and as a result the crystallinity fraction decreased from 47% (PEB homopolymer) to ∼23–39% (when 10 to 51% of δ-HL units were incorporated). The poly(EB-co-δ-HL)s showed a slightly deviated from random distribution of sequences (0.71 < R < 0.84) and were carefully characterized by NMR, GPC, DSC, WAXS and TGA measurements. In addition, their mechanical properties were tested at room temperature (21 °C) and at body temperature (37 °C), at the same time an in vitro degradation study was also carried out at 37 °C for 182 days in phosphate buffer solution. Moreover, cell culture studies were conducted with human dermal fibroblasts (HDFs) seeded on samples obtained from films of some poly(EB-co-δ-HL).These novel copolymers displayed upgraded thermal stability (they were completely degraded at high temperatures, around 475 °C), showed rapid crystallization from melt, good processing using thermoplastic techniques, very attractive mechanical properties at 21 and 37 °C (only those with a Tm higher than 50 °C) with improved flexibility and elongation at break values >900% and enhanced biodegradability (with hydrolytic degradation rates almost 4 times higher than that of poly(ε-caprolactone)). In addition, the cell culture studies of metabolic activity and cell morphology demonstrated that the materials employed in this work are not cytotoxic and provide a valid substrate for cells to attach to and proliferate despite their high residual catalyst content.
This set of properties makes them very interesting potential materials for use in medical applications when an elastomeric character is desirable, particularly, for the regeneration of soft tissues and for various clinical implants and devices. However, tests on animals and clinical trials should be carried out prior to their application in the biomedical field. On the other hand, significant improvements are necessary in the methodology of polymerization (search for more efficient catalysts) in order to shorten the reaction times and decrease the amount of metal catalyst, which after the synthesis procedure becomes part of the polymer chains. In addition, more detailed degradation studies, including at different pH, enzymatic degradation or composting, should be made if the ethylene brassylate-co-δ-hexalactone copolymers are also intended to be used as green polymers for other applications such as packaging.
Acknowledgements
The authors are thankful for funds from the Basque Government, Department of Education, Universities and Research (GIC12/161-IT-632-13) and the Spanish Ministry of Innovation and Competitiveness MINECO (MAT2013-45559-P). J. F. thanks the University of the Basque Country (UPV-EHU) for a pre-doctoral grant.References
- S. Ravi, D. Padmanabhan and V. R. Mandapur, Macrocyclic musk compounds: synthetic approaches to key intermediates for exaltolide, exaltone and dilactones, Journal of the Indian Institute of Science, 2001, 81, 299–312 CAS.
- S. Arctander, Perfume and Flavor Chemicals (Aroma Chemicals), S. Arctander, Montclair, New Jersey, 1969, vol. 1, p. 1264 Search PubMed.
- D. McGinty, C. S. Letizia and A. M. Api, Fragrance material review on ethylene brassylate, Food and Chemical Toxicology, 2011, 49, 174–182 CrossRef PubMed.
- S. Müller, H. Uyama and S. Kobayashi, Lipase-catalyzed ring-opening polymerization of cyclic diesters, Chem. Lett., 1999, 1317–1318 CrossRef.
- A. Pascual, H. Sardón, A. Veloso, F. Ruipérez and D. Mecerreyes, Organocatalyzed Synthesis of Aliphatic Polyesters from Ethylene Brassylate: A Cheap and Renewable Macrolactone, ACS Macro Lett., 2014, 3, 849–853 CrossRef CAS.
- M. Labet and W. Thielemans, Synthesis of polycaprolactone: a review, R. Soc. Chem., 2009, 38, 3484–3504 RSC.
- M. A. Woodruff and D. W. Hutmacher, The Return of a forgotten polymer: Polycaprolactone in the 21st century, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- J. Fernandez, A. Etxeberria and J. R. Sarasua, In vitro degradation studies and mechanical behavior of poly(ε-caprolactone-co-δ-valerolactone) and poly(ε-caprolactone-co-L-lactide) with random and semi-alternating chain microstructures, Eur. Polym. J., 2015, 71, 585–595 CrossRef CAS.
- J.-C. Chen, J. H. Li, J.-H. Liu and L.-Q. Xu, Amphiphilic poly(ethylene glicol)-b-poly(ethylene brassylate) copolymers: One-pot synthesis, self-assembly, and controlled drug release, Chin. Chem. Lett., 2015, 26, 1319–1321 CrossRef CAS.
- A. Pascual, H. Sardón, F. Ruipérez, R. Gracia, P. Sudam, A. Veloso and D. Mecerreyes, Experimental and Computational Studies of Ring-Opening Polymerization of Ethylene Brassylate Macrolactone and Copolymerization with ε-Caprolactone and TBD-Guanidine Organic Catalyst, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 552–561 CrossRef CAS.
- P. Pan and Y. Inoue, Polymorphism and isomorphism in biodegradable polyesters, Prog. Polym. Sci., 2009, 34, 605–640 CrossRef CAS.
- J. Fernández, A. Etxeberria and J. R. Sarasua, Crystallization and melting behavior of poly(ε-caprolactone-co-δ-valerolactone) and poly(ε-caprolactone-co-L-lactide) copolymers with novel chain microstructures, J. Appl. Polym. Sci., 2015, 45534, 902–907 Search PubMed.
- G. Ceccorulli, M. Scandola, A. Kumar, B. Kalra and R. A. Gross, Cocrystallization of random copolymers of ω-pentadecalactone and ε-caprolactone synthesized by lipase catalysis, Biomacromolecules, 2005, 6, 902–907 CrossRef CAS PubMed.
- S. W. Shalaby and A. Kafrawy, Synthesis and some properties of isomorphic copolymers of ε-caprolactone and 1,5-dioxepan-2-one, J. Polym. Sci., Polym. Chem. Ed., 1989, 27, 4423–4426 CrossRef CAS.
- J. P. L. Dwan’Isa, P. Lecomte, P. Dubois and R. Jérôme, Synthesis and characterization of random copolyesters of ε-caprolactone and 2-oxepane-1,5-dione, Macromolecules, 2003, 36, 2609–2615 CrossRef.
- S. A. Papadimitriou, G. Z. Papageorgiou and D. N. Bikiaris, Crystallization and enzymatic degradation of novel poly(ε-caprolactone-co-propylene succinate) copolymers, Eur. Polym. J., 2008, 44, 2356–2366 CrossRef CAS.
- T. F. Biazus, A. M. Cezaro, G. R. Borges, J. P. Bender, E. Franceschi, M. L. Corazza and J. V. Oliveira, Vapour pressure data of ε-caprolactone, δ-hexalactone, and γ-caprolactone, J. Chem. Thermodyn., 2008, 40, 437–441 CrossRef CAS.
- H. Kikuchi, H. Uyama and S. Kobayashi, Lipase-catalyzed enantioselective copolymerization of substituted lactones to optically active polyesters, Macromolecules, 2000, 33, 8971–8975 CrossRef CAS.
- S. Kobayashi, Enzymatic ring-opening polymerization of lactones by lipase catalyst: Mechanistic aspects, Macromol. Symp., 2006, 240, 178–185 CrossRef CAS.
- T. H. Parliament, W. W. Nawar and I. S. Fagerson, Delta-caprolactone in heated milk fat, J. Dairy Sci., 1965, 48, 615–616 CrossRef.
- J. Fernández, A. Etxeberria and J. R. Sarasua, Synthesis and Properties of ω-pentadecalactone-co-δ-hexalactone copolymers: A biodegradable thermoplastic elastomer as an alternative to poly(ε-caprolactone), RSC Adv., 2016, 6, 3137–3149 RSC.
- H. R. Kricheldorf, G. Behnken, G. Schwarz and J. A. Brockaert, High molecular weight poly(ε-caprolactone) by initiation with triphenyl bismuth, Macromol. Chem. Phys., 2008, 209, 1586–1592 CrossRef CAS.
- H. R. Kricheldorf, Syntheses of Biodegradable and Biocompatible Polymers by means of Bismuth Catalysts, Chem. Rev., 2009, 109, 5579–5594 CrossRef CAS PubMed.
- V. Rodilla, A. T. Miles, W. Jenner and G. M. Harcksworth, Exposure of cultured human proximal tubular cells to cadmium, mercury, zinc and bismuth: toxicity and metallothionein induction, Chem.-Biol. Interact., 1998, 115, 71–83 CrossRef CAS PubMed.
- Z. Guo and P. J. Sadler, Metalle in der Medizin, Angew. Chem., 1999, 111, 1610–1630 CrossRef.
- G. C. Briand and N. Burford, Bismuth compounds and preparations with biological or medicinal relevance, Chem. Rev., 1999, 99, 2601–2658 CrossRef CAS PubMed.
- H. R. Kricheldorf and G. Behnken, Copolymerization of Glycolide and L-lactide initiated with Bismuth(III) n Hexanoate or Bismuth Subsalicylate, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 795–800 CrossRef CAS.
- E. Prestch, T. Clerc, J. Seibl and W. Simon, Tables of Spectral Data for Structure Determination of Organic Compounds, Chemical Laboratory Practice, Springer Science & Business Media, Springer-Verlag Berlin Heidelberg GmbH, 2013 Search PubMed.
- I. R. Herbert, Statistical analysis of copolymer sequence distribution, in NMR Spectroscopy of Polymers, ed. R. N. Ibbet, Blackie Academic & Professional, London, 1993, ch. 2, pp. 50–79 Search PubMed.
- L. Wu and J. Ding, Effects of porosity and pore size on in vitro degradation of three-dimensional porous poly(D,L-lactide-co-glycolide) scaffolds for tissue engineering, J. Biomed. Mater. Res., 2005, 75, 767–777 CrossRef PubMed.
- J. Fernández, A. Larrañaga, A. Etxeberria and J. R. Sarasua, Effects of chain microstructures and derived crystallization capability on hydrolytic degradation of poly(L-lactide/ε-caprolactone) copolymers, Polym. Degrad. Stab., 2013, 98, 481–489 CrossRef.
- J. Fernández, A. Etxeberria and J. R. Sarasua, In vitro degradation study of biopolyesters using poly(lactide/δ-valerolactone) copolymers, Polym. Degrad. Stab., 2015, 112, 104–116 CrossRef.
- A. Larrañaga, P. Aldazabal, F. J. Martin and J. R. Sarasua, Hydrolytic degradation and bioactivity of lactide and caprolactone based sponge-like scaffolds loaded with bioactive glass particles, Polym. Degrad. Stab., 2014, 110, 121–128 CrossRef.
- P. Scherrer, Bestimmung der Grosse und der inneren Struktur von Kolloidteilchen mittels Rontgenstrahlen, Nachr Ges Wiss Gottingen, 1918, vol. 26, pp. 98–100 Search PubMed.
- M. de Geus, I. van der Meulen, B. Goderis, K. van Hecke, M. Doschu, H. van der Werff, C. E. Koning and A. Heise, Performance polymers from renewable monomers: high molecular weight poly(pentadecalactone) for fiber applications, Polym. Chem., 2010, 1, 525–533 RSC.
- M. L. Focarete, M. Scandola, A. Kumar and R. A. Gross, Physical characterization of poly(ω-pentadecalactone) synthesized by lipase-catalyzed ring opening polymerization, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 1721–1729 CrossRef CAS.
- J. Fernández, A. Etxeberria, A. Larrañaga Varga and J. R. Sarasua, Synthesis and characterization of ω-pentadecalactone-co-ε-decalactone copolymers: Evaluation of thermal, mechanical and biodegradatin properties, Polymer, 2015, 81, 12–22 CrossRef.
- L. C. Baxter, V. Frauchigher and M. Textor, ap Gwynn, I.; Richards, R.G. Fibroblast and osteoblast adhesion and morphology on calcium phosphate surfaces, Eur. Cells Mater., 2002, 4, 1–17 CAS.
- E. Tamariz and F. Grinnell, Modulation of fibroblast morphology and adhesion during collagen matrix remodeling, Mol. Biol. Cell, 2002, 11, 3915–3929 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01065b |
| This journal is © The Royal Society of Chemistry 2016 |