
Aggregation-induced emission in fluorophores containing a hydrazone structure and a central sulfone: restricted molecular rotation
Jing Lia,
Wen Yanga,
Weiqun Zhou*a,
Chunchun Lia,
Zhongqing Chengb,
Mengying Lib,
Liqun Xiec and
Youyong Lid
aSchool of Chemistry & Chemical Engineering and Material Science, Soochow University, 199 Ren’ai Road, Suzhou, 215123, People’s Republic of China. E-mail: wqzhou@suda.edu.cn
bSchool of Biology and Basic Medical Sciences, Soochow University, 199 Ren’ai Road, Suzhou, 215123, People’s Republic of China
cGold Mantis School of Architecture, Soochow University, 199 Ren’ai Road, Suzhou, 215123, People’s Republic of China
dFunctional Nano & Soft Materials Laboratory, Soochow University, 199 Ren’ai Road, Suzhou, 215123, People’s Republic of China
First published on 1st April 2016
Abstract
Four D–π–A–π–D structured 4,4′-di(E)-2-(4-4-alkoxyaniline)hydrazinyldiphenyl sulfones were synthesized and their structures were characterized using NMR, IR, and element analysis. These compounds displayed weak fluorescence emission in THF while their fluorescence was enhanced in a THF/water mixture solvent. Fluorescence microscope imaging showed the formation of the aggregate state. The mechanism of aggregation-induced emission (AIE) was supported by molecular dynamics calculations. The intermolecular hydrogen bonds between adjacent molecules inhibited the intramolecular rotations, preventing π–π stacking and causing AIE enhancement. Living cell imaging experiments opened up a new method for cell fluorescence staining.
1. Introduction
The main organic photoimaging materials are of great importance in the photochemical and applied chemical fields.1–5 Significant developments have taken place in the past few years leading to practical organic photoimaging materials. Outstanding fluorescent materials are often designed with large conjugated systems,6–10 so the conventional structural design strategy of organic fluorophores is to enlarge the extent of π-conjugation by melding more and more aromatic rings together. Although the bigger conjugate structure can produce high fluorescence in dilute solutions, the fluorescence can be usually quenched in concentrated solutions and the solid state. This phenomenon is usually called aggregation-caused fluorescence quenching (ACQ) because of π–π stacking and other non-radiative pathways.11 The ACQ effect has restricted the real-world applications of fluorescent materials.12,13 For example, as common organic materials are insoluble in water, the materials of the ACQ effect cannot be used as sensors to detect biological molecules in physiological buffers and monitor ions in river water.14,15 Since the luminescent materials as light-emitting devices must be fabricated in a solid film form, the ACQ effect materials cannot be used for the fabrication of efficient organic light-emitting diodes (OLEDs).16–24In 2003, Tang et al. synthesized TTPEPy25 with high fluorescence efficiency and a high AIE effect which overcame the ACQ effect. The appearance of AIE made it possible to apply the organic photoimaging materials in real-world applications.
It was realized that rational restricted intramolecular rotation (RIR) closed the possible non-radiative decay channels and activated radiative decay channels, which led to the strong fluorescence.26–28 Some typical examples of AIE systems have been developed such as hexaphenylsilole (HPS)29 with a propeller-shaped non-planar molecule, butterfly-shaped pyran derivatives,30 cyano-substituted biphenyl ethylene with a J-aggregation type spatial structure,31 and BODIPY dyes with a V-shaped structure.32 V-shaped compounds have twisted nonplanar structures that prevent π–π stacking and induce fluorescence enhancement in the aggregated states. For instance, V-shaped BODIPY32 is able to reduce close intermolecular interactions and correspondingly improve solid state emission of BODIPY dyes. The V-shaped pyridinium salt DMDPN33 has also exhibited a typical aggregation-induced emission behavior.
D–π–A–π–D type fluorescent materials have become an efficient way for the design of brilliant fluorescence materials for they emit stronger intramolecular charge transfer (ICT) fluorescence. For example, the V-shaped D–π–A–π–D compound BCPPM exhibited strong ICT fluorescence in the solid state.34 In this article, we synthesized four D–π–A–π–D structured V-shaped compounds of 4,4′-di(E)-2-(4-4-alkoxyaniline)hydrazinyldiphenyl sulfone (BAHDS1-4), where the sulfone unit served as the central acceptor core, two hydrazinyldiphenyl conjugated chains served as the π-bridge to extend the conjugation length, and methoxy, n-butoxy, tert-butoxy and 4-phenoxy were introduced as donor units. Larger substituent groups, such as n-butoxy, tert-butoxy and phenoxy, were introduced to restrict the intramolecular rotation and enhance fluorescence emission. The four compounds emitted weak fluorescence in THF, but the fluorescence gradually enhanced with the addition of non-soluble water in THF and presented the AIE effect.
2. Experimental section
2.1 Materials and instruments
Bis(4-chlorophenyl)sulphone, hydrazine hydrate (85%), 4-methoxybenzaldehyde, 4-n-butoxy benzaldehyde, 4-t-butoxy benzaldehyde, 4-methoxybenzaldehyde, THF, DMSO, acetone, acetonitrile and DMSO-D6 were purchased from J&K (CHINA). Melting points were measured using a Kofler melting point apparatus. IR spectra were obtained in KBr discs using a Nicolet 170SX FT-IR spectrometer. Elemental analyses were performed using a Yannaco CHNSO Corder MT-3 analyzer. 1H NMR and 13C NMR spectra were recorded using an INOVA 400 at 400 and 100 MHz, respectively, with TMS as the internal standard, DMSO-D6 as the deuterated solvent and chemical shifts reported in ppm. Mass spectra were recorded using a Finnigan MAT95 mass spectrometer. The absorption and fluorescence spectra were recorded using a CARY 50 UV-vis spectrophotometer and an FLS920 fluorescence spectrophotometer. The Quantum Yield (QY) of the as-synthesized BAHDS derivatives was calculated based on the following equation: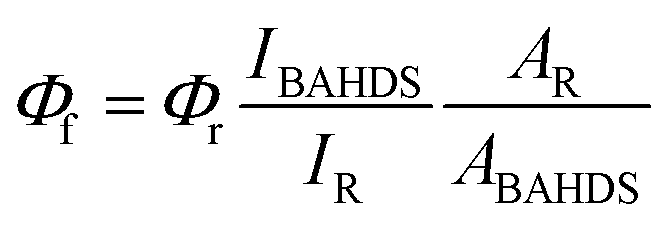 , where Φf is the fluorescence quantum yield, I is the intensity of the fluorescence spectra, and A is the optical density at the excitation wavelength. The QY was measured by taking anthracene as a standard for BAHDS. R referred to the standard anthracene with a known quantum yield. Film samples for the measurements of UV-vis and fluorescence spectroscopy were prepared by drop casting and subsequent spin-coating (2000 rpm, 30 s) from THF solution (100 mL) on a quartz cell (12.5 × 12.5 × 45 mm). Fluorescence microscope imaging of the aggregations was obtained using a Leica DM2500M. The cell images were gathered with the inverted fluorescence microscope (Olympus, IX71) and processed with Nikon AY software. Scanning electron microscope (SEM) images of the aggregations were obtained using a Hitachi S4800. All of the experiments were carried out at room temperature.
, where Φf is the fluorescence quantum yield, I is the intensity of the fluorescence spectra, and A is the optical density at the excitation wavelength. The QY was measured by taking anthracene as a standard for BAHDS. R referred to the standard anthracene with a known quantum yield. Film samples for the measurements of UV-vis and fluorescence spectroscopy were prepared by drop casting and subsequent spin-coating (2000 rpm, 30 s) from THF solution (100 mL) on a quartz cell (12.5 × 12.5 × 45 mm). Fluorescence microscope imaging of the aggregations was obtained using a Leica DM2500M. The cell images were gathered with the inverted fluorescence microscope (Olympus, IX71) and processed with Nikon AY software. Scanning electron microscope (SEM) images of the aggregations were obtained using a Hitachi S4800. All of the experiments were carried out at room temperature.
2.2 Cell culture and fluorescence imaging
A549 cells (human lung adenocarcinoma cells) were purchased from the Shanghai Institute of Cell Biology. The cells were cultured in Roswell Park Memorial Institute culture medium (RPMI-1640), supplemented with 6% calf serum, penicillin (100 U mL−1), streptomycin (100 × 10−6 g mL−1) and 2.5 × 10−4 mol L−1 L-glutamine at 37 °C in a 5% CO2 incubator. The cells were cultured in a 15 mm diameter cell culture dish for 2 days. The A549 cells were incubated with the aggregations at a final concentration of 50 μg mL−1 for 30 min, and washed with PBS buffer to remove extra cellular material. Then the dish was placed, to get long-term imaging, under an inverted fluorescence microscope (Olympus, IX71) at 450 nm (λ = 380 nm).2.3 Computational methods
The ground and excited states of molecules were optimized with Becke-3-Lee-Yang-Parr (B3LYP)35–37 by density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations at a level of 6-311G(d) with the Gaussian 09 program.38 TD-DFT combined with the SCRF method39 (CPCM, by THF as solvent) at the same level were applied to indicate the energies of the HOMO and LUMO. Vibrational frequency calculations were also performed to ensure the stability of the calculation result and to avoid virtual frequency.MD (molecular dynamics) simulations were performed using the discover module within Materials Studio 7.0 with the COMPASS force field for the studied molecules with an amorphous cell.40 They were carried out at a constant temperature and constant volume ensemble (NVT). The temperature was set at 300 K and kept constant using a nose thermostat.41 Pressure was set at 1 atm. Electrostatic energies were calculated using the vdW & Coulomb method with a 9.5 Å non-bonded cutoff. The total simulation time was 10.0 ps and each step took 1.0 fs.
2.4 Synthesis
The synthetic route is shown in Scheme 1. | ||
| Scheme 1 The synthetic route of BAHDS. | ||
BAHDS-1. White powder. Yield: 78%. mp: 259.6 °C. Anal. calc. formula: C28H26N4O3S. (%): C, 65.35; N, 10.89; H, 5.09. Found (%): C, 65.38; N, 10.72; H, 5.12. 1H-NMR (400 MHz, DMSO-D6) δ 10.79 (s, 2H), 7.94 (s, 2H), 7.68 (dd, J = 9.2 Hz, 8.8 Hz, 8H), 7.16 (d, J = 8.8 Hz, 4H), 7.01 (d, J = 8.8 Hz, 4H), 3.61 (s, 6H). 13C-NMR (100 MHz, DMSO-D6) δ 50.22 (CH3), 111.46 (Ph), 114.23 (Ph), 127.64 (Ph–C), 128.73 (Ph), 130.65 (Ph–S), 139.86 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 148.77 (Ph–N), 159.89 (Ph–O). FT-IR (ν, cm−1): 3304 (νN–H), 2930, 2837 (νC–H), 1598 (νCN), 1279, 1105 (νSO2), 1030 (νC–O).
N), 148.77 (Ph–N), 159.89 (Ph–O). FT-IR (ν, cm−1): 3304 (νN–H), 2930, 2837 (νC–H), 1598 (νCN), 1279, 1105 (νSO2), 1030 (νC–O).
BAHDS-2. White powder; yield: 74%. mp: 225.5 °C. Anal. calc. formula: C34H38N4O3S. (%): C, 68.20; N, 9.36; H, 6.40. Found (%): C, 68.32; N, 9.41; H, 6.38. 1H-NMR (400 MHz, DMSO-D6) δ 10.78 (s, 2H), 7.93 (s, 2H), 7.67 (dd, J = 9.2 Hz, 8.8 Hz, 8H), 7.15 (d, J = 8.8 Hz, 4H), 6.99 (d, J = 8.8 Hz, 4H), 4.02 (t, J = 6.4 Hz, 4H), 1.72 (m, 4H), 1.46 (m, 4H), 0.96 (t, J = 7.2 Hz, 6H) 13C-NMR (100 MHz, DMSO-D6) δ 13.70 (CH3), 18.72 (C–C), 30.71 (C–C), 67.24 (C–O), 111.44 (Ph), 114.69 (Ph), 127.52 (Ph–C), 128.72 (Ph), 130.62 (Ph–S), 139.89 (C
N), 148.77 (Ph–N), 159.35 (Ph–O). FTIR (ν, cm−1): 3328 (νN–H), 3092, 2962, 2879 (νC–H), 1593 (νCN), 1273, 1098 (νSO2), 1022 (νC–O).
BAHDS-3. White powder; yield: 73%. mp: 195.1 °C. Anal. calc. formula: C34H38N4O3S. (%): C, 68.20; N, 9.36; H, 6.40. Found (%): C, 68.27; N, 9.41; H, 6.38. 1H-NMR (400 MHz, DMSO-D6) δ 10.85 (s, 2H), 7.95 (s, 2H), 7.67 (dd, J = 9.2 Hz, 8.4 Hz, 8H), 7.16 (d, J = 8.8 Hz, 4H), 7.03 (d, J = 8.4 Hz, 4H), 1.35 (s, 18H). 13C-NMR (100 MHz, DMSO-D6) δ 28.56 (CH3), 78.47 (C–C), 111.53 (Ph), 123.53 (Ph), 127.00 (Ph–C), 128.76 (Ph), 129.84 (Ph), 130.79 (Ph–S), 139.64 (C
N), 148.70 (Ph–N), 155.90 (Ph–O). FTIR (ν, cm−1): 3290 (νN–H), 3115, 2978 (νC–H), 1593 (νCN), 1273, 1098 (νSO2), 1022 (νC–O).
BAHDS-4. Yellow powder; yield: 68%. mp: 178.4 °C. Anal. calc. formula: C38H30N4O3S. (%): C, 73.29; N, 9.00; H, 4.86. Found (%): C, 72.98; N, 9.11; H, 4.56. 1H-NMR (400 MHz, DMSO-D6) δ 10.89 (s, 2H), 7.98 (s, 2H), 7.73 (d, J = 9.2 Hz, 7.2 Hz, 8H), 7.45 (t, J = 8.0 Hz, 4H), 7.19 (m, 6H), 7.08 (t, J = 8.8 Hz, 8H). 13C-NMR (100 MHz, DMSO-D6) δ 111.46 (Ph), 118.93 (Ph), 123.81 (Ph), 127.91 (Ph–C), 128.77 (Ph), 130.33 (Ph), 130.90 (Ph–S), 139.86 (C
N), 148.77 (Ph–N), 159.89 (Ph–O). FTIR (ν, cm−1): 329 (δN–H), 3067, 3042 (δC–H), 1591 (δCN), 1273, 1105 (δSO2), 1061 (δC–O).
3. Result and discussion
3.1 Photo-physical properties
Fig. 1(a) shows UV absorption and fluorescence emission spectra of the BAHDS derivatives in THF. The four BAHDS derivatives have similar absorption and emission curves because of their similar molecular structures. The maximum absorption wavelength was located at 355–365 nm. The very weak fluorescence emissions were displayed under a 365 nm UV lamp, and the fluorescence quantum yield was lower than 1% (Table 1). The UV-vis spectra and fluorescent spectra of BAHDS-1 in different solvents were recorded and are shown in Fig. 1(b). A slight red-shift with the polarity increase of the solvents was observed in the UV spectra. The Stokes shift (Δλ) was between 55 and 60 nm. Both the moderate Stokes shift (Δλ = 55–60 nm) and the characteristics of the wavelength red-shift indicated the π–π* transition of the absorption.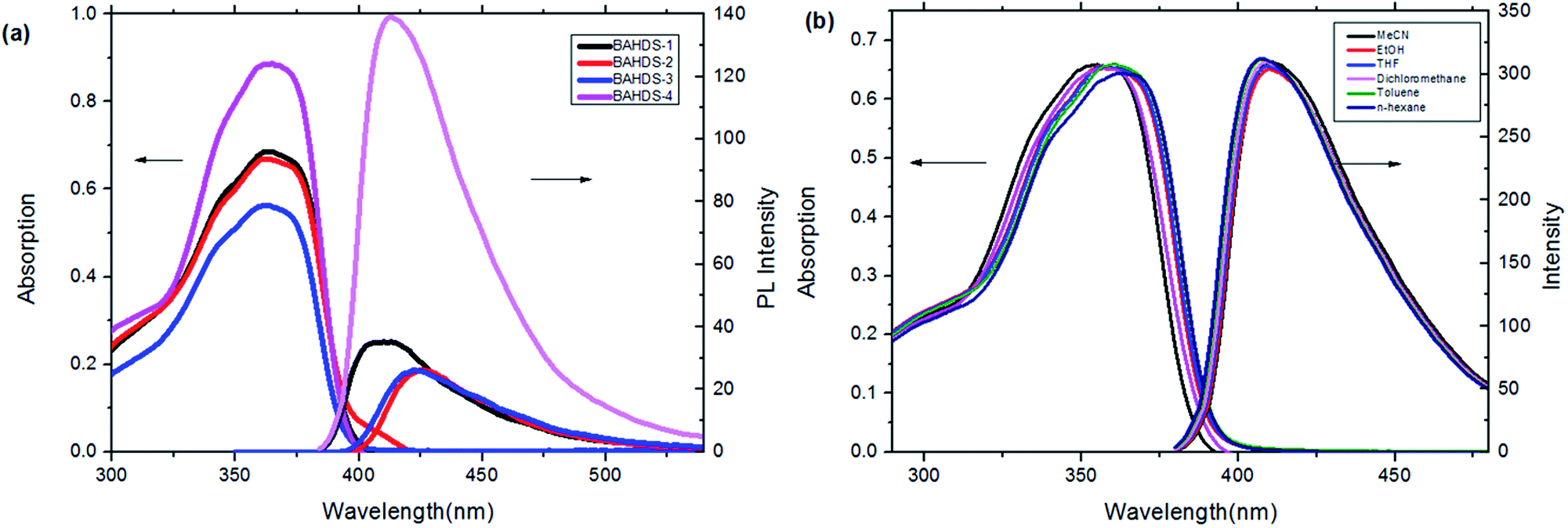 | ||
| Fig. 1 (a) UV-vis absorption and fluorescence emission of the BAHDS derivatives in THF (1 × 10−5 mol L−1). (b) UV-vis and fluorescence spectra of BAHDS-1 in different solvents (1 × 10−5 mol L−1). | ||
| THF | THF–water | Film | ||||||
|---|---|---|---|---|---|---|---|---|
| λab (nm) | λem (nm) | Φf (%) | λem (nm) | Φf (%) | Water (%) | I/I0a | λem (nm) | |
| a I/I0: the proportion of Φf in THF to Φf in THF–water. | ||||||||
| BAHDS-1 | 365 | 408 | 0.04 | 433 | 2.7 | 93 | 66.6 | 432 |
| BAHDS-2 | 364 | 423 | 0.03 | 433 | 6.9 | 93 | 228.4 | 433 |
| BAHDS-3 | 365 | 423 | 0.03 | 423 | 5.7 | 70 | 190.4 | 423 |
| BAHDS-4 | 363 | 413 | 0.16 | 438 | 22.4 | 90 | 140.0 | 437 |
Optimized molecular structures at the B3LYP/6-311G(d) level and molecular orbital amplitude plots of the HOMO and LUMO for BAHDS derivatives are depicted in Fig. 2. Because of their similar structures, BAHDS 1–4 have similar HOMOs and LUMOs. Take BAHDS-1 as an example: the hydrazine moiety has two sides conjugated to the adjacent two benzene rings consisting of a π conjugated plane separately. Two side π conjugated planes intersect at sulfone and form V-shaped D–π–A–π–D molecular structures (with sulfone as the central acceptor core, two hydrazinyldiphenyl conjugated chains as the π-bridge to extend the conjugation length, and methoxy, n-butoxy, tert-butoxy or 4-phenoxy as a donor, separately). The charges of the HOMO were focused on the hydrazone moiety and its nearby phenyl ring, while the charges of the LUMO were centred on the sulfone moiety, which indicated the intramolecular charge transition (ICT) of HOMO to LUMO.
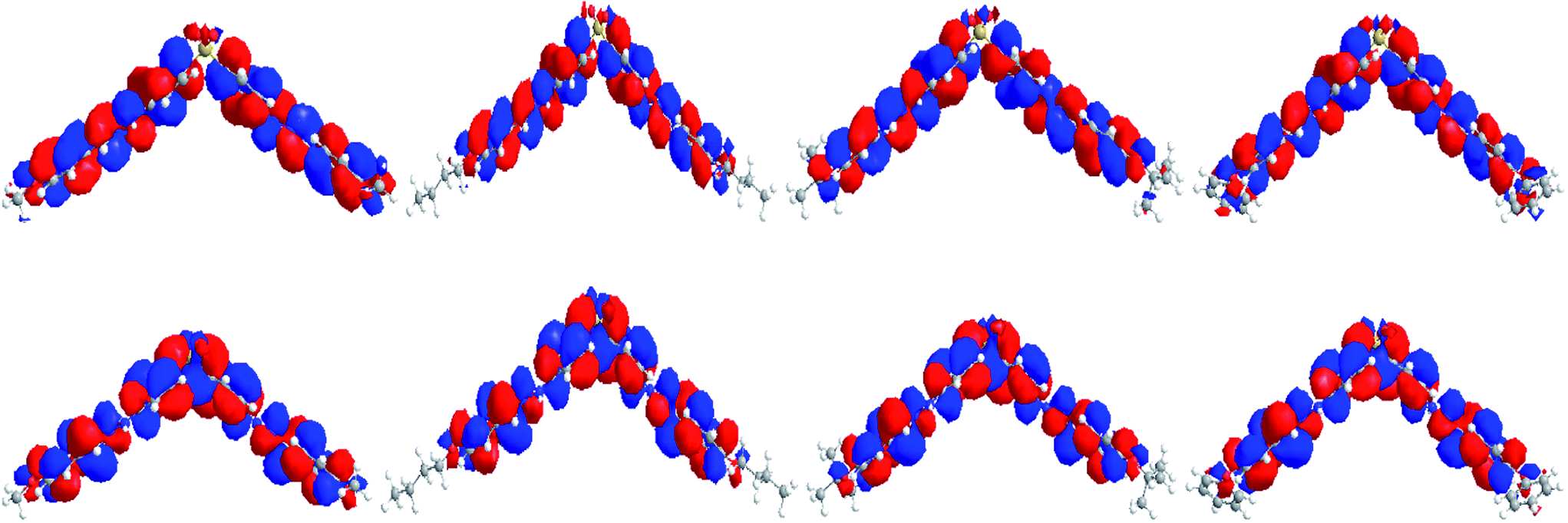 | ||
| Fig. 2 HOMOs (top) and LUMOs (bottom) of BAHDS-1, BAHDS-2, BAHDS-3 and BAHDS-4 (from left to right) computed using the B3LYP/6-311G(d) level of theory. | ||
Time-dependent density functional theory calculations (TD-DFT) were applied to indicate the energies of the HOMO and LUMO. Table 2 shows the energy gaps between the HOMOs and LUMOs from DFT prediction and UV absorption. From Table 2, the calculated energy gaps between the HOMO and LUMO were close to the corresponding UV absorption energies with a difference value of 0.15 eV, 0.18 eV, 0.19 eV, and 0.22 eV for BAHDS 1–4, respectively. The distinction between the calculations and experiments was in an acceptable range to indicate the significance of the DFT calculations.
| Experimental (eV) | Calculated (eV) | HOMO (eV) | LUMO (eV) | |
|---|---|---|---|---|
| BAHDS-1 | 3.40 (365 nm) | 3.25 | −5.40 | −1.68 |
| BAHDS-2 | 3.41 (364 nm) | 3.23 | −5.40 | −1.70 |
| BAHDS-3 | 3.40 (365 nm) | 3.21 | −5.36 | −1.69 |
| BAHDS-4 | 3.42 (363 nm) | 3.20 | −5.48 | −1.81 |
3.2 Aggregation-induced properties
A poor-solvent photoluminescence (PL) test, commonly used for studying the AIE phenomenon,42–44 was performed to explore the luminescence behaviour of the prepared BAHDS derivatives. As water was a poor solvent for BAHDS 1–4, the molecules would aggregate in THF/water mixtures with a high water content. Thus, the luminescence from such systems was primarily attributed to molecule aggregation, not the solute molecules. The AIE effect (I/I0) of the BAHDS derivatives is shown in Fig. 3 and Table 1. In Fig. 3, the changes in the UV spectra in THF/water mixtures with varying water volume fractions could confirm the formation of aggregates of BAHDS. BAHDS-1 exhibited an absorption peaks at 365 nm in pure THF solution (Fig. 3(a)). With an increase in the fw (water volume fraction) from 0% to 80%, the absorption peaks basically remained at the same positions. However, when the fw increased to 90%, the shoulder absorption peak at about 300 nm rose. Meanwhile, leveled-off tails appeared in the visible region when the fw was equal to 90%, indicating that the aggregates were probably of nanoscale sizes. When the fw ≤ 80%, the appearance of no leveled-off spectral tails in the long-wavelength region confirmed that the molecules dissolved as isolated species in the solvent mixture. A very weak fluorescence emission was observed in pure THF with Φf = 0.04% (Table 1). Similar to the UV spectra, the fluorescence spectra exhibited no obvious changes as the water content increased from 0% to 80%. A dramatic fluorescence enhancement was observed when the fw was equal to 90%. When the fw was equal to 93%, the fluorescence increased to a maximum value, the intensity was 66.6 times (Table 1) higher than in pure THF, and the fluorescence quantum yield increased to 2.7% (Table 1). In addition, the maximum emission wavelength showed a red shift of 24 nm, which indicated that the aggregates were probably of nanoscale sizes.45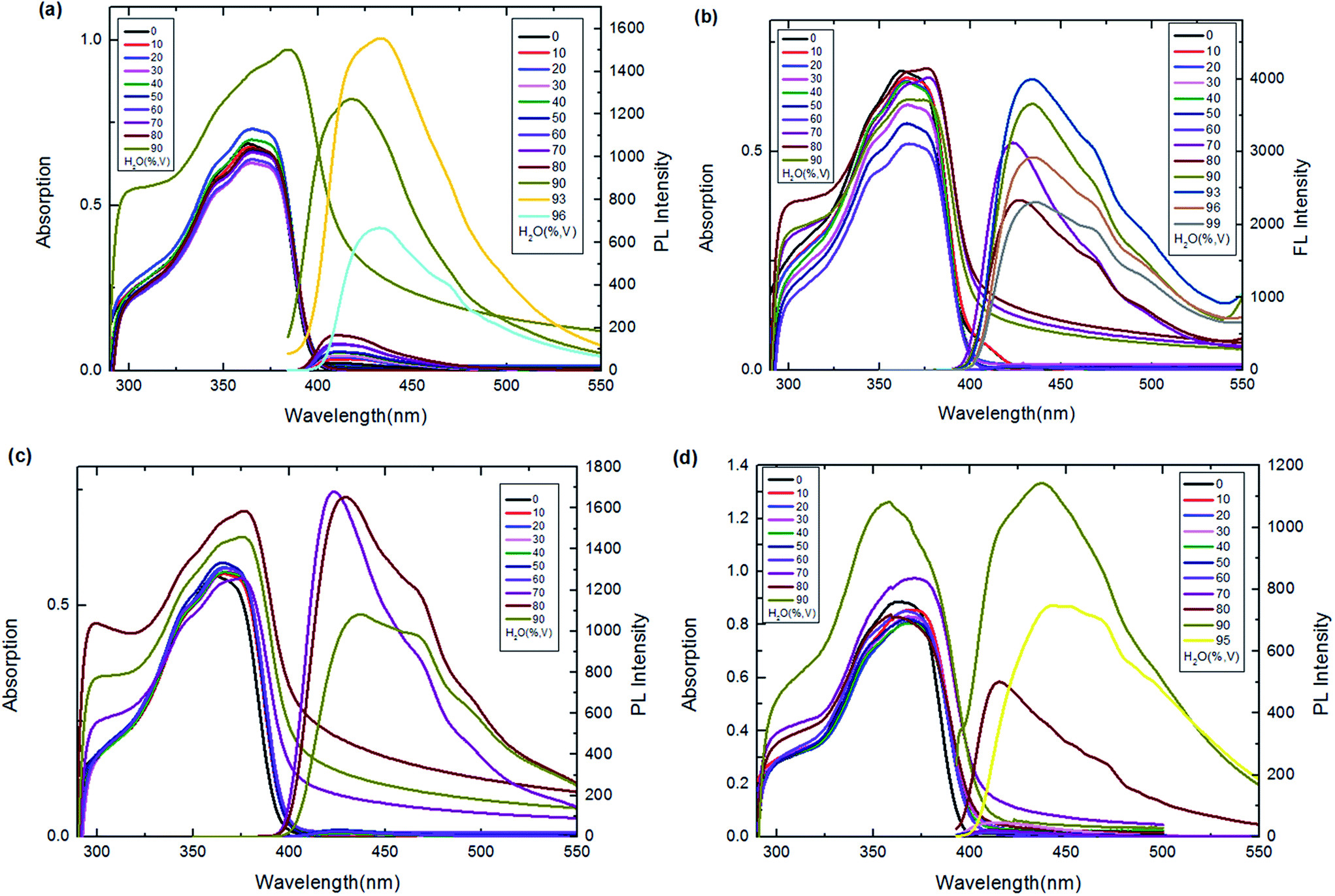 | ||
| Fig. 3 UV-vis and emission spectra of BAHDS-1 (a), BAHDS-2 (b), BAHDS-3 (c) and BAHDS-4 (d) in THF–water mixtures (5 × 10−5 mol L−1). | ||
Our strategy for increasing the steric hindrance of the substituted group was to introduce the larger alkoxy, n-butoxyl, tert-butoxyl and phenoxyl groups into the phenyl cycle of the molecules, which could prevent π–π stacking and was beneficial to the AIE effects of the BAHDS derivatives. As shown in Fig. 3(b)–(d), as the fw increased to 70%, the shoulder absorption peak appeared and the leveled-off tails appeared in the visible region, indicating the formation of aggregate states for BAHDS-2, 3, and 4. It can be found that both BAHDS-2 and BAHDS-3 exhibited outstanding fluorescence enhancement in the THF/water solutions where the fw > 70%. The maximum emission enhancements of 228.4-fold (fw = 93%), and 190.4-fold (fw = 70%) (Table 1) for BAHDS-2 and BAHDS-3, respectively, are larger than that of BAHDS-1 in THF/water (66.6-fold), and their fluorescence quantum yields were measured to be 6.9% and 5.7%. Moreover, the maximum emission wavelengths red shifted 10–20 nm.
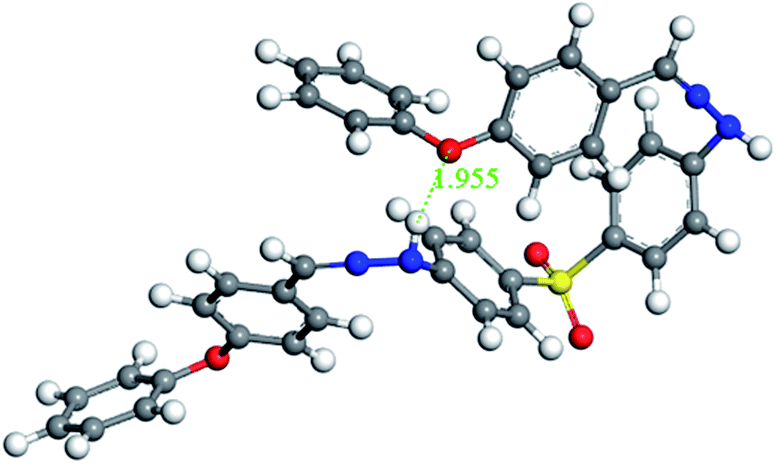
As shown in the MD calculations (Fig. 4), an intramolecular hydrogen bond interaction was found in BAHDS-4. It affected the vibration of BAHDS-4 and increased the rigidity of molecule, which was beneficial to the fluorescence enhancement in THF solution. As shown in Table 1, the fluorescence quantum yield of BAHDS-4 was 0.16% in THF. A further investigation of aggregation fluorescence in THF/water solutions showed that BAHDS-4 exhibited a large fluorescence enhancement when the fw was at 80%, as shown in Fig. 3(d). When the water fraction increased to 90%, the intensity reached the maximum, the emission was enhanced 140 times (fw = 90%) compared with BAHDS-4 in THF (Table 1), and the Φf increased to 22.4%, the largest in BAHDS 1–4. Additionally, the emitted wavelength red shifted from 422 nm for BAHDS-1 to 438 nm for BAHDS-4.
 | ||
| Fig. 4 Molecular dynamics calculation of BAHDS-4 in THF solution. | ||
As we can envisage, the steric hindrance relevant to the phenyl rings was gradually enhanced with increasing substituted groups. The steric hindrance of the substituted groups, n-butoxy, tert-butoxy and phenoxy was larger than that of methoxyl in the BAHDS system, and the motion or vibration of the phenyl rings was suppressed in the aggregation states, which led to the enhanced fluorescence. In comparison with BAHDS-2 and BAHDS-3, because of the p–π conjugation between oxygen and phenyl, the rigidity of the BAHDS-4 molecule probably improved as well, on account of the steric repulsion of the large groups, giving rising to fluorescence quantum yields boosting from 0.01 (BAHDS-1) to 0.16 (BAHDS-4) in THF solution (Table 1). Namely, by internal control through the introduction of alkoxyl groups, the PL properties of the BAHDS derivatives can be modulated. This indicated that steric congestion of the relevant substituted structure can fix the molecules and significantly inhibit the non-radiance annihilation process, leading to the high luminescence of BAHDS-4 even in dilute solution. Based on steric hindrance, by the introduction of para-alkoxylin to the BAHDS unit, the triggered RIR as the fundamental mechanism of the intriguing AIE phenomenon was directly verified through simple chemical modifications. Probably, the investigations on the AIE phenomenon can give us a definite understanding of the mechanism of AIE characteristics, which will be also beneficial for novel molecular design.
Fig. 5(a) displays SEM images of the BAHDS derivatives. In the SEM images, the diameter of the nanoscale sized BAHDS derivatives in THF/water was about 500 nm. BAHDS-1 and BAHDS-4 had a sphere shaped nanostructure, and BAHDS-2 and BAHDS-3 were needle like nanoscale structures. Fluorescence microscope images of the BAHDS derivatives under UV irradiation are shown in Fig. 5(b). The nanoscale particles of BAHDS 1–4 emitted the strongest bright blue fluorescence under illumination with a 365 nm UV lamp. The formation of an aggregation state could also be proven by the leveled-off tail stretching into the longer wavelength region in the film state, as shown in Fig. 5(c). Calculations of full-width half maximum (fwhm) revealed that the emission spectra of the BAHDS derivatives in the solid state were comparable and narrower, compared with the emission spectra obtained in THF/water. Formation of higher order aggregates in water or solution resulted in such narrower and higher intensity emission bands because of reduced intermolecular interactions. The packing efficiency and intermolecular interactions also contribute to enhanced luminescence.
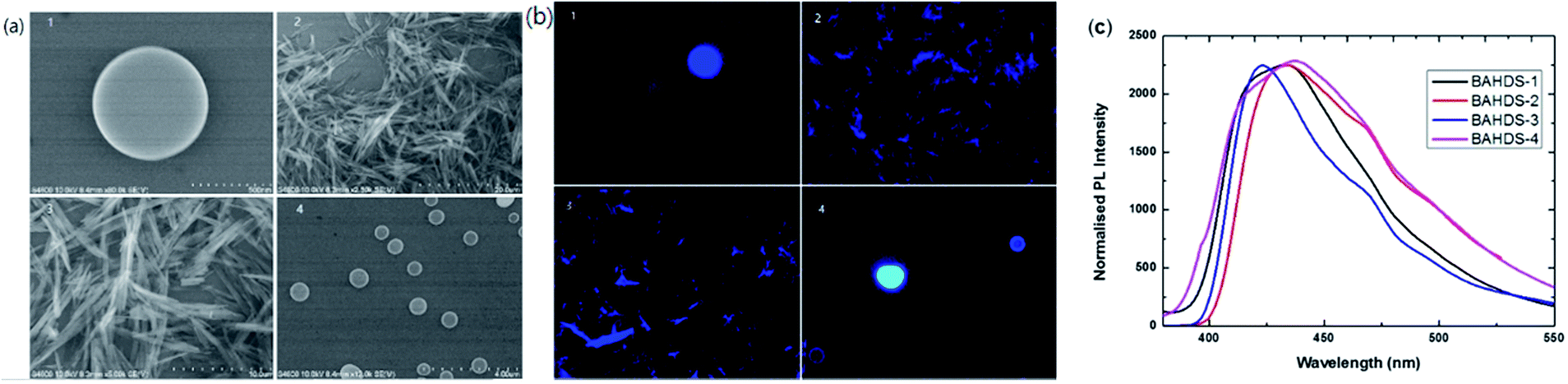 | ||
| Fig. 5 (a) SEM images of BAHDS-1 (1), BAHDS-2 (2), BAHDS-3 (3), and BAHDS-4 (4). (b) Fluorescence microscope images of BAHDS-1 (1), BAHDS-2 (2), BAHDS-3 (3), and BAHDS-4 (4) under UV irradiation. (c) Normalised PL spectra of the BAHDS derivatives in a film state. | ||
Fluorescence spectral measurements was determined every six hours for forty-eight hours. It is shown in Fig. 6(a) that with the increasing standing time of the BAHDS derivatives, few changes could be observed. It showed that strong aggregation induced fluorescence of the BAHDS derivatives could remain stable for a long time. Also, Fig. 6(b) indicates the excellent stability of the BAHDS derivatives in a large range of pH from 3 to 8.
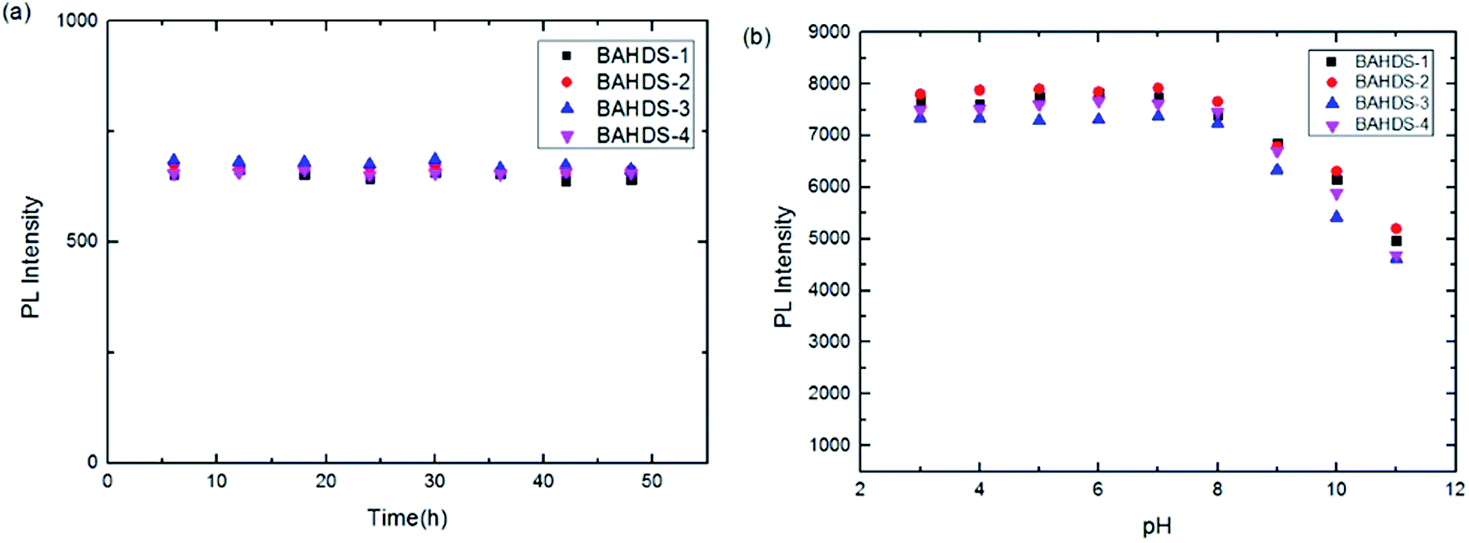 | ||
| Fig. 6 Normalized PL spectra of BAHDS 1–4 (10 μM in THF/water solvent) at different standing times (a) and different pHs (b). | ||
3.3 Mechanism of the AIE of the BAHDS derivatives
To find the links between AIE and the packing mode in the solid state, molecular dynamics calculations of BAHDS-1, BAHDS-2 and BAHDS-4 were performed. Fig. 7(a) shows the results of the molecular dynamics calculation of the packing of BAHDS-1. In Fig. 7, the main intermolecular interactions existed in O⋯H, and the distance between O and H was 2.774 Å and 2.467 Å, respectively (Fig. 7(a)). The intermolecular interactions forced a tortuosity of the molecules, which prevented the face-to-face packing mode, and caused AIE enhancement: I/I0 was 66.6 (Table 1). Fig. 7(b) shows the results of the molecular dynamics calculation of BAHDS-2. The intermolecular hydrogen bonds found in BAHDS-2 (Fig. 7(b)) were different from those found in BAHDS-1. The distances between O and H were 1.858 Å and 1.818 Å. The intermolecular hydrogen bonding interaction inhibited the intramolecular rotation,46–48 which benefited the AIE enhancement. Similar to in BAHDS-1, face-to-face stacking was not found. Both of the two reasons above increased the AIE effect (I/I0 = 228.4 (Table 1)). Because of the similarity of the structures, we did not calculate the packing mode of BAHDS-3. We held that the mechanism of AIE enhancement was similar to that of BAHDS-2 (I/I0 = 190.4). In the packing mode of BAHDS-4, intermolecular hydrogen bonding interactions were also found and the distances of O⋯H were 1.879 Å, 1.955 Å, 1.875 Å and 1.826 Å, respectively. Strong hydrogen bonds in BAHDS-4 held the structure of BAHDS-4 more securely, thus the bond vibration decreased and AIE increased (I/I0 = 140.0). | ||
| Fig. 7 Packing modes of BAHDS-1 (a), BAHDS-2 (b) and BAHDS-4 (c) computed by a MD method. | ||
3.4 Living cell imaging
Live cell imaging of A549 was obtained at 450 nm (λ = 380 nm) using an inverted fluorescence microscope. The cells displayed a bright blue fluorescence image. The fluorescence images are depicted in Fig. 8. The fluorescence and bright-field images revealed a high cell membrane permeability for BAHDS-1. | ||
| Fig. 8 Fluorescence images of A549 treated with 50 μg mL−1 BAHDS-1 under visible light (left) and UV irradiation (right). | ||
To confirm the formation of aggregates inside the cells, a small amount of BAHDS-1 was added to the cell culture fluid to simulate a cell environment, and a dynamic light scattering (DLS) measurement was conducted. As it is shown in Fig. 9, the average diameter of the particle was 391.5 nm, which indicated the formation of aggregates inside the cells. BAHDS-1 could probably permeate the cell membrane in the solution state, and aggregate inside the cell. These results demonstrated that BAHDS-1 was cell membrane permeable and can also be used for the imaging of cells.49–51
 | ||
| Fig. 9 DLS data for BAHDS-1 in the cell culture fluid. | ||
As for intracellular imaging applications, the cytotoxicity of the probe should be taken into consideration. A MTT assay was performed to evaluate the toxicity of BAHDS-1 to A549 cells. 8–16–24 h incubation was performed under the same conditions. The experimental data are expressed in Table 3. BAHDS-1 exhibited little cytotoxicity to the cells, and at a concentration of 20 μM, the cell viability was still more than 85%. This result demonstrates that BAHDS-1 has a good biocompatibility.
| Concentration of BAHDS-1 (×10−5 mol L−1) | Cell survival | ||
|---|---|---|---|
| 8 h | 16 h | 24 h | |
| 0 | 99% | 98% | 98% |
| 0.5 | 98% | 98% | 96% |
| 1 | 97% | 95% | 94% |
| 1.5 | 95% | 92% | 90% |
| 2 | 93% | 89% | 85% |
4. Conclusions
BAHDS derivatives were designed and synthesized, and their structures were characterized using NMR, IR, and element analysis. An investigation of their photo-physical properties displayed strong AIE effects of BAHDS in a THF/water mixture solvent. The maximum enhancement was 228.4 times that of in pure THF. Fluorescence microscope images showed blue fluorescence in a THF/water mixture solvent, which attributed to the formation of the aggregate state. The mechanism of aggregation induced emission (AIE) was supported by molecular dynamics calculations. The results depicted that the packing mode and intermolecular hydrogen bonds inhibited intramolecular rotation and prevented π–π stacking, which caused the AIE effect. Living cell imaging showed that the BAHDS derivatives had good cell permeability and distinct blue fluorescence in A549 living cells. They were successfully applied to live cell staining, which demonstrated their value for practical applications in physiological systems.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51079094) and the Natural Science Foundation of Jiangsu Province (No. BK2010215). The Project was funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- C. C. Coleman, A. B. Epstein and R. J. Ebert, J. Phys. Chem. Solids, 1993, 11, 1497–1500 CrossRef.
- A. M. Sarker, A. Mejiritski, B. R. Wheaton and D. C. Neckers, Macromolecules, 1997, 30, 2268–2273 CrossRef CAS.
- Y. M. Dong, D. Navarathne, A. Bolduc, N. McGregor and W. G. Skene, J. Org. Chem., 2012, 77, 5429–5433 CrossRef CAS PubMed.
- H. J. Cho, K. Seo, C. J. Lee, H. Yun and J. Y. Chang, J. Mater. Chem., 2003, 13, 986–990 RSC.
- A. Igarashi, K. Arimitsu, T. Seki and K. Ichimura, J. Mater. Chem., 2008, 18, 560–566 RSC.
- B. D. Lourdes, N. David Reinhoudt and C. C. Mercedes, Chem. Soc. Rev., 2007, 36, 993–1017 RSC.
- Y. C. Li, X. L. Li, X. Y. Cai, D. C. Chen, X. Liu, G. Z. Xie, Z. H. Wang, Y. C. Wu, C. C. Lo, A. Lien, J. B. Peng, Y. Cao and S. J. Su, J. Mater. Chem. C, 2015, 3, 6986–6996 RSC.
- H. S. P. Rao and A. Desai, RSC Adv., 2014, 4, 63642–63649 RSC.
- G. J. Chang, L. Yang, S. Y. Liu, R. X. Lin and J. S. You, Polym. Chem., 2015, 6, 697–702 RSC.
- Y. Takagi, T. Kunishi, T. Katayama, Y. Ishibashi, H. Miyasaka, M. Morimoto and M. Irie, Photochem. Photobiol. Sci., 2012, 11, 1661–1665 CAS.
- J. L. Geng, K. I. Li, D. Ding, X. H. Zhang, W. Qin, J. Z. Liu, B. Z. Tang and B. Liu, Small, 2012, 8, 3655 CrossRef CAS PubMed.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- C. D. Geddes and J. R. Lakopwicz, Advanced Concepts in Fluorescence Sensing, Springer, Norwell, 2005 Search PubMed.
- H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365–377 CrossRef CAS.
- X. L. Yang, G. J. Zhou and W. Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- X. L. Yang, X. B. Xu and G. J. Zhou, J. Mater. Chem. C, 2015, 3, 913–944 RSC.
- Y. H. Chen and D. G. Ma, J. Mater. Chem., 2012, 22, 18718–18734 RSC.
- U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471–1507 RSC.
- L. Duan, L. D. Hou, T. W. Lee, J. Qiao, D. Q. Zhang, G. F. Dong, L. D. Wang and Y. Qiu, J. Mater. Chem., 2010, 20, 6392–6407 RSC.
- B. Huang, Z. H. Yin, X. X. Ban, Z. M. Ma, W. Jiang, W. W. Tian, M. Yang, S. H. Ye, B. P. Lin and Y. M. Sun, J. Lumin., 2016, 172, 7–13 CrossRef CAS.
- Y. L. Lv, Y. X. Hu, J. H. Zhao, G. W. Zhao, Y. Qi, X. Li, H. J. Chi, Y. Dong, G. Y. Xiao and Z. S. Su, Opt. Mater., 2015, 49, 286–291 CrossRef CAS.
- G. W. Zhao, Y. X. Hu, H. J. Chi, Y. Dong, G. Y. Xiao, X. Li and D. Y. Zhang, Opt. Mater., 2015, 47, 173–179 CrossRef CAS.
- B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- B. Z. Tang, X. Zhan, G. Yu, P. P. S. Lee, Y. Liu and D. Zhu, J. Mater. Chem., 2001, 11, 2974 RSC.
- J. W. Chen, Z. L. Xie, J. W. Y. Lam, C. C. W. Law and B. Z. Tang, Macromolecules, 2003, 36, 1108–1117 CrossRef CAS.
- Z. J. Zhao, S. M. Chen, J. W. Y. Lam, P. Lu, Y. C. Zhong, K. S. Wong, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 2221–2223 RSC.
- H. Tong, Y. Hong and Y. Dong, J. Phys. Chem. B, 2007, 118, 2000–2007 CrossRef PubMed.
- B. K. An, S. K. Kwon and S. D. Jung, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- H. Xi, C. X. Yuan, Y. X. Li, Y. Liu and X. T. Tao, CrystEngComm, 2012, 14, 2087 RSC.
- C. X. Yuan, X. T. Tao, L. Wang, J. X. Yang and M. H. Jiang, J. Phys. Chem. C, 2009, 113, 6809–6814 CAS.
- X. F. Mei, G. X. Wen, J. W. Wang, H. M. Yao, Y. Zhao, Z. H. Lin and Q. D. Ling, J. Mater. Chem. C, 2015, 3, 7267 RSC.
- S. A. Svarovsky, R. H. Simoyi and S. V. Makarov, J. Phys. Chem. B, 2001, 105, 12634 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.02, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Oxford University Press, New York, 1987 Search PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- Z. Zhao, S. Chen, X. Shen, F. Mahtab, Y. Yu, P. Lu, J. W. Y. Lam, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 686 RSC.
- R. Katoh, S. Katoh, A. Furube, K. Tokumaru and M. Kotani, J. Phys. Chem. C, 2009, 113, 2961 CAS.
- Z. Zhao, S. Chen, J. W. Y. Lam, P. Lu, Y. Zhong, K. S. Wong, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 2221 RSC.
- M. Sarma, T. Chatterjee, S. Ghanta and S. K. Das, J. Org. Chem., 2012, 77, 432–444 CrossRef CAS PubMed.
- X. Q. Zhang, M. Y. Liu, B. Yang, X. Y. Zhang and Y. Wei, Colloids Surf., B, 2013, 112, 81–86 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- T. Y. Han, W. Wei, J. Yuan, Y. Duan, Y. P. Li, L. Y. Hu and Y. P. Dong, Talanta, 2016, 150, 104–112 CrossRef CAS PubMed.
- Y. L. Xu, W. Yang, J. Shao, W. Q. Zhou, W. Zhu and J. Xie, RSC Adv., 2014, 4, 15400 RSC.
- W. Yang, Z. Q. Cheng, Y. L. Xu, J. Shao, W. Q. Zhou, J. Xie and M. Y. Li, New J. Chem., 2015, 39, 7488 RSC.
- J. Cao, C. C. Zhao, X. Z. Wang, Y. F. Zhang and W. H. Zhu, Chem. Commun., 2012, 48, 9897–9899 RSC.
| This journal is © The Royal Society of Chemistry 2016 |