DOI:
10.1039/C6RA02405J
(Paper)
RSC Adv., 2016,
6, 25819-25828
Design, synthesis and biological evaluation of novel triazole-core reversal agents against P-glycoprotein-mediated multidrug resistance†
Received
26th January 2016
, Accepted 1st March 2016
First published on 2nd March 2016
Abstract
We designed and synthesized a novel series of P-glycoprotein (P-gp)-mediated multidrug resistance (MDR) inhibitors bearing a triazolphenethyl–tetrahydroisoquinoline scaffold through click chemistry. Then those synthesized compounds were tested on doxorubicin-resistant erythroleukemia K562/A02 cells in a series of experiments. The result is that most of the synthesized compounds exhibit more active MDR reversal activity than verapamil (VRP). However, among them, compound 11 exhibits less cytotoxicity (IC50s > 100 μM), but more potency than the known P-gp inhibitors WK-X-34 and VRP on increasing anticancer drug accumulation in K562/A02 cells. Moreover, this compound can not only maintain a longer chemo-sensitizing effect (>24 h) than VRP (<6 h) with reversibility, but also significantly reverses MDR in a dose-dependent manner. Therefore, considering the low intrinsic cytotoxicity but strong reversal activity in vitro, compound 11 may be a promising lead with a potent and safe resistance modulator in combinational cancer chemotherapy.
1 Introduction
Tumor chemotherapy is an important tool in the treatment of metastatic cancer. However, drug resistance is regarded as the major barrier to successful chemotherapy in cancer patients, a phenomenon known as multidrug resistance (MDR).1,2 The resistance to therapy is caused by the over-expression of membrane-associated transport proteins named ATP-binding cassette (ABC) transporters, including P-glycoprotein (P-gp/ABCB1), breast cancer resistance protein (BCRP/ABCG2), and multidrug resistance protein 1 (MRP/ABCC1), which has been recognized as resulting in the increased efflux of chemotherapeutic drugs from cancer cells.3,4 Among them, P-glycoprotein (P-gp) plays the most critical role in MDR6,5 which is a 170 kDa plasma membrane protein encoded by the MDR1 gene7.6 To overcome MDR, using specific inhibitors to direct inhibition of P-gp should be regarded as a suitable and attractive approach.7
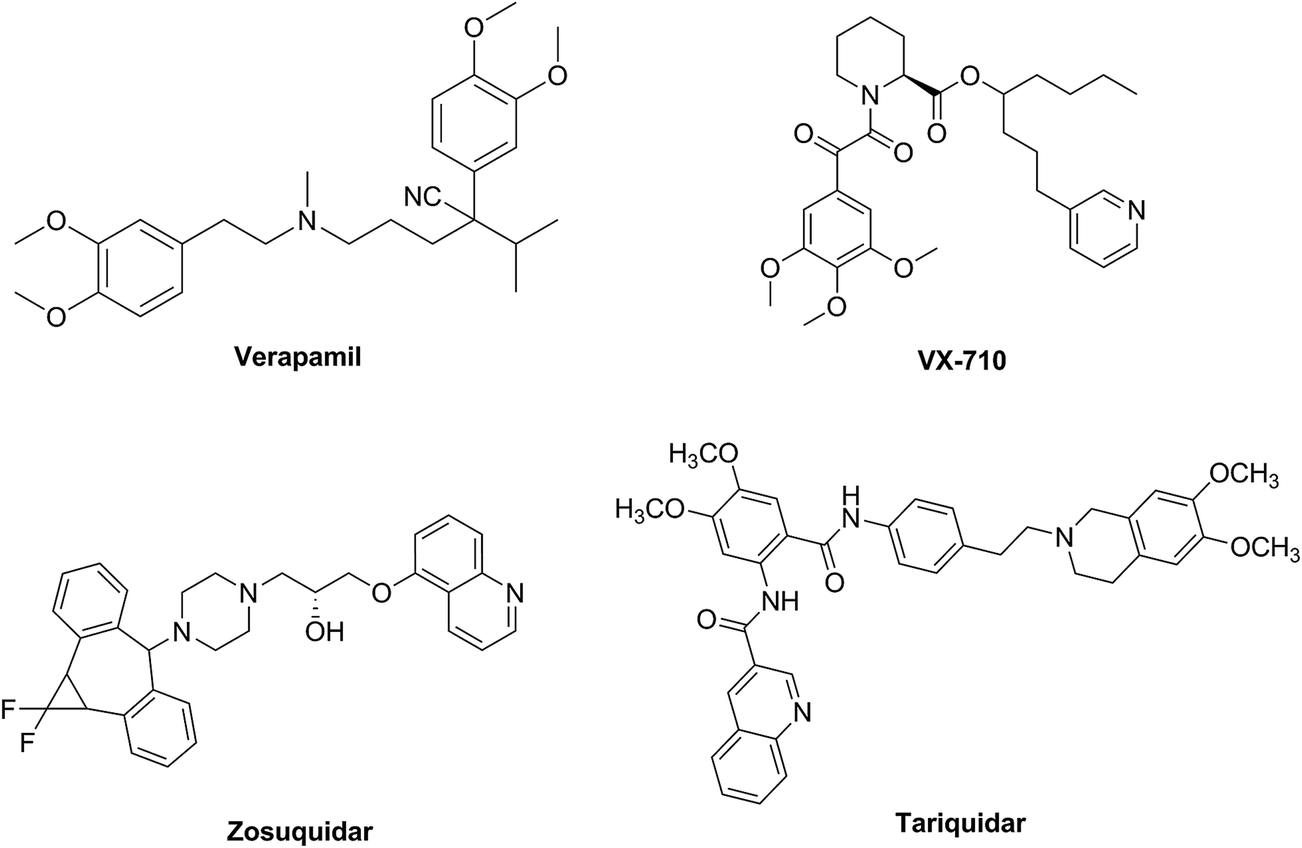
During the past three decades, in order to reverse the MDR in cancer cells, three generations of P-gp inhibitors have been developed such as verapamil,8 VX-710,9 zosuquidar (LY335979)10 and tariquidar (XR9576)11 (Fig. 1). However, because of many factors, none of the P-gp inhibitors has been approved for clinical application, such as unsatisfactory toxicity, low selectivity, poor potency or adverse pharmacokinetic interaction with anticancer drugs,12,13 which also lead to that, up to now, no P-gp inhibitor has been approved by regulatory agencies.14 Therefore, the discovery of safe and effective P-gp inhibitors still remains to be a huge challenge in this field.
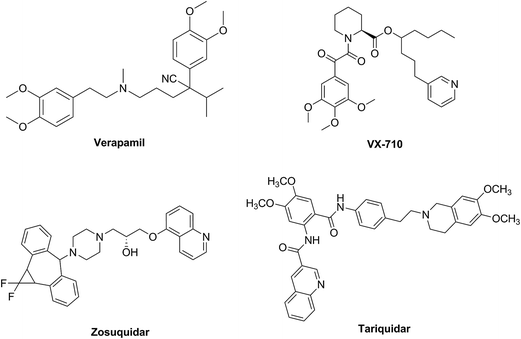 |
| | Fig. 1 Structures of verapamil, VX-710, zosuquidar and tariquidar. | |
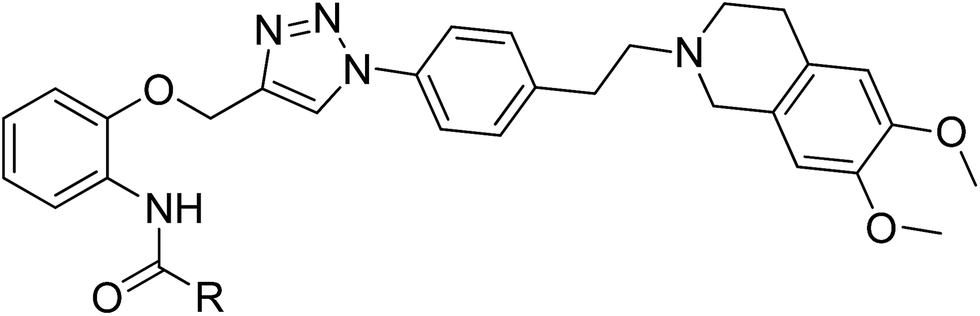
For designing novel P-gp inhibitors with high activity and low toxicity, we selected the tetrahydroisoquinolinethylphenylamine–based P-gp inhibitor WK-X-34 as a lead compound, which was designed and synthesized by researchers from the University of Bonn through CADD (Computer-Aided Drug Design) and methodical combinatorial chemistry. WK-X-34 is one of the third-generation of P-gp inhibitors, which not only exhibits little competitive effect against combination therapy drugs, but also shows relative high cytotoxicity (IC50 < 10 μM).15 Therefore, through click chemistry method, amide group is replaced by 1,2,3-triazole ring, which is a hydrophobic aromatic group, and can associate with biological targets through hydrogen bonding (Fig. 2).6,17 Click chemistry, which commonly employs the 1,3-dipolar cycloaddition combined with the synthetic design of triazoles and tetrazoles, has been widely applied in drug discovery.18 On the basis of principles above, we designed and synthesized a series of P-gp-mediated MDR reversal agents. Herein, we report the biological activity of newly synthesized P-gp-mediated MDR reversal agent.
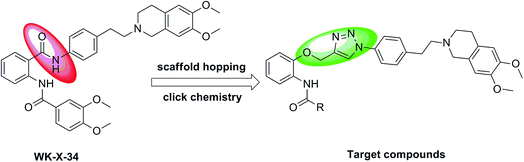 |
| | Fig. 2 Structure of WK-X-34 and design of the target compounds. | |
2 Chemistry
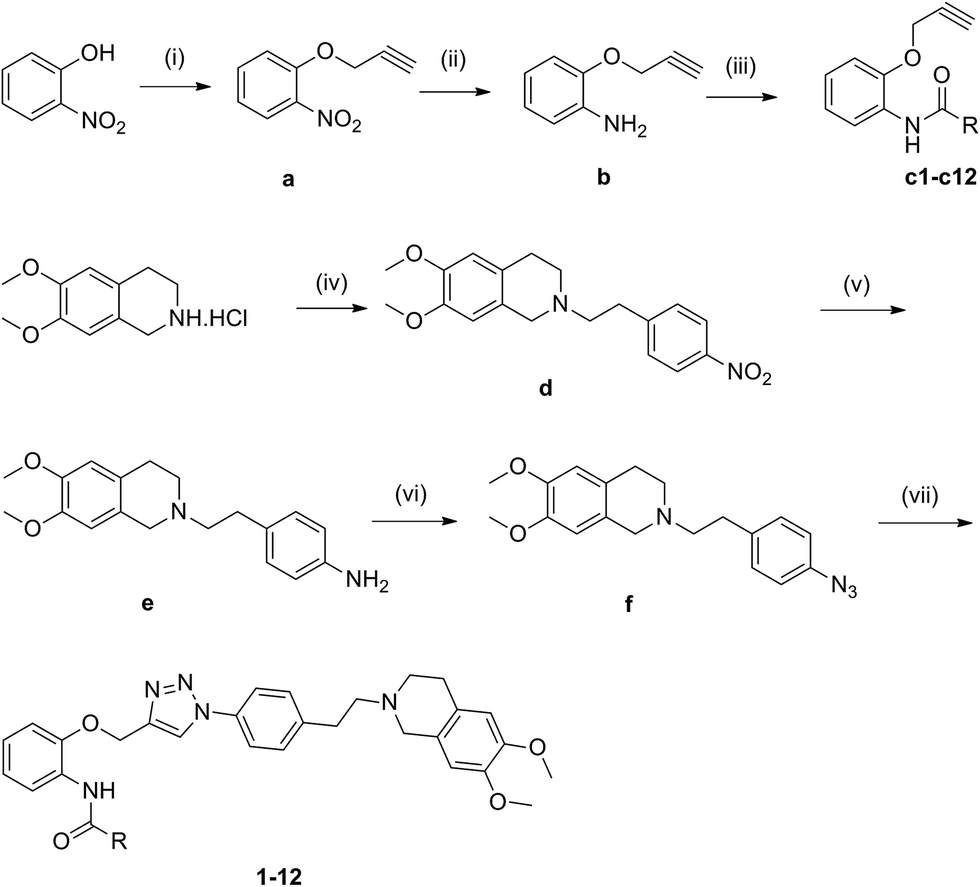
Compounds 1–12 were prepared starting from 2-nitrophenol and 3,4-dimethoxyphenethylamine, and the synthetic routes of target compounds were outlined in Scheme 1. Firstly, 2-nitrophenol and 3-bromo-1-propyne were refluxed in the mixture of acetone and potassium carbonate for 4 h affording compound a. Secondly, compound a reacted with ammonium chloride in the presence of iron powder in 80% ethanol affording compound b, which then reacted with freshly formed aromatic acyl chloride and triethylamine in dry dichloromethane to give compounds c1–c12. Compounds d and e were synthesized according to literature procedures with minor modification.16 Subsequently, compounds e reacted with sodium azide affording compound f. Last, compound f reacted with diverse alkyne c1–c12 through click chemistry to provide compounds 1–12. The structures of target compounds obtained were listed in Table 1.
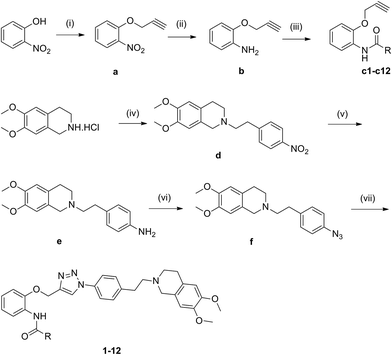 |
| | Scheme 1 Synthesis of the target compounds. Reagents and conditions: (i) 3-bromoprop-1-yne, K2CO3, acetone, reflux, 3 h; (ii) Fe/NH4Cl, 80% EtOH, reflux, 2.5 h; (iii) heterocyclic aromatic acyl chlorides, TEA/DCM, r.t., 24 h; or heterocyclic aromatic formic acids, EDC·HCl/DMAP/DCM, r.t., 48 h; (iv) 1-(2-bromoethyl)-4-nitrobenzene, K2CO3, acetonitrile, reflux, 17 h; (v) H2/Pd/C, DCM/EtOH, r.t., 24 h; (vi) NaNO2, 50% AcOH, 0–5 °C, 30 min; NaN3, 0–5 °C, 50 min; (vii) c1–c12, ascorbate sodium, CuSO4, 75% CH3OH, 24–48 h. | |
Table 1 Cytotoxicity of the target compounds against K562 and K562/A02 cell linesa
3 Results and discussion
3.1 Cytotoxicity of the target compounds
To identify ideal P-gp inhibitors reversing MDR at non-toxic concentrations, MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) assay was adopted to evaluate the intrinsic cytotoxicity of the target compounds against the human erythroleukemia K562 cells and the adriamycin (ADM)-resistant K562/A02 cells, which shows MDR due to P-gp overexpression (induced by ADM) and has been proved as useful in the quantification and characterization of MDR reversal by modulators of the MDR phenotype.19,20 We chose verapamil (VRP) as the positive control, which is a well-known classical P-gp inhibitor. As shown in Table 1, the majority of the target compounds displays low intrinsic cytotoxicity since their IC50 values are at a high micromolar level. As expected, K562/A02 cells are significantly resistant to ADR: K562/A02 (IC50 for ADM of 71.04 ± 3.2 μM) displaying about 151.9-fold higher than K562 cells (IC50 of 0.47 ± 0.03 μM). In contrast, compound 5 and 7 is found to exhibit high toxicity (IC50 < 25 μM) in both cell lines, VRP has weak cytotoxic effect toward K562 and K562/A02 cells with IC50 values 41.6 and 37.2 μM and WK-X-34 displaying a high level of toxicity toward K562 cells (IC50 of 6.9 ± 1.9 μM) but a weak toxicity toward K562/A02 cells (IC50 of 33.7 ± 2.7 μM). Based on the IC50s, most of target compounds possess low cytotoxicity in tested cell lines and are suitable candidates for the development of P-gp inhibitors.
3.2 Effects of the target compounds on reversing ADM resistance in K562/A02 cells
Based on the preliminary experiment, most of target compounds possess low cytotoxicity in tested cell lines. The reversal effects of target compounds on the MDR phenotype have been examined in K562/A02 cells at 0.5μM-concentrations by MTT assay. As the results summarized in Table 2, anticancer drug ADM alone displays little inhibitory effect on the survival of K562/A02 cells (IC50 of 71.04 ± 3.2 μM). However, the reversal fold (RF) data indicates that all target compounds and VRP increase the inhibitory effect of different doses. It reveals that all of the target compounds possess MDR reversal activity. Moreover, most of the target compounds exhibits more active MDR reversal activity than the classical P-gp inhibitor VRP, when co-administered with ADM at the same condition. Notably, compound 11 shows the strongest reversal activity among the synthesized compounds (IC50 of 1.76 ± 0.3 μM).
Table 2 ADM-resistance reversal activity of the target compounds 1–12 at 5 μM concentration in K562/A02 cellsa
| Compounds |
IC50 of ADM (μM) |
RF |
| The IC50 value was determined after exposure to a series of ADM concentration with different target compounds at 5 μM using K562/A02 cells. Reversal fold (RF) refers to fold-change in drug sensitivity. RF = (IC50 without modulator)/(IC50 with 5 μM modulator). 0.1% DMSO was added as solvent control for testing the P-gp modulating activity. |
| 1 |
13.36 ± 1.0 |
5.3 |
| 2 |
8.95 ± 0.5 |
7.9 |
| 3 |
4.76 ± 0.4 |
14.9 |
| 4 |
4.12 ± 0.3 |
17.2 |
| 5 |
13.42 ± 1.4 |
5.3 |
| 6 |
15.06 ± 0.9 |
4.7 |
| 7 |
4.4 ± 0.3 |
16.1 |
| 8 |
4.21 ± 0.2 |
16.9 |
| 9 |
16.92 ± 1.3 |
4.2 |
| 10 |
4.05 ± 0.2 |
17.5 |
| 11 |
1.76 ± 0.3 |
40.4 |
| 12 |
8.96 ± 0.7 |
7.9 |
| WK-X-34 |
2.31 ± 0.3 |
30.8 |
| VRP |
17.13 ± 1.7 |
4.1 |
| Control |
71.04 ± 3.2 |
1.0 |
3.3 Chemo-sensitizing effect of target compounds
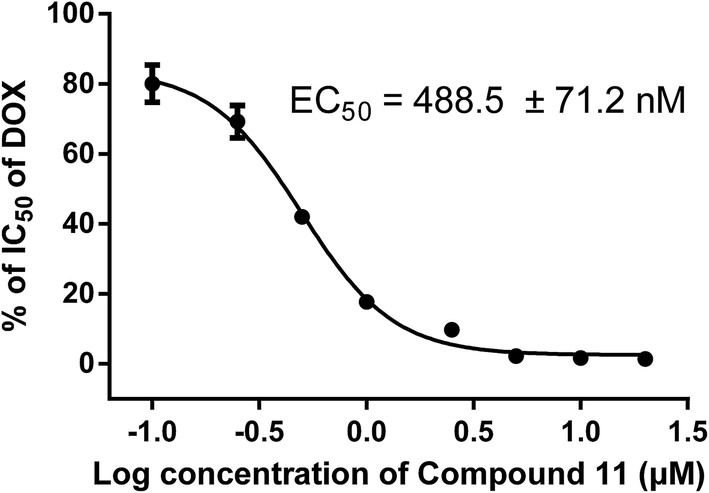
Based on the results of preliminary experiments, we selected the most potent compound 11 for further study. To further investigate the reversal potency and dose–response effects, the cytotoxicity of ADM against K562/A02 cells (P-gp-overexpression) was evaluated in the presence of the compound 11 at various concentrations (0.10 μM, 0.25 μM, 0.50 μM, 1.00 μM, 2.50 μM, 5.00 μM, 10.00 μM, 20.00 μM) by MTT assay.21 As the results summarized in Table 3, compound 11 showed apparent dose dependent activity and still exhibited potent MDR reversal activity (RF = 4.6) when the concentration decreased to 1.00 μM. Additionally, the EC50 value of compound 11 was 488.5 ± 71.2 nM (Fig. 3), which was analyzed by GraphPad Prism 5.0 software from the dose–response curves. Those results indicated that compound 11 significant potentiated the cytotoxicity of ADM in K562/A02 cells in a dose-dependent manner.
Table 3 Sensitization of compound 11 on reversing MDR towards K562/A02 cells at different concentrationsa
| Compounds 11 |
IC50 of ADM (μM) |
RF |
| Data were analyzed with GraphPad Prism 5.0 software and are the mean ± SD for 3 independent tests. The IC50 of ADM was 82.21 ± 3.7 μM. Reversal fold (RF), RF = (IC50 without modulator)/(IC50 with modulator). |
| 0.1 |
80.07 ± 9.3 |
1.0 |
| 0.25 |
69.21 ± 8.00 |
1.2 |
| 0.5 |
42.00 ± 1.16 |
2.0 |
| 1 |
17.75 ± 1.7 |
4.6 |
| 2.5 |
9.75 ± 0.25 |
8.4 |
| 5 |
2.26 ± 0.05 |
36.3 |
| 10 |
1.65 ± 0.03 |
49.9 |
| 20 |
1.41 ± 0.01 |
58.4 |
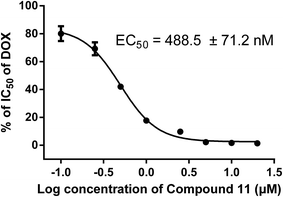 |
| | Fig. 3 EC50 for the compound 11 in K562/A02 cells. Various concentrations (0.10, 0.25.0.50, 1.00, 2.50, 5.00, 10.00, 20.00 μM) of compound 11 and ADM were added to 96-well plates and the cells were incubated for 48 h. The absorption values were measured with a Microplate Reader at 490 nm. Data were analyzed with GraphPad Prism 5.0 software and are the mean ± SD for 3 independent tests. | |
3.4 Duration of MDR reversal effect of compound 11 toward ADM in K562/A02 cells
It is known that an ideal P-gp inhibitor should possess a relatively long duration of action with reversibility for safe and effective therapy of P-gp-mediated MDR cancers.22 Therefore, we used VRP and WK-X-34 as positive controls to evaluate the duration of MDR reversal effect of compound 11. The experiment was executed as previous report with minor modification.21 K562/A02 cells were incubated with 5 μM of compound 11, VRP and WK-X-34 for 24 h, and then we washed compounds with PBS (phosphate buffered saline). At different time points (0, 6, 12 or 24 h), after removing the compounds, we added ADM in various concentrations to the culture, and incubated for additional 48 h. The results, examined by MTT assay, were shown in Table 4: after pre-incubation with VRP, WK-X-34 and compound 11, the IC50s of ADM towards K562/A02 cells were 19.32 μM, 2.32 μM, 2.11 μM respectively (no wash group). The MDR-reversing effect of VRP (RF = 1.6) waned immediately after its removal from the medium and totally disappeared after 6 h. In contrast, compound 11 and WK-X-34 showed a very strong reversal activity even after removed from the medium for 24 h, and the IC50s of ADM were 32.41 μM (RF = 2.5) and 12.91 μM (RF = 6.2), respectively. Moreover, compound 11 displayed potent chemo-sensitizing effect and persisted for longer time compared with the positive control VRP. The data also indicated that the MDR reversal effect of compound 11 towards K562/A02 cells was reversible.
Table 4 Duration of MDR reversal in K562/A02 cells after incubation and washout of P-gp inhibitora
| Treatment schedule |
IC50/ADM [μM] (RF) |
| Control |
VRP (5 μM) |
WK-X-34(5 μM) |
Compound 11(5 μM) |
| Numbers in parentheses, reversal fold (RF), RF = (IC50 without modulator)/(IC50 with modulator). Each experiment was carried out two to three times, and the values were presented as the mean ± standard error of mean. Nd: not determined. |
| No wash |
79.86 ± 2.25(1) |
19.32 ± 1.3(3.7) |
2.32 ± 0.3(34.4) |
2.11 ± 0.2(37.8) |
| Wash, 0 h |
Nd |
51.17 ± 6.3(1.6) |
3.71 ± 0.3(21.5) |
4.31 ± 0.3(18.5) |
| Wash, 6 h |
Nd |
Nd |
6.43 ± 0.2(12.4) |
9.42 ± 0.7(8.5) |
| Wash, 12 h |
Nd |
Nd |
9.65 ± 0.5(8.3) |
17.17 ± 1.6(4.7) |
| Wash, 24 h |
Nd |
Nd |
12.91 ± 0.7(6.2) |
32.41 ± 1.9(2.5) |
3.5 Accumulation of ADM
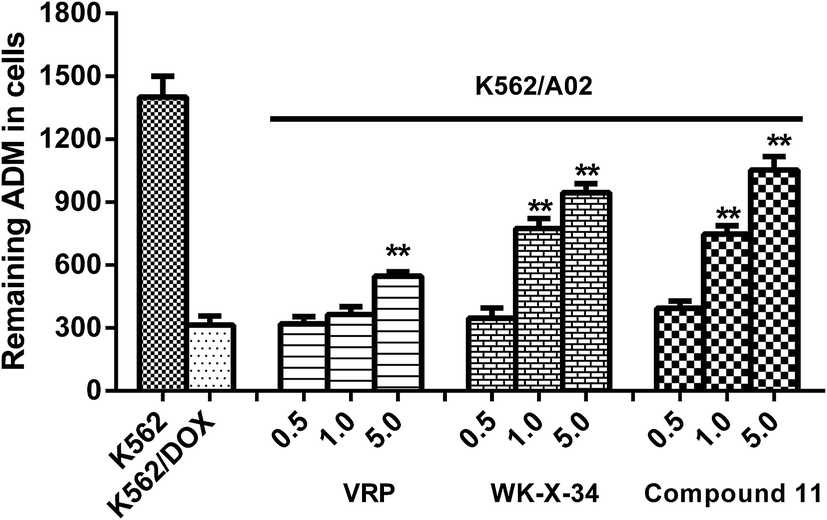
Furthermore, in order to investigate the compound 11 can reverse P-gp-mediated drug efflux, we selected ADM as a fluorescent probe, which was a fluorescent substrate of P-gp and could be used to monitor drug accumulation in cells,23 and measured the fluorescence intensity of ADM through spectrofluorometry according to the previous report with minor modification. As shown in Fig. 4, the P-gp inhibitor VRP and WK-X-34 were selected as positive controls. The ADM-sensitive K562 cells retained most of the fluorescence ADM, while the P-gp-overexpressing K562/A02 cells showed comparatively low intracellular level of ADM. The fluorescence intensity of ADM in K562/A02 cells, treated with compound 11, VRP or WK-X-34, was obviously increased in a dose-dependent manner. In comparison, compound 11 showed a higher level of ADM accumulation than VRP at the same dose, and has similar reversal activity compared with WK-X-34. The results indicated that compound 11 exhibited similar potency with WK-X-34 at different concentrations and more potent than VRP in inhibiting the ADM transport activity of P-gp.
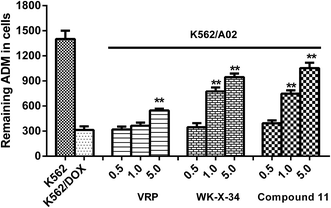 |
| | Fig. 4 Effect of compound 11 on intracellular ADM accumulation in K562/A02 cells. 0.1% DMSO was used as negative control. VRP and WK-X-34 were chosen as positive controls. The mean fluorescence intensity of retained intracellular ADM was estimated by spectrofluorometry. Data were expressed as means ± S.D. of 3 independent experiments. *P < 0.05, **P < 0.01 vs. untreated K562/A02 cells. | |
3.6 Inhibitory effect of compound 11 on P-gp efflux function
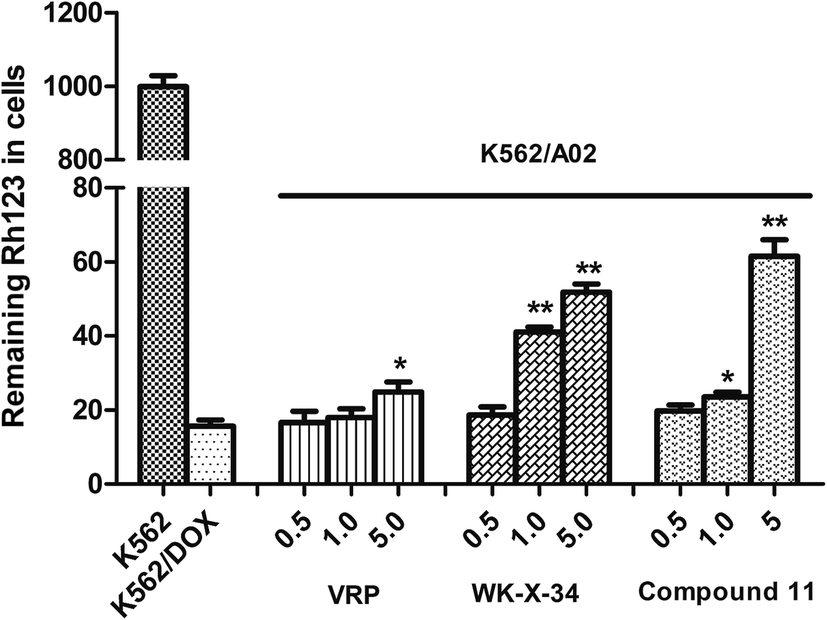
In order to further validate our assumption, we selected rhodamine-123 (Rh123) as another fluorescent probe, which was a well-known fluorescent substrate of P-gp and could be used to investigate whether compound 11 could reverse P-gp-mediated drug efflux by targeting P-gp. It is known that P-gp transporter can rapidly reduce Rh123 in the intracellular accumulation through an increase of Rh123 efflux from cells, which overexpresses on the adriamycin-selected K562/A02 cells. The level of Rh123 was measured by monitoring its fluorescence intensity through flow cytometry assay according to the method described in literatures with minor modification.24,25 As shown in Fig. 5, the P-gp inhibitor VRP and WK-X-34 were selected as positive controls. The adriamycin-sensitive K562 cells retained most of the fluorescence Rh123, while the P-gp-overexpressing K562/A02 cells showed comparatively low intracellular level of Rh123. In comparison, treated with compound 11, VRP or WK-X-34, the fluorescence intensity of Rh123 in K562/A02 cells was obviously increased in a dose-dependent manner. Particularly, compound 11 showed a higher level of Rh123 accumulation than VRP at the same dose, and had similar reversal activity compared with WK-X-34. The results indicated that compound 11 was targeting P-gp and could effectively block the drug efflux.
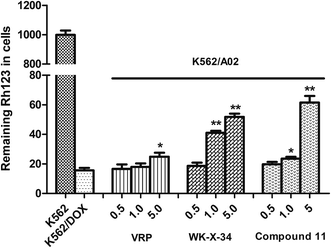 |
| | Fig. 5 Inhibitory effect of compound 11 on the efflux of Rh123. K562 or K562/A02 cells were incubated with 5 μM Rh123 for 60 min. Then various concentrations of compound 11, WK-X-34 or VRP (1.0, 2.5, 5 μM) was added and the cells were incubated for a further 90 min. Afterwards the cells were washed 3 times in ice-cold PBS. The mean fluorescence intensity of retained intracellular Rh123 was estimated by flow cytometry. Data were expressed as means ± S.D. of 3 independent experiments. *P < 0.05, **P < 0.01 vs. untreated K562/A02 cells. | |
4 Conclusions and future directions
In summary, we selected WK-X-34 as a lead compound, and twelve compounds were newly designed and synthesized based on the click chemistry. All synthesized compounds were evaluated in vitro for MDR chemo-sensitizing effects by using the classical P-gp inhibitor VRP as a positive control. Most of target compounds possessed low cytotoxicity and exhibited more active MDR reversal activity than VRP, especially for compound 11, which showed the strongest reversal activity and could effectively prevent the ADM efflux function of P-gp and increased the accumulation of ADM in K562/A02 cells. Therefore, compound 11 could be served as a promising candidate to develop P-gp-mediated MDR reversal modulator in cancer chemotherapy and further intensive investigations are still in progress.
5 Experimental section
5.1 General chemistry
All reagents and solvents were reagent grade and were used without further purification unless otherwise stated. Column chromatography was carried out on silica gel or alumina (200–300 mesh). Melting points were measured using a Mel-TEMP II melting point apparatus, which was uncorrected. All of the target compounds were analyzed by 1H NMR, 13C NMR (Bruker ACF-300Q, 300 MHz), MS (1100 LC/MSD spectrometer; Hewlett–Packard) and IR (Nicolet Impact 410); thin-layer chromatography (TLC) was performed on GF/UV 254 plates and the chromato-grams were visualized under UV light at 254 and 365 nm. Compounds d and e were prepared as previously described.16 The starting compound 2-nitrophenol was commercially available.
5.2 Synthesis of 1-nitro-2-(prop-2-yn-1-yloxy)benzene (a)
The mixture of 2-nitrophenol (11.2 g, 80 mmol), 3-bromoprop-1-yne (9.52 g, 80 mmol), and potassium carbonate (22.1 g, 160 mmol) in acetone (120 mL) was heated to reflux for 3 h. The reaction mixture was cooled to 0 °C followed by filtration and concentration to afford 12.6 g compound a. Yield: 89.1%. Yellow powder, mp: 66–68 °C.
5.3 Synthesis of 2-(prop-2-yn-1-yloxy)aniline (b)
The mixture of a (12.4 g, 70 mmol), ammonium chloride (18.7 g, 350 mmol) and iron powder (11.7 g, 210 mmol) in 80% ethanol (150 mL) was heated to reflux for 3.5 h. After cooling to room temperature, the mixture was adjusted with sodium carbonate to pH = 7–8, filtered with celite, and concentrated under vacuum to afford a brown residue. Dichloromethane (50 mL) was added into the residue and the resulted solution was washed by saturated aqueous Na2CO3 (30 mL) and brine (30 mL) in sequence. The organic layer was dried by anhydrous Na2SO4. The solvent was evaporated to afford 8.40 g compound b as brown oil. Yield: 81.7%.
5.4 General procedure for the preparation of c1–c12
Synthesis of c3, c6, c8: to the solution of compound b (0.74 g, 5 mmol) and triethylamine (0.84 g, 6 mmol) in dichloromethane (20 mL), heterocyclic aromatic acyl chloride (5 mmol) in dichloromethane (15 mL) was added at 0 °C, and then, the mixture was stirred at room temperature for 24 h. The reaction solution was washed by 3% hydrochloric acid (2 × 20 mL), saturated aqueous Na2CO3 (20 mL), and brine (20 mL) in sequence. The organic layer was dried by anhydrous Na2SO4. The solvent was evaporated to afford the desired product c3, c6 and c8. Synthesis of other compounds: to the solution of compound b (0.74 g, 5 mmol) and heterocyclic aromatic formic acid (5 mmol) in dichloromethane (20 mL), EDC·HCl (1.44 g, 7.5 mmol) in dichloromethane (15 mL) was added, and then, the mixture was stirred at room temperature for 24 h. The reaction solution was washed by 3% hydrochloric acid (2 × 20 mL), saturated aqueous Na2CO3 (20 mL), and brine (20 mL) in sequence. The organic layer was dried by anhydrous Na2SO4. Then, filtered and evaporated to afford the desired product compound c1–c12. Yield: 45.7–86.3%.
5.5 Synthesis of 2-(4-azidophenethyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (f)
Compound e (1.9 g, 6.1 mmol) was dissolved in 50% acetic acid (30 mL). To this solution, sodium nitrite (0.6 g, 8.5 mmol) was slowly added at −5 to 0 °C within 30 min. The solution was vigorously stirred at 0–5 °C for 50 min. Sodium azide (0.6 g, 9.2 mmol) was batch added into the reaction mixture at 0–5 °C. The resulting solution was stirred at 0–5 °C for 1 h followed by diluting with ice water (200 mL) and extracting with EtOAc (3 × 100 mL). The combined organic layer was washed with water (2 × 60 mL), saturated aqueous NaHCO3 (2 × 60 mL) and brine (2 × 50 mL), dried over anhydrous Na2SO4, filtered and concentrated to afford 1.2 g pink solid. Yield 58.2%. mp: 68–70 °C.
5.6 General procedure for the preparation of compounds 1–12
To the solution of c1–c12 (1 mmol) and f (1 mmol) in 75% methanol (40 mL), ascorbate sodium (30 mg) and CuSO4 (10 mg) were added, respectively. The reaction solution was stirred at room temperature for 24–48 h. After filtration, the solvent was evaporated and the crude product was purified by silica gel column chromatography using a mixture of ethylacetate/methanol (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as eluent to give the desire product with high purity. The purity of each tested compounds (>95%) was determined on a Waters UPLC/MS instrument using a Waters C18 column (1.7 μm 2.1 × 50 mm, with a flow rate of 0.2 mL min−1 and detection at 254 nm) employing a 10–90% acetonitrile/water/0.1% formic acid gradient.
:1, v/v) as eluent to give the desire product with high purity. The purity of each tested compounds (>95%) was determined on a Waters UPLC/MS instrument using a Waters C18 column (1.7 μm 2.1 × 50 mm, with a flow rate of 0.2 mL min−1 and detection at 254 nm) employing a 10–90% acetonitrile/water/0.1% formic acid gradient.
5.6.1 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)picolinamide (1). Yield 62.3%; white powder; mp: 164–165 °C. 1H NMR (300 MHz, DMSO-d6) δ: 10.52 (s, 1H, CONH), 8.94 (s, 1H, NCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 8.58 (d, J = 3.2 Hz, 1H, ArH), 8.45 (d, J = 7.5 Hz, 1H, ArH), 8.17 (d, J = 7.4 Hz, 1H, ArH), 8.03–8.11 (m, 1H, ArH), 7.78 (d, J = 7.8 Hz, 2H, ArH), 7.63 (d, J = 5.1 Hz, 1H, ArH), 7.49 (d, J = 7.7 Hz, 2H, ArH), 7.39 (d, J = 7.8 Hz, 1H, ArH), 7.15–7.05 (m, 2H, ArH), 6.64 (d, J = 7.5 Hz, 2H, ArH), 5.45 (s, 2H, OCH2), 3.70 (s, 6H, 2 × OCH3), 3.32 (s, 2H, ArCH2N), 2.90–2.71 (m, 8H, 4 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 152.7, 151.3, 148.2, 147.6, 146.7, 144.4, 139.4, 137.5, 134.8, 134.8, 134.0, 128.2, 127.7, 127.7, 126.8, 126.7, 126.5, 124.2, 122.1, 121.2, 120.3, 119.1, 112.8, 111.4, 108.0, 72.3, 59.7, 58.0, 56.3, 56.1, 33.4, 27.3; ESI-MS m/z: 590.7 [M + H]+; IR (KBr, cm−1) ν: 3321.63, 1682.10, 1253.90 (–CONH–), 2926.25, 1227.37 (–OCH3), 1601.04, 1533.16, 1519.15, 1454.96, 759.28, 689.13.
C), 8.58 (d, J = 3.2 Hz, 1H, ArH), 8.45 (d, J = 7.5 Hz, 1H, ArH), 8.17 (d, J = 7.4 Hz, 1H, ArH), 8.03–8.11 (m, 1H, ArH), 7.78 (d, J = 7.8 Hz, 2H, ArH), 7.63 (d, J = 5.1 Hz, 1H, ArH), 7.49 (d, J = 7.7 Hz, 2H, ArH), 7.39 (d, J = 7.8 Hz, 1H, ArH), 7.15–7.05 (m, 2H, ArH), 6.64 (d, J = 7.5 Hz, 2H, ArH), 5.45 (s, 2H, OCH2), 3.70 (s, 6H, 2 × OCH3), 3.32 (s, 2H, ArCH2N), 2.90–2.71 (m, 8H, 4 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 152.7, 151.3, 148.2, 147.6, 146.7, 144.4, 139.4, 137.5, 134.8, 134.8, 134.0, 128.2, 127.7, 127.7, 126.8, 126.7, 126.5, 124.2, 122.1, 121.2, 120.3, 119.1, 112.8, 111.4, 108.0, 72.3, 59.7, 58.0, 56.3, 56.1, 33.4, 27.3; ESI-MS m/z: 590.7 [M + H]+; IR (KBr, cm−1) ν: 3321.63, 1682.10, 1253.90 (–CONH–), 2926.25, 1227.37 (–OCH3), 1601.04, 1533.16, 1519.15, 1454.96, 759.28, 689.13.
5.6.2 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)isonicotinamide (2). Yield 72.9%; white powder; mp: 112–114 °C; 1H NMR (300 MHz, DMSO-d6) δ: 9.84 (s, 1H, CONH), 8.82 (s, 2H, ArH), 7.86 (s, 2H, ArH, NCHC), 7.73 (d, J = 8.2 Hz, 4H, ArH), 7.48 (d, J = 8.3 Hz, 2H, ArH), 7.38 (d, J = 8.1 Hz, 1H, ArH), 7.25 (t, J = 7.1 Hz, 1H, ArH),7.04 (d, J = 7.5 Hz, 1H, ArH), 6.64 (d, J = 8.0 Hz, 2H, ArH), 5.34 (s, 2H, OCH2), 3.70 (s, 6H, 2 × OCH3), 3.55 (s, 2H, ArCH2N), 2.93–2.70 (m, 8H, 4 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 164.7, 152.7, 149.7, 148.2, 146.7, 144.4, 140.8, 139.4, 134.8, 134.8, 134.0, 128.2, 127.7, 127.7, 126.8, 126.5, 124.2, 121.7, 121.7, 121.2, 120.3, 119.1, 112.8, 111.4, 108.3, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 33.4, 27.3; ESI-MS m/z: 591.4 [M + H]+; IR (KBr, cm−1) ν: 3410.09, 1666.55, 1257.70 (–CONH–), 2938.05, 1226.55 (–OCH3), 1599.35, 1540.60, 1519.05, 1452.51, 1129.59, 750.87.
5.6.3 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)furan-2-carboxamide (3). Yield 56.1%; brown powder; mp: 136–138 °C; 1H NMR (300 MHz, DMSO-d6) δ: 9.16 (s, 1H, CONH), 8.90 (s, 1H, NCHC), 7.97 (d, J = 7.9 Hz, 1H, ArH), 7.88 (s, 1H, ArH), 7.82 (d, J = 8.2 Hz, 2H, ArH), 7.52 (d, J = 8.3 Hz, 2H, ArH), 7.36 (d, J = 8.1 Hz, 1H, ArH), 7.29 (d, J = 3.3 Hz, 1H, ArH), 7.17 (t, J = 6.7 Hz, 1H, ArH), 7.01 (t, J = 7.7 Hz, 1H, ArH), 6.74 (s, 1H, ArH), 6.70–6.68 (m, 2H, ArH), 5, 39 (s, 2H, OCH2), 3.98 (s, 2H, ArCH2N), 3.72 (s, 6H, 2 × OCH3), 3.33–2.73 (m, 8H, 4 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 162.7, 152.7, 148.2, 148.2, 146.7, 144.4, 143.8, 139.4, 134.8, 134.8, 134.0, 128.2, 127.7, 127.7, 126.8, 126.5, 124.2, 121.2, 120.3, 119.1, 115.3, 112.8, 111.7, 111.4, 108.3, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 33.4, 27.3. ESI-MS m/z: 580.9 [M + H]+; IR (KBr, cm−1) ν: 3404.13, 1655.59, 1254.53 (–CONH–), 2932.15, 1227.88 (–OCH3), 1583.32, 1538.77, 1518.35, 1452.78, 1130.76, 755.12.
5.6.4 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)furan-3-carboxamide (4). Yield 59.0%; brown powder; mp: 143–145 °C; 1H NMR (300 MHz, DMSO-d6) δ:9.22 (s, 1H, CONH), 8.83 (s, 1H, NCHC), 8.38 (s, 1H, ArH), 7.78–7.72 (m, 4H, ArH), 7.49 (d, J = 8.4 Hz, 2H, ArH), 7.35 (d, J = 8.0 Hz, 1H, ArH), 7.19 (t, J = 7.2 Hz, 1H, ArH), 7.03–6.95 (m, 2H, ArH), 6.64 (d, J = 7.7 Hz, 2H, ArH), 5.32 (s, 2H, OCH2), 3.69 (s, 6H, 2 × OCH3), 3.55 (s, 2H, ArCH2N), 2.93–2.71 (m, 8H, 4 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 164.7, 152.7, 151.1, 148.2, 146.7, 144.8, 144.4, 139.4, 134.8, 134.8, 134, 128.8, 128.2, 127.7, 127.7, 126.8, 126.5, 124.2, 121.2, 120.3, 119.1, 112.3, 111.4, 108.3, 107.9, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 33.4, 27.3; ESI-MS m/z: 580.3 [M + H]+; IR (KBr, cm−1) ν: 3115.04, 1665.08, 1257.55 (–CONH–), 2938.05, 1227.76 (–OCH3), 1599.26, 1542.37, 1452.95, 1129.80, 754.01.
5.6.5 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-1H-indole-3-carboxamide (5). Yield 49.6%; brown powder; mp: 157–159 °C; 1H NMR (300 MHz, DMSO-d6) δ: 11.82 (s, 1H, NH), 8.90 (s, 1H, CONH), 8.85 (s, 1H, NCHC), 8.17–8.13 (m, 2H, ArH), 7.94 (d, J = 7.8 Hz, 1H, ArH), 7.73 (d, J = 8.1 Hz, 2H, ArH), 7.48 (s, 1H, ArH), 7.46–7.43 (m, 2H, ArH), 7.35 (d, J = 8.0 Hz, 1H, ArH), 7.16–6.94 (m, 4H, ArH), 6.65 (d, J = 7.4 Hz, 2H, ArH), 5.37 (s, 2H, OCH2), 3.70 (s, 6H, 2 × OCH3), 3.35 (s, 2H, ArCH2N), 3.18–2.72 (m, 8H, 4 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 164.7, 152.7, 148.2, 146.7, 144.4, 139.4, 138.6, 134.8, 134.8, 134.0, 130.7, 128.2, 128.2, 127.7, 126.8, 126.5, 124.2, 121.8, 121.7, 121.2, 120.3, 119.1, 112.8, 112.0, 111.4, 111.1, 108.3, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 33.4, 27.3; ESI-MS m/z: 629.4 [M + H]+; IR (KBr, cm−1) ν: 3439.53, 1654.51, 1244.75 (–CONH–), 2938.05, 1229.02 (–OCH3), 1518.43, 1449.65, 1129.30, 747.89.
5.6.6 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)benzofuran-2-carboxamide (6). Yield 43.9%; brown powder; mp: 185–186 °C; 1H NMR (300 MHz, DMSO-d6) δ: 9.55 (s, 1H, CONH), 8.93 (s, 1H, NCHC), 8.00 (s, 1H, ArH), 7.65 (s, 4H, ArH), 7.54 (m, 4H, ArH), 7.05 (s, 4H, ArH), 6.64 (s, 2H, ArH), 5.40 (s, 2H, OCH2), 3.69 (s, 6H, 2 × OCH3), 2.90 (s, 2H, ArCH2N), 2.71 (m, 8H, 4 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 162.7, 157.2, 152.7, 150.0, 148.2, 146.7, 144.4, 139.4, 134.8, 134.8, 134.0, 128.2, 127.7, 127.7, 126.9, 126.8, 126.5, 124.7, 124.2, 123.3, 121.2, 120.9, 120.3, 119.1, 112.8, 111.5, 111.4, 108.9, 108.3, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 33.4, 27.3; ESI-MS m/z: 630.5 [M + H]+; IR (KBr, cm−1) ν: 3392.40, 1681.31, 1255.42 (–CONH–), 2908.55, 1224.85 (–OCH3), 1538.34, 1518.73, 1454.83, 1134.29, 753.85.
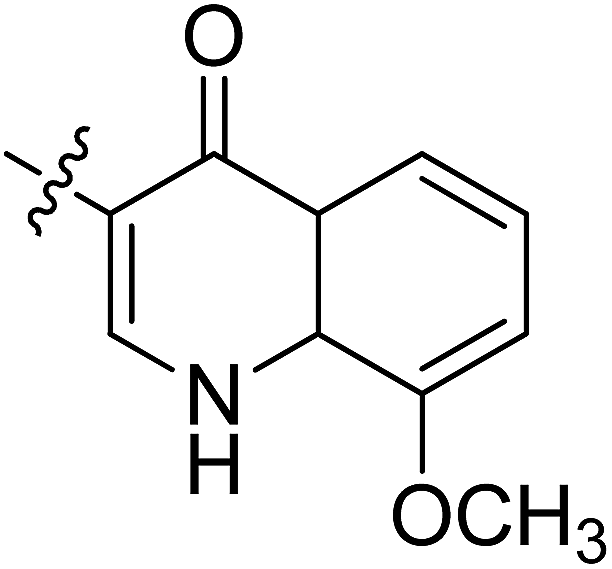
5.6.7 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxamide (7). Yield 61.9%; gray powder; mp: 139–141 °C; 1H NMR (300 MHz, DMSO-d6) δ:10.03 (s, 1H, CONH), 8.92 (s, 1H, NCHC), 8.87 (s, 1H, CH = C), 8.01 (d, J = 7.6 Hz, 1H, ArH), 7.78–7.69 (m, 4H, ArH), 7.46 (d, J = 8.2 Hz, 2H, ArH), 7.38–7.35 (m, 2H, ArH), 7.24 (t, J = 7.5, 1H, ArH), 7.06 (t, J = 7.6, 1H, ArH), 6.64 (d, J = 6.3 Hz, 2H, ArH), 5.33 (s, 2H, OCH2), 4.00 (s, 3H, OCH3), 3.70 (s, 6H, 2 × OCH3), 3.46 (s, 2H, ArCH2N), 2.94–2.71 (m, 8H, 4 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 174.5, 163.0, 156.7, 152.7, 148.2, 147.7, 146.7, 144.4, 139.4, 134.8, 134.8, 134.8, 134.0, 128.2, 127.7, 127.7, 126.8, 126.5, 124.2, 124.1, 121.2, 120.3, 119.1, 117.1, 112.8, 111.4, 108.3, 95.6, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 55.9, 54.0, 46.0, 33.4, 27.3; ESI-MS m/z: 686.7 [M + H]+; IR (KBr, cm−1) ν: 3445.43, 1670.84, 1270.60 (–CONH–), 2926.25, 1227.56 (–OCH3), 1519.42, 1452.95, 1118.97, 748.91.
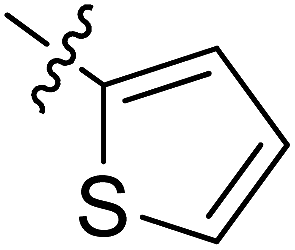
5.6.8 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)thiophene-2-carboxamide (8). Yield 76.9%; brown powder; mp: 160–162 °C; 1H NMR (300 MHz, DMSO-d6) δ: 9.57 (s, 1H, CONH), 8.84 (s, 1H, NCHC), 7.97 (d, J = 2.9 Hz, 1H, ArH), 7.83 (d, J = 4.9 Hz, 1H, ArH), 7.77 (d, J = 8.2 Hz, 2H, ArH), 7.70 (d, J = 7.7 Hz, 1H, ArH), 7.50 (d, J = 8.3 Hz, 2H, ArH), 7.36 (d, J = 8.1 Hz, 1H, ArH), 7.24–7.18 (m, 2H, ArH), 7.03 (d, J = 7.5 Hz, 1H, ArH), 6.70 (d, J = 9.4 Hz, 2H, ArH),5.35 (s, 2H, OCH2), 3.82 (s, 2H, ArCH2N), 3.72 (s, 6H, 2 × OCH3), 3.00–2.83 (m, 8H, 4 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 161.8, 152.7, 148.2, 146.7, 144.4, 139.4, 139.4, 134.8, 134.8, 134.0, 131.9, 130.3, 129.0, 128.2, 127.7, 127.7, 126.8, 126.5, 124.2, 121.2, 120.3, 119.1, 112.8, 111.4, 108.3, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 33.4, 27.3; ESI-MS m/z: 596.3 [M + H]+; IR (KBr, cm−1) ν: 3410.03, 1650.26, 1255.30 (–CONH–), 2926.25, 1228.89 (–OCH3), 1539.05, 1517.97, 1355.07, 1122.56, 743.12, 721.79.
5.6.9 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)thiophene-3-carboxamide (9). Yield 78.4%; brown powder; mp: 84–86 °C; 1H NMR (300 MHz, DMSO-d6) δ: 9.35 (s, 1H, CONH), 8.82 (s, 1H, NCHC), 8.34 (s, 1H, ArH), 7.78–7.72 (m, 3H, ArH), 7.65–7.59 (m, 2H, ArH), 7.48 (d, J = 8.2 Hz, 2H, ArH), 7.36 (d, J = 8.1 Hz, 1H, ArH), 7.20 (t, J = 7.1 Hz, 1H, ArH), 7.00 (t, J = 7.5 Hz, 1H, ArH), 6.64 (d, J = 7.6 Hz, 2H, ArH), 5.34 (s, 2H, OCH2), 3.70 (s, 6H, 2 × OCH3), 3.40 (s, 4H, 2 × CH2), 3.71 (s, 6H, 3 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 174.7, 152.7, 148.2, 146.7, 144.4, 139.4, 136.3, 134.8, 134.0, 129.0, 128.2, 127.7, 127.7, 127.6, 126.8, 126.6, 126.5, 124.2, 121.2, 120.3, 119.1, 112.8, 111.4, 108.3, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 33.4, 27.3; ESI-MS m/z: 597.1 [M + H]+; IR (KBr, cm−1) ν: 3091.45, 1658.70, 1257.22 (–CONH–), 2926.25, 1227.88 (–OCH3), 1533.09, 1517.75, 1128.25, 846.12, 751.56.
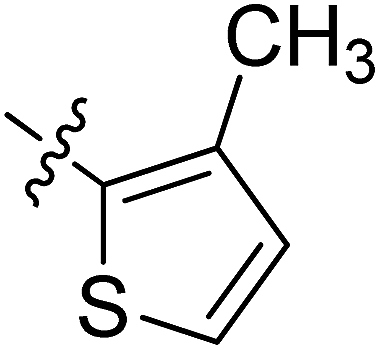
5.6.10 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-3-methylthiophene-2-carboxamide (10). Yield 52.4%; brown powder; mp: 163–165 °C; 1H NMR (300 MHz, DMSO-d6) δ: 8.92 (s, 1H, CONH), 8.83 (s, 1H, NCHC), 8.10 (d, J = 7.5 Hz, 1H, ArH), 7.78 (d, J = 7.3 Hz, 2H, ArH), 7.66 (d, J = 4.0 Hz, 1H, ArH), 7.49 (d, J = 7.1 Hz, 2H, ArH), 7.35 (d, J = 7.4 Hz, 1H, ArH), 7.17 (s, 1H, ArH), 7.04–6.99 (m, 2H, ArH), 6.64 (d, J = 6.9 Hz, 2H, ArH), 5.35 (s, 2H, OCH2), 3.93 (s, 6H, 2 × OCH3), 3.55 (s, 2H, CH2), 2.91 (s, 2H, CH2), 2.71 (s, 6H, 3 × CH2), 2.36 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 161.8, 152.7, 148.2, 146.7, 144.4, 139.4, 136.3, 135.0, 134.8, 134.8, 134.0, 129.2, 128.2, 127.7, 127.7, 126.8, 126.5, 124.5, 124.2, 121.2, 120.3, 119.1, 112.8, 111.4, 108.3, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 33.4, 27.3, 13.7; ESI-MS m/z: 610.8 [M + H]+; IR (KBr, cm−1) ν: 3427.73, 1638.00, 1251.86 (–CONH–), 2926.25, 1227.55 (–OCH3), 1536.27, 1518.12, 1125.48, 756.40, 743.31.
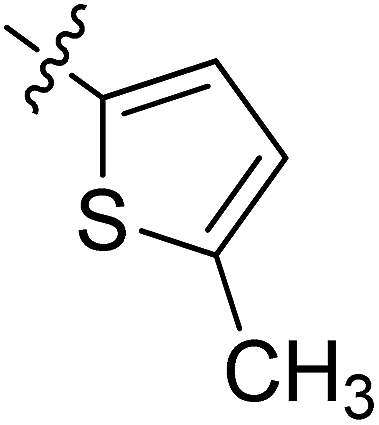
5.6.11 N-(2-((1-(4-(2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-methylthiophene-2-carboxamide (11). Yield 50.1%; brown powder; mp: 146–147 °C; 1H NMR (300 MHz, DMSO-d6) δ: 9.40 (s, 1H, CONH), 8.81 (s, 1H, NCHC), 7.74 (s, 4H, ArH), 7.49 (s, 2H, ArH), 7.34 (m, 1H, ArH), 7.20 (m, 1H, ArH), 7.01 (m, 1H, ArH), 6.88 (s, 1H, ArH), 6.65 (m, 2H, ArH), 5.34 (s, 2H, OCH2), 3.70 (s, 6H, 2 × OCH3), 3.56 (s, 2H, CH2), 2.90 (s, 2H, CH2), 2.71 (s, 6H, 3 × CH2), 2.46 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 161.8, 152.9, 152.7, 148.2, 146.7, 144.4, 139.4, 135.3, 134.8, 134.8, 134.0, 129.5, 128.5, 128.2, 127.7, 127.7, 126.8, 126.5, 124.2, 121.2, 120.3, 119.1, 112.8, 111.4, 108.3, 72.3, 59.7, 58.0, 56.3, 56.1, 33.4, 27.3, 14.5; ESI-MS m/z: 610.7 [M + H]+; IR (KBr, cm−1) ν: 3415.93, 1655.91, 1254.81 (–CONH–), 2932.15, 1227.35 (–OCH3), 1521.96, 1450.91, 1128.87, 738.78.
5.6.12 5-Chloro-N-(2-((1-(4-(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)thiophene-2-carboxamide (12). Yield 79.4%; brown powder; mp: 128–130 °C; 1H NMR (300 MHz, DMSO-d6) δ: 9.73 (s, 1H, CONH), 8.80 (s, 1H, NCHC), 7.86 (d, J = 2.8 Hz, 1H, ArH), 7.74 (d, J = 7.6 Hz, 2H, ArH), 7.61 (d, J = 7.5 Hz, 1H, ArH), 7.48 (d, J = 7.6 Hz, 2H, ArH), 7.37 (d, J = 7.9 Hz, 1H, ArH), 7.23 (t, J = 9.0 Hz, 2H, ArH), 7.02 (t, J = 7.5 Hz, 1H, ArH), 6.64 (d, J = 7.2 Hz, 2H, ArH), 5.33 (s, 2H, OCH2), 3.70 (s, 6H, 2 × OCH3), 3.56 (s, 2H, CH2), 2.90 (s, 2H, CH2), 2.71 (s, 6H, 3 × CH2); 13C NMR (75 MHz, DMSO-d6) δ: 161.8, 152.7, 148.2, 146.7, 144.4, 140.7, 139.4, 138.3, 135.9, 134.8, 134.8, 134.0, 129.8, 128.2, 127.7, 127.7, 126.8, 126.5, 124.2, 121.2, 120.3, 119.1, 112.8, 111.4, 108.3, 72.3, 59.7, 58.0, 56.3, 56.1, 56.1, 33.4, 27.3; ESI-MS m/z: 630.7 [M + H]+; IR (KBr, cm−1) ν: 3345.13, 1657.42, 1255.05 (–CONH–), 2926.25, 1229.30 (–OCH3), 1541.17, 1518.72, 1453.45, 1130.33, 745.36.
5.7 Materials and methods
5.7.1 Materials. Adriamycin (ADM), verapamil (VRP), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), and DMSO were purchased from Sigma-Aldrich (St. Louis, MO, USA). WK-X-34 with high purity was synthesized by our laboratory before.20 All other chemicals were molecular biology grade and obtained from Sigma-Aldrich or Thermo Fisher Scientific (Waltham, MA, USA).
5.7.2 Cell lines and cell culture. Human leukemia cell line K562 and its adriamycin-selected P-gp-overexpressing subline K562/A02 were kindly provided by Dr Yun-Man Li (Department of Physiology, China Pharmaceutical University, Nanjing, China). The cell lines were grown in RPMI-1640 medium containing 10% fetal bovine serum (FBS) and incubated at 37 °C in a humidified incubator with 5% CO2 in air growth. To maintain MDR phenotype, 1 mg mL−1 adriamycin was added to K562/A02 cultures and maintained in drug-free medium for 2 weeks before use. All experiments were performed with cells in exponential growth. To maintain MDR phenotype, 1 mg mL−1 adriamycin was added to the K562/A02 cultures and maintained in a drug-free medium for 2 weeks before use.
5.7.3 MTT assay. K562/A02 cells were incubated in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37 °C in a 5% CO2 humidified atmosphere. K562/A02 cells (1–2 × 104 cells per well) were seeded in 96-well plates. After 24 h incubation, cells were treated with various concentrations of ADM in absence or presence of target compounds for 48 h in an atmosphere of 95% air with 5% CO2 at 37 °C. Then, MTT was added directly to the cells. After additional incubation for 4 h at 37 °C, the absorbance at 570 nm was read on a microplate reader (Thermo Fisher Scientific). The IC50 values of the compounds for cytotoxicity were calculated by GraphPad Prism 5.0 software from the dose–response curves. Experiments were conducted in triplicates and repeated three times independently.
5.7.4 Duration of the MDR reversal. 1–2 × 104 K562/A02 cells per well were plated in 96-well plates and cultured overnight, then the cells were incubated for another 24 h with or without compound 11, VRP, WK-X-34 or PBS at the concentration of 5 μM before being washed 0 or 3 times with growth medium. Then, the cells were incubated for 0, 6, 12 or 24 h before the addition of various concentrations of ADM or vehicle. The incubation was continued for 48 h prior to the MTT analysis. The IC50 values of the compounds for cytotoxicity were calculated by GraphPad Prism 5.0 software from the dose–response curves.
5.7.5 Accumulation of ADM. ADM accumulation assay was performed according to the reported procedures with minor modification.26 In brief, 2 × 105 cells of K562 and K562/A02 were incubated with 20 μM ADM and different concentrations of compound 11 for 2.5 h at 37 °C. 0.1% DMSO was used as a negative control. VRP and WK-X-34 were used as positive controls. After incubation, the cells were washed with cold PBS and lysed with lysis buffer (0.75 M HCl, 0.2% Triton-X100 in isopropanol). The fluorescence level of ADM in the lysate was determined by fluorescence spectrophotometer (RF-5301 PC; Shimadzu) using an excitation and an emission wavelength pair of 460 and 610 nm. Accumulation was expressed as ADM fluorescent intensity (FI) per 2 × 105 cells. All incubations were carried out in triplicate and three independent experiments were performed.
5.7.6 Rhodamine123 efflux assay. K562 or K562/A02 cells were seeded into 24-well plates at 1.5 × 105/well and incubated with 5 μM Rh123 for 60 min before washing with ice PBS for three times. Then the cells were incubated with or without various concentrations of compound 11, WK-X-34 or VRP (1.0, 2.5, 5 μM) for another 90 min. Afterwards the cells were washed twice in ice-cold PBS. The mean fluorescence intensity of retained intracellular Rh123 was estimated by BD FACSCalibur flow cytometer.
Conflict of interest
All authors report no conflict of interest.
Acknowledgements
This work was supported by the National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2009ZX09102-033) and National Natural Science Foundation of China (NO. 81173088).
Notes and references
- T. Fojo and M. Menefee, Ann. Oncol., 2007, 18, v3–v8 CrossRef PubMed.
- P. D. Eckford and F. J. Sharom, Chem. Rev., 2009, 109, 2989–3011 CrossRef CAS PubMed.
- M. Susa, E. Choy, C. Yang, J. Schwab, H. Mankin, F. Hornicek and Z. Duan, J. Biomol. Screening, 2010, 15, 287–296 CrossRef CAS PubMed.
- G. Szakacs, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef CAS PubMed.
- A. Breier, L. Gibalova, M. Seres, M. Barancik and Z. Sulova, Anti-Cancer Agents Med. Chem., 2013, 13, 159–170 CrossRef CAS PubMed.
- B. I. Sikic, Oncology, 1999, 13, 183–187 CAS.
- C. Martelli, D. Alderighi, M. Coronnello, S. Dei, M. Frosini, B. Le Bozec, D. Manetti, A. Neri, M. N. Romanelli and M. Salerno, J. Med. Chem., 2009, 52, 807–817 CrossRef CAS PubMed.
- A. Mihályi, R. Gáspár, Z. Zalán, L. Lázár, F. Fülöp and P. A. de Witte, Anticancer Res., 2004, 24, 1631–1636 Search PubMed.
- F. Sharom, J. Membr. Biol., 1997, 160, 161–175 CrossRef CAS PubMed.
- P. Ruff, D. A. Vorobiof, J. P. Jordaan, G. S. Demetriou, S. D. Moodley, A. L. Nosworthy, I. D. Werner, J. Raats and L. J. Burgess, Cancer Chemother. Pharmacol., 2009, 64, 763–768 CrossRef CAS PubMed.
- J. Robert and C. Jarry, J. Med. Chem., 2003, 46, 4805–4817 CrossRef CAS PubMed.
- L. Zhang and S. Ma, ChemMedChem, 2010, 5, 811–822 CrossRef CAS PubMed.
- H. Y. Hung, E. Ohkoshi, M. Goto, K. Nakagawa-Goto and K. H. Lee, Bioorg. Med. Chem. Lett., 2012, 22, 7726–7729 CrossRef CAS PubMed.
- M. J. Henderson, M. Haber, A. Porro, M. A. Munoz, N. Iraci, C. Xue, J. Murray, C. L. Flemming, J. Smith, J. I. Fletcher, S. Gherardi, C. K. Kwek, A. J. Russell, E. Valli, W. B. London, A. B. Buxton, L. J. Ashton, A. C. Sartorelli, S. L. Cohn, M. Schwab, G. M. Marshall, G. Perini and M. D. Norris, J. Natl. Cancer Inst., 2011, 103, 1236–1251 CrossRef CAS PubMed.
- V. Jekerle, W. Klinkhammer, R. M. Reilly, M. Piquette-Miller and M. Wiese, Cancer Chemother Pharmacol, 2007, 59, 61–69 CrossRef CAS PubMed.
- R. Z. Pellicani, A. Stefanachi, M. Niso, A. Carotti, F. Leonetti, O. Nicolotti, R. Perrone, F. Berardi, S. Cellamare and N. A. Colabufo, J. Med. Chem., 2012, 55, 424–436 CrossRef CAS PubMed.
- M. Wijtmans, C. de Graaf, G. de Kloe, E. P. Istyastono, J. Smit, H. Lim, R. Boonnak, S. Nijmeijer, R. A. Smits, A. Jongejan, O. Zuiderveld, I. J. de Esch and R. Leurs, J. Med. Chem., 2011, 54, 1693–1703 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- X. Gu, Z. Ren, X. Tang, H. Peng, Q. Zhao, Y. Lai, S. Peng and Y. Zhang, Eur. J. Med. Chem., 2012, 51, 137–144 CrossRef CAS PubMed.
- B. Liu, Q. Qiu, T. Zhao, L. Jiao, J. Hou, Y. Li, H. Qian and W. Huang, Chem. Biol. Drug Des., 2014, 84, 182–191 CAS.
- X. Tang, X. Gu, Z. Ren, Y. Ma, Y. Lai, H. Peng, S. Peng and Y. Zhang, Bioorg. Med. Chem. Lett., 2012, 22, 2675–2680 CrossRef CAS PubMed.
- M. J. Newman, J. C. Rodarte, K. D. Benbatoul, S. J. Romano, C. Zhang, S. Krane, E. J. Moran, R. T. Uyeda, R. Dixon and E. S. Guns, Cancer Res., 2000, 60, 2964–2972 CAS.
- Y. Gong, Y. Wang, F. Chen, J. Han, J. Miao, N. Shao, Z. Fang and R. O. Yang, Leuk. Res., 2000, 24, 769–774 CrossRef CAS PubMed.
- Y.-Q. Shi, X.-J. Qu, Y.-X. Liao, C.-F. Xie, Y.-N. Cheng, S. Li and H.-X. Lou, Eur. J. Pharmacol., 2008, 584, 66–71 CrossRef CAS PubMed.
- B.-S. Ji, L. He and G.-Q. Liu, Life Sci., 2005, 77, 2221–2232 CrossRef CAS PubMed.
- K.-F. Chan, I. L. Wong, J. W. Kan, C. S. Yan, L. M. Chow and T. H. Chan, J. Med. Chem., 2012, 55, 1999–2014 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02405j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.