
Microwave-assisted one-pot synthesis of steroid–quinoline hybrids and an evaluation of their antiproliferative activities on gynecological cancer cell lines†
Ádám Baji‡
a,
András Gyovai‡b,
János Wölflinga,
Renáta Minoricsb,
Imre Ocsovszkic,
István Zupkó*b and
Éva Frank*a
aDepartment of Organic Chemistry, University of Szeged, Dóm tér 8., Szeged H-6720, Hungary. E-mail: frank@chem.u-szeged.hu; Fax: +36-62-544200; Tel: +36-62-544275
bDepartment of Pharmacodynamics and Biopharmacy, University of Szeged, Eötvös u. 6., Szeged H-6720, Hungary. E-mail: zupko@pharm.u-szeged.hu
cDepartment of Biochemistry, University of Szeged, Dóm tér 9, Szeged H-6720, Hungary
First published on 10th March 2016
Abstract
Novel D- and A-ring-fused quinolines in the estrone and 5α-androstane series were efficiently synthesized from the corresponding β-chlorovinyl aldehydes with different arylamines in DMF under microwave irradiation. The rates of the one-pot catalyst-free syntheses and the yields of the desired products were found to be affected significantly by the electronic and steric character of the substituents on the anilines and the different reactivities of rings D and A of the sterane skeleton. All the synthesized compounds were tested in vitro on human cervical (C33A, HeLa and SiHa) and breast (MCF-7, MDA-MB-231, MDA-MB-361 and T47D) cancer cell lines in order to investigate their antiproliferative activities in vitro. Evidence of cell cycle blockade and apoptosis induction was obtained for the most effective compound 14c by means of flow cytometry, caspase-3 activity determination and microscopic techniques.
1. Introduction
Hybrid compounds, which combine two or more structural entities in a single molecule, at least one part being a biologically active natural product, hold great promise for the design of novel pharmaceutical agents with better selectivity and diminished toxicity.1 Steroids are attractive building blocks for such hybrids in view of their widespread natural occurrence, their rigid sterane skeleton with a range of functionalization, their broad spectrum of bioactivity and their specific ability to penetrate cell membranes.2 Several molecular hybrids or ‘chimeras’ derived from steroids and other molecules through the domain integration of key functional elements or via covalent linkages were reported earlier.3 Among these strategies, the introduction of different heterocyclic systems either connected to or condensed with the sterane core offer an excellent possibility through which to modify the physicochemical properties of the parent compound4 and to optimize certain pharmacokinetic features. Hybridization of steroids often allows modulation of the binding ability and therefore the biological activity by altering the effect of the original molecule through action on another biological target.5 Consequently, in recent years considerable attention has been devoted to the synthesis and pharmacological evaluation of different ring A- and D-fused steroidal heterocycles containing pyrazol(in)e,6 (is)oxazol(id)ine,7 thiazole,8 pyri(mi)dine9 or triazole10 building blocks, a number of which have been reported to possess cell-growth-inhibitory, antibacterial, anti-inflammatory and other beneficial effects.In spite of the fact that quinolines and their derivatives are known to be very common structural motifs in various natural products, pharmacologically active synthetic compounds and clinically relevant drugs (Fig. 1),11 the incorporation of these moieties into the steroid backbone is rather rare12 and the pharmacological effects of these derivatives have not been deeply investigated. In this respect, Boruah et al. recently reported the synthesis of some quinolines condensed to ring D of the androstane and ring A of the cholestane skeleton, and tested their antimicrobial activities.13
 | ||
| Fig. 1 Structures of some quinoline-based pharmacologically active compounds. | ||
Since a number of sex-hormone-derived heterocyclic steroids exert cell-growth-inhibitory effects on malignant cell lines of diverse origins by disturbing the normal cell cycle and inducing apoptosis,8,14 and since a number of natural and synthetic quinoline derivatives are also well-known anticancer agents,15 integration of the quinoline and the sterane scaffolds may be of interest in a search for novel hybrid molecules with antiproliferative activity. In view of the absence of sufficient information concerning the exact mode of action of steroidal heterocycles due to the complexity of the apoptotic mechanisms and the different targets that may be affected, random searches still remain effective for the identification of new lead compounds.
Quinolines can be obtained by various synthetic routes, including the Skraup, Combes, Conrad–Limpach, Doebner–Miller, Friedlander, Povarov, Camps or Niementowski methods, all of which involve an initial and intermolecular reaction of an aniline derivative with a carbonyl-containing compound or a precursor thereof.16 Despite their versatility, these reactions suffer from certain disadvantages, such as a multistep character, or the need for harsh conditions, large amounts of promoters, expensive additives or difficult work-up procedures. Accordingly, there have been continuous efforts to develop clean and rapid novel protocols for the construction of quinoline-based structures. In recent years, considerable attention has been focused on the application of β-halovinyl aldehydes, which are easily accessible from α-methylene ketones by use of the Vilsmeier–Haack reagent, as useful synthons for the synthesis of polycyclic azaarenes.17 As regards the regioselective access to 2,3-disubstituted quinolines, β-halovinyl aldehydes react with 2 equivalents of anilines to afford N-aryleneaminoimine hydrohalogenide intermediates, which then undergo thermolysis in solution18 or under solvent-free conditions.13 On the other hand, the selective arylamination of β-chloro- or β-bromovinyl aldehydes can occur at position β on use of an equimolar amount of aniline derivative in the presence of a palladium catalyst in basic medium without any trace of imine formation.19 These results led several groups to develop a two-step procedure for the synthesis of N-arylaminovinyl aldehydes through a cross-coupling reaction and subsequent acid-catalyzed cyclization to quinolines. The one-pot version of the metal-catalyzed reaction has also been investigated, but a long reaction time proved to be needed for sufficient conversion under conventional heating,20 or by-products were formed under microwave conditions.21 Imines, readily available from β-halovinyl aldehydes under modified reaction conditions, were also found to be cyclized to quinolines.18,20 Despite the great potential of β-halovinyl aldehydes as organic synthons, their application in steroid chemistry is yet to be investigated.
In a continuation of our research on the synthesis of novel sex-hormone-derived heterocyclic steroids with potential antiproliferative activity, we now report a microwave-assisted catalyst-free one-pot approach to novel molecular hybrids integrating steroidal and substituted quinoline structural elements. All of the synthesized compounds were tested in vitro on three cervical and four breast cancer cell lines in order to determine their cell-growth-inhibitory potency. The most effective compound was subjected to additional in vitro experiments on T47D cells, including flow cytometric cell cycle analysis and fluorescence microscopy, in order to characterize the mechanism of its action.
2. Results and discussion
2.1. Synthetic studies
For the preparation of ring D-fused quinoline derivatives, estrone 3-methyl ether (1) was used as starting material. The simultaneous addition of POCl3 and DMF to an ice-cold solution of 1 in CHCl3 and subsequent reflux for 4 h afforded a β-chlorovinyl aldehyde (2), together with a 17-chloro derivative (3)23 as by-product (Table 1). The microwave-assisted solvent-free reaction of 2 with aniline (4a, 2 equiv.) at 140 °C for 10 min was first attempted, according to the method reported for the synthesis of similar ring D-fused quinolines in the androstane series by Boruah et al.,13 but the TLC control of the reaction indicated the presence of several compounds in the mixture, and the purification procedure proved extremely difficult. Consequently, the reaction was repeated with 1 equiv. of 4a in DMF, which was earlier found to be a suitable solvent for such reactions,18,22 and the solution was irradiated at 140 °C for 10 min. In this case, a significant conversion of 2 was observed by TLC, but an additional 10 min was needed for the disappearance of the starting material. Besides the main spot, however, the presence of a considerable amount of polar compounds could be noticed, and these were not transformed on further lengthening of the reaction time. After chromatographic purification, the desired ring D-fused quinoline 5a was obtained in a yield of 53% (Table 1, entry 1), which could not be improved either by the application of 4a in a 2-fold excess in solution or by prolonging the reaction time. Similar transformations of 2 with aniline 4b or 4c containing an electron-donating group (EDG) in the para position on the aromatic moiety likewise furnished the corresponding product 5b or 5c in moderate yield, but within 10 min (Table 1, entries 2 and 3). Interestingly, o-toluidine was not sufficiently reactive even at a longer (30 min) irradiation time, which suggested the steric hindrance of the ortho-CH3 substituent on the ring-closure reaction. At the same time, similar transformations also failed when anilines with an electron-withdrawing group (EWG), such as a halogen or nitro substituent, were used and only certain intermediates could be detected by TLC, which underwent decomposition during the purification process.
The different behavior of partially unsaturated rings A and D of the sterane core under identical reaction conditions has often been observed previously, which may be attributed to the higher rigidity and sterically more hindered character of the five-membered ring D, mainly due to the presence of the angular methyl group on C-13, as compared with the more flexible six-membered ring A.24 6-Methoxy-1-tetralone (6) was therefore used for further experiments as a model of the six-membered ring A (Table 2). β-Chlorovinyl aldehyde25 7 was prepared similarly to described for the synthesis of 2 with the Vilsmeier–Haack reagent, and was separated from the 4-chloro-1,2-dihydro-7-methoxynaphthalene by-product (8).26 The reactions of 7 with anilines (4a–f) containing either EDGs or EWGs resulted in the formation of benz[c]acridine derivatives (9a,13 9b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 and 9c–f) in varying yields, depending on the steric and electronic character of the substituents on the aromatic moiety. The possibility of the application of a lower temperature (120 °C) and a shorter reaction time to achieve the complete conversion of 7 indicated the higher reactivity of 7 relative to that of steroidal β-chlorovinyl aldehyde 2. Moreover, the substituent effect was found to be more pronounced in these cases. Thus, EDG-substituted anilines (4b and 4c) favored the reaction in comparison with unsubstituted 4a, and the corresponding products 9b and 9c were obtained in high yields after purification when a 2 min irradiation time was used (Table 2, entries 2 and 3). The isolated yields of the desired products (9e and 9f), however, were only moderate, even when the heating period was extended to 10 min when anilines containing electron-withdrawing halogens (4e and 4f) were reacted with 7 (Table 2, entries 5 and 6). During the reaction of 7 with 2-chloroaniline 4d, the additive electron-withdrawing and steric effects of the ortho substituent hampered the formation of 9d, resulting in a yield of only 35% within longer reaction time (Table 2, entry 4).
13 and 9c–f) in varying yields, depending on the steric and electronic character of the substituents on the aromatic moiety. The possibility of the application of a lower temperature (120 °C) and a shorter reaction time to achieve the complete conversion of 7 indicated the higher reactivity of 7 relative to that of steroidal β-chlorovinyl aldehyde 2. Moreover, the substituent effect was found to be more pronounced in these cases. Thus, EDG-substituted anilines (4b and 4c) favored the reaction in comparison with unsubstituted 4a, and the corresponding products 9b and 9c were obtained in high yields after purification when a 2 min irradiation time was used (Table 2, entries 2 and 3). The isolated yields of the desired products (9e and 9f), however, were only moderate, even when the heating period was extended to 10 min when anilines containing electron-withdrawing halogens (4e and 4f) were reacted with 7 (Table 2, entries 5 and 6). During the reaction of 7 with 2-chloroaniline 4d, the additive electron-withdrawing and steric effects of the ortho substituent hampered the formation of 9d, resulting in a yield of only 35% within longer reaction time (Table 2, entry 4).
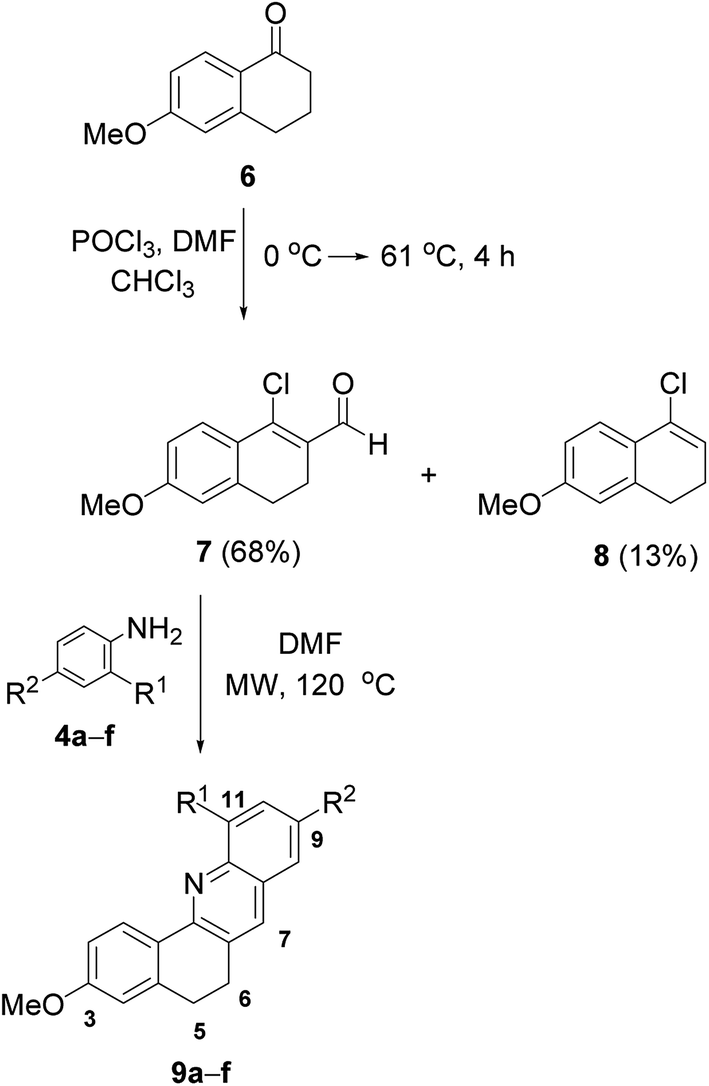
These results led us next to investigate similar reactions on ring A of the sterane skeleton. For this purpose, 17β-acetoxy-5α-dihydrotestosterone (10) was reacted with the Vilsmeier–Haack reagent in refluxing CHCl3 for 4 h, but bis-formylation occurred under these conditions to afford compound 1227 as main product (Table 3, method A), together with the minor formation of the desired β-chlorovinyl aldehyde 11.28 However, this latter compound could be obtained in 78% yield by performing the reaction at 25 °C for 2 h (Table 3, method B), and its reactions with differently substituted aniline derivatives (4a–i) were then carried out. The outcomes of the microwave-assisted transformations were quite similar to those observed for the reactions of 7. Anilines with EDGs (4b, 4c and 4i) readily underwent cyclization with 11 within 2 min to furnish ring A-fused quinolines (13b, 13c and 13i + 13i′) in excellent yields (Table 3, entries 2, 3 and 9), although the ortho position of the substituent in 4h had a certain steric effect resulting in a somewhat diminished yield of the corresponding quinoline 13h. The presence of EWGs on the aromatic moiety in 4e–g led to moderate yields of 13e–g (Table 3, entries 5–7), and for 4d with an ortho-Cl substituent the yield of 13d was further reduced (entry 4). Furthermore, two regioisomeric quinolines (13g and 13g′ or 13i and 13i′) were found to be produced in a ratio of about 3:1 when m-chloroaniline 4g or m-toluidine 4i was applied (Table 3, entries 7 and 9). The failure of quinoline formation was observed in the attempted reaction of 4-nitroaniline 4j, containing a strong EWG, with 11, and the isolated product was identified by NMR spectroscopy as being the imino–enol form of a β-arylaminovinyl aldehyde 15 (Table 3). A similar result was reported previously for the reaction between 3-nitroaniline and 3-chloro-2-methylbut-2-enal.29 In order to find evidence for the advantage of microwave irradiation, the reaction of 11 with 4b (1 equiv.) under conventional heating (120 °C, DMF) was also carried out. However, this latter method required a longer time (4 h instead of 2 min) for the formation of the corresponding quinoline 13b, with a lower yield (75% instead of 91%). The ring A-fused quinolines (13a–h and 13g′) were finally deacetylated in alkaline MeOH to give the 17-OH analogs (14a–h and 14g′), with the exception of 13i and 13i′, which could not be separated by column chromatography (Table 3). The structures of all the synthesized compounds were confirmed by 1H and 13C NMR measurements indicating the presence of the characteristic signals of the newly-formed quinoline ring in the aromatic range of the related spectra as compared with the corresponding β-chlorovinyl aldehydes.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Anilinea | R1 | R2 | R3 | Reaction time (min) | Product(s) | Yieldb (%) |
| a Equimolar amount.b After purification by column chromatography.c The 3:1 mixture of 13i and 13i′ determined by 1H NMR could not be separated by column chromatography. |
|||||||
| 1 | 4a | H | H | H | 5 | 13a | 76 |
| 2 | 4b | H | H | CH3 | 2 | 13b | 91 |
| 3 | 4c | H | H | OMe | 2 | 13c | 92 |
| 4 | 4d | Cl | H | H | 20 | 13d | 43 |
| 5 | 4e | H | H | Cl | 10 | 13e | 59 |
| 6 | 4f | H | H | Br | 10 | 13f | 60 |
| 7 | 4g | H | Cl | H | 10 | 13g + 13g′ | 46 + 16 |
| 8 | 4h | CH3 | H | H | 5 | 13h | 73 |
| 9 | 4i | H | CH3 | H | 2 | 13i + 13i′ | 90c |

The reactions of β-chlorovinyl aldehydes such as 2, 7, or 11 with aniline derivative 4 always lead to 2,3-disubstituted quinolines such as 5, 9 or 13 instead of the 3,4-disubstituted regioisomers 16 (Scheme 1), regardless of the intermediately formed species which depend on the applied conditions.18,29 Thus, transformations with 1 equiv. of aniline were earlier proposed to proceed through 1-chlorovinyl(N-aryl)imine A via a rather complex intermolecular cyclization process involving tetrahydropyrimidine intermediates to afford 2,3-disubstituted quinolines.22 However, imine A is more likely to undergo initial hydrolysis due to the inadvertent presence of moisture in the DMF, to generate a small amount of the aromatic primary amine 4, which in a subsequent reaction with A leads to the formation of N-arylenaminoimine hydrochloride B.18 This latter compound can cyclize through intermediate C to give the heterocyclic product 5, 9 or 13 in varying yield depending on the steric and electronic character of the substituent R in the aromatic moiety. Thus, EDGs facilitate the reaction of B to C, while the presence of EWGs is unfavorable from the aspect of the ring-closure step. The strong electron-withdrawing effect of the NO2 group disables the cyclization and the N-arylenaminoimine intermediate B is rather converted to aldehyde 15, which is more stable in its tautomeric imino–enol form.29 The presence of an ortho substituent on the aromatic amine and the vicinity of functional groups to the reaction center as well as the conformational flexibility of the affected sterane ring may exert significant influence on the quinoline formation.
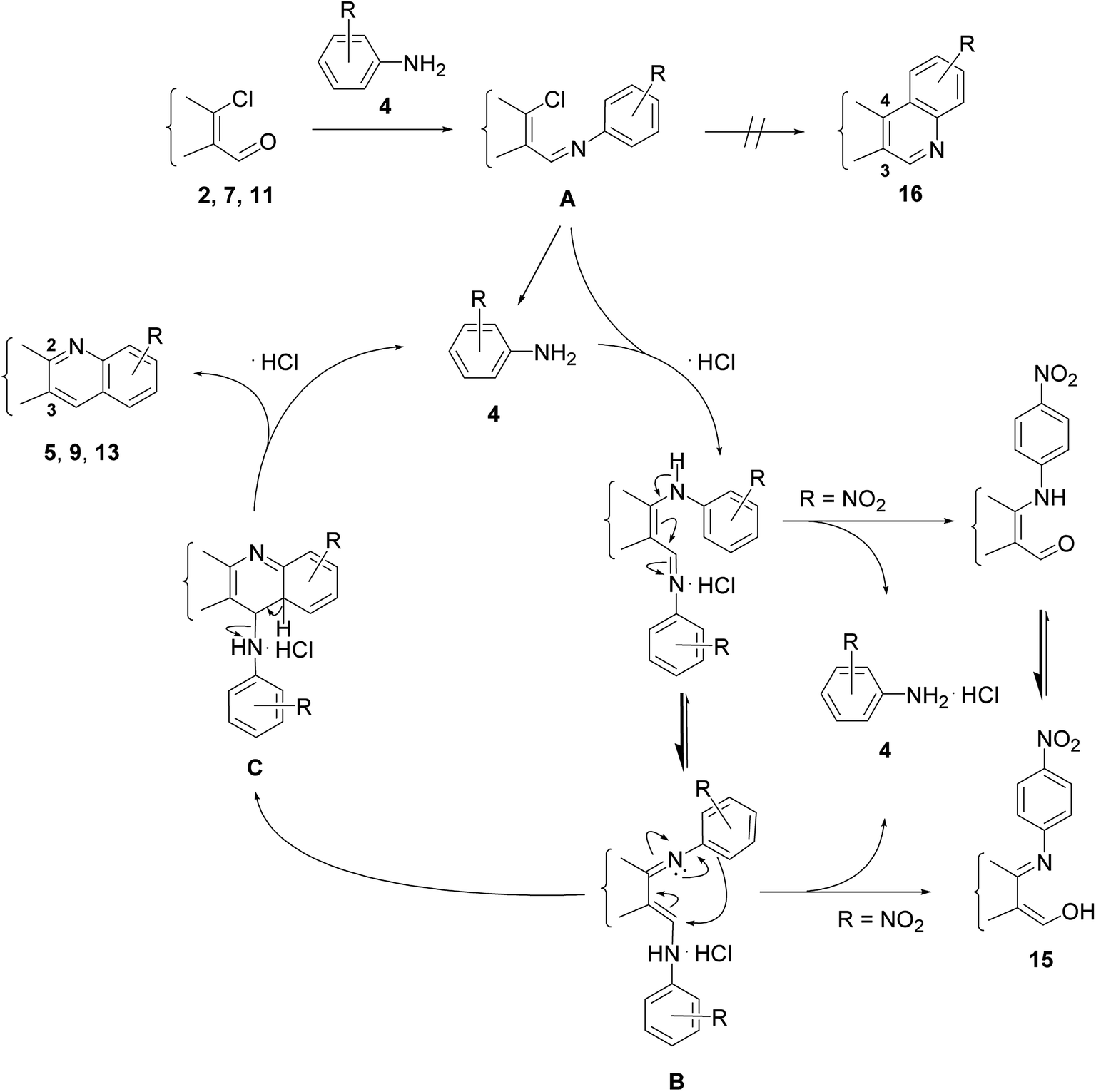 | ||
| Scheme 1 Explanation of substituent effects on the basis of the proposed mechanisms. | ||
2.2. Pharmacological studies
| Compound | Conc. (μM) | Growth inhibition% ± SEM [calculated IC50 value (μM)] | ||||||
|---|---|---|---|---|---|---|---|---|
| C33A | HeLa | SiHa | MCF7 | MDA-MB-231 | MDA-MB-361 | T47D | ||
| a Compounds eliciting less than 20% inhibition of proliferation were considered ineffective and the exact results are not given, for simplicity. n.d.: not determined. | ||||||||
| 5a | 10 | —a | 30.8 ± 1.2 | — | 26.0 ± 1.4 | — | — | — |
| 30 | 47.7 ± 0.5 | 59.5 ± 1.1, [21.2] | — | 51.2 ± 1.7, [29.6] | 37.3 ± 2.1 | 29.9 ± 1.9 | 37.2 ± 0.7 | |
| 5b | 10 | — | — | — | 21.9 ± 0.4 | — | 25.4 ± 1.3 | 32.9 ± 2.9 |
| 30 | 35.8 ± 1.0 | 46.5 ± 0.8 | — | 46.1 ± 0.6 | 29.5 ± 2.1 | 41.1 ± 0.7 | 48.1 ± 1.8 | |
| 5c | 10 | — | 28.3 ± 1.5 | — | 27.1 ± 1.9 | — | — | 32.0 ± 1.9 |
| 30 | 40.6 ± 1.0 | 43.9 ± 0.8 | 24.6 ± 1.7 | 57.0 ± 1.5, [24.1] | 45.0 ± 1.8 | 47.2 ± 3.7 | 63.0 ± 2.2, [18.9] | |
| 9a | 10 | — | — | — | — | — | — | — |
| 30 | — | — | — | 31.5 ± 2.2 | — | — | — | |
| 9b | 10 | — | — | 24.7 ± 1.4 | — | — | — | — |
| 30 | — | — | — | — | — | — | — | |
| 9c | 10 | — | — | — | — | — | — | — |
| 30 | — | 26.7 ± 1.9 | — | 35.6 ± 1.1 | — | — | — | |
| 9d | 10 | — | — | — | — | — | — | — |
| 30 | — | — | 25.6 ± 1.0 | 21.3 ± 2.5 | — | — | — | |
| 9e | 10 | — | 25.9 ± 2.3 | — | 35.3 ± 2.2 | — | — | — |
| 30 | — | 96.6 ± 0.4, [12.3] | — | 40.2 ± 2.1 | — | — | — | |
| 9f | 10 | — | — | — | — | — | — | — |
| 30 | — | 89.4 ± 0.7, [13.1] | 25.0 ± 2.6 | 26.4 ± 1.7 | — | 21.1 ± 2.8 | — | |
| 13a | 10 | — | — | — | — | — | — | — |
| 30 | — | 28.1 ± 1.2 | — | 38.6 ± 1.4 | — | — | — | |
| 13b | 10 | 37.1 ± 2.5 | — | — | — | — | — | — |
| 30 | 61.0 ± 1.8, [16.2] | — | — | 38.7 ± 2.8 | — | 27.5 ± 1.9 | — | |
| 13c | 10 | — | — | — | — | — | — | — |
| 30 | 38.9 ± 1.3 | 42.5 ± 1.1 | 35.2 ± 0.7 | 47.5 ± 0.7 | 71.2 ± 0.9, [21.5] | 28.5 ± 1.2 | 49.9 ± 1.1 | |
| 13d | 10 | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. |
| 30 | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | |
| 13e | 10 | — | 38.5 ± 0.7 | 28.0 ± 0.8 | 25.9 ± 0.9 | — | 45.2 ± 0.8 | — |
| 30 | 32.0 ± 2.5 | 34.0 ± 0.9 | 43.0 ± 1.5 | 37.0 ± 0.7 | 24.4 ± 2.7 | 66.0 ± 0.9, [12.6] | 48.2 ± 1.1 | |
| 13f | 10 | — | — | — | 28.3 ± 1.7 | — | 33.7 ± 1.7 | — |
| 30 | 57.9 ± 2.4, [26.5] | 48.0 ± 2.7 | 65.9 ± 0.5, [23.6] | 51.5 ± 2.2, [27.9] | 48.0 ± 1.1 | 68.2 ± 0.5, [16.8] | 57.0 ± 0.8, [20.8] | |
| 13g | 10 | 37.9 ± 1.7 | 46.9 ± 0.7 | — | 36.1 ± 2.4 | — | — | — |
| 30 | 46.1 ± 1.7 | 51.6 ± 0.6, [20.3] | 31.3 ± 2.0 | 62.9 ± 0.9, [17.6] | 30.3 ± 0.9 | 59.9 ± 1.5, [16.8] | 48.6 ± 1.5 | |
| 13g′ | 10 | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. |
| 30 | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | |
| 13h | 10 | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. |
| 30 | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | |
| 14a | 10 | 24.3 ± 1.2 | — | — | — | — | — | 24.1 ± 0.6 |
| 30 | 93.6 ± 0.2, [11.8] | — | 47.8 ± 1.0 | 91.3 ± 0.2, [18.7] | 44.2 ± 0.9 | 48.0 ± 1.6 | 89.5 ± 0.6, [12.4] | |
| 14b | 10 | 43.2 ± 0.5 | — | — | — | — | — | — |
| 30 | 71.0 ± 1.0, [20.1] | — | 49.4 ± 1.5 | — | 37.9 ± 3.1 | 47.7 ± 1.6 | 31.5 ± 1.9 | |
| 14c | 10 | 44.8 ± 1.8 | — | — | 20.3 ± 1.4 | — | — | 47.3 ± 1.4 |
| 30 | 78.8 ± 0.9, [10.2] | — | 35.3 ± 2.4 | 73.6 ± 0.7, [19.1] | 41.3 ± 1.0 | 45.7 ± 1.1 | 80.4 ± 0.4, [10.2] | |
| 14d | 10 | — | — | — | — | 26.6 ± 1.8 | 42.7 ± 1.4 | — |
| 30 | 53.6 ± 0.7, [15.4] | 48.6 ± 1.6 | 35.4 ± 0.7 | 51.8 ± 0.6, [29.9] | 63.0 ± 2.0, [18.7] | 73.1 ± 0.3, [12.8] | 59.7 ± 1.3, [27.1] | |
| 14e | 10 | 27.1 ± 0.5 | — | — | — | — | 29.6 ± 1.5 | 22.4 ± 1.6 |
| 30 | 86.4 ± 0.6, [12.2] | — | 47.5 ± 2.1 | 79.8 ± 1.6, [24.9] | 43.7 ± 1.0 | 47.1 ± 0.7 | 61.0 ± 1.1, [23.4] | |
| 14f | 10 | 48.6 ± 2.5 | — | — | — | — | 23.1 ± 2.1 | — |
| 30 | 86.9 ± 2.5, [12.3] | — | 38.1 ± 0.6 | 64.7 ± 1.0, [23.3] | 31.9 ± 1.9 | 40.1 ± 1.5 | 62.2 ± 0.4, [18.5] | |
| 14g | 10 | — | — | — | — | — | — | — |
| 30 | 74.7 ± 0.7, [18.6] | — | 39.9 ± 2.3 | 62.0 ± 1.3, [24.2] | 76.0 ± 0.8, [22.4] | 40.8 ± 1.9 | 48.8 ± 0.9 | |
| 14g′ | 10 | — | — | — | — | — | — | — |
| 30 | 80.4 ± 0.4, [20.5] | — | 43.5 ± 1.2 | 58.8 ± 1.0, [16.4] | 44.2 ± 0.4 | 47.9 ± 1.4 | 65.5 ± 1.4, [24.0] | |
| 14h | 10 | 24.0 ± 0.7 | 22.9 ± 2.0 | — | — | 26.5 ± 1.5 | 46.2 ± 0.7 | — |
| 30 | 63.5 ± 0.3, [20.0] | 31.31 ± 0.3 | 30.7 ± 0.2 | 42.5 ± 0.9 | 34.4 ± 1.9 | 75.2 ± 0.9, [10.5] | 46.8 ± 1.4 | |
| Cisplatin | 10 | 43.5 ± 1.8 | 42.6 ± 2.3 | 88.6 ± 0.5 | 66.9 ± 1.8 | — | 67.5 ± 1.0 | 51.0 ± 2.0 |
| 30 | 74.0 ± 2.3, [3.69] | 99.9 ± 0.3, [12.43] | 90.2 ± 1.8, [7.84] | 96.8 ± 0.4, [5.78] | 71.5 ± 1.2, [19.13] | 87.8 ± 1.1, [3.74] | 55.0 ± 1.5, [9.78] | |
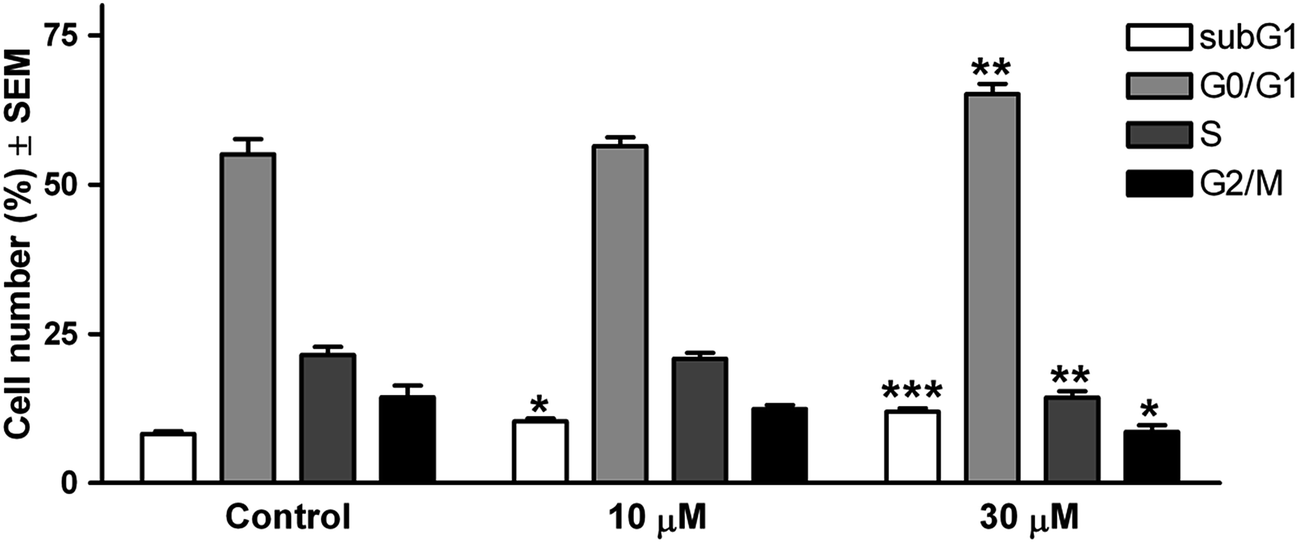 | ||
| Fig. 2 Effects of 14c on the cell cycle phase distribution of T47D cells determined by flow cytometry after incubation for 48 h. *, ** and *** indicate p < 0.05, p < 0.01 and p < 0.001 as compared with the untreated control cells. N = 6. | ||
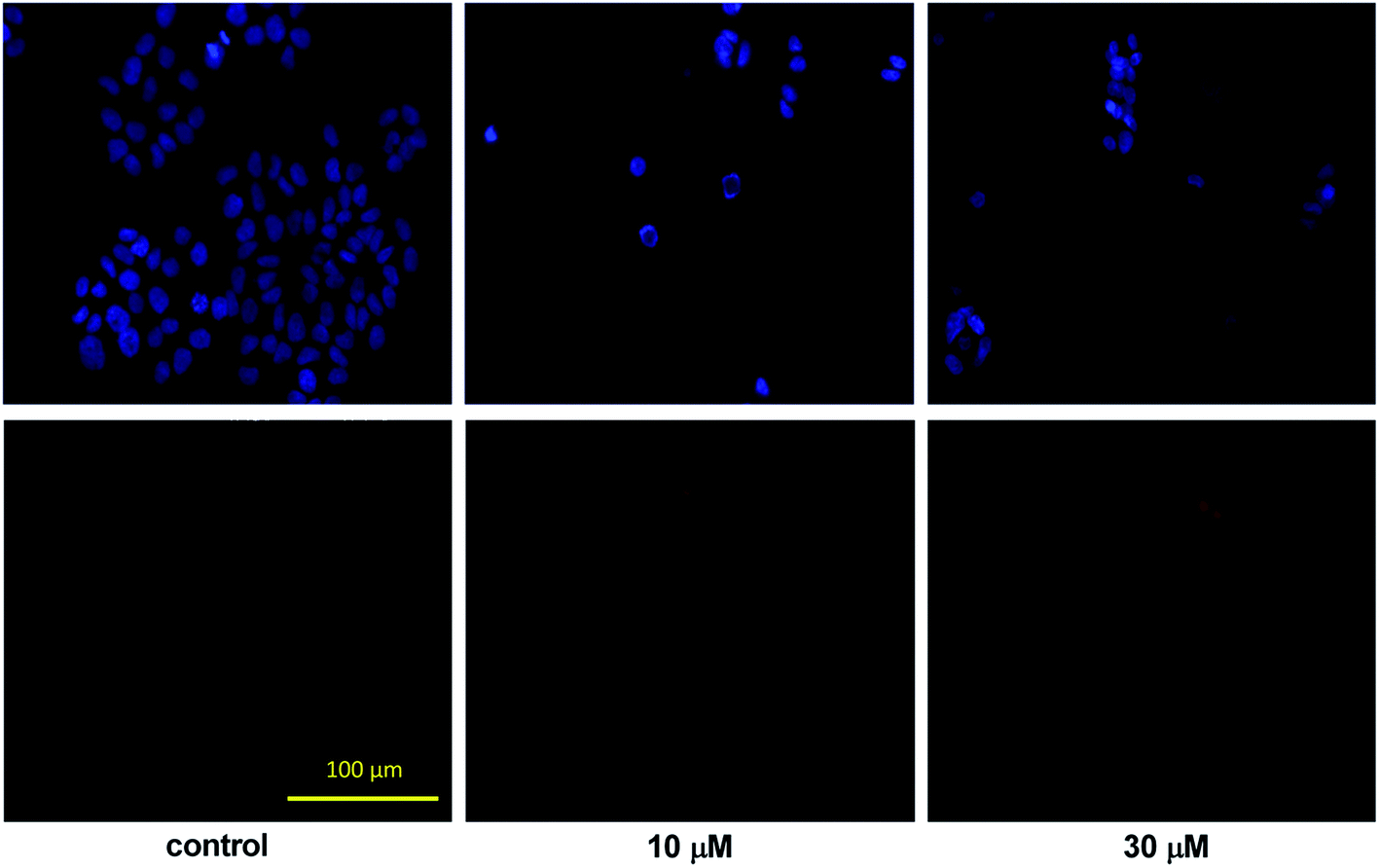 | ||
| Fig. 3 Fluorescent double-stained T47D cells treated with 10 or 30 μM 14c. The upper and lower panels indicate the fluorescence of Hoechst 33258 and PI, respectively. | ||
For a quantitative description, intact, apoptotic and necrotic cells were counted in all conditions. The ratio of intact cells demonstrated a concentration-dependent decrease after the treatment, and the proportion of apoptotic cells was increased significantly even at the lower concentration (Fig. 4). The tendency was more pronounced in the presence of 30 μM 14c and the ratio of the necrotic cells was also significantly elevated. These findings suggest that 14c has the capacity to induce apoptosis in a concentration-dependent manner.
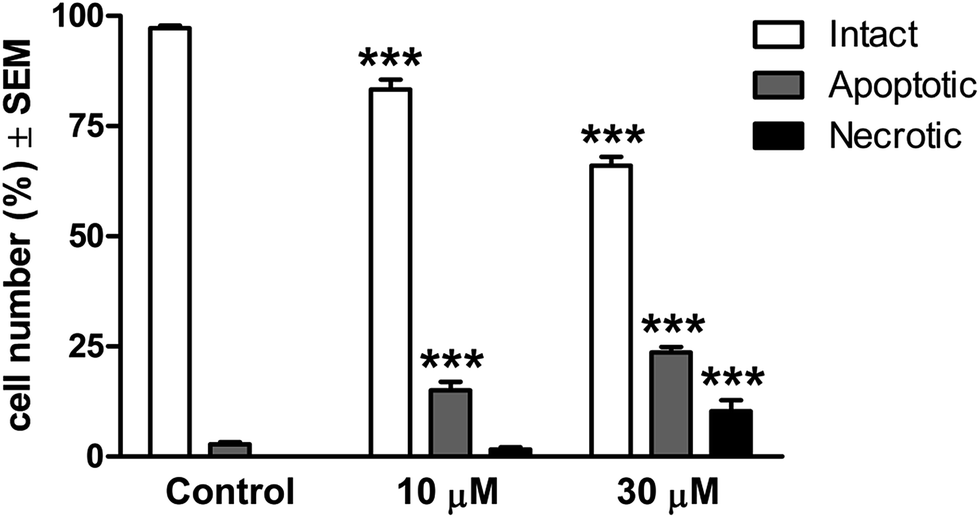 | ||
| Fig. 4 Qualitative evaluation of fluorescent double staining of T47D cells treated with 10 or 30 μM 14c. *** indicates p < 0.001 as compared with the untreated control cells. | ||
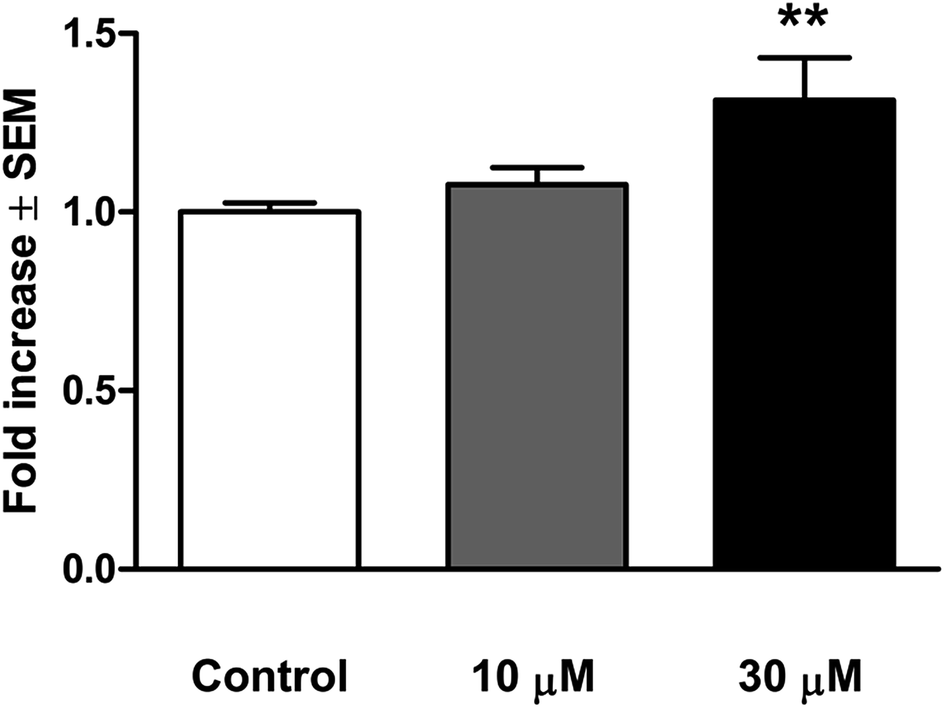 | ||
| Fig. 5 Activation of caspase-3 after 48 h incubation with 14c. ** indicates p < 0.01 as compared with the untreated control cells. | ||
3. Conclusions
In summary, novel ring D- and A-modified steroids containing a fused quinoline scaffold were prepared from β-chlorovinyl aldehydes of estrone 3-methyl ether and 17β-acetoxy-5α-dihydrotestosterone, respectively, with differently substituted anilines under microwave conditions. The yields of the heterocyclic products were observed to depend on the substitution pattern of the arylamines and also on the different number of members of rings D and A. Thus, a shorter reaction time and a lower temperature were sufficient for the conversion of 17β-acetoxy-3-chloro-5α-androst-2-ene-2-carbaldehyde than for that of 17-chloro-3-methoxyestra-1,3,5(10),16-tetraene-16-carbaldehyde especially with anilines containing EDGs, indicating the higher reactivity of the six-membered ring A as compared with that of the five-membered ring D. The presence of EWGs and/or ortho substituents was found to hamper the cyclization step of the reactions, resulting in sluggish or moderate yields of the desired quinolines. The experimental findings can be explained by assuming that the reaction occurs through an N-arylenaminoimine intermediate, the ring-closure of which can be affected by electronic and steric factors. As concerns the pharmacological profile of the presented molecules, most of them exerted weak or modest antiproliferative activities against the utilized panel of human adherent cell lines. The most potent agents, some of the ring A-fused quinolines, exhibited IC50 values in the low micromolar range, i.e. comparable to those of the reference agent cisplatin. The apoptosis-inducing capacity of a selected molecule was evidenced by morphological and biochemical approaches. The described results and the structure–activity relationships can be utilized to design further anticancer drug candidates with steroid skeletons.4. Experimental
4.1. General
All solvents were distilled prior to use. Reagents and materials were obtained from commercial suppliers and were used without purification. The reactions under microwave irradiation were carried out with a CEM Corporation Focused Microwave System, Model Discover SP. The reactions were monitored by TLC on Kieselgel-G (Merck Si 254 F) layers (0.25 mm thick). The spots were detected by spraying with 5% phosphomolybdic acid in 50% aqueous phosphoric acid. Flash chromatography: Merck silica gel 60, 40–63 μm. Melting points (mp) were determined on an SMS Optimelt digital apparatus. Elementary analysis data were determined with a PerkinElmer CHN analyzer model 2400. NMR spectra were obtained at room temperature with a Bruker DRX 500 instrument. Chemical shifts are reported in ppm (δ scale), and coupling constants (J) in Hz. For the determination of multiplicities, the J-MOD pulse sequence was used. Automated flow injection analyses were performed by using an HPLC/MSD system. The system comprised an Agilent 1100 micro vacuum degasser, a quaternary pump, a micro-well plate autoinjector and a 1946A MSD equipped with an electrospray ion source (ESI) operated in positive ion mode. The ESI parameters were: nebulizing gas N2, at 35 psi; drying gas N2, at 350 °C and 12 L min−1; capillary voltage (VCap) 3000 V; fragmentor voltage 70 V. The MSD was operated in scan mode with a mass range of m/z 60–620. Samples (0.2 μL) with automated needle wash were injected directly into the solvent flow (0.3 mL min−1) of CH3CN/H2O 70:30 (v/v) supplemented with 0.1% formic acid. The system was controlled by Agilent LC/MSD Chemstation software.
4.2. General procedure for the preparation of β-chlorovinyl aldehydes 2, 7 and 11
To an ice-cold solution of estrone 3-methyl ether (1, 5.69 g, 20.0 mmol), 6-methoxy-1-tetralone (6, 3.52 g, 20.0 mmol) or 17β-acetoxy-5α-dihydrotestosterone (10, 6.64 g, 20.0 mmol) in CHCl3 (120 mL), POCl3 (30 mL) and DMF (30 mL) were simultaneously added dropwise and the solution was stirred for 10 min at 0 °C. The mixture was allowed to reach room temperature and then refluxed for 4 h (for 1, 6 and 10, method A) or stirred at 25 °C for 2 h (in the case of 10, method B). After completion of the reaction, the mixture was poured into ice and extracted with CH2Cl2 (3 × 50 mL), and the organic phase was washed with brine. The combined organic layer was dried over anhydrous Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography.Substrate: 1. After purification with hexane/CH2Cl2 (25
:75) as eluent, 2 (4.43 g, 67%) and 3 (ref. 23) (0.91 g, 15%) were obtained as white solids.
2. Mp 129–131 °C; 1H NMR (500 MHz, CDCl3): δH = 1.00 (s, 3H, 18-CH3), 1.44 (m, 1H), 1.57–1.68 (overlapping m, 3H), 1.74 (m, 1H), 1.95 (m, 1H), 2.00 (m, 1H), 2.16 (m, 1H), 2.32 (m, 1H), 2.46 (m, 1H), 2.65 (dd, 1H, J = 14.7 Hz, J = 6.4 Hz), 2.90 (m, 2H, 6-CH2), 3.78 (s, 3H, 3-OMe), 6.65 (d, 1H, J = 2.1 Hz, 4-H), 6.73 (dd, 1H, J = 8.5 Hz, J = 2.1 Hz, 2-H), 7.19 (d, 1H, J = 8.5 Hz, 1-H), 10.02 (s, 1H, 16-CHO); 13C NMR (125 MHz, CDCl3): δC = 15.2 (C-18), 25.9 (CH2), 27.0 (CH2), 28.2 (CH2), 29.5 (CH2), 33.0 (CH2), 37.2 (CH), 44.0 (CH), 51.0 (C-13), 53.0 (CH), 55.2 (3-OMe), 111.6 (C-2), 113.8 (C-4), 125.9 (C-1), 131.8 (C-10), 136.4 (C-16), 137.7 (C-5), 157.6 (C-3), 162.4 (C-17), 188.0 (16-CHO); ESI-MS: 331 [M + H]+; anal. calcd for C20H23ClO2 C, 72.61; H, 7.01. Found: C, 72.80; H, 7.12.
4.3. General procedure for the preparation of steroidal quinolines (5 and 13) and benz[c]acridines (9)
Steroidal or nonsteroidal β-chlorovinyl aldehyde (2, 11 or 7, 1.00 mmol) and (susbstituted)aniline (1.00 mmol) were dissolved in DMF (3 mL) and the solution was irradiated in a closed vessel at 140 °C or 120 °C for the appropriate time. After completion of the reaction, the mixture was poured into water (10 mL) and the precipitate was filtered off. The crude product was purified by column chromatography.:30. Yield: 104 mg (yellowish solid); mp 143–145 °C; 1H NMR (500 MHz, CDCl3): δH = 1H NMR (CDCl3, 500 MHz): δ 2.98 (m, 2H) and 3.11 (m, 2H): 5-CH2 and 6-CH2, 3.88 (s, 3H, 3-OMe), 6.78 (d, 1H, J = 2.4 Hz, 4-H), 6.97 (dd, 1H, J = 8.6 Hz, J = 2.4 Hz, 2-H), 7.33 (t, 1H, J = 7.6 Hz, 9-H), 7.62 (d, 1H, J = 7.6 Hz, 8-H), 7.74 (d, 1H, J = 7.6 Hz, 10-H), 7.86 (s, 1H, 7-H), 8.63 (d, 1H, J = 8.6 Hz, 1-H); 13C NMR (125 MHz, CDCl3): δC = 28.5 and 28.6 (C-5 and C-6), 55.3 (3-OMe), 112.8 and 113.2: C-2 and C-4, 125.4 and 125.9: C-8 and C-9, 127.5 (C-7a), 128.4 and 128.7: C-1 and C-10, 128.7 (C-12), 130.8 (C-6a), 133.3 (C-11), 133.8 (C-7), 141.2 (C-4a), 143.6 (C-11a), 153.9 (C-12a), 161.2 (C-3); ESI-MS: 296 [M + H]+; anal. calcd for C18H14ClNO C, 73.10; H, 4.77. Found: C, 73.25; H, 4.86.:90. Yield: 160 mg (yellowish solid); mp 122–124 °C; 1H NMR (500 MHz, CDCl3): δH = 2.96 (m, 2H) and 3.08 (m, 2H): 5-CH2 and 6-CH2, 3.87 (s, 3H, 3-OMe), 6.79 (d, 1H, J = 2.4 Hz, 4-H), 6.96 (dd, 1H, J = 8.6 Hz, J = 2.4 Hz, 2-H), 7.55 (dd, 1H, J = 8.9 Hz, J = 2.2 Hz, 10-H), 7.67 (d, 1H, J = 2.2 Hz, 8-H), 7.76 (s, 1H, 7-H), 8.02 (d, 1H, J = 8.9 Hz, 11-H), 8.48 (d, 1H, J = 8.6 Hz, 1-H); 13C NMR (125 MHz, CDCl3): δC = 28.5 and 28.8 (C-5 and C-6), 55.3 (3-OMe), 112.9 and 113.1 (C-2 and C-4), 125.5 (C-8), 127.3 (C-7a), 127.8 (C-1), 128.0 (C-6a), 129.4 (C-10), 130.6 (C-11), 131.0 and 131.1 (C-9 and C-12), 132.5 (C-7), 141.2 (C-4a), 148.4 (C-11a), 153.6 (C-12a), 161.1 (C-3); ESI-MS: 296 [M + H]+; anal. calcd for C18H14ClNO C, 73.10; H, 4.77. Found: C, 72.98; H, 4.69.:80. Yield: 211 mg (orange solid); mp 122–124 °C; 1H NMR (500 MHz, CDCl3): δH = 2.96 (m, 2H) and 3.09 (m, 2H): 5-CH2 and 6-CH2, 3.87 (s, 3H, 3-OMe), 6.78 (d, 1H, J = 2.3 Hz, 4-H), 6.96 (dd, 1H, J = 8.6 Hz, J = 2.3 Hz, 2-H), 7.68 (dd, 1H, J = 8.9 Hz, J = 1.9 Hz, 10-H), 7.75 (s, 1H, 7-H), 7.85 (d, 1H, J = 1.9 Hz, 8-H), 7.96 (d, 1H, J = 8.9 Hz, 11-H), 8.48 (d, 1H, J = 8.6 Hz, 1-H); 13C NMR (125 MHz, CDCl3): δC = 28.2 and 28.4 (C-5 and C-6), 55.0 (3-OMe), 112.6 and 112.8 (C-2 and C-4), 118.9 (C-9), 127.5 (C-1), 128.2 (C-7a), 128.5 (C-8), 130.3 (C-11), 130.6 (C-6a), 131.6 (C-10), 132.1 (C-7), 134.5 (C-12), 140.9 (C-4a), 147.1 (C-11a), 153.4 (C-12a), 160.8 (C-3); ESI-MS: 340 [M + H]+; anal. calcd for C18H14BrNO C, 63.55; H, 4.15. Found: C, 63.75; H, 4.10.:90. Yield: 317 mg (white solid); mp 226–228 °C; 1H NMR (500 MHz, CDCl3): δH = 0.80 (s, 3H, 18-CH3), 0.82 (s, 3H, 19-CH3), 0.88 (m, 1H), 0.98 (m, 1H), 1.07 (m, 1H), 1.23 (m, 1H), 1.29–1.55 (overlapping m, 5H), 1.66–1.82 (overlapping m, 6H), 2.05 (s, 3H, Ac-CH3), 2.17 (m, 1H), 2.60 (d, 1H, J = 16.0 Hz, one of 1-H2), 2.79 (dd, 1H, J = 18.5 Hz, J = 12.5 Hz, one of 4-H2), 2.97 (d, 1H, J = 16.0 Hz, the other of 1-H2), 3.08 (dd, 1H, J = 18.5 Hz, J = 5.5 Hz, the other of 4-H2), 4.62 (t, 1H, J = 8.4 Hz, 17-H), 7.42 (t-like m, 1H, 6′-H), 7.60 (t-like m, 1H, 7′-H), 7.69 (d, 1H, J = 8.0 Hz, 5′-H), 7.79 (s, 1H, 4′-H), 7.97 (d, 1H, J = 8.4 Hz, 8′-H); 13C NMR (125 MHz, CDCl3): δC = 11.6 (C-19), 12.0 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 23.5 (CH2), 27.5 (CH2), 28.6 (CH2), 31.1 (CH2), 35.3 (C-10), 35.3 (CH), 36.9 (CH2), 37.3 (CH2), 42.2 (CH), 42.5 (C-13), 43.5 (CH2), 50.7 (CH), 53.5 (CH), 82.8 (C-17), 125.5 (C-6′), 126.8 (C-5′), 127.2 (C-4a′), 128.2 and 128.5 (C-7′ and C-8′), 130.0 (C-3′), 135.8 (C-4′), 146.6 (C-8a′), 158.3 (C-2′), 171.2 (Ac-CO); ESI-MS: 418 [M + H]+; anal. calcd for C28H35NO2 C, 80.53; H, 8.45. Found: C, 80.34; H, 8.30.:95. Yield: 393 mg (white solid); mp 247–249 °C; 1H NMR (500 MHz, CDCl3): δH = 0.79 (s, 3H, 18-CH3), 0.82 (s, 3H, 19-CH3), 0.86 (m, 1H), 0.97 (m, 1H), 1.05 (m, 1H), 1.22 (m, 1H), 1.28–1.54 (overlapping m, 5H), 1.64–1.82 (overlapping m, 6H), 2.05 (s, 3H, Ac-CH3), 2.17 (m, 1H), 2.49 (s, 3H, 6′-CH3), 2.58 (d, 1H, J = 16.0 Hz, one of 1-H2), 2.77 (dd, 1H, J = 18.3 Hz, J = 12.8 Hz, one of 4-H2), 2.95 (d, 1H, J = 16.0 Hz, the other of 1-H2), 3.06 (dd, 1H, J = 18.3 Hz, J = 4.7 Hz, the other of 4-H2), 4.62 (t-like m, 1H, 17-H), 7.44 (d, 1H, J = 8.2 Hz, 7′-H), 7.44 (s, 1H, 5′-H), 7.71 (s, 1H, 4′-H), 7.88 (d, 1H, J = 8.2 Hz, 8′-H); 13C NMR (125 MHz, CDCl3): δC = 11.6 (C-19), 12.0 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 21.5 (6′-CH3), 23.5 (CH2), 27.5 (CH2), 28.6 (CH2), 31.1 (CH2), 35.3 (C-10), 35.3 (CH), 36.9 (CH2), 37.1 (CH2), 42.2 (CH), 42.5 (C-13), 43.5 (CH2), 50.7 (CH), 53.5 (CH), 82.8 (C-17), 125.6 (C-5′), 127.2 (C-4a′), 127.7 (C-8′), 130.0 (C-3′), 131.0 (C-7′), 135.3 (C-6′), 135.4 (C-4′), 146.4 (C-8a′), 157.2 (C-2′), 171.2 (Ac-CO); ESI-MS: 432 [M + H]+; anal. calcd for C29H37NO2 C, 80.70; H, 8.64. Found: C, 80.55; H, 8.82.:95. Yield: 412 mg (white solid); mp 255–257 °C; 1H NMR (500 MHz, CDCl3): δH = 0.79 (s, 3H, 18-CH3), 0.81 (s, 3H, 19-CH3), 0.86 (m, 1H), 0.96 (m, 1H), 1.06 (m, 1H), 1.21 (m, 1H), 1.28–1.54 (overlapping m, 5H), 1.64–1.82 (overlapping m, 6H), 2.05 (s, 3H, Ac-CH3), 2.17 (m, 1H), 2.57 (d, 1H, J = 16.0 Hz, one of 1-H2), 2.74 (dd, 1H, J = 18.2 Hz, J = 12.7 Hz, one of 4-H2), 2.93 (d, 1H, J = 16.0 Hz, the other of 1-H2), 3.05 (dd, 1H, J = 18.2 Hz, J = 5.4 Hz, the other of 4-H2), 3.89 (s, 3H, 6′-OMe), 4.62 (t, 1H, J = 8.7 Hz, 17-H), 6.95 (d, 1H, J = 2.6 Hz, 5′-H), 7.26 (dd, 1H, J = 9.1 Hz, J = 2.6 Hz, 7′-H), 7.68 (s, 1H, 4′-H), 7.86 (d, 1H, J = 9.1 Hz, 8′-H); 13C NMR (125 MHz, CDCl3): δC = 11.6 (C-19), 12.0 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 23.5 (CH2), 27.5 (CH2), 28.6 (CH2), 31.1 (CH2), 35.3 (C-10), 35.3 (CH), 36.9 (CH2), 37.0 (CH2), 42.2 (CH), 42.5 (C-13), 43.5 (CH2), 50.7 (CH), 53.5 (CH), 55.4 (6′-OMe), 82.8 (C-17), 104.3 (C-5′), 121.3 (C-7′), 128.0 (C-4a′), 129.6 (C-8′), 130.3 (C-3′), 134.7 (C-4′), 146.5 (C-8a′), 155.5 and 157.1 (C-2′ and C-6′), 171.2 (Ac-CO); ESI-MS: 448 [M + H]+; anal. calcd for C29H37NO3 C, 77.82; H, 8.33. Found: C, 77.65; H, 8.47.:95. Yield: 267 mg (white solid); mp 288–291 °C; 1H NMR (500 MHz, CDCl3): δH = 0.79 (s, 3H, 18-CH3), 0.82 (s, 3H, 19-CH3), 0.88 (m, 1H), 0.98 (m, 1H), 1.08 (m, 1H), 1.23 (m, 1H), 1.29–1.55 (overlapping m, 5H), 1.64–1.82 (overlapping m, 6H), 2.05 (s, 3H, Ac-CH3), 2.17 (m, 1H), 2.59 (d, 1H, J = 16.2 Hz, one of 1-H2), 2.76 (dd, 1H, J = 18.4 Hz, J = 12.9 Hz, one of 4-H2), 2.97 (d, 1H, J = 16.2 Hz, the other of 1-H2), 3.05 (dd, 1H, J = 18.4 Hz, J = 5.2 Hz, the other of 4-H2), 4.62 (t, 1H, J = 8.5 Hz, 17-H), 7.53 (dd, 1H, J = 8.9 Hz, J = 1.9 Hz, 7′-H), 7.66 (d, 1H, J = 1.9 Hz, 5′-H), 7.70 (s, 1H, 4′-H), 7.90 (d, 1H, J = 8.9 Hz, 8′-H); 13C NMR (125 MHz, CDCl3): δC = 11.8 (C-19), 12.0 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 23.5 (CH2), 27.5 (CH2), 28.5 (CH2), 31.1 (CH2), 35.3 (C-10), 35.3 (CH), 36.9 (CH2), 37.2 (CH2), 42.1 (CH), 42.5 (C-13), 43.5 (CH2), 50.7 (CH), 53.4 (CH), 82.7 (C-17), 125.4 (C-5′), 127.8 (C-4a′), 129.4 and 129.8 (C-7′ and C-8′), 131.1 (2C, C-6′ and C-3′), 134.8 (C-4′), 144.9 (C-8a′), 158.7 (C-2′), 171.2 (Ac-CO); ESI-MS: 453 [M + H]+; anal. calcd for C28H34ClNO2 C, 74.40; H, 7.58. Found: C, 74.25; H, 7.43.:95. Yield: 298 mg (white solid); mp 295–297 °C; 1H NMR (500 MHz, CDCl3): δH = 0.79 (s, 3H, 18-CH3), 0.82 (s, 3H, 19-CH3), 0.88 (m, 1H), 0.98 (m, 1H), 1.08 (m, 1H), 1.22 (m, 1H), 1.28–1.55 (overlapping m, 5H), 1.64–1.82 (overlapping m, 6H), 2.05 (s, 3H, Ac-CH3), 2.17 (m, 1H), 2.59 (d, 1H, J = 16.2 Hz, one of 1-H2), 2.75 (dd, 1H, J = 18.5 Hz, J = 12.7 Hz, one of 4-H2), 2.97 (d, 1H, J = 16.2 Hz, the other of 1-H2), 3.04 (dd, 1H, J = 18.5 Hz, J = 5.4 Hz, the other of 4-H2), 4.62 (t-like m, 1H, 17-H), 7.65 (dd, 1H, J = 9.0 Hz, J = 2.0 Hz, 7′-H), 7.69 (s, 1H, 5′-H), 7.82 (d, 1H, J = 9.0 Hz, 8′-H), 7.83 (s, 1H, 4′-H); 13C NMR (125 MHz, CDCl3): δC = 11.7 (C-19), 12.1 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 23.5 (CH2), 27.5 (CH2), 28.5 (CH2), 31.1 (CH2), 35.3 (C-10), 35.3 (CH), 36.9 (CH2), 37.3 (CH2), 42.1 (CH), 42.5 (C-13), 43.5 (CH2), 50.7 (CH), 53.4 (CH), 82.7 (C-17), 119.2 (C-6′), 128.3 (C-4a′), 128.8 (C-5′), 130.0 (C-8′), 131.1 (C-3′), 131.9 (C-7′), 134.7 (C-4′), 145.2 (C-8a′), 158.9 (C-2′), 171.2 (Ac-CO); ESI-MS: 498 [M + H]+; anal. calcd for C28H34BrNO2 C, 67.74; H, 6.90. Found: C, 67.89; H, 6.75.:98. Yields: 208 mg (13g) and 72 mg (13g′) as white solids. 13g: mp 258–260 °C; 1H NMR (500 MHz, CDCl3): δH = 0.78 (s, 3H, 18-CH3), 0.81 (s, 3H, 19-CH3), 0.86 (m, 1H), 0.96 (m, 1H), 1.06 (m, 1H), 1.22 (m, 1H), 1.29–1.55 (overlapping m, 5H), 1.64–1.82 (overlapping m, 6H), 2.05 (s, 3H, Ac-CH3), 2.17 (m, 1H), 2.56 (d, 1H, J = 16.1 Hz, one of 1-H2), 2.76 (dd, 1H, J = 18.5 Hz, J = 12.7 Hz, one of 4-H2), 2.94 (d, 1H, J = 16.1 Hz, the other of 1-H2), 3.05 (dd, 1H, J = 18.5 Hz, J = 5.4 Hz, the other of 4-H2), 4.62 (t, 1H, J = 8.6 Hz, 17-H), 7.37 (dd, 1H, J = 8.7 Hz, J = 1.8 Hz, 6′-H), 7.61 (d, 1H, J = 8.7 Hz, 5′-H), 7.76 (s, 1H, 4′-H), 7.96 (dd, 1H, J = 1.8 Hz, 8′-H); 13C NMR (125 MHz, CDCl3): δC = 11.7 (C-19), 12.0 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 23.5 (CH2), 27.5 (CH2), 28.5 (CH2), 31.1 (CH2), 35.3 (C-10), 35.3 (CH), 36.9 (CH2), 37.3 (CH2), 42.1 (CH), 42.5 (C-13), 43.4 (CH2), 50.7 (CH), 53.4 (CH), 82.7 (C-17), 125.5 (C-4a′), 126.5 (C-6′), 127.3 (C-8′), 128.0 (C-5′), 130.4 (C-3′), 134.2 (C-7′), 135.6 (C-4′), 146.9 (C-8a′), 159.5 (C-2′), 171.2 (Ac-CO); ESI-MS: 453 [M + H]+; anal. calcd for C28H34ClNO2 C, 74.40; H, 7.58. Found: C, 74.24; H, 7.65. 13g′: mp 262–265 °C; 1H NMR (500 MHz, CDCl3): δH = 0.80 (s, 3H, 18-CH3), 0.82 (s, 3H, 19-CH3), 0.89 (m, 1H), 0.97 (m, 1H), 1.08 (m, 1H), 1.23 (m, 1H), 1.28–1.55 (overlapping m, 5H), 1.66–1.83 (overlapping m, 6H), 2.05 (s, 3H, Ac-CH3), 2.18 (m, 1H), 2.63 (d, 1H, J = 16.2 Hz, one of 1-H2), 2.78 (dd, 1H, J = 18.5 Hz, J = 12.7 Hz, one of 4-H2), 3.06 (d, 1H, J = 16.2 Hz, the other of 1-H2), 3.10 (dd, 1H, J = 18.5 Hz, J = 5.4 Hz, the other of 4-H2), 4.63 (t, 1H, J = 8.6 Hz, 17-H), 7.50 (m, 2H, 6′-H and 7′-H), 7.90 (dd, 1H, J = 5.9 Hz, J = 3.1 Hz, 8′-H), 8.20 (s, 1H, 4′-H); 13C NMR (125 MHz, CDCl3): δC = 11.7 (C-19), 12.1 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 23.5 (CH2), 27.5 (CH2), 28.5 (CH2), 31.1 (CH2), 35.3 (CH), 35.3 (C-10), 36.9 (CH2), 37.1 (CH2), 42.1 (CH), 42.5 (C-13), 43.6 (CH2), 50.7 (CH), 53.4 (CH), 82.7 (C-17), 125.3 (C-4a′), 125.5, 127.5 and 128.2 (C-6′, C-7′ and C-8′), 130.4 (C-5′), 131.3 (C-3′), 132.6 (C-4′), 147.2 (C-8a′), 159.2 (C-2′), 171.2 (Ac-CO); ESI-MS: 453 [M + H]+; anal. calcd for C28H34ClNO2 C, 74.40; H, 7.58. Found: C, 74.58; H, 7.70.:95. Yield: 393 mg (white solid); mp 262–265 °C; 1H NMR (500 MHz, CDCl3): δH = 0.81 (s, 3H, 18-CH3), 0.82 (s, 3H, 19-CH3), 0.87 (m, 1H), 0.98 (m, 1H), 1.06 (m, 1H), 1.22 (m, 1H), 1.29–1.55 (overlapping m, 5H), 1.66–1.82 (overlapping m, 6H), 2.06 (s, 3H, Ac-CH3), 2.18 (m, 1H), 2.58 (d, 1H, J = 16.0 Hz, one of 1-H2), 2.78 (s, 3H, 8′-CH3), 2.82 (dd, 1H, J = 18.2 Hz, J = 12.7 Hz, one of 4-H2), 2.97 (d, 1H, J = 16.0 Hz, the other of 1-H2), 3.12 (dd, 1H, J = 18.2 Hz, J = 3.8 Hz, the other of 4-H2), 4.62 (t-like m, 1H, 17-H), 7.31 (t-like m, 1H, 6′-H), 7.45 (d, 1H, J = 6.8 Hz, 7′-H), 7.54 (d, 1H, J = 8.1 Hz, 5′-H), 7.75 (s, 1H, 4′-H); 13C NMR (125 MHz, CDCl3): δC = 11.7 (C-19), 12.0 (C-18), 18.0 (8′-CH3), 20.8 (CH2), 21.2 (Ac-CH3), 23.5 (CH2), 27.5 (CH2), 28.6 (CH2), 31.2 (CH2), 35.2 (C-10), 35.4 (CH), 36.9 (CH2), 37.6 (CH2), 42.3 (CH), 42.5 (C-13), 43.5 (CH2), 50.7 (CH), 53.5 (CH), 82.8 (C-17), 124.9 and 125.2 (C-5′ and C-6′), 127.1 (C-4a′), 128.5 (C-7′), 129.6 (C-3′), 132.1 (C-8′), 135.9 (C-4′), 136.0 (C-8a′), 157.2 (C-2′), 171.2 (Ac-CO); ESI-MS: 432 [M + H]+; anal. calcd for C29H37NO2 C, 80.70; H, 8.64. Found: C, 80.87; H, 8.52.:1 mixture of 13i and 13i′ could not be separated by column chromatography. Yield: 388 mg (white solid). 13i (assigned from the spectra of the regioisomeric mixture); 1H NMR (500 MHz, CDCl3): δH = 0.77 (s, 3H, 18-CH3), 0.81 (s, 3H, 19-CH3), 0.85 (m, 1H), 0.96 (m, 1H), 1.04 (m, 1H), 1.22 (m, 1H), 1.29–1.55 (overlapping m, 5H), 1.64–1.80 (overlapping m, 6H), 2.04 (s, 3H, Ac-CH3), 2.15 (m, 1H), 2.51 (s, 3H, 7′-CH3), 2.55 (d, 1H, J = 16.0 Hz, one of 1-H2), 2.75 (dd, 1H, J = 18.3 Hz, J = 12.3 Hz, one of 4-H2), 2.93 (d, 1H, J = 16.0 Hz, the other of 1-H2), 3.05 (dd, 1H, J = 18.5 Hz, J = 5.8 Hz, the other of 4-H2), 4.61 (t-like m, 1H, 17-H), 7.25 (d, 1H, J = 8.0 Hz, 6′-H), 7.57 (d, 1H, J = 8.0 Hz, 5′-H), 7.73 (s, 1H) and 7.75 (s, 1H): 4′-H and 8′-H; 13C NMR (125 MHz, CDCl3): δC = 11.6 (C-19), 12.0 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 21.8 (7′-CH3), 23.5 (CH2), 27.5 (CH2), 28.6 (CH2), 31.1 (CH2), 35.3 (C-10), 35.3 (CH), 36.9 (CH2), 37.3 (CH2), 42.1 (CH), 42.5 (C-13), 43.4 (CH2), 50.7 (CH), 53.4 (CH), 82.8 (C-17), 125.3 (C-4a′), 126.4 (C-6′), 127.2 (C-8′), 127.8 (C-5′), 129.1 (C-3′), 135.5 (C-4′), 138.6 (C-7′), 146.9 (C-8a′), 158.1 (C-2′), 171.2 (Ac-CO); ESI-MS: 432 [M + H]+; anal. calcd for C29H37NO2 C, 80.70; H, 8.64. Found: C, 80.59; H, 8.74.:98. Yield: 326 mg (orange solid); >270 °C (decomp.); 1H NMR (500 MHz, CDCl3): δH = 0.81 (s, 3H), 0.82 (s + m, 4H): 18-H3, 19-H3 and 1H, 0.94 (m, 1H), 1.06 (m, 1H), 1.14–1.25 (overlapping m, 2H), 1.32 (m, 1H), 1.41 (m, 2H), 1.46–1.54 (overlapping m, 2H), 1.58–1.68 (overlapping m, 3H), 1.72 (m, 1H), 1.79 (m, 1H), 2.04 (s, 3H, Ac-CH3), 2.09–2.18 (overlapping m, 2H, one of 4-H2 and 1H), 2.22 (d, 1H, J = 14.7 Hz, one of 1-H2), 2.35 (dd, 1H, J = 19.2 Hz, J = 5.6 Hz, the other of 4-H2), 2.42 (d, 1H, J = 14.7 Hz, the other of 1-H2), 4.61 (t-like m, 1H, 17-H), 7.03 (d, 2H, J = 9.1 Hz, 2′-H and 6′-H), 7.09 (d, 1H, J = 11.3 Hz, 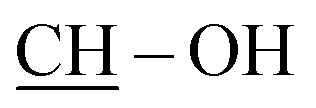 ), 8.18 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 11.98 (d, 1H, J = 11.3 Hz, OH); 13C NMR (125 MHz, CDCl3): δC = 11.5 (C-19), 12.0 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 23.5 (CH2), 27.5 (CH2), 28.2 (CH2), 31.0 (CH2), 35.3 (CH), 35.7 (C-10), 36.8 (CH2), 41.7 (CH), 42.3 (C-13), 42.4 (C-4), 42.5 (C-1), 50.7 (CH), 53.1 (CH), 82.7 (C-17), 108.2 (C-2), 112.5 (C-3), 114.7 (2C, C-2′ and C-6′), 126.0 (2C, C-3′ and C-5′), 139.4 (
), 8.18 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 11.98 (d, 1H, J = 11.3 Hz, OH); 13C NMR (125 MHz, CDCl3): δC = 11.5 (C-19), 12.0 (C-18), 20.8 (CH2), 21.2 (Ac-CH3), 23.5 (CH2), 27.5 (CH2), 28.2 (CH2), 31.0 (CH2), 35.3 (CH), 35.7 (C-10), 36.8 (CH2), 41.7 (CH), 42.3 (C-13), 42.4 (C-4), 42.5 (C-1), 50.7 (CH), 53.1 (CH), 82.7 (C-17), 108.2 (C-2), 112.5 (C-3), 114.7 (2C, C-2′ and C-6′), 126.0 (2C, C-3′ and C-5′), 139.4 ( ), 142.2 (C-1′), 146.2 (C-4′), 171.2 (Ac-CO); ESI-MS: 503 [M + Na]+; anal. calcd for C28H36N2O5 C, 69.98; H, 7.55. Found: C, 70.14; H, 7.69.
), 142.2 (C-1′), 146.2 (C-4′), 171.2 (Ac-CO); ESI-MS: 503 [M + Na]+; anal. calcd for C28H36N2O5 C, 69.98; H, 7.55. Found: C, 70.14; H, 7.69.4.4. General procedure for the synthesis of 17-deacetylated ring A-fused quinolines (14)
The 17β-acetoxy derivative (13, 0.30 mmol) was dissolved in MeOH (10 mL), and KOH (56 mg, 1.00 mmol) was added. The mixture was stirred for 2 h at room temperature, and then diluted with water. The resulting precipitate was filtered off, washed with water and dried.4.5. Determination of antiproliferative activities
Antiproliferative effects were determined in vitro against a panel of human cell lines of gynecological origin, including three cervical (HeLa, SiHa and C33A) and four breast cancer cell lines (MCF7, T47D, MDA-MB-361 and MDA-MB-231). All these cell lines were purchased from the European Collection of Cell Cultures (ECCAC, Salisbury, UK) with the exceptions of SiHa and C33A, which were obtained from LGC Standards GmbH, Wesel, Germany. The cells were cultivated in minimal essential medium (Lonza Ltd, Basel, Switzerland) supplemented with 10% fetal bovine serum, 1% non-essential amino acids and an antibiotic–antimycotic mixture. Near-confluent cancer cells were seeded onto a 96-well microplate (5000 cells per well except in the cases of C33A and MDA-MB-361, which were seeded at 10000 per well) and, after overnight standing, new medium (200 μL) containing the tested compound at 10 or 30 μM was added. After incubation for 72 h at 37 °C in humidified air containing 5% CO2, the living cells were assayed by the addition of 5 mg mL−1 MTT solution (20 μL). MTT was converted by intact mitochondrial reductase and precipitated as blue crystals during a 4 h contact period. The medium was then removed and the precipitated formazan crystals were dissolved in DMSO (100 μL) during a 60 min period of shaking at 25 °C. Finally, the reduced MTT was assayed at 545 nm, using a microplate reader; wells with untreated cells were utilized as controls.30 When compounds elicited at least 50% growth inhibition at 30 μM, the assays were repeated with a set of dilutions, sigmoidal dose–response curves were fitted to the determined data and the IC50 values (the concentration at which the extent of cell proliferation was half that of the untreated control) were calculated by means of GraphPad Prism 4.0 (GraphPad Software, San Diego, CA, USA). All in vitro experiments were carried out on two microplates with at least five parallel wells. Stock solutions of the tested substances (10 mM) were prepared in DMSO. The highest DMSO content of the medium (0.3%) did not have any substantial effect on the cell proliferation. Cisplatin (Ebewe Pharma GmbH, Unterach, Austria) was used as reference agent.
4.6. Cell cycle analysis
The subsequent experiments were carried out only with one of the most effective compounds, 14c. In order to characterize the cell cycle phase distribution, the cellular DNA content was determined by means of flow cytometry. T47D cells were seeded onto six-well plates (250000 cells per well) and treated with the selected compound (10 μM or 30 μM) for 48 h. Untreated cells served as control. After the incubation, the medium was removed and the cells were washed in phosphate-buffered saline (PBS) and harvested using trypsin. The collected cells were washed in PBS and the pellets were resuspended in ice-cold 70% EtOH. The fixed cells were stained with PI solution (1 mL of DNA staining buffer containing PI, ribonuclease-A, Triton-X and sodium citrate) for 1 h at room temperature in the dark, and then analyzed by flow cytometry (Partec CyFlow, Partec GmbH, Münster). Cells belonging in the various phases of the cell cycle (subG1, G1, S and G2/M) were determined by ModFit LT 3.3.11 software (Verity Software House, Topsham, ME, USA).
4.7. Morphological changes and double staining
Morphological changes and extents of apoptosis induction by the tested compound were detected by Hoechst 33258 and PI double staining using fluorescent microscopy. T47D cells were seeded onto 96-well plates (5000 cells per well) and treated with two concentrations of 14c for 48 h. Untreated cells were used as control. After the treatment, fluorescent dye solution (containing 5 and 3 μg mL−1 Hoechst 33258 and PI, respectively) were added to each well and the plate was stored in humidified air for 1 h. The cells were then examined with a fluorescent microscope (Nikon ECLIPSE 146 TS100, Nikon Instruments Europe, Amstelveen, The Netherlands) equipped with a Digital Sight Camera System, including appropriate filters for Hoechst 33258 and PI.33 At least six fields were recorded in the case of each condition and the cells were counted. The ratio of intact, early apoptotic and late apoptotic/necrotic cells were determined and analyzed statistically.4.8. Determination of caspase-3 activity
In order to evince apoptosis, the activity of caspase-3 was determined by colorimetric assay (Sigma-Aldrich Ltd, Budapest, Hungary) according to the manufacturer's instructions. Briefly, T47D cells were treated with two concentrations (10 μM or 30 μM) of 14c for 48 h in cell culture flasks. After incubation, the cells were scraped, washed, lysed and centrifuged, and the supernatants were then transferred into new tubes. Lysates were diluted in order to achieve equal protein concentrations, determined by the BCA method. The reactions were performed on 96-well plates; the samples of lysates and substrate were added into wells in the presence or the absence of caspase-3 inhibitor. After the incubation, the activity of caspase-3 was measured at 405 nm by using ELISA reader (Stat Fax-2100, Awareness Technologies Inc., Palm City, FL, USA). Measured data were evaluated as fold increase compared with the caspase-3 activity of control samples.4.9. Statistical analysis
Statistical evaluation of the in vitro results was performed by one-way analysis of variance followed by the Dunnett posttest, using GraphPad Prism 4 (GraphPad Software; San Diego, CA, USA) software. The mean values and standard deviation were calculated in all experiments.Acknowledgements
Financial support by the Hungarian Scientific Research Fund (OTKA K-109107 and K-109293) is gratefully acknowledged. The work of Á. Baji was supported by a PhD Fellowship of the Talentum Fund of Richter Gedeon Plc.References
- J. Coro, H. Rodríguez, D. G. Rivera, M. Suárez, D. Molero, M. Á. Herranz, R. Martínez-Álvarez, S. Filippone and N. Martin, Eur. J. Org. Chem., 2009, 4810 CrossRef CAS.
- J. Balogh, R. Skoda-Földes, K. Vazdar and I. Habuš, J. Organomet. Chem., 2012, 703, 51 CrossRef CAS.
- G. Mehta and V. Singh, Chem. Soc. Rev., 2002, 31, 324 RSC; L. F. Tietze, G. Schneider, J. Wölfling, A. Fecher, T. Nöbel, S. Petersen, I. Schuberth and C. Wulff, Chem.–Eur. J., 2000, 6, 3755 CrossRef CAS; N. S. Vatmurge, B. G. Hazra, V. S. Pore, F. Shirazi, P. S. Chavan and M. V. Deshpande, Bioorg. Med. Chem. Lett., 2008, 18, 2043 CrossRef PubMed.
- C. Jianguo, L. Liang, G. Chunfang, X. Qi and H. Yanmin, Progr. Chem., 2014, 26, 320 Search PubMed.
- M. Decker, Curr. Med. Chem., 2011, 18, 1464 CrossRef CAS PubMed.
- É. Frank, Z. Mucsi, I. Zupkó, B. Réthy, G. Falkay, G. Schneider and J. Wölfling, J. Am. Chem. Soc., 2009, 131, 3894 CrossRef CAS PubMed; G. Mótyán, I. Zupkó, R. Minorics, G. Schneider, J. Wölfling and É. Frank, Mol. Diversity, 2015, 19, 511 CrossRef PubMed; M. M. Abdalla, M. A. Al-Omar, M. A. Bhat, A. G. E. Amr and A. M. Al-Mohizea, Int. J. Biol. Macromol., 2012, 50, 1127 CrossRef PubMed; W. S. Laitonjam, T. S. Rajkumar and B. S. Chingakham, Steroids, 2002, 67, 203 CrossRef PubMed.
- C. Camoutsis, J. Heterocycl. Chem., 1996, 33, 539 CrossRef CAS; É. Frank, Z. Mucsi, M. Szécsi, I. Zupkó, J. Wölfling and G. Schneider, New J. Chem., 2010, 34, 2671 RSC; G. Mótyán, Z. Kádár, D. Kovács, J. Wölfling and É. Frank, Steroids, 2014, 87, 76 CrossRef PubMed.
- B.-L. Zhang, E. Zhang, L.-P. Pang, L.-X. Song, Y.-F. Li, B. Yu and H.-M. Liu, Steroids, 2013, 78, 1200 CrossRef CAS PubMed.
- J. Z. Yan, J. Li and G. W. Rao, Steroids, 2007, 72, 736 CrossRef CAS PubMed; C. Wang, H. Xu, Z. Xie, X. Wang, Z. Zhang and Q. Sun, Steroids, 2010, 75, 1033 CrossRef PubMed; L. H. Huang, Y. F. Zheng, C. J. Song, Y. G. Wang, Z. Y. Xie, Y. W. Lai, Y. Z. Lu and H. M. Liu, Steroids, 2012, 77, 367 CrossRef PubMed.
- M. N. Sakač, A. R. Gakovic, J. J. Csanádi, E. A. Djurendić, O. Klisurić, V. Kojić, G. Bogdanović and K. M. Penov Gaši, Tetrahedron Lett., 2009, 50, 4107 CrossRef.
- S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchai and H. D. Patel, RSC Adv., 2014, 4, 24463 RSC; V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157 CrossRef CAS PubMed.
- A. Magyar, J. Wölfling, M. Kubas, J. A. Cuesta Seijo, M. Sevvana, R. Herbst-Irmer, P. Forgó and G. Schneider, Steroids, 2004, 69, 301 CrossRef CAS PubMed; L. M. R. Antinarelli, A. M. L. Carmo, F. R. Pavan, C. Q. F. Leite, A. D. Da Silva, E. S. Coimbra and D. B. Salunke, Org. Med. Chem. Lett., 2012, 2, 16 CrossRef PubMed; J. Wölfling, É. Frank, G. Schneider and L. F. Tietze, Eur. J. Org. Chem., 1999, 3013 CrossRef.
- S. Gogoi, K. Shekarrao, A. Duarah, T. C. Bora, S. Gogoi and R. C. Boruah, Steroids, 2012, 77, 1438 CrossRef CAS PubMed.
- É. Frank and G. Schneider, J. Steroid Biochem. Mol. Biol., 2013, 137, 301 CrossRef CAS PubMed; I. Zupkó, J. Molnár, B. Réthy, R. Minorics, É. Frank, J. Wölfling, J. Molnár, I. Ocsovszki, Z. Topcu, T. Bitó and L. G. Puskás, Molecules, 2014, 19, 2061 CrossRef PubMed; R. Minorics, T. Szekeres, G. Krupitza, P. Saiko, B. Giessrigl, J. Wölfling, É. Frank and I. Zupkó, Steroids, 2011, 76, 156 CrossRef PubMed.
- C.-H. Tseng, C.-C. Tzeng, C.-L. Yang, P.-J. Lu, H.-L. Chen, H.-Y. Li, Y.-C. Chuang, C.-N. Yang and Y.-L. Chen, J. Med. Chem., 2010, 53, 6164 CrossRef CAS PubMed; C.-H. Tseng, C.-C. Tzeng, C.-L. Yang, P.-J. Lu, H.-L. Chen, C.-Y. Chen, C.-N. Yang and Y.-L. Chen, Mol. Diversity, 2013, 17, 781 CrossRef PubMed; R. K. Arafa, G. H. Hegazy, G. A. Piazza and A. H. Abadi, Eur. J. Med. Chem., 2013, 63, 826 CrossRef PubMed; D. D. N’Da and P. J. Smith, Med. Chem. Res., 2014, 23, 1214 CrossRef; N. Jiang, X. Zhai, T. Li, D. Liu, T. Zhang, B. Wang and P. Gong, Molecules, 2012, 17, 5870 CrossRef PubMed; F.-S. Chang, W. Chen, C. Wang, C.-C. Tzeng and Y.-L. Chen, Bioorg. Med. Chem., 2010, 18, 124 CrossRef PubMed.
- S. Madapa, Z. Tusi and S. Batra, Curr. Org. Chem., 2008, 12, 1116 CrossRef CAS; V. V. Kouznetsov, L. Y. Vargas Mendez and C. M. Mendez Gomez, Curr. Org. Chem., 2005, 9, 141 CrossRef.
- B. C. Sekhar, S. R. Ramadas and D. V. Ramana, Heterocycles, 2000, 53, 741 Search PubMed; A. Soibgui, A. Gaucher, J. Marrot, F. Bourdreux, F. Aloui, B. B. Hassine and D. Prim, Tetrahedron, 2014, 70, 3042 CrossRef.
- P. Karthikeyan, A. M. Rani, R. Saiganesh, K. K. Balasubramanian and S. Kabilan, Tetrahedron, 2009, 65, 811 CrossRef CAS.
- S. Hesse and G. Kirsch, Tetrahedron, 2005, 61, 6534 CrossRef CAS; K. S. Some and J. K. Ray, Tetrahedron Lett., 2007, 48, 5013 CrossRef.
- C. S. Cho, H. B. Kim, W. X. Ren and N. S. Yoon, Appl. Organomet. Chem., 2010, 24, 817 CrossRef CAS; K. R. Prasad and M. Darbarwar, Org. Prep. Proced. Int., 1995, 27, 547 CrossRef.
- K. Shekarrao, P. P. Kaishap, V. Saddanapu, A. Addlagatta, S. Gogol and R. C. Boruah, RSC Adv., 2014, 4, 24001 RSC.
- R. Bera, G. Dhananjaya, S. N. Singh, B. Ramu, S. U. Kiran, P. R. Kumar, K. Mukkanti and M. Pal, Tetrahedron, 2008, 64, 582 CrossRef CAS.
- J. Pan, X. Wang, Y. Zhang and S. L. Buchwald, Org. Lett., 2011, 13, 4974 CrossRef CAS PubMed.
- Z. Kádár, J. Molnár, G. Schneider, I. Zupkó and É. Frank, Bioorg. Med. Chem., 2012, 20, 1396 CrossRef PubMed; Z. Kádár, Á. Baji, I. Zupkó, T. Bartók, J. Wölfling and É. Frank, Org. Biomol. Chem., 2011, 9, 8051 Search PubMed.
- A. L. LaFrate, J. R. Gunther, K. E. Carlson and J. A. Katzenellenbogen, Bioorg. Med. Chem., 2008, 16, 10075 CrossRef CAS PubMed.
- A. Lilienkampf, M. P. Johansson and K. Wähälä, Org. Lett., 2003, 5, 3387 CrossRef CAS PubMed.
- O. Hara, S. Isao, H. Eiichi, S. Saji, M. Kimura, T. Ono and T. Temma, 2012, Patent WO2012026316 A1.
- G. Jones and S. P. Stanforth, in Organic Reactions, ed. L. E. Overman, 2000, vol. 56 Search PubMed.
- J. M. F. Gagan and D. Lloyd, J. Chem. Soc., 1970, 2488 Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
- I. Vermes, C. Haanen and C. Reutelingsperger, J. Immunol. Methods, 2000, 243, 167 CrossRef CAS PubMed.
- E. Mernyák, J. Huber, J. Szabó, G. Schneider, A. Hetényi, L. Márk, G. Maász, Á. Berényi, I. Kovács, R. Minorics, I. Zupkó and J. Wölfling, Steroids, 2013, 78, 1021 CrossRef PubMed.
- D. Ribble, N. B. Goldstein, D. A. Norris and Y. G. Shellman, BMC Biotechnol., 2005, 5, 12 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03910c |
| ‡ Authors (Á. Baji and A. Gyovai) contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |