DOI:
10.1039/C6RA04090J
(Paper)
RSC Adv., 2016,
6, 58529-58540
Novel highly conductive ferroferric oxide/porous carbon nanofiber composites prepared by electrospinning as anode materials for high performance Li-ion batteries†
Received
15th February 2016
, Accepted 30th May 2016
First published on 10th June 2016
Abstract
In this paper, ferroferric oxide (Fe3O4) nanoparticles/porous carbon nanofiber (Fe3O4/PCNFs) composites were successfully fabricated by electrospinning and subsequent calcination. The composites were characterized by X-ray diffraction, thermogravimetric analysis, scanning electron microscopy and transmission electron microscopy to analyze the structure, composition and morphology. The electrochemical performance was evaluated by coin-type cells vs. metallic lithium. The results indicated that Fe3O4/PCNFs composites exhibited high reversible capacity and good capacity retention. The discharge capacity was maintained at 717.2 mA h g−1 at 0.5 A g−1 after 100 cycles. The excellent performances of Fe3O4/PCNFs composites are attributed to good crystallinity and uniformly dispersive Fe3O4 nanoparticles, and a porous carbon shell with high conductivity. The carbon coating buffered the tremendous volumetric changes between Fe3O4 nanoparticles and Fe atoms in the charge/discharge processes and kept the structure integrity of Fe3O4 nanoparticles. Porous carbon nanofibers prepared by the unique calcination process improved the conductivity of composites and provided free space for migration of lithium ions. The preparation strategy is expected to be applicable to the preparation of other transition metal oxide materials as superior anode materials for lithium-ion batteries.
1 Introduction
Pollutant haze and the greenhouse effect have become severe challenges to the environment, which are closely connected to excessive use of fossil fuels.1–3 Rechargeable lithium ion batteries are tools of energy exchange between chemical and electric energy,3,4 which can store energy from clean energy sources.5 As rechargeable lithium ion batteries have the characteristics of high energy density, long lifespan, are environmentally friendly and show fast charge–discharge rates,6,7 they have been widely applied to mobile phones, digital cameras and laptops, etc.7 However, hybrid electric vehicles and electric vehicles require Li-ion batteries to have higher energy density and rate capability3,8 to match the performance of internal combustion vehicles.3
Natural graphite seems to be the most promising candidate for the anode material in lithium-ion batteries because of its numerous advantages.9,10 Graphite and graphitized carbon as anode materials in lithium-ion batteries have been used in many commercial products on the market.9 Modification of carbonaceous anode materials which has been a research focus11 can greatly improve the materials’ electrochemical performance. Composites of carbon nanomaterials and sulfides12–16/metal oxides17–21 have been intensively studied for efficient energy storage. Transition metal oxides (MO) (Fe2O3, Fe3O4, NiO, CoO, Co3O4, Cu2O, CuO, RuO2 and Cr2O3 etc.) have much higher theoretical capacities (∼1000 mA h g−1) than graphite based on the conversion between MO and M.17 However, pure transition metal oxides as anodes in lithium ion batteries often have poor cycling performance owing to the collapse of lattice structure of the original crystal over several discharge/charge cycles as a result of the tremendous volume changes.18,19 A number of research methods concerning transition metal oxides as anodes in lithium ion batteries have been carried out to improve this deficiency and excellent electrochemical performances have been obtained by constructing nanostructured materials, hollow nanostructures, hybrid nanostructures, etc. Carbon coating is the most widely used modification technique for transition metal oxides. On the one hand, carbon coating on the surfaces of metal oxide nanoparticles can reduce the side reactions of the solid electrolyte interface (SEI) at the interface between the metal oxide and electrolyte.19 On the other hand, good electrical conductivity of carbon can make up for the poor conductivity of metal oxides and promote electron transport. In addition, carbon coating as elastic buffer layers/supports20 can confine the position of metal oxides and prevent the agglomeration and cracking of the crystal structure, which can enhance the cycle stability of the electrode. In addition, nanostructured electrode materials have some special characteristics, such as large proportion of surface atoms, small size, etc. which lead to higher electrode/electrolyte contact area, shorter path lengths for Li+ transport and higher charge/discharge rates.21 So carbon coating nanostructured metal oxides as anodes in lithium ion batteries would strengthen the cycle stability and improve the high-rate charge–discharge performance. Iron oxides have the characteristics of low price, environment-friendliness and high abundance, which make them attractive anodes for high performance lithium-ion batteries.20
A large number of Fe3O4/carbon nanocomposites22–25 and Fe2O3/carbon nanocomposites26–28 have been fabricated and investigated as electrode materials for lithium ion batteries. Carbon coating iron oxide nanoparticles can lead to 0D nanospheres,29,30 1D nanowires,31,32 2D nanoflakes33,34 and 3D structures of porous carbon foam loading iron oxide.35,36 Electrospinning is one of the carbon coating methods and has been used to fabricate 1D hybrid carbon coated iron oxide nanofiber composites,37–41 1D iron oxide nanofibers42,43 and 1D carbon nanofibers for Li ion batteries.44,45 The method can embed iron oxides into a conductive carbon by subsequent heat treatment and effectively enhance the electrochemical properties. Zhang et al.37 fabricated Fe2O3–carbon composite nanofibers as durable anode materials for lithium ion batteries. The cell exhibits a reversible capacity of 820 mA h g−1 at a current rate of 0.2C even after 100 cycles. Chaudhari et al..42 synthesized hollow-structured α-Fe2O3 nanofibers by a simple electrospinning technique and subsequent calcination at 500 °C for 4 h in air. The hollow fiber anodes showed a high reversible capacity of 1293 mA h g−1 at a current density of 60 mA g−1 (0.06C). Kim et al..44 fabricated high-purity carbon nanofiber webs by a combination of electrospinning 10 wt% polyacrylonitrile (PAN) polymer–dimethyl formamide (DMF) solution and thermal treatment. Nanofiber webs thermally treated at 1000 °C in Ar have a higher reversible capacity of 450 mA h g−1, than for samples heated at 700 or 2800 °C, at a discharge current density of 30 mA g−1.
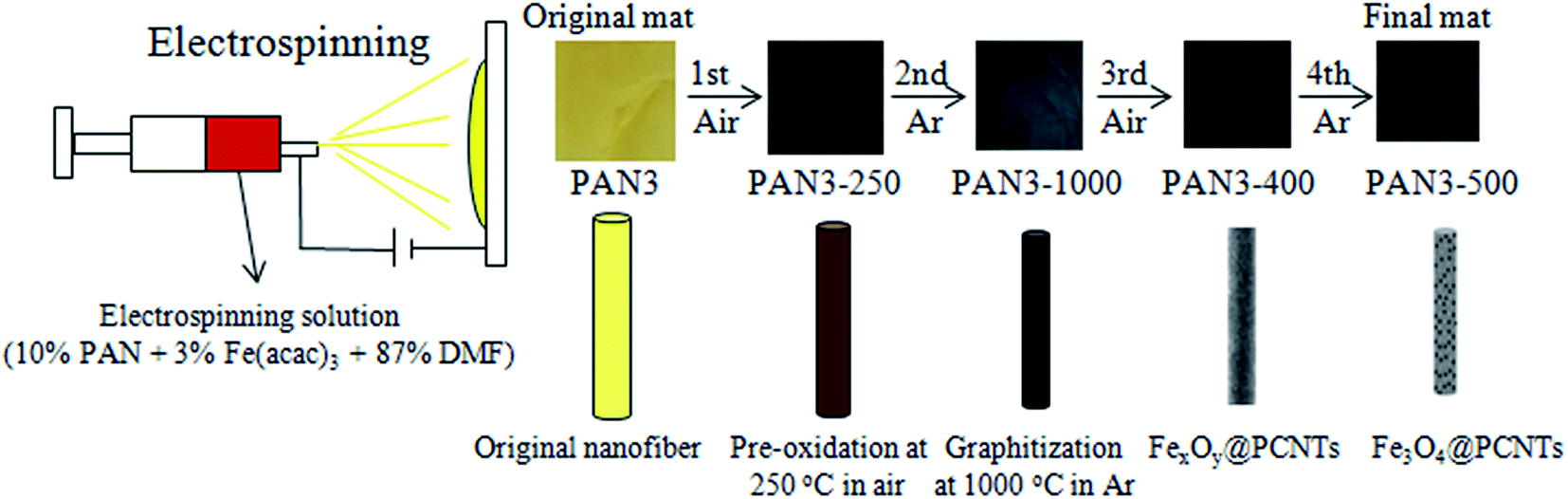
Here, 1D Fe3O4 nanoparticles/porous carbon nanofiber (Fe3O4/PCNFs) composites are fabricated by electrospinning a 10 wt% PAN and 3 wt% Fe(acac)3 DMF solution and subsequent thermal treatment (Fig. 1). The innovative thermal treatment is comprised of four calcination processes for the first time. First, the electrospun nanofibers were pre-oxidized at 250 °C in air to maintain their morphologies. Second, carbonization was conducted at 1000 °C in argon to obtain highly conductive graphitized carbon. Third, oxidation was continued at 400 °C in an air flow to change Fe atoms and Fe C compounds into iron oxides and remove some graphitized carbon of the nanofibers to form porous structures. Carbothermic reduction was finally conducted at 500 °C in Ar to change Fe2O3 nanoparticles into highly conductive Fe3O4 nanoparticles with better crystal structure and to further increase the porosity of the nanofibers. It should be noted that the flow rate of air and calcination equipment have very significant effects on the content of carbon and types of iron oxides in the third calcination step. The excellent electrochemical properties of the novel highly conductive Fe3O4/PCNFs composite anode is evident from the high capacity of 717.2 mA h g−1 after 100 cycles at 0.5 A g−1.
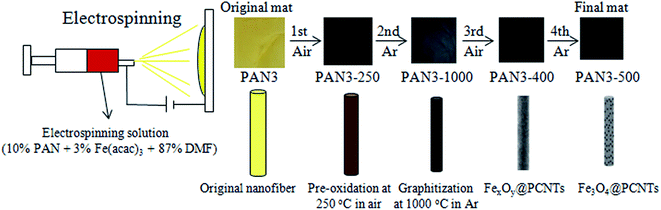 |
| | Fig. 1 Schematic illustration of the process of electrospinning and products of different calcination conditions. | |
2 Experimental section
2.1 Fabrication of Fe3O4/PCNFs composites
PAN with an average molecular weight of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was purchased from Aladdin. Analytic grade ferric acetylacetonate (Fe(acac)3) and DMF were obtained from Sinopharm Chemical Reagent Co., Ltd. None of the reagents were further purified before use.
000 was purchased from Aladdin. Analytic grade ferric acetylacetonate (Fe(acac)3) and DMF were obtained from Sinopharm Chemical Reagent Co., Ltd. None of the reagents were further purified before use.
In a typical process, 1 g PAN and 0.3 g Fe(acac)3 were added into 8.70 g DMF and formed a 10 g mixture, followed by vigorous stirring at room temperature for at least 24 h. The obtained homogeneous dispersion was used as the Fe3O4/PCNFs composite precursor solution for electrospinning. The precursor solution was loaded into a 10 mL plastic syringe connected to a blunt-tip needle with a inner diameter of 0.33 mm. The distance between the needle tip and collector was 15 cm. The injection flow rate of the solution was set to 0.1 mm min−1 (about 1 mL h−1) driven by a pump. A total voltage power of 15 kV was applied between the needle (12 kV) and a plate (−3 kV) covered with aluminum foil. Electrospinning was proceeded with a commercial setup (Beijing Ucalery Technology Development Co., Ltd, SS-2535DC). The environmental humidity of electrospinning was 45 ± 10%, and the temperature was 25 ± 5 °C. The electrospun nanofibers were first preoxidized at 250 °C for 120 min with a heating rate of 1 °C min−1 in air atmosphere in a tube furnace (Hefei Ke Jing Materials Technology Co., Ltd., OTF-1200x Φ 50) to maintain the fibrous morphology, and then carbonized at 1000 °C for 120 min with a heating rate of 5 °C min−1 in argon atmosphere to obtain highly conductive graphitized carbon. Then the composite nanofibers were calcined at 400 °C for 3 h with a heating rate of 5 °C min−1 in air atmosphere to obtain the Fe2O3 and Fe3O4 nanoparticle-loaded porous carbon nanofibers (FexOy/PCNFs) composites. It should be noted that the flow of air and the calcination time both have a profound influence on the compositions and morphology of the FexOy/PCNFs composites because it is likely that graphitized carbon is completely oxidized to CO2 and FexOy transforms into pure Fe2O3 by much longer time calcination and higher air flow rate at 400 °C in air. Finally, Fe3O4/PCNFs composites were prepared by further calcination at 500 °C for 2 h with a heating rate of 5 °C min−1 in argon atmosphere.
For comparison, a homogeneous dispersion containing 1.00 g PAN and 9.00 g DMF was also prepared, and then electrospinning and calcination using the same procedures was also carried out. In addition, the preoxidized nanofibers were calcined at 700 or 900 °C in Ar to observe electrical conductivity of calcinated nanofibers at different calcination temperatures. For ease of description, the above products are denoted as indicated in Table 1.
Table 1 Prepared samples with different compositions and calcination temperaturesa
| Sample |
Fe(acac)3/g |
PAN/g |
DMF/g |
T1 (atm) |
T2 (atm) |
T3 (atm) |
T4 (atm) |
| atm = atmosphere. |
| PAN0-250 |
0.00 |
1.00 |
9.00 |
250 °C (air) |
— |
— |
— |
| PAN0-700 |
0.00 |
1.00 |
9.00 |
250 °C (air) |
700 °C (Ar) |
— |
— |
| PAN0-900 |
0.00 |
1.00 |
9.00 |
250 °C (air) |
900 °C (Ar) |
— |
— |
| PAN0-1000 |
0.00 |
1.00 |
9.00 |
250 °C (air) |
1000 °C (Ar) |
— |
— |
| PAN0-400 |
0.00 |
1.00 |
9.00 |
250 °C (air) |
1000 °C (Ar) |
400 °C (air) |
— |
| PAN0-500 |
0.00 |
1.00 |
9.00 |
250 °C (air) |
1000 °C (Ar) |
400 °C (air) |
500 °C (Ar) |
| PAN3 |
0.30 |
1.00 |
8.70 |
— |
— |
— |
— |
| PAN3-250 |
0.30 |
1.00 |
8.70 |
250 °C (air) |
— |
— |
— |
| PAN3-1000 |
0.30 |
1.00 |
8.70 |
250 °C (air) |
1000 °C (Ar) |
— |
— |
| PAN3-400 |
0.30 |
1.00 |
8.70 |
250 °C (air) |
1000 °C (Ar) |
400 °C (air) |
— |
| PAN3-500 |
0.30 |
1.00 |
8.70 |
250 °C (air) |
1000 °C (Ar) |
400 °C (air) |
500 °C (Ar) |
2.2 Materials characterizations
The crystal structures of as-prepared materials were characterized using powder X-ray diffraction (XRD) on a D8 Advance instrument with lynxEye and SolX (Bruker AXS, WI, USA) with a Cu-Kα radiation source between 10 and 80°. The morphologies were characterized using field-emission scanning electron microscopy (SEM) (S-4800II, Hitachi, Japan) and field emission transmission electron microscopy (TEM) (Tecnai 12, Philips, Holland) in the Test Center of Yangzhou University. A SEM (Zeiss Supra™ 55, Sapphire Carl Zeiss Group, Germany) and a TEM (JEM-2100, JEOL, Japan) in College of Chemistry and Chemical Engineering of Yangzhou University were also used to characterize the morphologies. High-resolution TEM (HRTEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) were conducted using a FEI Tecnai G2 F30 STWIN (USA) operating at 200 kV. Thermogravimetric analysis (TGA) (Pyris 1 TGA, PerkinElmer, USA) was performed in an air atmosphere up to 800 °C at a heating rate of 10 °C min−1. Raman spectra were recorded with a laser Raman spectrometer (In Via, Renishaw, UK) at 532 nm wavelength. The magnetic measurements of PAN3-400 and PAN3-500 were performed on a vibrating sample magnetometer (VSM) (EV7, ADE, USA). The electrical conductivities of the carbon nanofibers were measured by a direct voltage–current method (SZT-2A, Suzhou Tong-Chang Electronics Company Ltd, CHN). Nitrogen physisorption–desorption measurements at 100 °C were performed by a surface area and porosity analyzer (ASAP 2020 HD88, Micromeritics, USA). Brunauer–Emmett–Teller (BET) analyses were performed using software to characterize the surface properties of the porous carbon nanofibers. X-Ray photoelectron spectroscopy (XPS) measurements were conducted with an Al-Kα (1486.8 eV) X-ray source (ESCALAB 250Xi, Thermo Fisher Scientific, USA).
2.3 Electrochemical measurements
The calcined flexible PAN0-500, PAN3-400 and PAN3-500 mats were cut into electrodes with size Φ 16 mm, which were assembled into lithium ion batteries by attaching onto a current collector copper foil with 10 wt% PVDF which dissolved in 1-methyl-2-pyrrolidone (NMP) as binder. The electrodes were first dried in a vacuum drying oven at 80 °C for 12 h. The PAN0-500, PAN3-400 or PAN3-500 electrodes were about 2 mg. Then coin cells were assembled with metallic lithium as the counter/reference electrode, 1 M LiPF6 in ethylene carbonate (EC), diethyl carbonate (DMC) and ethyl methyl carbonate (EMC) (1:1:1 by volume) as electrolyte, and Celgard 2400 polypropylene as separator in an high-purity argon-filled glovebox (VAC-Omni, OMNI-LAB, Vacuum atmospheres company, CA). Cyclic voltammetry (CV) measurements were performed using an electrochemical workstation (CHI660 E, Chenghua, CHN) at a scan rate of 0.1 mV s−1 between 0.01 and 3.0 V. Galvanostatic charge (lithium extraction) and discharge (lithium insertion) cycling of the cells was carried out using a battery test system (CT-3008W, Xinwei, CHN) at different current densities of 0.05, 0.1, 0.2, 0.5 and 1.0 A g−1 between 0.01 and 3 V (vs. Li+/Li) to observe ratio performances, and at a current density of 0.5 A g−1 to study cycle stability. Electrochemical impedance spectroscopic (EIS) measurements were carried out on an electrochemical analyzer (Autolab, Ecochemie, NL). All the current densities and capacities in this study were calculated on the basis of the weight of the integral maps.
Finally, the morphology of the PAN3-500 anode was observed by S-4800II SEM and Tecnai 12 field emission TEM after 100 charge/discharge cycles at 0.5 A g−1.
3 Results and discussion
3.1 Preparation process
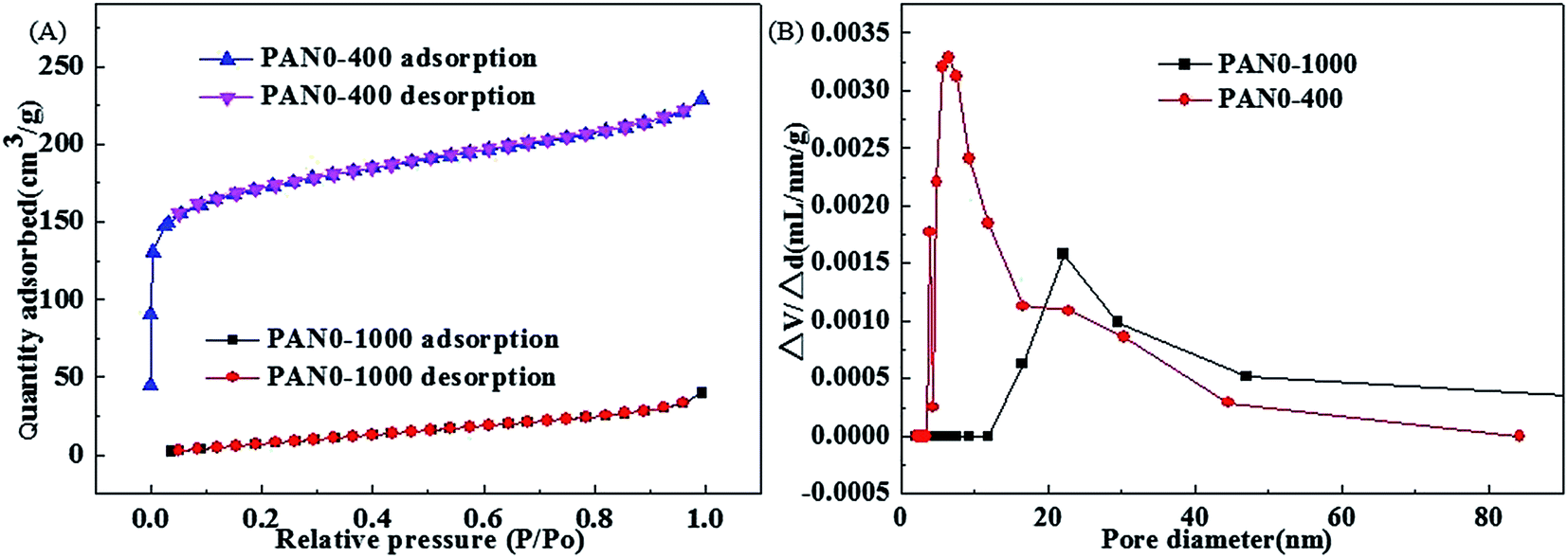
The overall synthesis procedures of Fe3O4/PCNFs composites are schematically illustrated in Fig. 1. PAN and Fe(acac)3 were dissolved in DMF to form a uniform viscous reddish-brown electrospinning solution by stirring for 24 h. Electrospinning was the carried out using the conditions specified in the Experimental section. The original electrospinning nanofibers (PAN3) films are yellow. Wide brown slices (PAN3-250) are obtained by pre-oxidation of PAN3 films at 250 °C in air. Black graphitized nanofiber (PAN3-1000) composites were prepared by calcination of PAN3-250 films at 1000 °C in Ar. Black porous graphitized carbon coated FexOy nanofibers (PAN3-400) sheets were then obtained by calcining PAN3-1000 at 400 °C in air. Finally, black Fe3O4/PCNFs (PAN3-500) were obtained by calcining PAN3-400 in Ar at 500 °C. The pieces show continuous macroscopic area shrinkages by pre-oxidation and graphitization, which can be demonstrated from microscopic characterization of SEM and TEM images (see below). PAN3-400 flakes show an obvious weight loss and are more fluffy than PAN3-1000 pieces. A portion of the carbon will be oxidized to CO2 at 400 °C and a large amount of pores will be formed in air atmosphere, which is demonstrated by the BET results of PAN0-1000 and PAN0-400 as shown in Fig. 2.
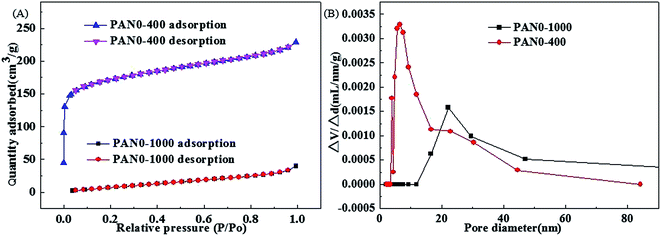 |
| | Fig. 2 Nitrogen adsorption–desorption isotherms of PAN0-1000 and PAN0-400 (A) and pore size distributions of PAN0-1000 and PAN0-400 (B). | |
3.2 Materials characterization
It is well known that high temperature is advantageous to the graphitization of nanofibers. The pure PAN0-250 was calcinated at temperatures of 700, 900 and 1000 °C under Ar. Table 2 shows the conductivities of carbon nanofibers with various calcination temperatures and BET results of PAN0-1000 and PAN0-400. PAN0-1000 has the highest conductivity with a value of 476.2 μS cm−1, which indicates that the graphitization degree increases with the carbonization temperature from 700 to 1000 °C. However, the GCNFs have small specific surface area. In order to increase the specific surface area of graphitized carbon nanofibers, the carbon nanofibers were subsequently calcined at 400 °C under an air atmosphere. Fig. 2(A) shows the nitrogen adsorption–desorption isotherms of PAN0-1000 and PAN0-400. The BET specific surface areas of PAN0-1000 and PAN0-400 are calculated to be 40.6 and 542.6 m2 g−1, respectively; i.e. after further calcination at 400 °C in air, the specific surface area of PAN0-400 was increased by 12.4 times relative to PAN0-1000. Fig. 2(B) displays the distributions of the pore sizes of PAN0-1000 and PAN0-400. Most pore diameters of PAN0-1000 range from 15 to 50 nm with the main peak centered at ∼22 nm. The pore diameters of PAN0-400 have a relatively larger variation range of 3.5–50 nm with most pore diameters of PAN0-400 in the range from 3.5 to 23 nm and a main peak centred at ∼6.4 nm. As shown in Table 2, it is also of note that the conductivity of PAN0-400 is also much higher than for PAN0-250 calcined at 700 °C (PAN0-700). The optimal calcination procedure (T2 = 1000 °C (Ar), T3 = 400 °C (air)) is subsequently applied in the preparation of the follow-up composites of nanofibers described in this paper.
Table 2 Conductivity of carbon nanofibers for different calcination processes and BET test results
| Sample |
PAN0-700 |
PAN0-900 |
PAN0-1000 |
PAN0-400 |
| Conductivity (μS cm−1) |
1.1 |
217.4 |
476.2 |
37.5 |
| BET (m2 g−1) |
— |
— |
40.6 |
542.6 |
The morphology and the diameter of the electrospun nanofibers of PAN0 and PAN3 are compared. It is found that there is no distinct difference between pure PAN nanofibers and composite nanofibers as shown in Fig. S1,† indicating that the electrospinning solution together with operation conditions of electrospinning in this experiment are appropriate to obtain uniform nanofibers. Fig. 3 shows SEM images of PAN3-250, PAN3-1000, PAN3-400 and PAN3-500. All nanofibers exhibit homogeneously distributed diameters. Fig. S2† shows the diameter distributions of PAN3-400 and PAN3-500. The mean sizes of PAN3-400 and PAN3-500 are 250 and 160 nm, respectively. More interestingly, when the carbon nanofibers was successively calcined at 500 °C under Ar atmosphere, the average diameter of PAN3-500 decreased obviously comparing with that of PAN3-400, which was due to the consumption of carbon in the ferric iron reduction reaction. In the control experiment, the average diameter of PAN0-500 shows no distinct decrease relative to that of PAN0-400 as shown in Fig. S3.†
 |
| | Fig. 3 SEM images of PAN3-250 (a), PAN3-1000 (b), PAN3-400 (c) and PAN3-500 (d). | |
As shown in Fig. 4(a) and (c), PAN3-400 and PAN3-500 have homogeneous morphologies. However, the porosities distinctly increase with continuous calcination. As can be seen from Fig. 4(b) and (d), there are many tiny pores and particles in PAN3-400 and PAN3-500. The areas of darker colour of PAN3-400 and PAN3-500 would correspond to iron oxide nanoparticles and the lighter colour areas would be carbon and tiny pores of them, while the little white spots should be pores. The above-mentioned iron oxide nanoparticles, carbon and pores can be verified by the XRD patterns in Fig. 8(A), Raman spectra in Fig. 8(E) and BET values in Table 2, respectively. What should be noted is that in the third calcination process, both flow of air and the applied apparatus for PAN3-1000 have great influence on the integral morphology of the nanofibers and the respective carbon and iron oxide content. Fig. S4† shows two different equipments used in the third calcination process. Fig. 5 shows SEM and TEM images of the product calcined using apparatus in Fig. S4(b)† in the third calcination process (T3). Fig. 5(a) and (b) reveal obviously different morphologies comparing with Fig. 3(c) and 4(b), respectively, in which the degree of air contact of PAN3-1000 in the third calcination process is responsible for the results. Additionally, a larger percentage shrinkage in area would occur or red brown product is generated at higher temperature or higher flow rate of air. In this paper, the various experiments using a series of air flows and different equipment in the third calcination process will not be further discussed. Here, we concentrate on calcination using the apparatus in Fig. S4(a).†
 |
| | Fig. 4 Typical TEM image of PAN3-400 (a) and its partial magnification (b), representative TEM image of PAN3-500 (c) and its partial magnification (d). | |
 |
| | Fig. 5 SEM (a) and TEM (b) images of the product using calcination equipment in Fig. 4(b) in the third calcination process. | |
Fig. 6 shows HRTEM images of PAN3-500. The clear shell lattice fringes with d-spacing of 0.30 nm in the HRTEM image (Fig. 6(c)) are in good agreement with the (220) plane of cubic Fe3O4. Fig. 6(d) demonstrates clear shell lattice fringes with average d-spacing of 0.34 nm, corresponding to the (002) plane of hexagonal graphite though the lattice structures are seldom observed. As shown in Table 2, the conductivity of PAN0-400 is reduced significantly compared with PAN0-1000. The lattice structure of graphitized carbon nanofibers will be destroyed to a great extent in the calcination processes at 400 °C in air for 3 h and the subsequent carbon thermal reduction at 500 °C for 2 h.
 |
| | Fig. 6 Typical structure of PAN3-500 (a), partial enlargement of a cross-section of PAN3-500 (b), the boxed region of part (b) – the observed crystallites in the HRTEM image corresponding to Fe3O4 (c), and seldom observed HRTEM image of lattice structure porous carbon of PAN3-500 (d). | |
The PAN3-500 composites are further characterized by the element maps from EDX spectroscopy. The distributions of C, O and Fe are shown in Fig. 7(c)–(e), respectively. The Fe and O elements establish the existence of iron oxide. All elements are distributed uniformly in the nanofibers and confirm the composition of the PAN3-500 composites.
 |
| | Fig. 7 HAADF-STEM images of PAN3-500 (a) and partial enlargement (b), element mapping of C (c), O (d) and Fe (e). | |
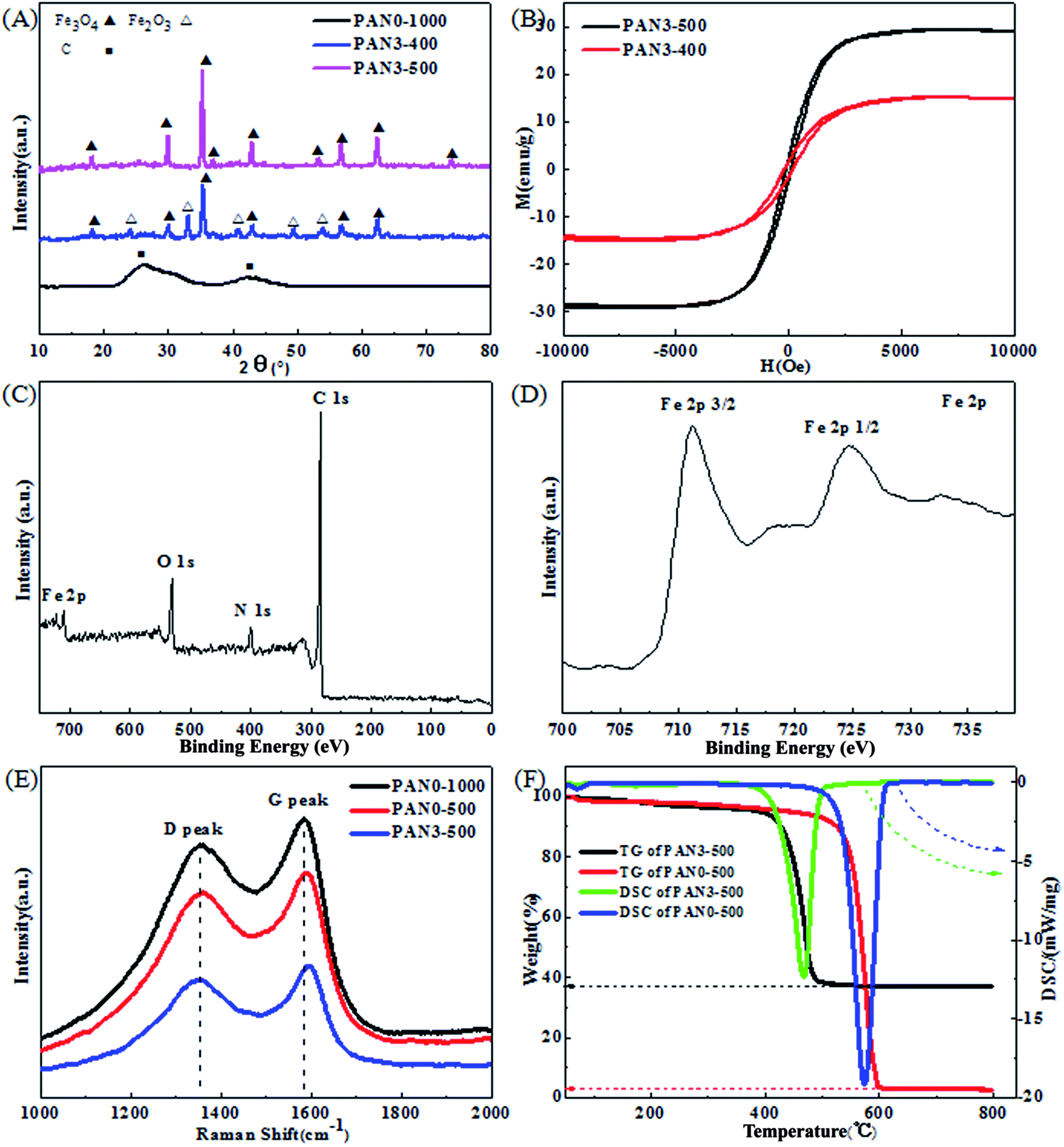
Fig. 8(A) shows the XRD patterns of PAN0-1000, PAN3-400 and PAN3-500. The identified diffraction peaks of PAN0-1000 nanofibers can be clearly assigned to hexagonal graphite (JCPDS, card 41-1487). The peak at 2θ = 26.38 can be indexed to the (002) lattice plane of hexagonal graphite, which indicates that the precursor-PAN has changed into highly conductive graphitic carbon. The peaks at 2θ = 24.14, 33.15, 35.61, 40.85, 49.48, 54.09, 62.45 and 63.99° can be indexed to (012), (104), (110), (113), (024), (116), (214) and (300) lattice planes of hexagonal Fe2O3 (JCPDS, card 33-0664), respectively. The peaks at 2θ = 18.27, 30.10, 35.42, 43.05, 56.94 and 62.52° can be indexed to (220), (311), (400), (511) and (440) lattice planes of cubic Fe3O4 (JCPDS, card 19-0629), respectively. In order to obtain pure Fe3O4 in the nanofibers, PAN3-400 composites were calcined at 500 °C in Ar. Under the inert atmosphere, carbon acts as a reducing agent and Fe2O3 transforms into Fe3O4. No impurity peaks from other iron oxides were observed, implying the high purity of Fe3O4 in the nanofibers. The intense diffraction peaks of Fe3O4 in PAN3-500 demonstrate the better crystalline form of Fe3O4 than that in PAN3-400. Also, the strong diffraction peak of graphitized carbon disappeared in XRD patterns of PAN3-400 and PAN3-500, which indicates the structures of graphitized carbon are destroyed to a great extent. The hysteresis curves of as-prepared PAN3-400 and PAN3-500 are shown in Fig. 8(B). The saturated magnetizations of PAN3-400 and PAN3-500 are about 15.0 and 29.2 emu g−1, respectively. The results of hysteresis curves are consistent with their compositions shown by XRD results. The surface electronic state and composition of PAN3-500 were further investigated by XPS analysis as presented in Fig. 8(C) and (D). Fig. 8(C) reveals the existence of C, N, O and Fe elements in PAN3-500. The existence of N of PAN3-500 in Fig. 8(C) should be attributable to nitrogen-containing groups of PCNFs originating from the PAN raw material. Fig. 8(D) shows the high resolution Fe2p spectrum. The two main peaks located at 711.0 and 725.7 eV correspond to Fe2p3/2 and Fe2p1/2, respectively. The results show the Fe3+ and Fe2+ states coexist in PAN3-500, which further confirmed the iron oxide in PAN3-500 is Fe3O4.
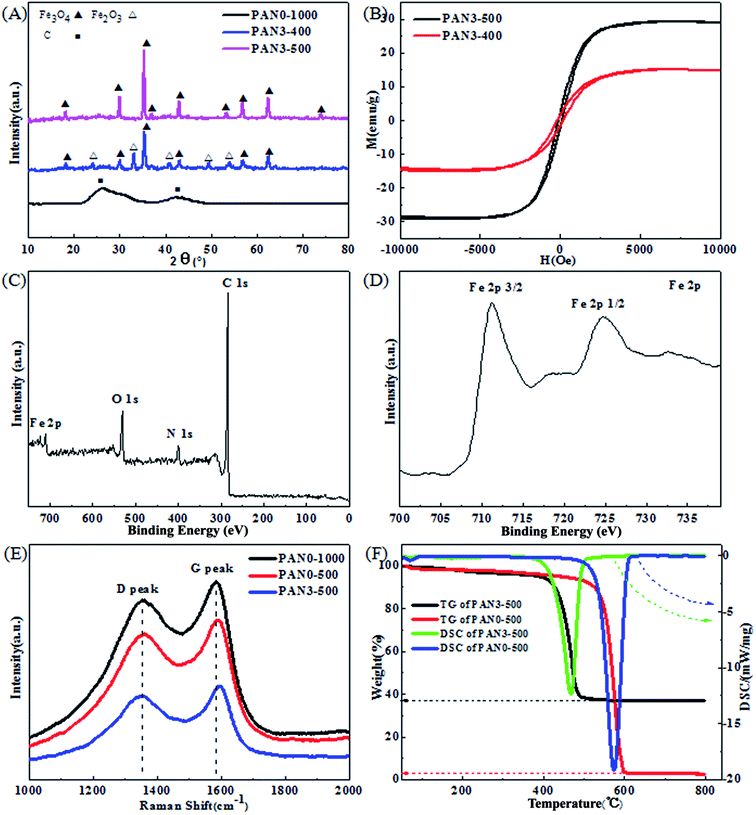 |
| | Fig. 8 XRD patterns of PAN0-1000, PAN3-400 and PAN3-500 (A), hysteresis loops of PAN3-400 and PAN3-500 (B), XPS survey spectrum of PAN3-500 (C) and high resolution Fe2p spectrum (D), Raman spectra of PAN0-1000, PAN0-500 and PAN3-500 (E), and TG-DSC curves of PAN0-500 and PAN3-500 (F). | |
Fig. 8(E) shows the Raman spectra of PAN0-1000, PAN0-500 and PAN3-500. All samples show a strong D (disorder) peak at about 1360 cm−1 and G (graphite) peak at about 1590 cm−1. The intensity ratios R (ID/IG) of PAN0-1000, PAN0-500 and PAN3-500 are 0.96, 0.99 and 0.99, respectively. The large values of R indicate the high degree of disordered and defective carbon. From Fig. 8(F), the differential scanning calorimetry (DSC) and thermogravimetry (TG) curves of PAN0-500 indicate that the reaction is exothermic and the weight loss is mainly between 550 and 600 °C. However, the TG curve of PAN3-500 reveals that the weight loss is mainly in the temperature range of 400–490 °C. It can be speculated that the graphitized carbon structures of PAN3-500 are destroyed more substantially than in PAN0-500 due to the reaction of Fe2O3 and carbon, and the exothermic reaction of Fe3O4 oxidizing into Fe2O3 promotes the oxidation reaction of porous carbon at lower temperature. The residual proportion of PAN3-500 is 36.9 wt%. According to the results of TGA tests, the original weight of carbon and Fe3O4 in PAN3-500 are calculated to be 66.0 and 34.0 wt%, respectively.
3.3 Electrochemical performance
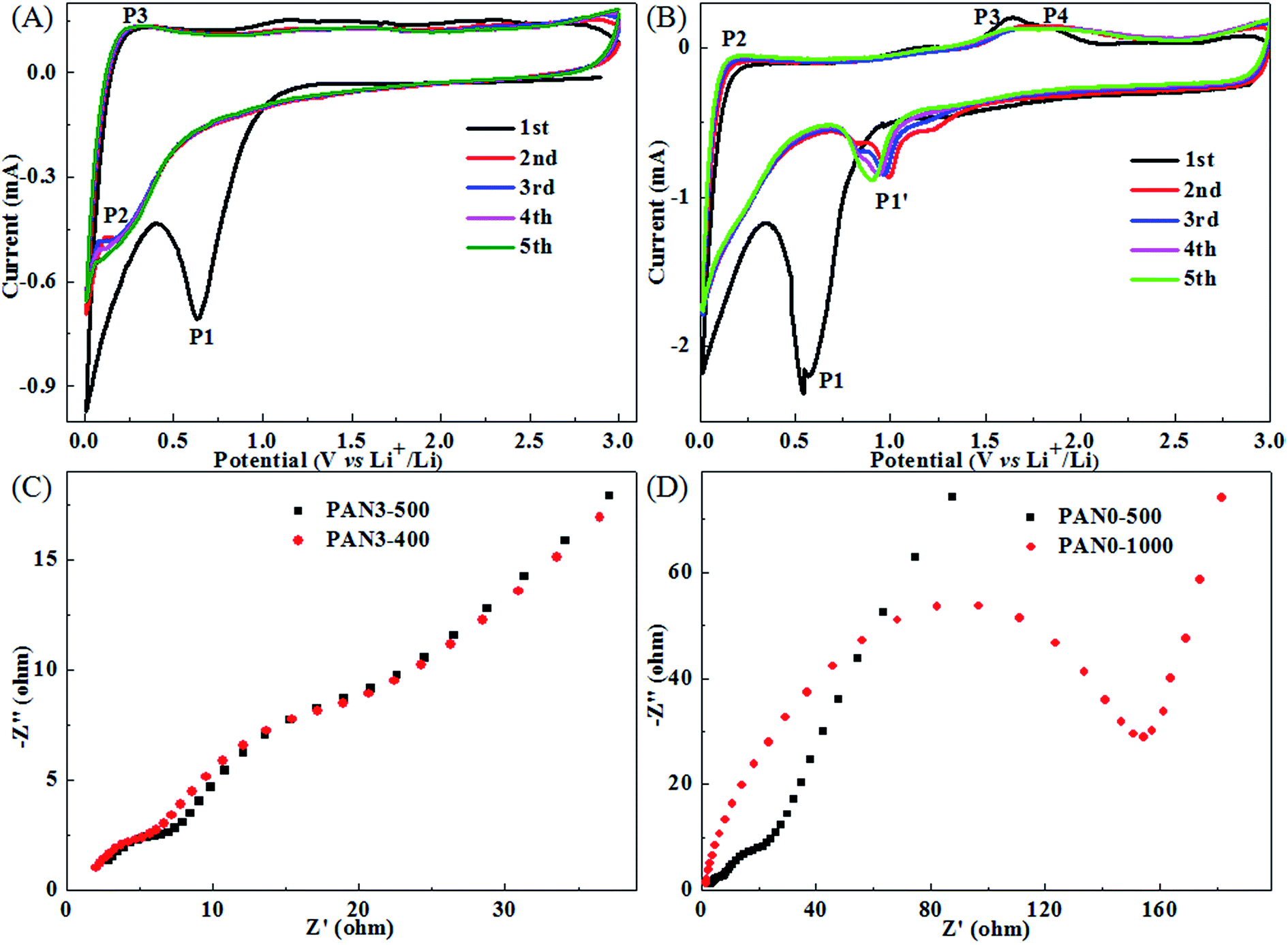
Porous carbon in the final mat PAN3-500 is sufficient to form a stable framework and has very good flexibility, as shown in Fig. S5.† CV measurements were carried out to investigate the electrochemical reactions of PAN3-500 in the range of 3.0 to 0.01 V at a scan rate of 0.1 mV s−1 at room temperature. It is seen in Fig. 9(A) that PAN0-500 exhibits a cathodic peak at about 0.63 V which represents the formation of the SEI film and the decomposition of the electrolyte46 and two broad anodic peaks at about 1.16 and 2.11 V attributed to irreversible reactions with electrolyte. In the subsequent cycles, the peak P2 at about 0.18 V and the peak P3 at about 0.25 V should correspond to the insertion and extraction of lithium ions from the graphitized structures of porous carbon nanofibers, respectively. Subsequently, the peak P1 at about 0.63 V disappears, indicating a stable and complete SEI film has formed on the surface of carbon. As shown in Fig. 9(B), at the first cycle, the PAN3-500 electrode exhibits a clear cathodic peak. The peak P1 at about 0.54 V might be due to the initial lithium insertion into the Fe3O4 to form Li2Fe3O4, given by eqn (1)33,47 and the reduction reaction of Li2Fe3O4 to Fe0, along with the formation of amorphous Li2O, given by eqn (2), as well as the formation of SEI film33,41,47 and the decomposition of the electrolyte. The peak P2 should correspond to the extraction of lithium ions from the graphitized structures of porous carbon nanofibers and the peak at about 1.20 V should correspond to irreversible reactions with electrolyte such as the peak at about 1.16 V of PAN0-500 in Fig. 9(A). The continuous broad peaks P3 and P4 at about 1.64 and 1.82 V correspond to the oxidation reactions of Fe0 to Fe2+ and Fe2+ to Fe3+, respectively.33,36,47 The two electrochemical reactions of Fe0 to Fe2+ and Fe2+ to Fe3+ with nearby peak potentials and the irreversible reactions could be responsible for the phenomenon that the peaks P3 and P4 merged into a more broadened peak in subsequent cycles. The total reaction is given by eqn (3).33,41,47| | |
Fe3O4 + 2Li+ + 2e → Li2Fe3O4
| (1) |
| | |
Li2Fe3O4 + 6Li+ + 6e → 3Fe0 + 4Li2O
| (2) |
| | |
3Fe0 + 4Li2O → Fe3O4 + 8Li+ + 8e
| (3) |
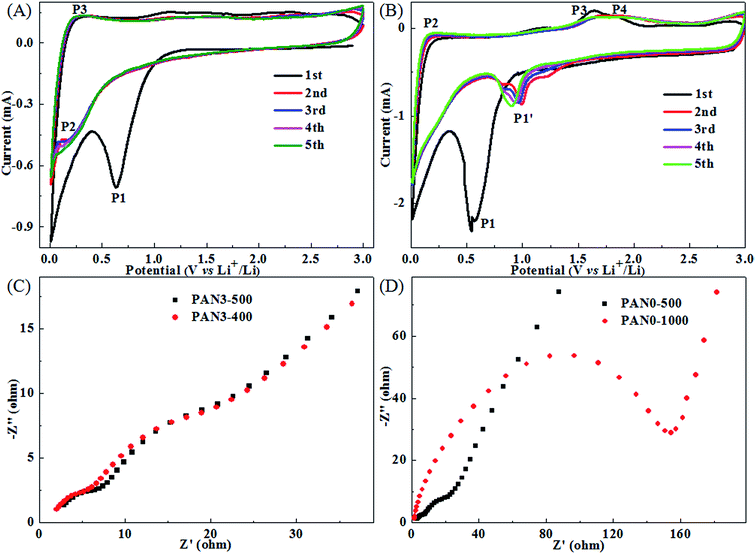 |
| | Fig. 9 CV measurements of PAN0-500 (A), PAN3-500 (B) cycled between 0.01 and 3 V at a scan rate of 0.1 mV s−1, Nyquist plots of PAN3-500 and PAN3-400 (C), PAN0-500 and PAN0-1000 (D). | |
EIS measurements were carried out at open circuit potential with an AC voltage amplitude of 5.0 mV in a frequency range from 100 kHz to 0.01 Hz to more fully understand the electrochemical performance of PAN3-500 in comparison with PAN3-400, PAN0-500 and PAN0-1000. Fig. 9(C) and (D) display the Nyquist plots of PAN3-500 and PAN3-400 electrodes, PAN0-1000 and PAN0-500 electrodes after 1 cycle at 0.5 A g−1. From Fig. 9(C), the Nyquist plots of PAN3-500 and PAN3-400 have similar profiles, which consist of two semicircles, as well as a line, respectively. The two semicircles and the line from high frequency to low frequency are related to the resistance of SEI, charge-transfer resistance on the electrolyte/electrode interface and the solid-state diffusion resistance of Li ion in the electrode, respectively.13,15 From Fig. 9(D), it is clearly seen that the radius of the semicircle for PAN0-500 in the medium frequency region is much smaller than that of PAN0-1000, indicating the porous structured PAN0-500 has a much lower electron-transfer resistance than graphitized PAN0-1000. The PAN3-500, PAN3-400 and PAN0-500 electrodes have remarkably small electron-transfer resistance compared with that of PN0-1000. The results indicate the greatly diminished charge-transfer resistance at the electrode/electrolyte interface should be due to the porous structure in PCNFs of PAN3-500, PAN3-400 and PAN0-500, which can greatly improve the diffusion of lithium ions as well as the transfer of electrons for better electrochemical performance.12–16
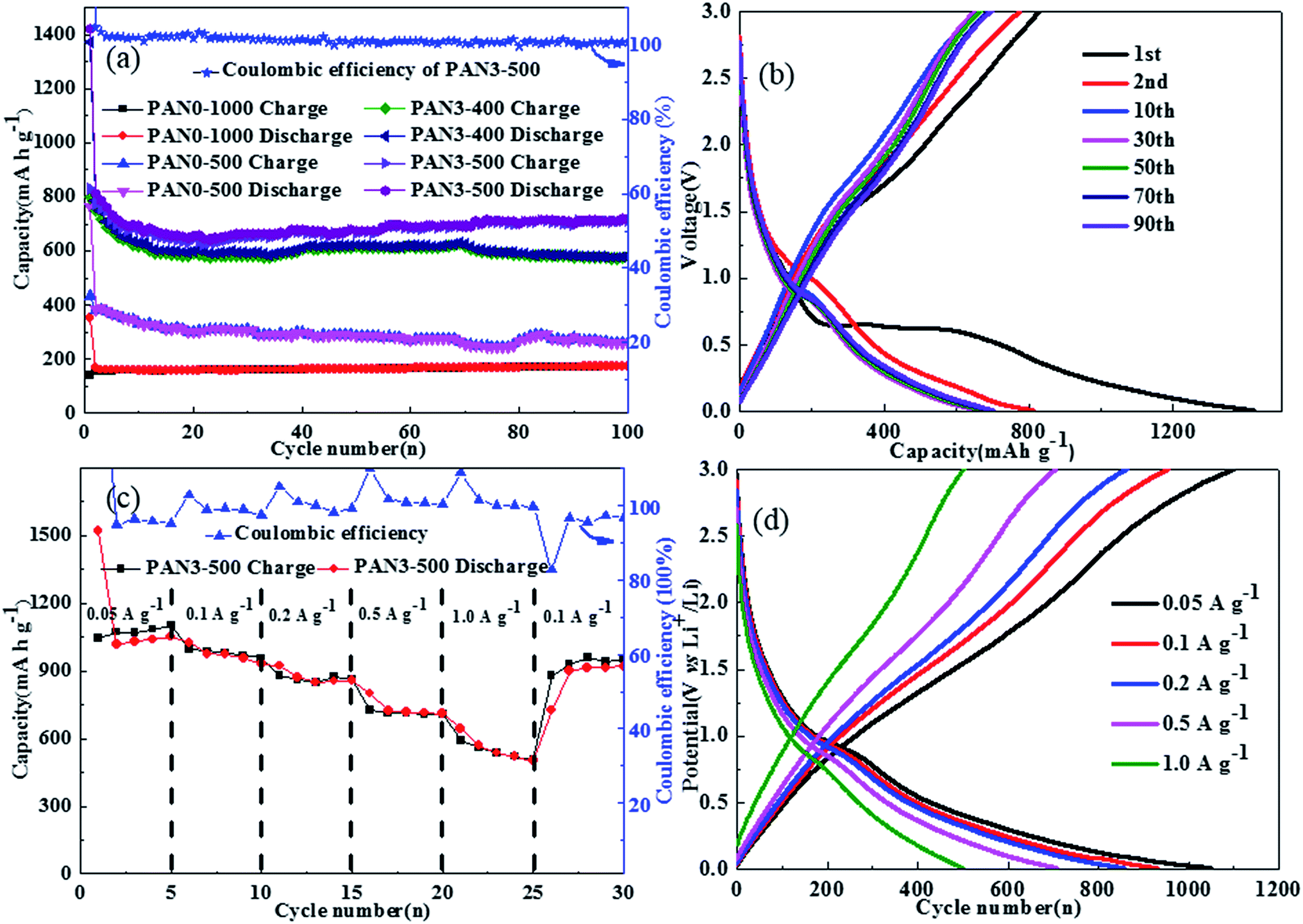
As the proportion of carbon in PAN3-500 is 66.0 wt%, the specific capacitance of the composites is calculated as approximately 560 mA h g−1 based on the theoretical capacities of Fe3O4 (926 mA h g−1) and graphite (372 mA h g−1). Fig. 10(a) shows the charge–discharge cycle performance of PAN0-1000, PAN0-500, PAN3-400 and PAN3-500 at 0.5 A g−1 in the range of 3.0 to 0.01 V. In the 100th cycle, the discharge capacity of PAN0-1000, PAN0-500, PAN3-400 and PAN3-500 were 173.1, 261.1, 579.3 and 717.2 mA h g−1, respectively. The test results show that the capacities of composite nanofibers are obviously higher than that of carbon nanofibers. The high capacity of PAN3-500 is attributed to the higher porosity of the porous carbon, the carbon coating, and the good crystallinity of Fe3O4. The discharge capacities of PAN3-500 in the 1st, 2nd, 10th, 30th, 50th, 70th and 90th cycles were 1422.1, 811.7, 672.9, 665.0, 674.6, 692.7 and 700.6 mA h g−1, respectively, indicating that PAN3-500 had a high capacity and remarkable capacity retention. The SEM images of PAN3-500 after 100 charge–discharge cycles and one discharge cycle are shown in Fig. 11 and the diameter distribution of PAN3-500 after 100 charge–discharge cycles and one discharge cycle is shown in Fig. S6.† The mean diameter of PAN3-500 after 100 charge–discharge cycles and one discharge cycle is 180 nm. The results show that the mean diameter of PAN3-500 after 100 charge–discharge cycles and one discharge cycle is the same as the original PAN3-500, which demonstrates that PAN3-500 essentially shows no volume change in the charging–discharging process at a current density of 0.5 A g−1. The presence of small pores which can afford space for inserted lithium should be responsible for this phenomenon. Fig. 10(b) shows the detailed charge and discharge curves of PAN3-500 at different cycle numbers. As shown in Fig. 10(c), the rate capabilities of PAN3-500 were investigated at various rates from 0.05 to 1.0 A g−1 to further evaluate the electrochemical performance. When the densities were 0.05, 0.1, 0.2, 0.5 and 1.0 A g−1, the PAN3-500 retained high specific capacities of 1050.6, 931.1, 857.0, 709.0 and 501.6 mA h g−1 in the 5th cycle, respectively. On taking back to 0.1 A g−1, the capacity still achieved 919.3 mA h g−1. Fig. 10(d) shows the charge/discharge curves of PAN3-500 at different current densities, which display an approximately symmetrical shape, indicating good reversibility of Li+ insertion/extraction.
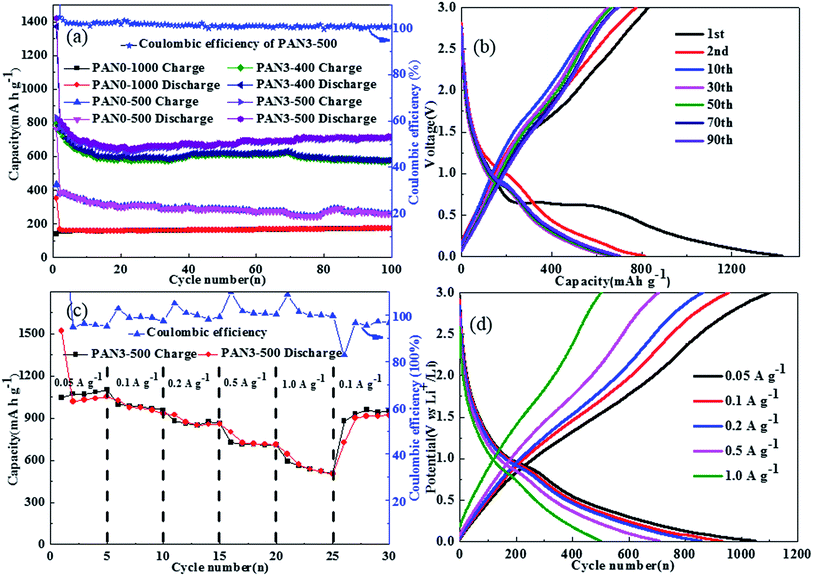 |
| | Fig. 10 The cycling performance of PAN0-1000, PAN0-500, PAN3-400 and PAN3-500 at a current density of 0.5 A g−1 (a) and relevant charge and discharge curves of PAN3-500 (b), charge–discharge performance of PAN3-500 at various current rates (c) and relevant charge and discharge curves (d). | |
 |
| | Fig. 11 SEM image of PAN3-500 after 100 charge–discharge cycles and one discharge cycle (a) and relevant magnification (b). | |
It is worth noting that the electrochemical performance of PAN3-500 in this work is comparable to most of the electrospinning and calcined products, such as Fe2O3 nanofibers,42,43 carbon nanofibers40–44 and iron oxides/CNFs composites37–41 reported in previous literature, as shown in Table 3.
Table 3 Comparison of the retaining capacity of FexOy nanofibers, carbon nanofibers or FexOy/CNFs composites prepared by electrospinninga
| Nanocomposite |
Precursors |
Calcination temperature |
Capacitance/mA h g−1 |
Ref. |
| PS = polystyrene, PVP = polyvinylpyrrolidone, PCFs = porous carbon fibers. |
| Fe2O3-carbon |
PAN–Fe(acac)3 |
At 500 °C for 3 h in air |
820 (0.2C) |
37 |
| α-Fe2O3-CNFs |
PAN–FeCl3 |
At 600 °C for 8 h in Ar |
600 (50 mA g−1) |
38 |
| α-Fe2O3 nanorods |
PVP–Fe(acac)3 |
At 500 °C for 5 h in air |
1095 (0.05C) |
39 |
| C/Fe3O4 |
PAN–Fe(acac)2 |
At 600 °C for 10 h in Ar |
1096 (0.2 A g−1) |
40 |
| |
|
At 700 °C for 10 h in Ar |
300 (0.2 A g−1) |
40 |
| Fe3O4@PCFs |
PAN–PS–Fe3O4 |
At 600 °C for 2 h in Ar |
541 (2.0 A g−1) |
41 |
| Hollow Fe2O3 nanofibers |
PVP–Fe(acac)3 |
At 500 °C for 4 h in air |
1293 (0.06 A g−1) |
42 |
| Porous Fe2O3 nanotubes |
PVP–Fe(acac)3 |
At 500 °C for 3 h in air |
987.7 (0.2 A g−1) |
43 |
| Carbon nanofibers |
PAN |
At 700 °C in Ar |
275 (0.03 A g−1) |
44 |
| |
|
At 1000 °C in Ar |
450 (0.03 A g−1) |
44 |
| |
|
At 2800 °C in Ar |
140 (0.03 A g−1) |
44 |
| Carbon nanofiber |
PAN |
At 800 °C for 1 h in Ar |
407 (0.15 A g−1) |
45 |
| |
|
At 1300 °C for 1 h in Ar |
239 (0.15 A g−1) |
45 |
| Fe3O4/PCNFs |
PAN–Fe(acac)3 |
At 1000 °C for 2 h in Ar, |
|
|
| |
|
then 400 °C for 3 h in air, |
579.3 (0.5 A g−1) |
This work |
| |
|
then 500 °C for 2 h in Ar |
717.2 (0.5 A g−1) |
This work |
4 Conclusions
In summary, we synthesized Fe3O4/porous carbon nanofibers by electrospinning and subsequent unique calcination processes. Graphitized carbon was obtained by calcination at 1000 °C in Ar. Porous structured carbon was obtained by further calcination at 400 °C in air, which had much higher porosity than graphitized carbon. Fe3O4/porous carbon nanofibers were then prepared via the carbon-thermal reduction process by a final calcination at 500 °C in Ar. The porous carbon coated nano-sized Fe3O4 nanofiber composites had excellent performance as anodes for lithium ion batteries. The reversible capacity was higher than for FexOy/porous carbon nanofibers composites and carbon nanofibers. Fe3O4/porous carbon nanofibers retained a reversible capacity of 717.2 mA h g−1 at 0.5 A g−1 after 100 cycles. At 0.05 A g−1, the composites delivered a reversible capacity as high as 1050.6 mA h g−1. The porous carbon nanofibers of coated Fe3O4 nanoparticles have good conductivity, which is useful for electronic transmission and fast diffusion of lithium in the charge–discharge process. Additionally, carbon can buffer the volume changes between nano-sized Fe3O4 nanoparticles and Fe atoms in the charging and discharging process and facilitate the transition reactions in situ to maintain the crystal structures. The combination of conductive porous carbon nanofibers and nano-sized Fe3O4 are responsible for the higher capability and good cycling stability. We believe much higher capacity Fe3O4/porous carbon nanofibers can be fabricated by more appropriate calcination conditions using further optimised calcination apparatus, temperatures and flow rate of air. Importantly, this multistep method of preparing Fe3O4/porous carbon nanofibers is novel.
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
The authors appreciate the Test Center of Yangzhou University for TEM and HRTEM measurements and the financial support from the National Natural Science Foundation of China (grant no. 21273195 and 20901065), the Project Funded by the Natural Science Foundation of Education Committee of Jiangsu Province (12KJB150023), the Scientific Innovation Planing Program for Academic Graduates of Jiangsu Ordinary Universities (KYLX_1336), the Cooperation Fund from Yangzhou Government and Yangzhou University (Grant no. 2012038-9) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
- V. Ramanathan and G. Carmichael, Global and Regional Climate Changes Due to Black Carbon, Nat. Geosci., 2008, 1, 221–227 CrossRef CAS.
- V. Ramanathan and Y. Feng, Air Pollution, Greenhouse Gases and Climate Change: Global and Regional Perspectives, Environment, 2009, 43, 37–50 CAS.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Electrical Energy Storage for Transportation-approaching the Limits of, and Going Beyond, Lithium-ion Batteries, Energy Environ. Sci., 2012, 5, 7854–7863 Search PubMed.
- M. S. Dresselhaus and I. L. Thomas, Alternative Energy Technologies, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Electrical Energy Storage for the Grid: a Battery of Choices, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- R. Van Noorden, The Rechargeable Revolution: A Better Battery, Nature, 2014, 507, 26–28 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, Lithium Batteries: Status, Prospects and Future, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Electrodes with High Power and High Capacity for Rechargeable Lithium Batteries, Science, 2006, 311, 977–980 CrossRef CAS PubMed.
- M. Yoshio, H. Wang, K. Fukuda, Y. Hara and Y. Adachi, Effect of Carbon Coating on Electrochemical Performance of Treated Natural Graphite as Lithium-ion Battery Anode Material, J. Electrochem. Soc., 2000, 147, 1245–1250 CrossRef CAS.
- M. Yoshio, H. Wang, K. Fukuda, T. Umeno, T. Abe and Z. Ogumi, Improvement of Natural Graphite as a Lithium-ion Battery Anode Material, from Raw Flake to Carbon-Coated Sphere, J. Mater. Chem., 2004, 14, 1754–1758 RSC.
- Y. P. Wu, E. Rahm and R. Holze, Carbon Anode Materials for Lithium Ion Batteries, J. Power Sources, 2003, 114, 228–236 CrossRef CAS.
- L. S. Zhang, W. Fan and T. X. A. Liu, Flexible Free-standing Defect-rich MoS2/graphene/carbon Nanotube Hybrid Paper as a Binder-free Anode for high-performance Lithium Ion Batteries, RSC Adv., 2015, 5, 43130–43140 RSC.
- Y. E. Miao, Y. Huang, L. Zhang, W. Fan, F. Lai and T. Liu, Electrospun Porous Carbon Nanofiber@MoS2 Core/sheath Fiber Membranes as Highly Flexible and Binder-free Anodes for Lithium-ion Batteries, Nanoscale, 2015, 7(25), 11093–11101 RSC.
- L. S. Zhang, Y. P. Huang, Y. F. Zhang, W. Fan and T. X. Liu, Three-Dimensional Nanoporous Graphene-Carbon Nanotube Hybrid Frameworks for Confinement of SnS2 Nanosheets: Flexible and Binder-Free Papers with Highly Reversible Lithium Storage, ACS Appl. Mater. Interfaces, 2015, 7(50), 27823–27830 Search PubMed.
- L. S. Zhang, Y. P. Huang, Y. F. Zhang, H. H. Gu, W. Fan and T. X. Liu, Flexible Electrospun Carbon Nanofiber@NiS Core/Sheath Hybrid Membranes as Binder-Free Anodes for Highly Reversible Lithium Storage, Adv. Mater. Interfaces, 2016, 3 DOI:10.1002/admi.201500467.
- L. S. Zhang, W. Fan, W. W. Tjiu and T. X. Liu, 3D Porous Hybrids of Defect-rich MoS2/graphene Nanosheets with Excellent Electrochemical Performance as Anode Materials for Lithium Ion Batteries, RSC Adv., 2015, 5, 34777–34787 RSC.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nano-sized Transition-metal Oxides as Negative-electrode Materials for Lithium-ion Batteries, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- M. J. Armstrong, C. O’Dwyer, W. J. Macklin and J. D. Holmes, Evaluating the Performance of Nanostructured Materials as Lithium-ion Battery electrodes, Nano Res., 2014, 7, 1–62 CrossRef CAS.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanostructured Metal Oxide-based Materials as Advanced Anodes for Lithium-ion Batteries, Nanoscale, 2012, 4, 2526–2542 RSC.
- L. Zhang, H. B. Wu and X. W. Lou, Iron-oxide-based Advanced Anode Materials for Lithium-ion Batteries, Adv. Energy Mater., 2014, 4, 1032–1039 Search PubMed.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nanostructured Materials for Advanced Energy Conversion and Storage Devices, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- T. Zhu, J. S. Chen and X. W. Lou, Glucose-assisted One-pot Synthesis of FeOOH Nanorods and Their Transformation to Fe3O4@ Carbon Nanorods for Application in Lithium Ion Batteries, J. Phys. Chem. C, 2011, 115, 9814–9820 CrossRef CAS.
- S. Jin, H. Deng, D. Long, X. Liu, L. Zhan and X. Liang, et al., Facile Synthesis of Hierarchically Structured Fe3O4/carbon Micro-flowers and Their Application to Lithium-ion Battery Anodes, J. Power Sources, 2011, 196, 3887–3893 CrossRef CAS.
- T. Yoon, C. Chae, Y. K. Sun, X. Zhao, H. H. Kung and J. K. Lee, Bottom-up in situ Formation of Fe3O4 Nanocrystals in a Porous Carbon Foam for Lithium-ion Battery Anodes, J. Mater. Chem., 2011, 21, 17325–17330 RSC.
- J. E. Lee, S. H. Yu, D. J. Lee, D. C. Lee, S. I. Han and Y. E. Sung, et al., Facile and Economical Synthesis of Hierarchical Carbon-coated Magnetite Nanocomposite Particles and Their Applications in Lithium Ion Battery Anodes, Energy Environ. Sci., 2012, 5, 9528–9533 Search PubMed.
- G. Zhou, D. W. Wang, P. X. Hou, W. Li, N. Li and C. Liu, et al., A Nanosized Fe2O3 Decorated Single-walled Carbon Nanotube Membrane as a High-performance Flexible Anode for Lithium Ion Batteries, J. Mater. Chem., 2012, 22, 17942–17946 RSC.
- S. L. Chou, J. Z. Wang, D. Wexler, K. Konstantinov, C. Zhong and H. K. Liu, et al., High-surface-area α-Fe2O3/Carbon Nanocomposite: One-step Synthesis and its Highly Reversible and Enhanced High-rate Lithium Storage Properties, J. Mater. Chem., 2010, 20, 2092–2098 RSC.
- Y. Li, C. Zhu, T. Lu, Z. Guo, D. Zhang and J. Ma, et al., Simple Fabrication of a Fe2O3/Carbon Composite for Use in a High-performance Lithium Ion Battery, Carbon, 2013, 52, 565–573 CrossRef CAS.
- P. Lv, H. Zhao, Z. Zeng, J. Wang, T. Zhang and X. Li, Facile Preparation and Electrochemical Properties of Carbon Coated Fe3O4 as Anode Material for Lithium-ion Batteries, J. Power Sources, 2014, 259, 92–97 CrossRef CAS.
- Z. Zeng, H. Zhao, J. Wang, P. Lv, T. Zhang and Q. Xia, Nanostructured Fe3O4@C as Anode Material for Lithium-ion Batteries, J. Power Sources, 2014, 248, 15–21 CrossRef CAS.
- T. Muraliganth, A. V. Murugan and A. Manthiram, Facile Synthesis of Carbon-decorated Single-crystalline Fe3O4 Nanowires and their Application as High Performance Anode in Lithium Ion Batteries, Chem. Commun., 2009, 7360–7362 RSC.
- Y. Wu, Y. Wei, J. Wang, K. Jiang and S. Fan, Conformal Fe3O4 Sheath on Aligned Carbon Nanotube Scaffolds as High-performance Anodes for Lithium Ion Batteries, Nano Lett., 2013, 13, 818–823 CrossRef CAS PubMed.
- C. He, S. Wu, N. Zhao, C. Shi, E. Liu and J. Li, Carbon-encapsulated Fe3O4 Nanoparticles as a High-rate Lithium Ion Battery Anode Material, ACS Nano, 2013, 7, 4459–4469 CrossRef CAS PubMed.
- Y. Su, S. Li, D. Wu, F. Zhang, H. Liang and P. Gao, et al., Two-dimensional Carbon-coated Graphene/metal Oxide Hybrids for Enhanced Lithium Storage, ACS Nano, 2012, 6, 8349–8356 CrossRef CAS PubMed.
- T. Yoon, C. Chae, Y. K. Sun, X. Zhao, H. H. Kung and J. K. Lee, Bottom-up in situ Formation of Fe3O4 Nanocrystals in a Porous Carbon Foam for Lithium-ion Battery Anodes, J. Mater. Chem., 2011, 21, 17325–17330 RSC.
- E. Kang, Y. S. Jung, A. S. Cavanagh, G. H. Kim, S. M. George and A. C. Dillon, et al., Fe3O4 Nanoparticles Confined in Mesocellular Carbon Foam for High Performance Anode Materials for Lithium-ion Batteries, Adv. Funct. Mater., 2011, 21, 2430–2438 CrossRef CAS.
- X. Zhang, H. Liu, S. Petnikota, S. Ramakrishna and H. J. Fan, Electrospun Fe2O3–carbon Composite Nanofibers as Durable Anode Materials for Lithium Ion Batteries, J. Mater. Chem. A, 2014, 2, 10835–10841 RSC.
- L. Ji, O. Toprakci, M. Alcoutlabi, Y. Yao, Y. Li and S. Zhang, et al., α-Fe2O3 Nanoparticle-loaded Carbon Nanofibers as Stable and High-capacity Anodes for Rechargeable lithium-ion Batteries, ACS Appl. Mater. Interfaces, 2012, 4, 2672–2679 Search PubMed.
- C. T. Cherian, J. Sundaramurthy, M. Kalaivani, P. Ragupathy, P. S. Kumar and V. Thavasi, et al., Electrospun α-Fe2O3 Nanorods as a Stable, High Capacity Anode Material for Li-ion Batteries, J. Mater. Chem., 2012, 22, 12198–12204 RSC.
- L. Wang, Y. Yu, P. C. Chen, D. W. Zhang and C. H. Chen, Electrospinning Synthesis of C/Fe3O4 Composite Nanofibers and Their Application for High Performance Lithium-ion Batteries, J. Power Sources, 2008, 183, 717–723 CrossRef CAS.
- X. Qin, H. Zhang, J. Wu, X. Chu, Y. B. He and C. Han, et al., Fe3O4 Nanoparticles Encapsulated in Electrospun Porous Carbon Fibers with a Compact Shell as High-performance Anode for Lithium Ion Batteries, Carbon, 2015, 87, 347–356 CrossRef CAS.
- S. Chaudhari and M. Srinivasan, 1D Hollow α-Fe2O3 Electrospun Nanofibers as High Performance Anode Material for Lithium Ion Batteries, J. Mater. Chem., 2012, 22, 23049–23056 RSC.
- H. G. Wang, Y. Zhou, Y. Shen, Y. Li, Q. Zuo and Q. Duan, Fabrication, Formation Mechanism and the Application in Lithium-ion Battery of Porous Fe2O3 Nanotubes via Single-spinneret Electrospinning, Electrochim. Acta, 2015, 158, 105–112 CrossRef CAS.
- C. Kim, K. S. Yang, M. Kojima, K. Yoshida, Y. J. Kim and Y. A. Kim, et al., Fabrication of Electrospinning-derived Carbon Nanofiber Webs for the Anode Material of Lithium-ion Secondary Batteries, Adv. Funct. Mater., 2006, 16, 2393–2397 CrossRef CAS.
- Y. T. Peng and C. T. Lo, Effect of Microstructure and Morphology of Electrospun Ultra-small Carbon Nanofibers on Anode Performances for Lithium Ion Batteries, J. Electrochem. Soc., 2015, 162, A1085–A1093 CrossRef CAS.
- M. Chen, C. Yu, S. Liu, X. Fan, C. Zhao, X. Zhang and J. Qiu, Micro-sized Porous Carbon Spheres with Ultra-high Rate Capability for Lithium Storage, Nanoscale, 2015, 7, 1791–1795 RSC.
- J. Z. Wang, C. Zhong, D. Wexler, N. H. Idris, Z. X. Wang, L. Q. Chen and H. K. Liu, Graphene-encapsulated Fe3O4 Nanoparticles with 3D Laminated Structure as Superior Anode in Lithium Ion Batteries, Chem.–Eur. J., 2011, 17, 661–667 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data including SEM images of PAN0 and PAN3, SEM images of PAN3-400 (a), PAN3-500 (c) and relevant diameter distributions of PAN3-400 (b), PAN3-500 (d), SEM images of PAN0-400, PAN0-500 and relevant diameter distributions, two different equipments in the third calcination process, picture of flexible PAN3-500 electrodes, SEM image of PAN3-500 after 100 charge–discharge cycles and one discharge cycle and relevant diameter distribution. See DOI: 10.1039/c6ra04090j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.