
Anatase TiO2 nanoparticle coating on porous COK-12 platelets as highly active and reusable photocatalysts
L. H. Wee*a,
M. Meledinab,
S. Turnerb,
K. Custersa,
S. Kerkhofsa,
S. P. Sreea,
E. Gobechiyaa,
C. E. A. Kirschhocka,
G. Van Tendeloob and
J. A. Martensa
aCentre for Surface Chemistry and Catalysis, KU Leuven, Celestijnenlaan 200f, Heverlee, B3001, Leuven, Belgium. E-mail: likhong.wee@kuleuven.be; Tel: +32 16 37 67 48
bElectron Microscopy for Materials Science, University of Antwerp, Groenenborgerlaan 171, B2020, Antwerp, Belgium
First published on 5th May 2016
Abstract
Nanoscale TiO2 photocatalysts are widely used for biomedical applications, self-cleaning processes and wastewater treatments. The impregnation/deposition of TiO2 nanoparticles is indispensable for facile handling and separation as well as the improvement of their photocatalytic performance. In the present study, ordered mesoporous COK-12 silica thin platelets with a high-aspect-ratio and rough surfaces are demonstrated as a potential nanoporous support for homogeneous TiO2 nanoparticle coatings with high loading up to 16.7 wt%. The photocatalytic composite of COK-12 platelets and TiO2 nanoparticles is characterized in detail by HRSEM, SAXS, XRD, N2 physisorption analysis, solid-state UV-vis spectroscopy, HAADF-STEM, EDX analysis, and electron tomography. HAADF-STEM-EDX and electron tomography studies reveal a homogeneous dispersion of nanosized TiO2 nanoparticles over COK-12 platelets. The final composite material with anatase TiO2 nanoparticles that demonstrate a blueshifted semiconductor band gap energy of 3.2 eV coated on a highly porous COK-12 support shows exceptional photocatalytic catalytic activity for photodegradation of organic dyes (rhodamine 6G and methylene blue) and an organic pollutant (1-adamantanol) under UV light radiation, outperforming the commercial P25 TiO2 (Degussa) catalyst.
Introduction
Titanium dioxide (TiO2) is one of the most promising semiconductors for photocatalytic degradation of organic pollutants, oxidation reactions, and hydrogen production owing to its high photocatalytic activity, high chemical and photostability, water insolubility and non-toxicity.1,2 The high efficiency of commercially available TiO2 (P25 Degussa) is well known for environmental remediation processes such as in the treatment of exhaust gas3 and wastewater.4 The photocatalytic activity of TiO2 is governed by various parameters such as particle size, crystalline phase and reactive surface area.5–8 For instance, anatase TiO2 nanoparticles with particle sizes of less than 10 nm are of particular interest due to their excellent photocatalytic activity as a result of quantum size effects.9 However, the effectiveness of TiO2 in commercial applications is still limited by its low surface area and nanoparticle agglomeration. Moreover, the separation and recovery of these nanoparticles from the reaction medium via standard techniques such as filtration and centrifugation is very difficult.In order to overcome these limitations, impregnation of TiO2 nanoparticles into various solid supports has been investigated. For instance, the ordered mesoporous silica materials like MCM-41 (ref. 10) and SBA-15 (ref. 11) having high surface area and uniform pore size distribution are ideal supports for excellent dispersion and spatial confinement of photoactive nanoparticles.12–19 Over the years, different methods have been developed for the preparation of silica supported TiO2 nanoparticles such as direct synthesis,20–24 incipient wetness impregnation,25–27 grafting28,29 and atomic layer deposition (ALD).30–32 One of the early reports about successful incorporation of anatase nanoparticles into the pores of hexagonally ordered mesoporous material MCM-41 was reported by Xu and Langford via an incipient impregnation method.17 Hydrolysis of titanium isopropoxide precursor and its condensation inside the porous structure of silica, followed by thermal treatment also resulted in the successful impregnation of anatase TiO2 nanoparticles with particle sizes ranging from 3–7 nm inside the mesopores of SBA-15. Precise control of the TiO2 incorporation into the matrix of the silica host has been reported via various fabrication techniques. Manipulation of the synthesis parameters, in order to obtain a uniform dispersion of TiO2 into the mesocages of the ordered mesoporous silica aiming to optimise its performance remains a great scientific challenge, especially in the search for a promising supported photoactive catalyst. Despite huge efforts, immobilised TiO2 is generally less photoactive compared than the commercial P25 Degussa catalyst. Often only a small fraction of the TiO2 is successfully incorporated into the silica host matrix or large particles/aggregates on the external surface of the silica after the calcination process, reducing the catalytic activity. Moreover, the transport of molecules to and from the active sites of the catalyst via diffusion through the catalyst pore system is regulated by the pore morphology of the support material. Therefore, fine tuning of the ordered mesoporous silica pore size and pore length in the nanometer scale range is of great scientific interest.33–42 The preparation of ordered mesoporous silica with desired particle size and shape in order to improve its practical applications in adsorption,43 separation44 and catalysis have been documented.45,46 For example, Kubo and Kosuge reported the advantages of SBA-15 morphology, length and shape control for longer breakthrough time and sharp breakthrough in dynamic adsorption measurement of gaseous toluene.43 Fajula and co-workers demonstrated the pseudomorphic transformation synthesis of MCM-41 with controlled morphology for potential fast separation processes in chromatography.44 In the field of catalysis, Arribas and co-workers reported on the impact of the SBA-15 pore length on the uniform distribution of bimetallic Ru–Co catalysts in order to improve the catalytic performance of Fischer–Tropsch synthesis.45 The same group also demonstrated the efficiency of amino functionalized SBA-15 hexagonal platelets in the heterogeneous liquid phase reactions of Knoevenagel and Claisen–Schmidt condensations.46 Obviously, short mesochannels play an important role in easy diffusion and rapid mass transfer of the substrates and the reaction products.45,46
Coating of TiO2 on either dense (e.g. glass, stainless steel, quartz)47,48 or porous supports (e.g. activated carbon, zeolites, polymer membrane)49–51 is an alternative option to improve the recovery of the catalyst and its catalytic performance. Supported catalysts are fabricated by spreading the metal precursor solution onto the surface of a support by various techniques such as spin-coating,52 sol–gel,53 or chemical vapour deposition54 followed by a thermal treatment. However, such synthesis methods often result in the formation of large particles as direct control over the crystallization is difficult. Recently, some of us have reported facile room temperature and quasi-neutral pH synthesis of P6m ordered mesoporous silica nanoplatelets having a thickness of 200–300 nm coined COK-12 (material of Centrum voor Oppervlatechemie en Katalyse no. 12) via spontaneous precipitation of sodium silicate and citrate/citric acid buffered P123 triblock copolymer solutions.55 More importantly, this simplified synthesis method also enables large scale production of COK-12 ordered mesoporous material via a continuous process, achieving a high synthesis yield of 30 g min−1.56 Inspired by the high-aspect-ratio and high surface roughness of COK-12 due to its submicron-sized thin platelet morphology, COK-12 is exploited in the present study as a porous support for coating of anatase TiO2 nanoparticles. Taking into account the deposition of TiO2 nanocatalysts with high loading on a highly porous mesostructural support, this composite material shows outstanding photocatalytic performance for photodegradation of organic dyes (methylene blue and rhodamine 6G) and organic pollutants (1-adamantanol), and outperforms the commercial P25 (Degussa) catalyst under UV light irradiation.
Experimental
Materials
Pluronic triblock copolymer P123 (BASF Corporation), citric acid monohydrate (≥99%, Sigma-Aldrich), trisodium citrate dihydrate (≥99%, Sigma-Aldrich), sodium silicate solution (extra pure, Merck), titanium(IV) isopropoxide (99.999% trace metals basis, Sigma-Aldrich), absolute ethanol (analytical grade, Fisher Scientific), Milli-Q water (18.2 MΩ cm).Preparation of TiO2–COK-12 platelets
COK-12 was synthesized according to the recipe reported by Jammaer et al.55 For a typical synthesis, 4 g of pluronic triblock copolymer P123 was dissolved in 107.5 g of distilled water and 3.6 g of citric acid monohydrate and 2.5 g of trisodium citrate dehydrate were added. The solution was stirred for 24 h at room temperature. Another solution containing 10.4 g of sodium silicate in 30 g of distilled water was prepared and added to the surfactant solution. The mixed solution was stirred for 10 min and left static at room temperature for 24 h. The solid was filtered and washed with distilled water and dried at room temperature. The material was calcined at 550 °C for 8 h with a heating rate of 1 °C min−1. For the synthesis of the TiO2-COK-12 composite material, 200 mg of calcined COK-12 was added to 1 M of titanium(IV) isopropoxide ethanolic solution and stirred for 10 min. The suspension was left static at room temperature for 24 h. The material was recovered via centrifugation at 4000 rpm and the ethanol solution was discarded. The sediment was dried at room temperature and followed by calcination at 550 °C (1 °C min−1) for 8 h.Characterization
COK-12 and TiO2–COK-12 platelets were characterized using a Nova Nano scanning electron microscope (NanoSEM450, FEI, Eindhoven), HAADF-STEM and EDX (FEI Tecnai Osiris and Titan “cubed” microscopes), N2 adsorption (Micromeritics Tristar 3000), solid state UV-Vis NIR spectroscopy (Cary Series Varian spectrophotometer), a powder X-ray diffractometer (XRD, STOE STADI MP) diffractometer with a linear position sensitive detector (PSD) (6° 2θ window) and SAXS (SAXSess mc2, Anton Paar GmbH, Graz, Austria instrument). For the XRD measurements, the sample was finely ground and filled into a 0.5 mm capillary (Hilgenberg) and the measurements were performed in Debye–Scherrer mode at room temperature using Cu Kα1 radiation with λ = 1.54056 Å selected by means of a Ge(111) monochromator in the region 2θ = 0 to 60.5°, with a step width of 0.5°, internal PSD resolution 0.01°, and a steptime of 100 s. For SAXS measurements, the sample was loaded into a vacuum-tight rotor cell and measured using line-collimated Cu Kα radiation and an image plate detector. Measurements were performed for 5 min at room temperature (25 °C). Diffraction patterns were normalized to incident beam intensity. Background subtraction and correction for instrumental broadening was performed using the SAXSquant software. The scattering of the empty rotor cell was subtracted as background. HAADF-STEM imaging was performed on a FEI Titan “cubed” microscope, operated at 120 kV acceleration voltage. The convergence semi-angle used was 21 mrad, the inner ADF detection angle was 85 mrad. EDX mapping was carried out on a FEI Tecnai Osiris microscope, operated at 200 kV and equipped with a wide solid angle “super-X” EDX detector. The convergence semi-angle used was 10 mrad, the inner ADF detection angle was 68 mrad.Photocatalysis
The photocatalytic tests were carried out in a Luzchem photoreactor equipped with rotating sample carousel and 14 cool white fluorescent light tubes (Sylvania, 8 watts, 12′′), 8 of which were positioned at the top and 3 on both side of the reactor compartment. Air-saturated aqueous solutions (5 mL) of rhodamine 6G, methylene blue or 1-adamantanol (200 ppm) to which 10 mg of TiO2–COK-12 platelets or 2 mg of TiO2 P25 (Degussa) was added into the closed Pyrex test tubes (10 mL volume), each provided with a magnetic stirring bar. To exclude any possible influence of the temperature on the degradation of the organic compound, the temperature in the reactor was set at 35 °C using a thermostat. Prior to UV-A light illumination, the suspension was strongly magnetically stirred for 1 h in the dark to achieve an adsorption/desorption equilibrium. The UV light irradiation was turned on for 3 h. After reaction, the catalyst was separated from the aqueous solution by centrifugation. The percentage of methylene blue and rhodamine 6G degradation were determined by a UV-Vis spectrophotometry (TECAN Infinite M200 PRO, with Tecan i-control software) whereas the concentration of 1-adamantanol was analyzed by a gas chromatography (Shimadzu GC-2014, 60 m long HP-1 capillary column (Agilent) equipped with an flame ionization detector (FID) detector).Results and discussion
The morphology and surface property of calcined COK-12 and COK-12 after TiO2 nanoparticle coating (TiO2–COK-12) are revealed by HRSEM as presented in Fig. 1. The COK-12 material has a particle size of about 800 nm and is present as aggregates of hexagonal lamellae. The average thickness of the platelets is ca. 200 nm (Fig. 1a). The HRSEM images in Fig. 1b and d reveal the closely packed slit-like pore structure running in a parallel orientation to the short dimension along the z-axis of the particle forming a cylindrical pore-mouth opening with rough surfaces. After TiO2 coating, the hexagonal lamellae morphology of COK-12 is transformed into hexagonal prisms with a slightly increased thickness. The mesostructural ordering of the calcined COK-12 and TiO2–COK-12 platelets is further characterized by SAXS as presented in Fig. 2. The SAXS pattern of the calcined COK-12 exhibits four well-resolved diffraction peaks in the low-angle region that can be indexed in P6m hexagonal space group symmetry as (10), (11) and (20). The presence of an additional (21) reflection at higher angle reveals the high quality of the long range ordering. The d-spacing of COK-12 of 9.28 nm corresponds to a unit cell parameter a = 10.7 nm (Table 1). The long range ordering and excellent textural uniformity of the COK-12 mesostructure material after deposition of TiO2 are preserved as evidenced by the SAXS pattern, although a slight shift of the first diffraction peak is noted (Fig. 2a, insert). A slight decrease of the d-spacing value to 8.79 nm corresponding to a unit cell parameter of 10.4 nm is observed (Table 1) due to a structural shrinkage after TiO2 deposition, which is in full agreement with the HRSEM results (Fig. 1c and d). The deposition causes a global contraction of the mesoporous silica particles.57 | ||
| Fig. 1 HRSEM images of (a and b) calcined COK-12 and (c and d) TiO2–COK-12 platelets viewed at different magnifications. | ||
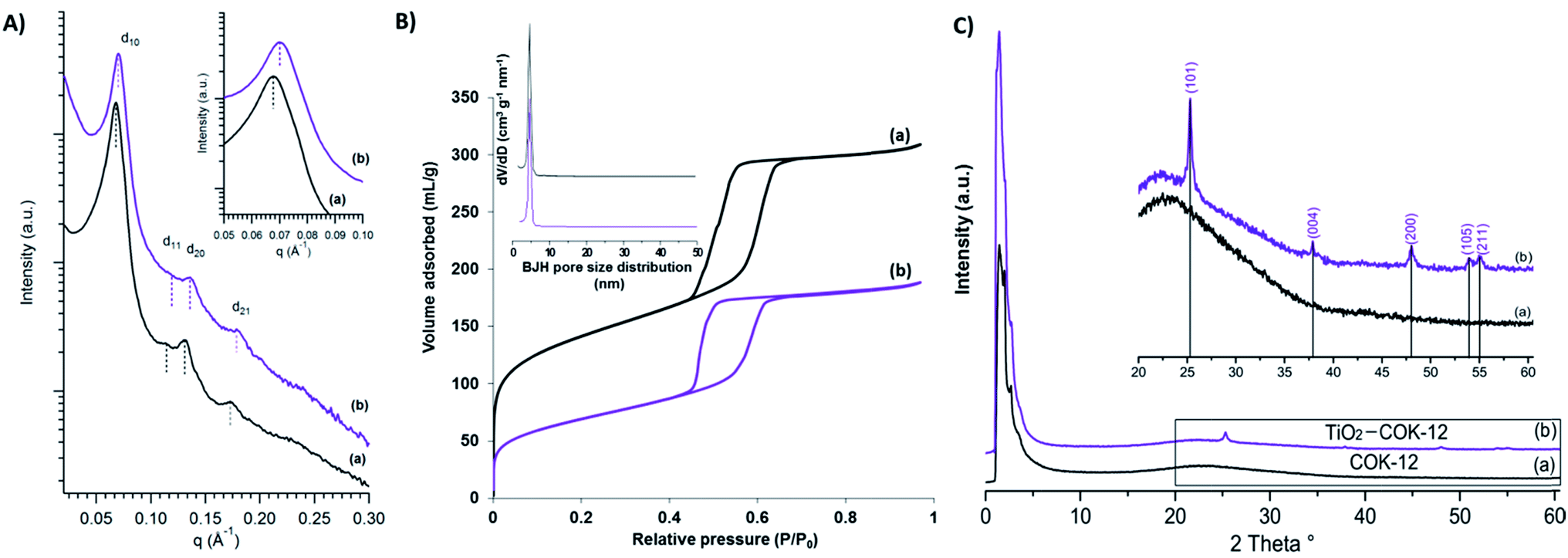 | ||
| Fig. 2 (A) SAXS patterns of (a) calcined COK-12 and (b) TiO2–COK-12 platelets. The insert in (A) shows the corresponding enlarged view of the d10 peak. (B) N2 adsorption–desorption isotherms of (a) calcined COK-12 and (b) TiO2–COK-12 platelets. The insert in (B) shows the corresponding BJH pore size distribution. (C) XRD patterns of (a) COK-12 and (b) TiO2–COK-12 platelets. The insert in (C) shows the enlarged region for the indexation of the TiO2 crystalline phase. | ||
| Samples | d10a (nm) | ab (nm) | BET surface area (cm2 g−1) | Langmuir surface area (cm2 g−1) | Pore volume (cm3 g−1) | Pore diameterc (nm) |
|---|---|---|---|---|---|---|
| a d10 spacing of calcined material determined from SAXS.b Unit cell parameter calculated from SAXS.c Pore diameter determined from BJH method. | ||||||
| COK-12 | 9.28 | 10.7 | 478 | 546 | 0.32 | 4.9 |
| TiO2-COK-12 | 8.79 | 10.4 | 244 | 256 | 0.23 | 4.7 |
N2 adsorption–desorption isotherms of calcined COK-12 and TiO2-COK-12 platelets are presented in Fig. 2b. Both the adsorption–desorption branches reveal Type IV isotherm with an H1 hysteresis loop, typical characteristics of an ordered mesoporous material having open tubular pores (Fig. 2b). The steep adsorption and desorption branches confirm the narrow pore size distribution. COK-12 has an average pore diameter of 4.9 nm according to the Barrett–Joyner–Halenda (BJH) pore size distribution (Fig. 2b, insert). The structural and textural properties of the COK-12 before and after deposition of TiO2 are summarised in Table 1. The Brunauer–Emmett–Teller (BET) surface area and pore volume of COK-12 are 478 cm2 g−1 and 0.32 cm3 g−1, respectively. After deposition of TiO2, it is noted that the pore volume and pore diameter of COK-12 has decreased (Table 1), most probably due to pore blocking of TiO2 nanoparticles on the surface of COK-12.58–60 A similar phenomenon has been reported in earlier publications.58–60 Nevertheless, a considerable BET surface area and pore volume of 244 m2 g−1 and 0.23 cm3 g−1, respectively, are preserved. The wide-angle XRD pattern of TiO2–COK-12 shows five reflection peaks at d101 = 3.51, d004 = 2.37, d200 = 1.89, d105 = 1.69 and d211 = 1.66 Å, which are assigned to the anatase phase of TiO2 (Fig. 2c, insert). According to the Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) analysis, the Ti atomic concentration amounted to 10 wt%, corresponding to a loading of TiO2 on COK-12 of ca. 16.7 wt%. Such a loading is comparable to other recently reported TiO2 composite photocatalysts such as TiO2–SiO2,61 TiO2–ZnFe2O4,62 TiO2–zeolite,63 TiO2–polymer64 and TiO2–graphene65 composites.
The successful coating of TiO2 nanoparticles on the surface of COK-12 is further confirmed by HAADF-STEM, EDX mapping and electron tomography. The HAADF-STEM overview image and corresponding EDX elemental maps for Ti and Si in Fig. 3a and b prove the presence of a Ti-containing coating at the surface and hexagonal pore mouths of COK-12. The thickness of the nanoparticle coating, as measured from the high magnification HAADF-STEM images (Fig. 3c and d) is between 5 and 10 nm. Whereas the average diameter of the TiO2 nanoparticles ranges from 4–6 nm according to the magnified HAADF-STEM images (Fig. 3f). It should be mentioned that no large TiO2 agglomerates are observed. The HAADF-STEM micrographs of TiO2-COK-12 also confirm the hexagonal long range ordering of the mesostructures. The results clearly show that the TiO2 coating process and the consecutive thermal treatment do not destroy the mesostructural of COK-12 host. The estimated pore were conducted to study the elemental composition and distribution of TiO2 throughout the COK-12 platelets. Elemental mapping was performed using STEM-EDX. The results are presented in Fig. 4b and e. The maps suggest a distribution of part of the titania material within the COK-12, but do indeed indicate a clear enrichment of titania at the COK-12 surface. Due to the two-dimensional nature of the electron micrographs, it is impossible to ascertain the three-dimensional location of the TiO2 nanoparticles from these micrographs. Thus, in order to confirm the three dimensional position of these TiO2 nanoparticles, electron tomography was performed on the sample. By combining HAADF-STEM projection images and an iterative SIRT tomographic reconstruction, a three dimensional representation of the composite material is obtained. Fig. 4e and f are slices through the tomographic reconstruction tomographic volume. The reconstructed volume clearly shows the presence of high contrast titania material at the COK-12 surface, confirming the coating of the material. The favourable formation of TiO2 nanoparticles on the outer surface of COK-12 is due to its with one-dimensional (1D) mesoporosity which hinders the penetration of the titanium precursor into the mesopores.31
 | ||
| Fig. 3 (a) HAADF-STEM overview showing TiO2 nanoparticle coated COK-12 material (b) corresponding EDX elemental maps showing Si in red and Ti in green (c) HAADF-STEM image of a COK-12 platelet imaged perpendicular to the pore direction. The arrow indicates TiO2 material at the pore mouths. (d–f) HAADF-STEM image of a COK-12 platelet imaged along the pore direction. Bright contrast features in the image correspond to the TiO2 nanoparticles (example indicated by arrows). | ||
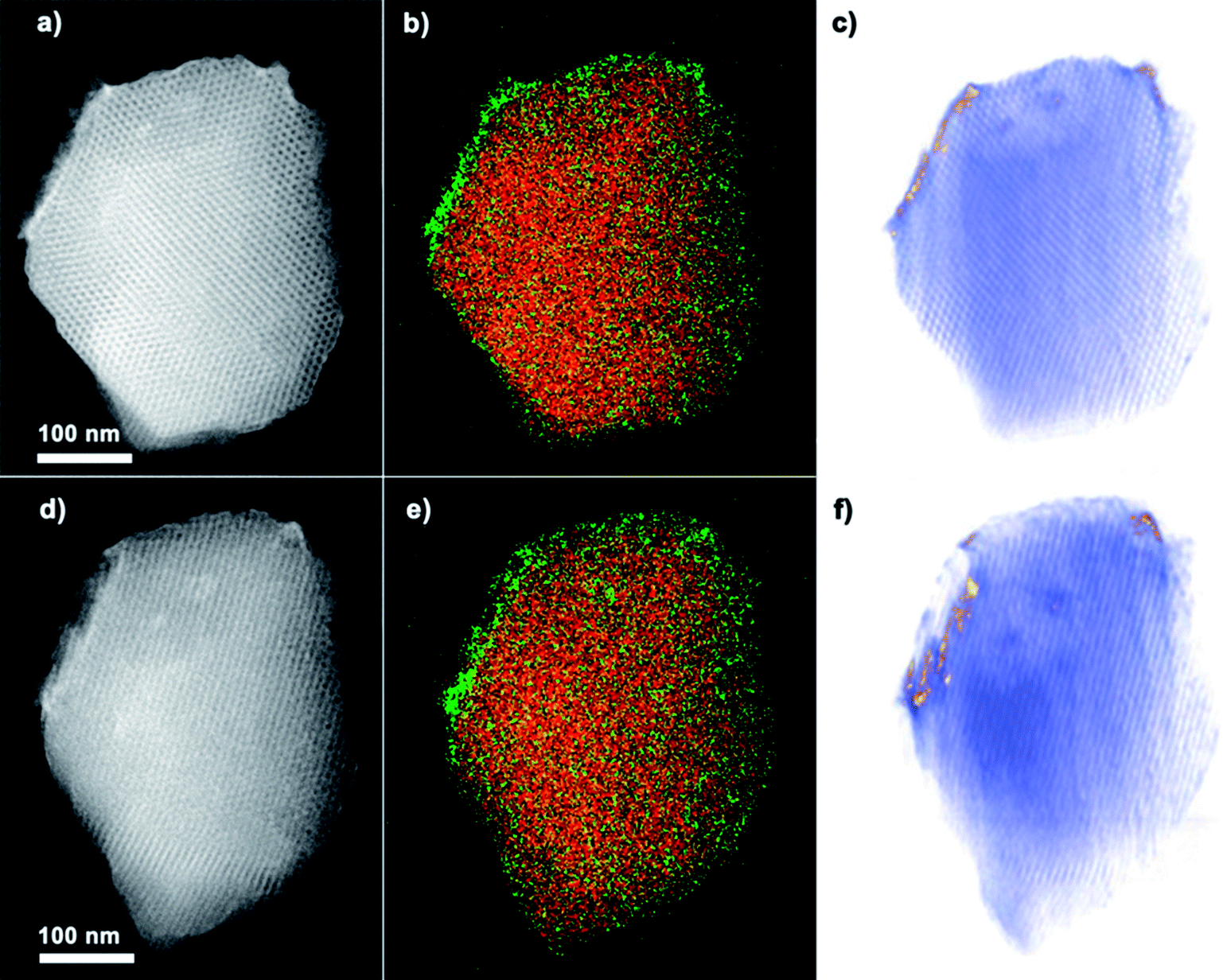 | ||
| Fig. 4 (a and d) HAADF-STEM overview images of a Ti–COK-12 crystal viewed from two distinct directions. (b and e) EDX elemental maps acquired along the same crystal orientations showing Si in red and Ti in green. (c and f) Tomographically reconstructed volume showing the silica porous framework in blue and the titania nanoparticles in gold. | ||
The optical properties of TiO2–COK-12 platelets were investigated by solid-state UV-vis spectroscopy. According to the UV spectrum (Fig. 5a), absorption bands at 230 and 270 nm are assigned to tetrahedral and octahedral coordination of TiO2, respectively. No absorption is observed from the COK-12 parent material as expected. The absorbance above 300 nm is indicative of the presence of TiO2 nanoparticles. The band gap energy of TiO2 was estimated from a Tauc plot of (α)1/2 versus photo energy (hν). The intercept of the tangent to the plot can give an approximation of the band gap energy for the in-direct band gap of TiO2. Extrapolation of the straight line to the photon energy axis derived a semiconductor band gap of 3.2 eV (Fig. 5b). The larger band gap of the TiO2 nanoparticles when compared to the reported value of 2.96 eV for bulk anatase TiO2 material can be attributed to a quantum size effect.9 A similar value was reported for a single crystal anatase TiO2 nanomaterial.66
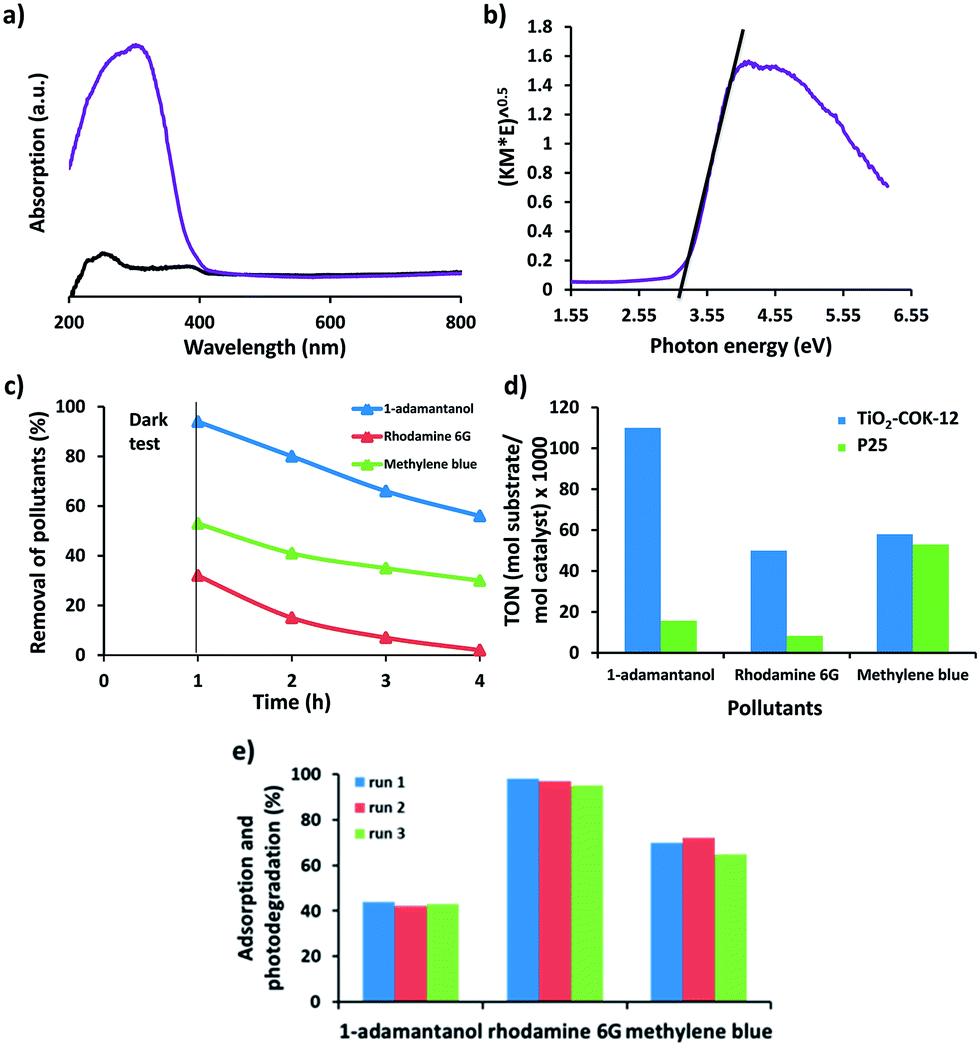 | ||
| Fig. 5 (a) Solid-state UV-vis absorption spectra of COK-12 (black curve) and TiO2–COK-12 platelets (purple curve). (b) Tauc plot of (F(R)hν)1/2 versus hν for TiO2–COK-12 coating. (c) Adsorption and photodegradation kinetics of 1-adamantanol, rhodamine 6G and methylene blue under UV light illumination over TiO2–COK-12 platelets. Conditions: 10 mg of photocatalyst in 5 mL of an aqueous solution of 1-adamantanol, rhodamine 6G or methylene blue (200 ppm) at 35 °C. (d) Turnover number (TON) of the TiO2-COK-12 in comparison to the commercial P25 TiO2 reference catalyst in the photodegradation of 1-adamantanol, rhodamine 6G and methylene blue. TON = mol of substrate degraded/mol of TiO2. The mol of substrate degraded is calculated by subtracting the mol of substrate adsorbed during the test for 1 h in the dark from the mol of substrate degraded at the end of the photocatalytic test. (e) Recycling tests for photodegradation of 1-adamantanol, rhodamine 6G and methylene blue in the presence of TiO2–COK-12 catalyst under UV light. After each run, the catalyst was washed three time with distilled water and dried in an oven at 60 °C for the next use. Conditions: 10 mg of TiO2–COK-12 in 5 mL of 200 ppm aqueous solution of 1-adamantanol, rhodamine 6G or methylene blue at 35 °C. The calculated standard deviations of three tests were <5%. | ||
The preparation of an efficient TiO2 photoactive catalyst that can photodegrade a broad range of organic molecules is highly demanding for applications in wastewater remediation. The photocatalytic degradation mechanism of the irradiated TiO2 is a well-known process. During photocatalysis, TiO2 is irradiated with light resulting in photon absorption and excitation of an electron from the valence band to the conduction band, thereby generating an electron–hole pair. The electron–hole pair can undergo recombination and dissipate the excess energy through nonradiative mechanisms, generating different reactive oxygen species such as the superoxide radicals or hydroperoxide radicals. These reactive oxygen species induce the degradation of organic pollutants, whereas the positive holes in the valence band will oxidize surface adsorbed water generating hydroxyl radicals to oxidise the organic pollutants.67
Recently, Patzke and Seeger reported the deposition of TiO2 nanoparticles on silicone nanofilaments for efficient photocatalytic decomposition of methylene blue under UV irradiation.68 TiO2–graphene composite photocatalysts have also attracted a considerable attention for photocatalytic applications.69–71 In the present study, COK-12 platelets coated with TiO2 nanoparticles were evaluated for the photodegradation of two model organic dyes: methylene blue and rhodamine 6G as well as a mimic of off-flavor compounds in water,72 1-adamantanol under UV light irradiation. Commercial P25 TiO2 (Degussa) was used as a reference catalyst for comparison. The photocatalytic degradation of dye and pollutant were monitored by analysing the concentration of the target compound after UV irradiation. However, we should take into account that adsorption of the pollutants on the surface of the photocatalysts also causes a decrease in its concentration in the solution. In order to discriminate adsorption from photodegradation, dark tests were performed prior to UV light illumination. The suspension was magnetically stirred for 1 h in the dark to achieve an adsorption/desorption equilibrium. The remaining concentration of dye in the solution after centrifugation and separation of the catalyst was determined and followed by 3 h of UV irradiation to determine the photocatalytic activity of the materials. The adsorption and photodegradation of 1-adamantanol, rhodamine 6G and methylene blue are presented in Fig. 5c. Our TiO2 coated COK-12 material absorbs a high amount of rhodamine 6G and methylene blue up to 6.8 mgrhodamine 6G mgCOK-12−1 and 5.3 mgmethylene blue mgCOK-12−1, respectively. Only a negligible amount of 1-adamantanol is absorbed by COK-12. The turnover number (TON), defined as mol of substrate degraded per mol of TiO2, is calculated in order to allow accurate evaluation of the photocatalytic activity based on of the amount of TiO2 present in the photocatalytic reactions. The amount (mol) of substrate degraded is calculated by subtracting the amount of substrate adsorbed during the test for 1 h in the dark from the amount of substrate degraded at the end of the photocatalytic test (Fig. 5d). The results confirm that the TiO2–COK-12 catalyst outperforms the commercial P25 TiO2 catalyst for all three organic pollutants. Promisingly, the TiO2–COK-12 catalyst gives a higher TON value of 50 for the photodegradation of rhodamine 6G versus a TON of 8 achieved for the commercial P25 TiO2 catalyst under similar reaction conditions. The TiO2–COK-12 catalyst also shows a higher TON value for the photodegradation of 1-adamantanol (TON = 110) compared to the commercial P25 TiO2 catalyst (TON = 16). We compared the photodegradation activity of our catalyst with other recently reported composite catalyst such as TiO2@zeolite 4A or bifunctional hybrid photocatalyst such as ZnO@graphene oxide for the photodegradation of methylene blue under UV radiation. Our catalyst outperformed the commercial P25 Degussa catalyst whereas the TiO2–zeolite 4A catalyst only showed a comparable photocatalytic activity to the commercial P25 catalyst for the photodegradation of methylene blue under UV light.63 Another reported TiO2–zeolite 4A catalyst was able to photodegrade methylene blue up to 0.1 mgmethylene blue mgTiO2–zeolite 4A−1 after 100 min under oxygen rich environment.73 We also compared our catalyst to a recently reported bifunctional hybrid ZnO@graphene oxide photocatalyst.74 It could photodegrade 0.028 mgmethylene blue mgZnO@graphene oxide−1 versus 0.1078 mgmethylene blue mgTiO2–COK-12−1 achieved by our TiO2–COK-12 catalyst after 2 h. Therefore, our catalyst is more photoactive than references. Under UV light irradiation, TiO2 yields electron–hole pairs.75 The electrons react with the dissolved oxygen to generate superoxide radical anions, (O2˙−) which finally yield hydroxyl radicals (OH˙) via the formation of hydroperoxy radicals (HO˙2). At the same time, holes are trapped by the water molecules at the surface of the catalyst to produce hydroxyl radicals (OH˙).75 The formation of active OH˙ radicals will subsequently degrade the organic pollutant to CO2 and H2O. The enhanced photocatalytic activity of the TiO2–COK-12 catalyst is due to the highly exposed nanosized anatase phase polymorph coatings at the surface of the COK-12. The combination of photoefficient titania and mesopores of the COK-12 carrier material concentrating the dye molecules in the vicinity of the photocatalytic sites may explain the superior behaviour. In previous work we showed that geosmin, an aqueous pollutant presence at very low concentration can be efficiently eliminated by combining an adsorbent with titania photocatalytic.50 More importantly, this hybrid catalyst can be reused for at least three runs without a significant drop in photocatalytic activity as demonstrated in Fig. 5e.
Conclusions
We have demonstrated the manipulation of COK-12 platelets as an ideal support for anatase TiO2 nanoparticle coatings. A combined HAADF-STEM and electron tomography study provided detailed insight into the particle size, location and distribution of the TiO2 nanoparticles on the surface of COK-12 at a nanometer scale. This supported catalyst showed improved photocatalytic degradation of various organic pollutants namely 1-adamantanol, rhodamine 6G and methylene blue under UV light irradiation, and outperformed the commercial P25 TiO2 catalyst. The results open up the opportunity for future fabrication of other nanocrystalline metal oxide such as ZnO and Fe2O3 which are of great interest for other advanced applications such as chemical sensing and light-induced drug release. Moreover, the highly exposed anatase TiO2 nanoparticles on the surface of COK-12 would be ideally suited for decoration with Ag or Au noble-metal plasmonic nanostructures, aiming for efficient conversion of solar energy in photovoltaic and photocatalytic devices.Acknowledgements
L. H. W. and S. T. thanks the FWO-Vlaanderen for a postdoctoral research fellowships under contract number (12M1415N) and (G004613N), respectively. J. A. M gratefully acknowledge financial supports from Flemish Government (Long-term structural funding-Methusalem). Collaboration among universities was supported by the Belgium Government (IAP-PAI networking).References
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- N. Bowering, G. S. Walker and P. G. Harrison, Appl. Catal., B, 2006, 62, 208 CrossRef CAS.
- F. Han, V. S. R. Kambala, M. Srinivasan, D. Rajarathnam and R. Naidu, Appl. Catal., A, 2009, 359, 25 CrossRef CAS.
- A. Testino, I. R. Bellobono, V. Buscaglia, C. Canevali, M. D'Arienzo, S. Polizzi, R. Scotti and F. Morazzoni, J. Am. Chem. Soc., 2007, 129, 3564 CrossRef CAS PubMed.
- Z. Zhang, C. C. Wang, R. Zakaria and J. Y. Ying, J. Phys. Chem. B, 1998, 102, 10871 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- H. Lin, C. P. Huang, W. Li, C. Ni, S. Ismat Shah and Y. H. Tseng, Appl. Catal., B, 2006, 68, 1 CrossRef CAS.
- N. Satoh, T. Nakashima, K. Kamikura and K. Yamamoto, Nat. Nanotechnol., 2008, 3, 106 CrossRef CAS PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS PubMed.
- K. Inumaru, T. Kasahara, M. Yasui and S. Yamanaka, Chem. Commun., 2005, 2131 RSC.
- Y. J. Acosta-Silva, R. Nava, V. Hernández-Morales, S. A. Macías-Sánchez, M. L. Gómez-Herrera and B. Pawelec, Appl. Catal., B, 2011, 110, 108 CrossRef CAS.
- S. Perathoner, P. Lanzafame, R. Passalacqua, G. Genti, R. Schlögl and D. S. Su, Microporous Mesoporous Mater., 2006, 90, 347 CrossRef CAS.
- G. Li and X. S. Zhao, Ind. Eng. Chem. Res., 2006, 45, 3569 CrossRef CAS.
- M. J. López-Muñoz, R. van Grieken, J. Aquado and J. Marugán, Catal. Today, 2005, 101, 307 CrossRef.
- Y. Xu and C. H. Langford, J. Phys. Chem. B, 1997, 101, 3115 CrossRef.
- N. B. Lihitkar, M. K. Abyaneh, V. Samuel, R. Pasricha, S. W. Gosavi and S. K. Kulkarni, J. Colloid Interface Sci., 2007, 314, 310 CrossRef CAS PubMed.
- K. Moller and T. Bein, Chem. Mater., 1998, 10, 2950 CrossRef CAS.
- W. H. Zhang, J. Lu, B. Han, M. Li, J. Xiu, P. Ying and C. Li, Chem. Mater., 2002, 14, 3413 CrossRef CAS.
- B. L. Newalkar, J. Olanrewaju and S. Komarneni, Chem. Mater., 2001, 13, 552 CrossRef CAS.
- V. Zeleňák, V. Hornebecq, S. Mornet, O. Schäf and P. Llewellyn, Chem. Mater., 2006, 18, 3184 CrossRef.
- F. Bérubé, F. Kleitz and S. Kaliaguine, J. Phys. Chem. C, 2008, 112, 14403 Search PubMed.
- R. van Grieken, J. Aguado, M. J. López-Muñoz and J. Marugán, J. Photochem. Photobiol., A, 2002, 148, 315 CrossRef CAS.
- P. Wu and T. Tatsumi, Chem. Mater., 2002, 14, 1657 CrossRef CAS.
- Z. Luan, E. M. Maes, P. A. W. van der Heide, D. Zhou, R. S. Czernuszewicz and L. Kevan, Chem. Mater., 1999, 11, 3680 CrossRef CAS.
- L. Zhao and J. Yu, J. Colloid Interface Sci., 2006, 304, 84 CrossRef CAS PubMed.
- B. J. Aronson, C. F. Blanford and A. Stein, Chem. Mater., 1997, 9, 2842 CrossRef CAS.
- M. S. Morey, S. O'Brien, S. Schwarz and G. D. Stucky, Chem. Mater., 2000, 12, 898 CrossRef CAS.
- A. K. Chandiran, A. Yella, M. Stefik, L. P. Heiniger, P. Comte, M. K. Nazeeruddin and M. Grätzel, ACS Appl. Mater. Interfaces, 2013, 5, 3487 CAS.
- S. Pulinthanathu Sree, J. Dendooven, J. Jammaer, K. Masschaele, D. Deduytsche, J. D'Haen, C. E. A. Kirschhock, J. A. Martens and C. Detavernier, Chem. Mater., 2012, 24, 2775 CrossRef CAS.
- S. Pulinthanathu Sree, J. Dendooven, K. Masschaele, H. M. Hamed, S. Deng, S. Bals, C. Detavernier and J. A. Martens, Nanoscale, 2013, 5, 5001 RSC.
- A. Sayari, B. H. Han and Y. Yang, J. Am. Chem. Soc., 2004, 126, 14348 CrossRef CAS PubMed.
- X. Ji, K. T. Lee, M. Monjauze and L. F. Nazar, Chem. Commun., 2008, 36, 4288 RSC.
- D. Zhao, J. Sun, Q. Li and G. D. Stucky, Chem. Mater., 2000, 12, 275 CrossRef CAS.
- H. I. Lee, J. K. Kim, G. D. Stucky, Y. Shi, C. Pak and J. M. Kim, J. Mater. Chem., 2010, 20, 8483 RSC.
- K. Kosuge, T. Sato, N. Kikukawa and M. Takemori, Chem. Mater., 2004, 16, 899 CrossRef CAS.
- S. Y. Chen, C. Y. Tang, W. T. Chuang, J. J. Lee, Y. L. Tsai, J. C. C. Chan, C. Y. Lin, Y. C. Liu and S. Cheng, Chem. Mater., 2008, 20, 3906 CrossRef CAS.
- P. Linton and V. Alfredsson, Chem. Mater., 2008, 20, 2878 CrossRef CAS.
- H. Zhang, J. Sun, D. Ma, X. Bao, A. Klein-Hoffmann, G. Weinberg, D. Su and R. Schlögl, J. Am. Chem. Soc., 2004, 126, 7440 CrossRef CAS PubMed.
- H. Zhang, J. Sun, D. Ma, G. Weinberg, D. S. Su and X. Bao, J. Phys. Chem. B, 2006, 110, 25908 CrossRef CAS PubMed.
- B. C. Chen, H. P. Lin, M. C. Chao, C. Y. Mou and C. Y. Tang, Adv. Mater., 2004, 16, 1657 CrossRef CAS.
- S. Kubo and K. Kosuge, Langmuir, 2007, 23, 11761 CrossRef CAS PubMed.
- A. Galarneau, J. Iapichella, K. Bonhomme, F. D. Renzo, P. Kooyman, O. Terasaki and F. Fajula, Adv. Funct. Mater., 2006, 16, 1657 CrossRef CAS.
- G. Prieto, A. Martínez, R. Murciano and M. A. Arribas, Appl. Catal., A, 2009, 367, 146 CrossRef CAS.
- S. E. A. Prasetyanto and S.-E. Park, Appl. Catal., A, 2008, 350, 244 CrossRef.
- J. M. Herrmann, H. Tahiri, Y. Ait-Ichou, G. Lassaletta, A. R. González-Elipe and A. Fernández, Appl. Catal., B, 1997, 13, 219 CrossRef CAS.
- G. Helsc and J. Deubener, Sol. Energy, 2012, 86, 831 CrossRef.
- G. L. Puma, A. Bono, D. Krishnaiah and J. G. Collin, J. Hazard. Mater., 2008, 157, 209 CrossRef PubMed.
- L. H. Wee, N. Janssens, J. Vercammen, L. Tamaraschi, L. C. J. Thomassen and J. A. Martens, J. Mater. Chem. A, 2015, 3, 2258 CAS.
- S. Meng, J. Mansouri, Y. Ye and V. Chen, J. Membr. Sci., 2014, 450, 48–59 CrossRef CAS.
- A. Lewkowicz, A. Synak, B. Grobelna, P. Bojarski, R. Bogdanowicz, J. Karczewski, K. Szczodrowski and M. Behrendt, Opt. Mater., 2014, 36, 1739 CrossRef CAS.
- A. Lewkowicz, P. Bojarski, A. Synak, B. Grobelna, I. Akopowa, I. Gryczyński and L. Kułak, J. Phys. Chem. C, 2012, 116, 12304 CAS.
- C. W. Dunnill, A. Kafizas and I. P. Parkin, Chem. Vap. Deposition, 2012, 18, 89 CrossRef CAS.
- J. Jammaer, A. Aerts, J. D'Haen, J. W. Seo and J. A. Martens, J. Mater. Chem., 2009, 19, 8290 RSC.
- J. Jammaer, T. S. van Erp, A. Aerts, C. E. A. Kirschhock and J. A. Martens, J. Am. Chem. Soc., 2011, 133, 13737 CrossRef CAS PubMed.
- R. C. Cammarata, K. Sieradzki and F. Spaepen, J. Appl. Phys., 2000, 87, 1227 CrossRef CAS.
- K. Schrijnemakers, N. R. E. N. Impens and E. F. Vansant, Langmuir, 1999, 15, 5807 CrossRef CAS.
- B. J. Aronson, C. F. Blanford and A. Stein, Chem. Mater., 1997, 9, 2842 CrossRef CAS.
- Z. Luan, E. M. Maes, P. A. W. van der Heide, D. Zhao, R. S. Czernuszewicz and L. Kevan, Chem. Mater., 1999, 11, 3680 CrossRef CAS.
- M. Brigante and P. C. Schulz, J. Hazard. Mater., 2011, 192, 1597 CrossRef CAS PubMed.
- P. P. Hankare, R. P. Patil, A. V. Jadhav, K. M. Garadkar and R. Sasikala, Appl. Catal., B, 2011, 107, 333 CrossRef CAS.
- D. I. Petkowicz, S. B. C. Pergher, C. D. S. da Silva, Z. N. da Rocha and J. H. Z. dos Santos, Chem. Eng. J., 2010, 158, 505 CrossRef CAS.
- C. R. Crick, J. C. Bear, A. Kafizas and I. P. Parkin, Adv. Mater., 2012, 24, 3505 CrossRef CAS PubMed.
- Y. Zhang, Z. R. Tang, X. Fu and Y. J. Xu, ACS Nano, 2010, 4, 7303 CrossRef CAS PubMed.
- L. Kavan, M. Grätzel, S. E. Gillert, C. Klemenz and H. J. Scheel, J. Am. Chem. Soc., 1996, 118, 6716 CrossRef CAS.
- S. Banerjee, S. C. Pillai, P. Falaras, K. E. O'Shea, J. A. Byrne and D. D. Dionysiou, J. Phys. Chem. Lett., 2014, 5, 2543 CrossRef CAS PubMed.
- G. R. Meseck, R. Kontic, G. R. Patzke and S. Seeger, Adv. Funct. Mater., 2012, 22, 4433 CrossRef CAS.
- N. Zhang, M. Q. Yang, S. Liu, Y. Sun and Y. J. Xu, Chem. Rev., 2015, 115, 10307 CrossRef CAS PubMed.
- S. Liu, Z. R. Tang, Y. Sun, J. C. Colmenares and Y. J. Xu, Chem. Soc. Rev., 2015, 44, 5053 RSC.
- N. Zhang and Y. J. Xu, CrystEngComm, 2016, 18, 24 RSC.
- L. H. Wee, M. Meledina, S. Turnerb, K. Custers, S. Kerkhofs, G. van Tendeloo and J. A. Martens, J. Mater. Chem. A, 2015, 3, 19884 CAS.
- R. Nagarjuna, S. Roy and R. Ganesan, Microporous Mesoporous Mater., 2015, 211, 1 CrossRef CAS.
- M. Azarang, A. Shuhaimi, R. Yousefi and S. P. Jahromi, RSC Adv., 2015, 5, 21888 RSC.
- P. Wilhelm and D. Stephan, J. Photochem. Photobiol., A, 2007, 185, 19 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |