
Photothermal conversion upon near-infrared irradiation of fluorescent carbon nanoparticles formed from carbonized polydopamine†
Sung Han Kim‡
a,
Shazid Md. Sharker‡b,
Haeshin Leeb,
Insik In*ac,
Kang Dae Lee*d and
Sung Young Park*ae
aDepartment of IT Convergence, Korea National University of Transportation, Chungju 380-702, Republic of Korea
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Republic of Korea
cDepartment of Polymer Science and Engineering, Korea National University of Transportation, Chungju 380-702, Republic of Korea. E-mail: in1@ut.ac.kr
dDepartment of Otolaryngology-Head and Neck Surgery, College of Medicine Kosin University, Busan, 602-702, Republic of Korea. E-mail: kdlee@ns.kosinmed.or.kr
eDepartment of Chemical and Biological Engineering, Korea National University of Transportation, Chungju 380-702, Republic of Korea. E-mail: parkchem@ut.ac.kr
First published on 22nd June 2016
Abstract
Fluorescence and photothermal conversion mediated by near-infrared radiation (NIR) is reported for carbonized polydopamine nanoparticles. Carbonized polydopamine demonstrated excitation-dependent fluorescence emission, together with NIR-responsive photothermal conversion properties. The concentration-dependent photothermal heating from carbonized fluorescent carbon nanoparticles-polydopamine (FNP-pDA) induces hyperthermal killing of both cancer cell lines and bacteria in vitro. Although most of the dopamine moieties of polydopamine become dehydrated upon carbonization, the remaining dopamine-hydroxyl groups can confer adhesive properties. These fluorescent coatings are compatible with many substrates, and the surface passivation of FNP-pDA with polyethylene glycol improves quantum yield and extends fluorescence lifetimes. The novel infrared-responsive photothermal and fluorescent carbon nanoparticles reported here show promise for a range of potential biomedical and research applications.
Introduction
Fluorescent carbon nanoparticles (FNPs) or carbon dots (CDs) have attracted considerable interest from researchers owing to their unique size-dependent properties.1 They are now being extensively investigated for various potential applications, such as in vitro and in vivo bio-imaging, drug delivery, photothermal therapy, and surface patterning ink. The lower toxicity profile of CDs compared to traditional quantum dots (QDs), opens up an impressive range of opportunities in various areas of biomedicine and optoelectronics.2,3 Structurally, CDs are usually composed of an outer shell of carboxylic acids, or other functional groups, and an inner core that gradually transitions to a different carbon scaffold, similar to graphite, that contains either delocalized electrons or increasing sp2 hybridization. Water soluble and readily surface functionalized CDs have now been manufactured, paving the way for significantly expanded use of these promising materials in future research applications.4–8Materials made from polydopamine (pDA), containing catechol and amine functional groups, have been widely studied in surface chemistry research where they have found use as adhesion layers. pDA is possessed of several distinctive materials-independent coating features; including biocompatibility, antibacterial surface properties, bio-mineralization potential and corrosion resistance.9 Recently, dopamine was used as a carbon source for the synthesis of yolk–shell carbon spheres. In this process, self-polymerized dopamine adhered followed carbonization, using a silica template that was eventually removed.10 Furthermore, carbonized pDA exhibit a graphite-like nanostructure giving distinctive D- and G-bands during Raman spectroscopy. It has been speculated that the 15 nm thicknesses of the 40–50 stacking layers in carbonized pDA offer substantial potential to the field of optoelectronics.11
Carbon-based materials, such as nanoscale reduced graphene oxide and carbon nano-tubes, have recently been applied in photothermal therapy (PTT), because of the ability of these materials to absorb light from the ultraviolet (UV) to near infrared (NIR) ends of the electromagnetic spectrum and convert it into heat through non-radiative decay.12,13 Although broad NIR absorption of carbon-based materials shows greater photothermal conversion efficiency, these carbon derivatives remain limited by poor colloidal stability, which requires surface modification/passivation using a stabilizing agent such as polyethylene glycol (PEG), or lipid. Furthermore carbon derived FNPs have similarly poor photoluminescence properties because of a surface ionic state that can become passivated following strong emission.14
In this report, we explored the multi-color photoluminescence properties of carbonized pDA both in solution, as well as in a coated surface in which carbonized pDA exhibits concentration-dependent photothermal heating in response to NIR irradiation. Passivation of the surface on carbonized pDA with PEG molecules terminated with amine groups, permitted distinguishable bright fluorescence. We also established that our material has promising antifouling properties that allow cultured cells to detach from coated substrate.
Experimental
Materials and characterization
Trizma base, Trizma HCl, [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT), dopamine hydrochloride, PEG-diamine (Mn 1500), concentrated H2SO4 were purchased from Sigma Aldrich, Korea. Penicillin-streptomycin, fetal bovine serum (FBS), 0.25% (w/v) Trypsin, 0.05% (w/v) EDTA solution and RPMI-1640 medium were purchased from Gibco BRL (Carlsbad, CA, USA).1H NMR spectra were recorded using a Bruker Advance 400 MHz spectrometer with deuterium oxide (D2O) and deuterium oxide (D2O) as the solvent. The UV-vis spectra were recorded using an Optizen 2020UV; Mecasys Co. XPS spectra were obtained using an Omicrometer ESCALAB (Omicrometer, Taunusstein, Germany) and photoluminescence (PL) spectra were obtained on a L550B luminescence spectrometer from Perkin Elmer. Static water contact angles were measured using a DO3210 (KRUSS Ltd., Germany), and the X-ray diffraction were recorded using an XRD Bruker AXS ADVANCE D-8. Using an infrared camera (NEC Avio, Thermo Tracer TH9100), particle size was measured with dynamic light scattering (DLS) (Zetasizer Nano, Malvern-Germany) and transmission electron microscopy (FEI, Netherlands). The NIR laser was 808 nm (PSU-III-LRD, CNI Optoelectronics Tech. Co. LTD, China). Raman spectra were investigated using a Laser Raman spectrophotometer (NRS-3200 Jasco, Japan).
Fluorescence lifetimes were measured using a NanoLED laser light source (Horiba Jobin Yvon NanoLog spectrophotometer) at 375 nm for the excitation, and the data were fitted by a multi-exponential decay model. The samples for the fluorescence lifetimes measurements were prepared by dissolving FNP-pDA and surface passivation of FNP-pDA in an aqueous solution at very low concentrations (1.0 mg mL−1). Quinine sulphate (QY 55 (%) at 354 nm excitation) was used as a reference standard to measure quantum yield, as adapted from a published report elsewhere.15
Synthesis of FNP-polydopamine (FNP-pDA)
The synthesis method was adopted from a similar report that could be found could be found elsewhere.12 In details, dopamine 1 g was dissolving in 5 mL of Tris buffered solution (10 mM, pH = 8.5) and stirred at room temperature for 12 h. The formation of polydopamine (pDA) was confirmed by visual evaluation of dark gray color. Then 10 mL of H2SO4 (36 N) was added and reacted for another 10 min at room temperature. Afterward 90 mL of NaOH (4 N) was added to neutralize the acid. At the end of synthesis, the purification was carried out through dialysis (molecular weight cutoff of 1000) against water and, finally freeze-dried to obtain the products (FNP-pDA).Surface passivation of FNP-polydopamine with diamine-terminated poly(ethylene glycol) (surface passivation of FNP-pDA)
FNP-(polydopamine) (0.3 g) was dissolved in 30 mL of distilled water in a 250 mL flask. This was followed by adding of diamine-terminated oligomeric poly(ethylene glycol) (0.6 g), the mixture was stirred vigorously to form a homogenous solution. Then the mixture was transferred to hydrothermal reactor and heated at a constant temperature of 120 °C for 72 h in an inert N2 atmosphere. At the end of reaction, the solution was allowed to cool at room temperature and, centrifuged 4000 rpm for 10 min to collected PEG passivated FNP-pDA. The purification was performed through dialysis (molecular weight cutoff of 3500) 24 h against water and, finally freeze-dried to obtain the PEG passivated products FNP (surface passivation of FNP-pDA).MTT assay
Cytotoxicity was measured using the [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] MTT assay method. Here, 200 μL aliquots of KB (oral carcinoma) and MDCK (Madin–Darby Canine Kidney) cells, at a density of 2 × 105 cells per mL, were placed in each well of a 96-well plate. The cells were then incubated for 24 h at 37 °C in a humidified 5% CO2 atmosphere. To determine the cellular viability, a stock solution of FNP-pDA was dissolved in RPMI medium at the concentration of 10 mg mL−1, after which the stock solution was diluted to 0.001 mg mL−1. The media was removed and the cells were treated with different concentrations of the FNP-pDA. The cells were then incubated as described above for another 24 h. The media containing FNP-pDA was then replaced with 180 μL fresh medium and 20 μL of a stock solution containing 15 mg of MTT in 3 mL PBS, after which the cells were incubated for another 4 h. Finally, the medium was removed and 200 μL MTT solubilizing agents was added to the cells. Then the absorbance was measured at 570 nm wavelength using a microplate reader in shaking mood (Filter Max F3, Multi-Mode Micriplate Reader, Molecular Device). Relative cell viability was measured by comparison with the control wells absorbance containing only cells.In vitro photothermal cytotoxic assay
The photothermal cytotoxicity of FNP-pDA was evaluated on KB and MDCK cells. First, 200 μL of the cells at a density of 2 × 105 cells per mL was placed in each well of a 96-well plate. The cells were then incubated for 24 h at 37 °C in a humidified 5% CO2 atmosphere. To assess the in vitro photothermal effects of FNP-pDA, the culture media were replaced with media containing FNP-pDA at concentrations of 10 mg mL−1 to 0.001 mg mL−1. After replacement of the media, the cells were incubated in 96-well plates at 37 °C for 30 min, and then irradiated with an 808 nm laser at the power density of 2 W cm−2 for 5 min. The cells were then incubated for another 24 h. The viability and proliferation of KB and MDCK cells were evaluated by the MTT assay method.Calcein AM and propidium iodide cell staining assay
Imaging of the photothermal cytotoxicity was carried out through a calcein AM and propidium iodide (PI) co-staining method. In details, the MDCK and KB cells were incubated for 30 min at 37 °C in 8-well plates containing FNP-pDA at a concentration of 1 mg mL−1. At the end of the incubation time, the cells were irradiated with a laser (808 nm, 2 W cm−2) for 5 min. The cells were then stained with both calcein AM (calcein acetoxymethyl ester) and PI (propidium iodide). Finally, an LSM510 confocal laser scan microscope (Carl Zeiss, Germany) was used at 10× magnification for imaging of the stained (live/dead) cells.Results and discussion
An attractive feature of CDs and derivatized fluorescent carbon nanoparticles (FNPs) is their dominant emission wavelength, which provides a versatile platform for tackling the challenges facing the next generation of biomaterials for bioimaging and PTT under various formulations.5,16–18The synthetic routes reported for producing FNPs from pDA employ controlled carbonization in a strongly acidic environment for a predetermined period.12 This synthetic strategy was fixed to retain dopamine moieties on the prepared FNPs. These approaches produce FNPs that display characteristic fluorescent properties, and they were improved by including a surface passivation step using PEG, which resulted in improved photo-luminescent properties. A detailed account of the experimental processes involved is provided in Scheme 1.
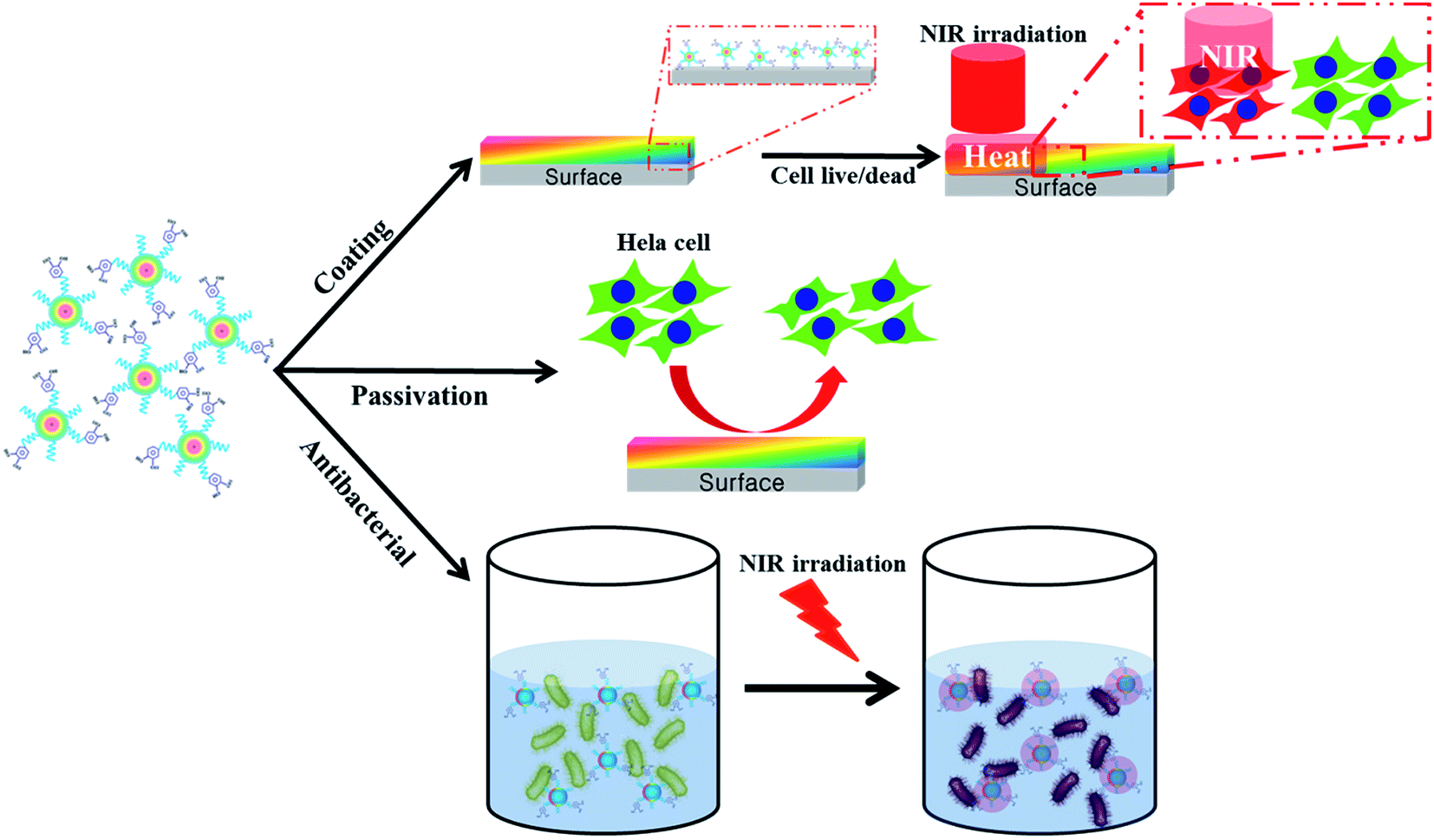 | ||
| Scheme 1 Illustration of the preparation and application of FNP-polydopamine (FNP-pDA) obtained from polydopamine (pDA). | ||
Absorption analysis from the UV to the NIR provided information about the chemical nature of pDA, FNP-pDA and the effects of surface passivation. It has been shown that an absorption maximum at 280 nm is mainly attributed to pDA. Although FNP-pDA obtained through carbonization had the same absorption band at 280 nm, the intensity was moderately decreased, which indicated a functional decline of the dopamine component.9 However, the intensity of the characteristically broad absorption between the visible range and the NIR (600–1000 nm) was similar to that of other carbonized nanoparticles, including other photothermal and hyperthermal agents.12 FT-IR analysis showed two distinctive broad peaks of high intensity at around 3400 cm−1 and 3100 cm−1, which might result from surface active N–H/O–H stretching vibrations of pDA. At the same time, the amide bond (N–H) shearing band (1510 cm−1), the aromatic ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration band (1442 cm−1) and the C–N stretching band (1292 cm−1), which are all characteristic of pDA, appeared in the FT-IR spectrum (Fig. S1†).19 Moreover, the appearance of high intensity, broad signals from C–H bonds at around 2800 cm−1 is consistent with the presence of surface-passivated PEG. 1H-NMR spectroscopy revealed an aromatic proton peak (6.5–6.7 ppm), which is characteristic of pDA in FNP-pDA (Fig. S2†).9
C vibration band (1442 cm−1) and the C–N stretching band (1292 cm−1), which are all characteristic of pDA, appeared in the FT-IR spectrum (Fig. S1†).19 Moreover, the appearance of high intensity, broad signals from C–H bonds at around 2800 cm−1 is consistent with the presence of surface-passivated PEG. 1H-NMR spectroscopy revealed an aromatic proton peak (6.5–6.7 ppm), which is characteristic of pDA in FNP-pDA (Fig. S2†).9
Carbonized materials often contain a mixture of sp2 and sp3 hybridized carbon atoms. The opto-electronic properties of such carbonized materials are strongly influenced by the number and spatial distribution of double bonds, and sites of delocalized electrons. Since the optical bandgap depends in part on the size, shape, and fraction of the sp2-hybridized species; tunable fluorescence may be achieved by modulating the number and distribution of such species.5 Following the above principle, the excitation-dependent emission profiles of FNP-pDA and surface-passivated FNP-pDA were evaluated at a fixed concentration. As we can see in Fig. 1c and d, the expected excitation-dependent fluorescence emission was seen for FNP-pDA, whereas its source precursor (pDA) did not show these properties. Moreover, when compared to FNP-pDA (1.33% QY blue and 4.77% QY green), it is clear that surface-passivated FNPs have not only retained similar fluorescence profiles, but have even higher intensity peaks. As demonstrated by the excitation-dependent fluorescence profiles, upon changing the surface ionic state of FNP-pDA with amine capped PEG, an increased QY was measured. This feature is indeed attractive, since it establishes that the brightness of carbon derived FNPs can be significantly increased (2.90% QY blue and 8.80% QY green). The fluorescence lifetimes (τ) of FNP-pDA and surface-passivated FNP-pDA are 5.96 ns and 8.13 ns, respectively, a finding which also demonstrates the sustainability of these materials for a range of envisioned applications in opto-electronics (Fig. S3†).14
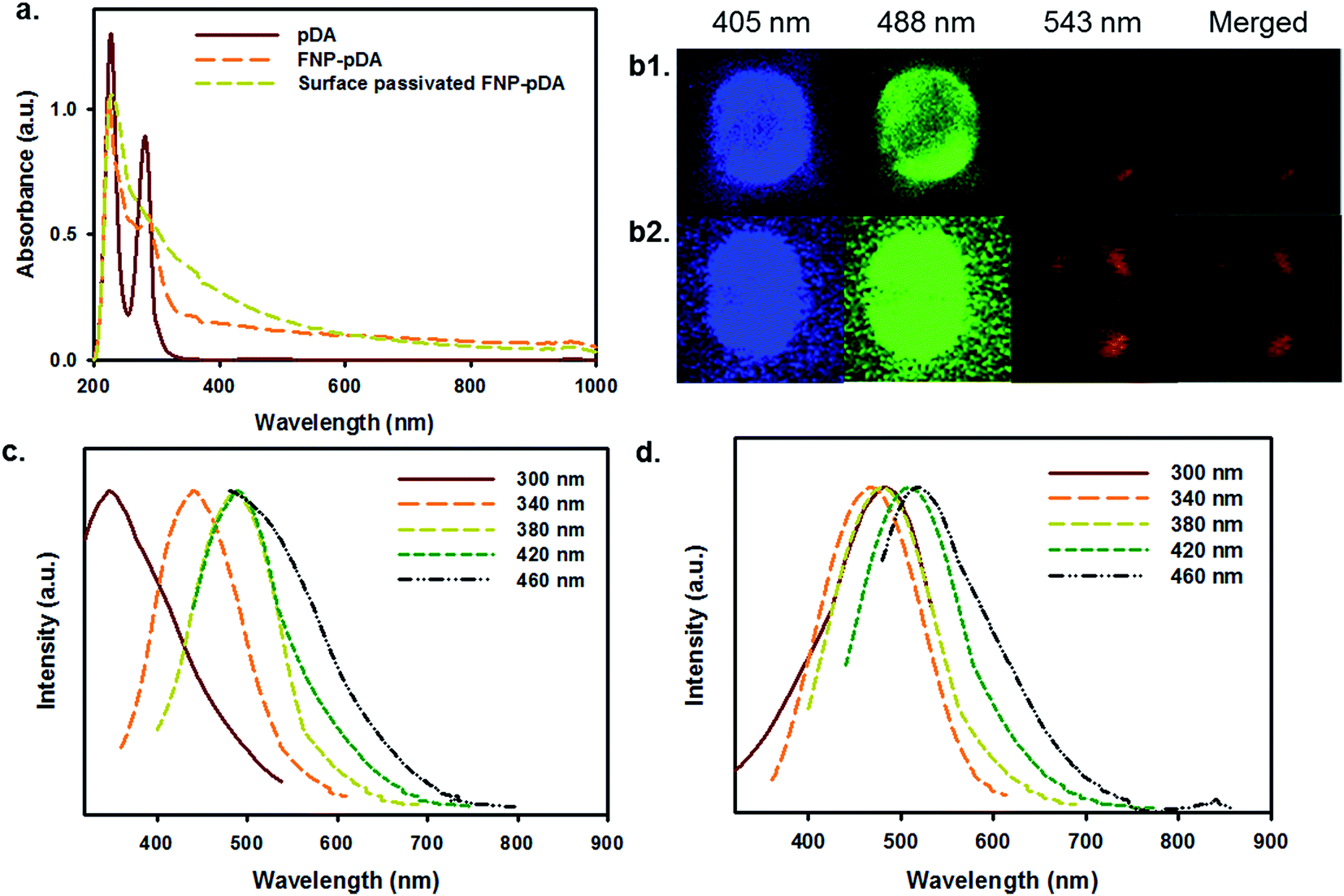 | ||
| Fig. 1 (a) UV-vis-NIR absorption spectra of the polydopamine (pDA), FNP-polydopamine (FNP-pDA) and surface passivated FNP-pDA. An illuminated of confocal laser scan microscope (CLSM) image of the aqueous solutions of (b1) FNP-pDA and (b2) surface passivated FNP-pDA in 405 nm, 488 nm and 543 nm wavelength, respectively. The normalized fluorescence emission spectra of (c) FNP-pDA and (d) surface passivated FNP-pDA at different excitation wavelengths (300–460 nm). | ||
Our transmission electron microscopy (TEM) measurements revealed that the resulting particles exhibited a spherical shape with an average diameter of 15 ± 2 nm, and that the lattice separations (0.308 nm) were consistent with those of graphitic carbon structures (Fig. 2a).8 The formation of surface-passivated FNP-pDA results in almost same lattice separations (0.308 nm) for what appears to be surface coated dark carbon materials (Fig. 2b). The particle size distributions of both FNP-pDA and surface-passivated FNP-pDA in the aqueous phase were evaluated by DLS. The size dispersion of the FNP-pDA was between 12 and 19 nm with an average size of 15.69 nm. In contrast, PEG-passivated FNP-pDA diameter sizes were much larger, ranging between 26 and 42 nm, with an average size of 34.7 nm. It seems likely that the pronounced differences in size between the two types of FNP reflect the behaviour of the PEG chains on particles surface.
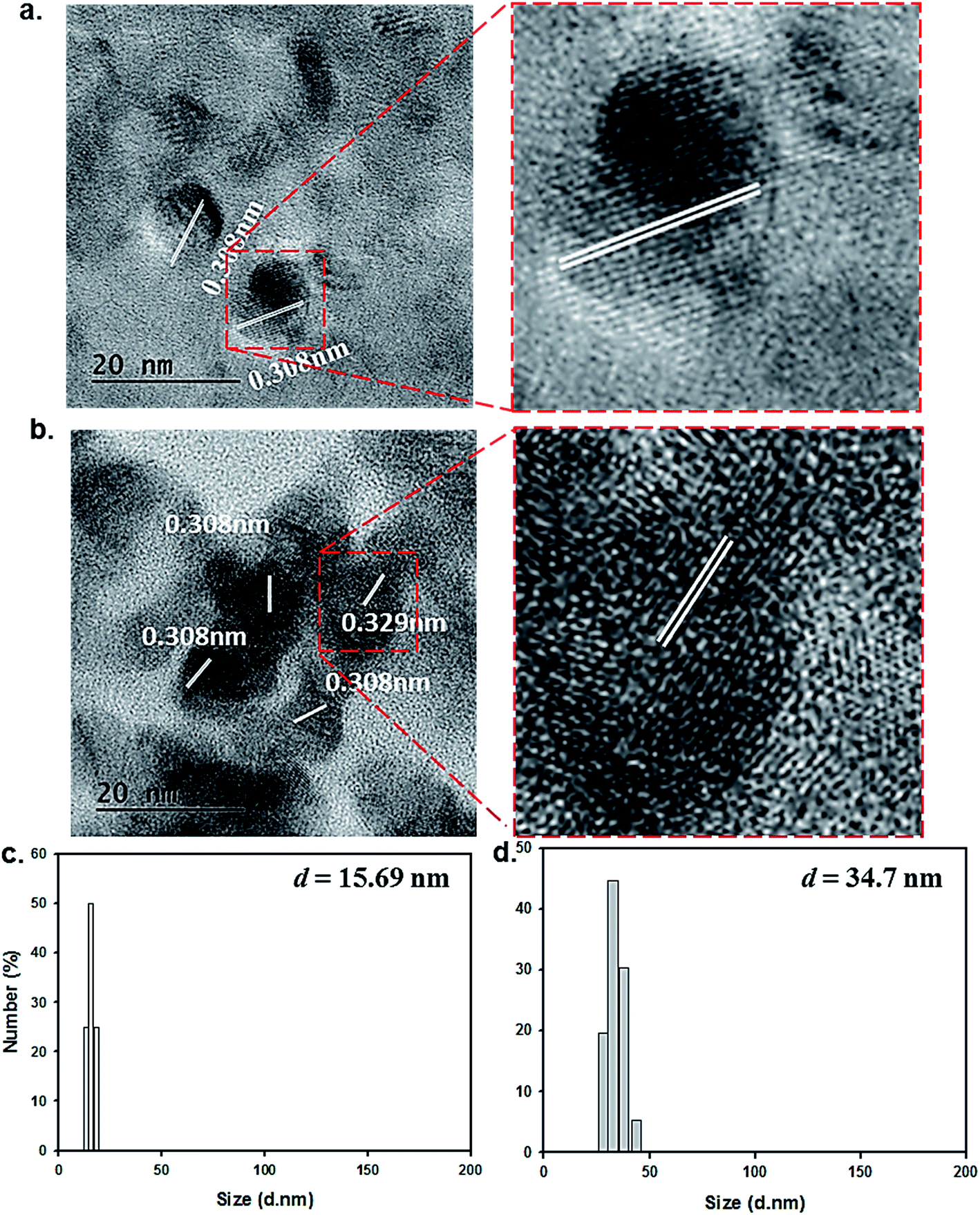 | ||
| Fig. 2 TEM image and corresponding lattice distance of (a) FNP-pDA and (b) surface passivated FNP-pDA. The right panel has shown larger magnifying TEM images of (a) and (b), respectively. The DLS measurements for particles size distribution in aqueous state of (c) FNP-pDA and (d) surface passivated FNP-pDA. | ||
An apparent G-band at 1590 cm−1 and D-band at 1360 cm−1 were observed in the Raman spectrum of both FNP-pDA and surface-passivated FNP-pDA (Fig. 3a), implying the presence of both sp2 and sp3 hybridized carbons in these nanoparticles.9 However such a Raman active bands are not seen for the native pDA precursor. The intensity ratio [ID/IG], which is often used to correlate the structural constituency of carbonized nanoparticles, also indicated different ratios of the sp2 and sp3 forms of carbon. Therefore, we concluded from the intensity of the D/G bands that pDA derived FNP-pDA and modified surface-passivated FNP-pDA were mainly mixture of sp2/sp3 form of carbons.13 Additionally, the characteristic X-ray powder diffraction (XRD) patterns (2θ) of surface-passivated FNP-pDA were observed at around 19°, 23° and 27°, due to the size increase wrought by PEG.19 The XRD pattern of FNP-pDA had decreased crystallinity relative to pDA, which may be accounted for by the increased variation in oriented carbon–carbon bonds arising from the mixture of sp2 and sp3 hybridized carbon atoms in these nanoparticles (Fig. 3b).
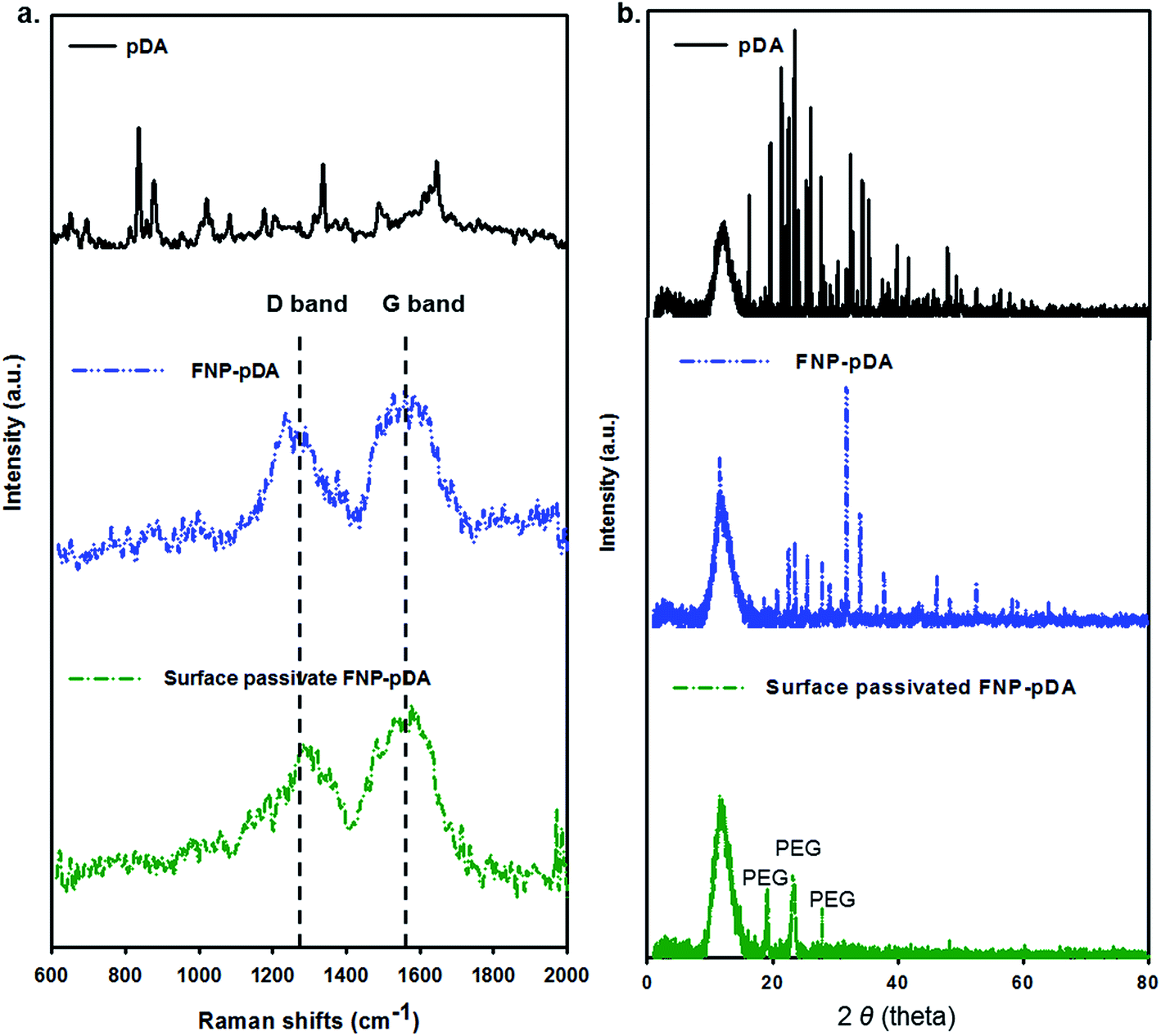 | ||
| Fig. 3 (a) The Raman shift (cm−1) of G and D bands of pDA, FNP-pDA and surface passivated FNP-pDA, respectively. (b) X-ray diffraction (XRD) pattern in the 2θ range (1–80°) of polydopamine (pDA), FNP-pDA and surface passivated FNP-pDA, respectively. | ||
Owing to intrinsic absorption in the NIR region, carbon nanomaterials such as carbon nanotubes (CNTs) and graphene oxide (GO) have recently received much attention as efficient materials for photothermal cancer therapy (PTT).13,20 Carbonization of pDA broadens the NIR absorption bands. Furthermore XRD and Raman spectroscopy demonstrate the presence of sp2 hybridized carbon, similar to graphene oxide. Since NIR irradiation of FNP-pDA results in rapid photothermal heating (5 mg generates 45 °C), whereas native pDA remains insensitive to NIR radiation. Surface-passivated FNP-pDA also exhibits photothermal heating, but does so to a slightly lower extent than FNP-pDA (Fig. 4 and S4†). Temperature generation in these materials is als1o concentration-dependent. These findings suggest that the NIR absorption-dependent photothermal heating seen in our materials shows promise for several photothermal applications.
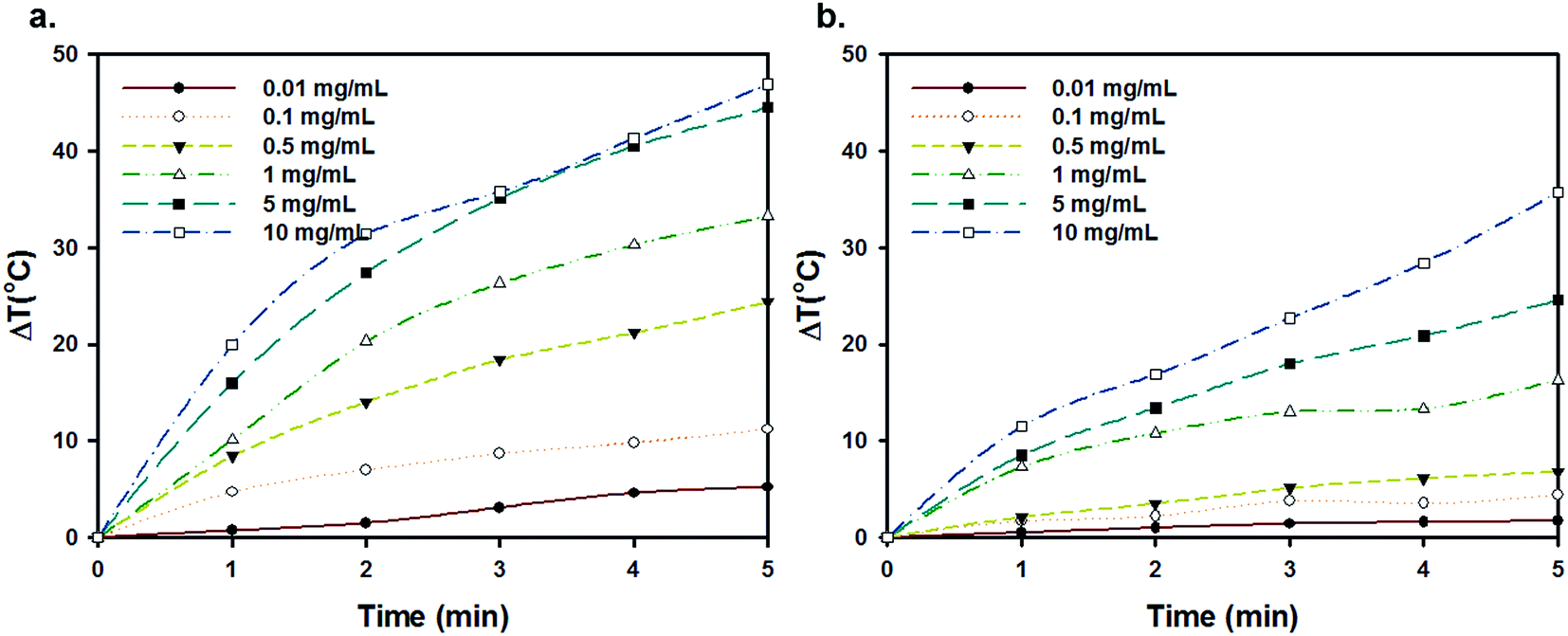 | ||
| Fig. 4 The concentration dependent photothermal conversion of (a) FNP-pDA and (b) surface passivated FNP-pDA in response to NIR light irradiation. The NIR laser was 808 nm and 2W cm−2 power intensity. | ||
Compositionally, the carbonized materials contained of a mixture of sp2 and sp3 bonds and, photoelectric properties of such materials largely depend on the p states of the sp2 fraction. The strong localization of p and p* electronic levels of the sp2 domain lie within s and s* states, that essentially works as luminescence centers or chromophores.5 Those localized confined sp2 π-electron have ascribed photoluminescence, which cover the near infrared (NIR), visible and blue spectral ranges.6 Since the NIR light has long penetration ability with minimum tissue absorption, the infrared responsive carbon nanoparticles should allow monitoring of theragnosis from deep tissue.7 Moreover, the lattice of carbonized FNPs might also dissipating absorbed light (fluorescence) energy by non-radiative pathways such as vibrational relaxation that can be converted light into local heat influencing photothermal conversion (Fig. 5).5,8
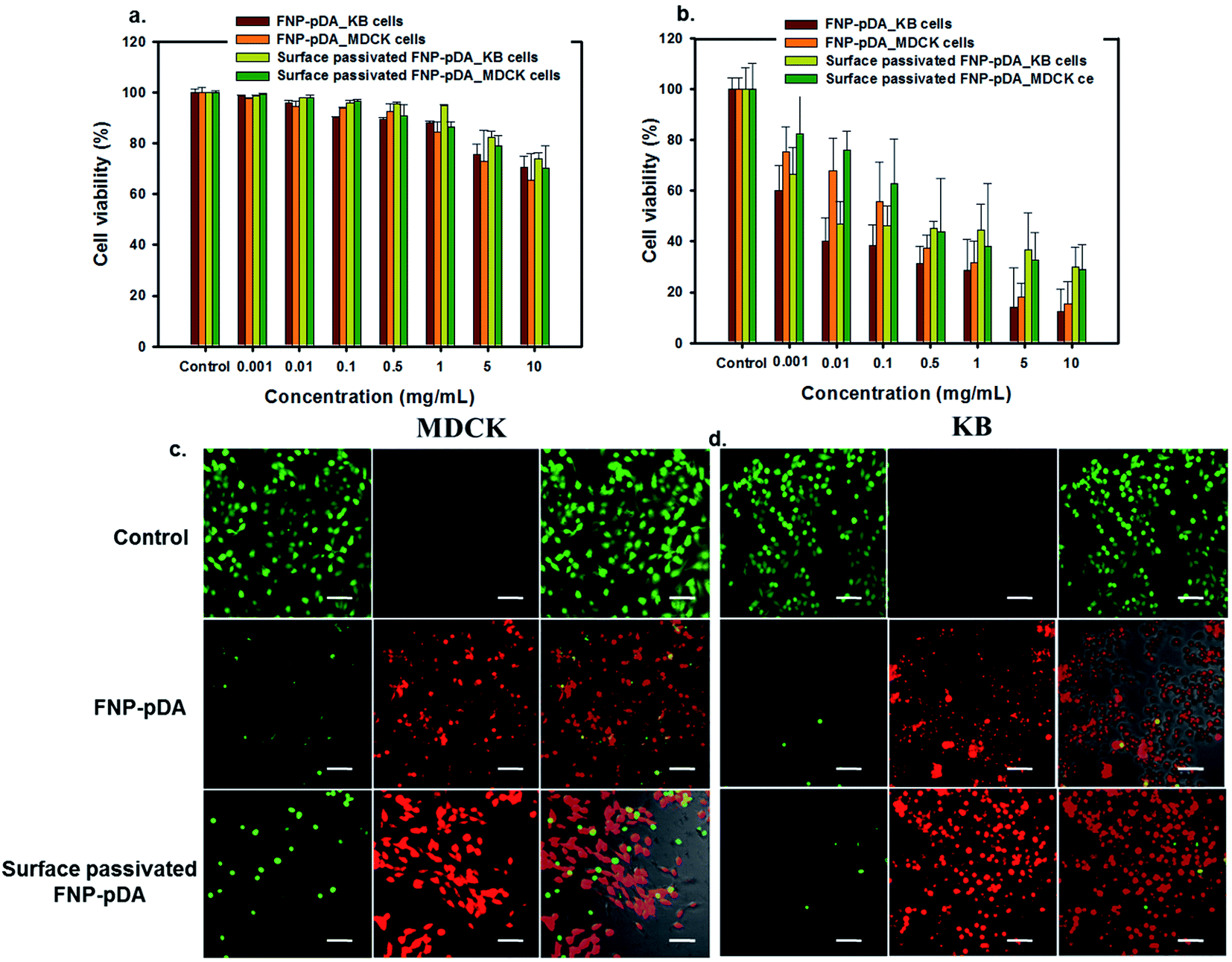 | ||
| Fig. 5 The MTT assay used for the concentration-dependent in vitro biocompatibility study on KB and MDCK cells treated with FNP-pDA and surface passivated FNP-pDA. (a) In vitro biocompatibility and (b) photothermal cytotoxicity of FNP-pDA and surface passivated FNP-pDA at different concentrations (0.001–10 mg mL−1) after NIR irradiation. Calcein AM and propidium iodide co-stained live/dead cell fluorescence imaging of FNP-pDA and surface passivated FNP-pDA treated (c) MDCK and (d) KB cells. NIR irradiation was carried out for 5 min with an 808 nm laser at a power intensity of 2 W cm−2. | ||
PTT has been used to locally generate the temperatures required for vascular damage in the tumor environment. The ability to generate heat specifically in tumor tissues, avoiding surrounding compartments, is required for effective PTT.21 From a materials perspective, the characteristic absorption in the NIR window enables FNP-pDA to generate photothermal heating, which resulted in concentration-dependent in vitro killing of KB and MDCK cells. Cells viability assays using MTT showed a concentration-dependent decrease in the growth of cells. Treatment of cells with 1 mg mL−1 FNP-pDA resulted in 35% killing in response to 5 min NIR (808 nm) irradiation. The cell killing efficiency decreased moderately in the PEG-passivated FNP-pDA treated groups, due to lower absorption in NIR window. However, the viability of cell growth appeared unchanged in the control groups (unexposed to NIR radiation), suggesting that the toxicity of these nanoparticles stems predominantly from photothermal heating and not from direct chemical toxicity. To establish photothermal cytotoxicity further, FNP-pDA and surface-passivated FNP-pDA treated cells were studied by examining stained cells under confocal microscopy. Both NIR-irradiated FNP-pDA and surface-passivated FNP-pDA killed cells efficiently, as reflected in the number of dead cells (red color). In contrast, an increased number of live cells (green color) were observed in the control group, which were not exposed to NIR irradiation. Since the NIR absorption intensity and photothermal conversion efficiency of FNP-pDA increase in direct proportion to the concentration used, it is conceivable that the usefulness of FNP-pDA for hyperthermal tumor ablation could be optimized by adjusting the dosing and irradiation protocols during treatment.
To test the effect of photothermal treatment on bacterial cells, Staphylococcus aureus (Gram-positive) and Escherichia coli (Gram-negative) were premixed with FNP-pDA and surface-passivated FNP-pDA. NIR radiation (808 nm) was then used to irradiate the S. aureus and E. coli culture plates. Photothermal killing of bacteria was confirmed after 24 h by counting colonies. We found that bacterial killing efficiency depends on nanoparticle concentration. Fig. 6 shows the percentage of bacteria killed after 5 min NIR exposure; and illustrates that killing efficiency increased with sample concentration.22 However, the FNP-pDA treated agent exhibits more prominent antibacterial efficiency compared with surface-passivated FNP-pDA. After 5 min of NIR treatment with nanoparticles at a concentration of 0.5 mg mL−1, almost 80% of bacteria were killed in the FNP-pDA treated group; while in the group treated with surface-passivated FNP-pDA, only 60% were killed under identical experimental conditions. The weak photothermal conversion of surface-passivated FNP-pDA resulted in only moderate toxicity against the bacteria tested. However, FNP-pDA generated and released heat more efficiently, inducing irreversible bacterial damage. Moreover, when 1 mg of FNP-pDA was used against bacteria, 99.9% of bacteria were killed in a very short period, for both species (Fig. S5 and S6†). We conclude that we have demonstrated that our materials can also kill certain bacteria effectively, via a photothermal mechanism.
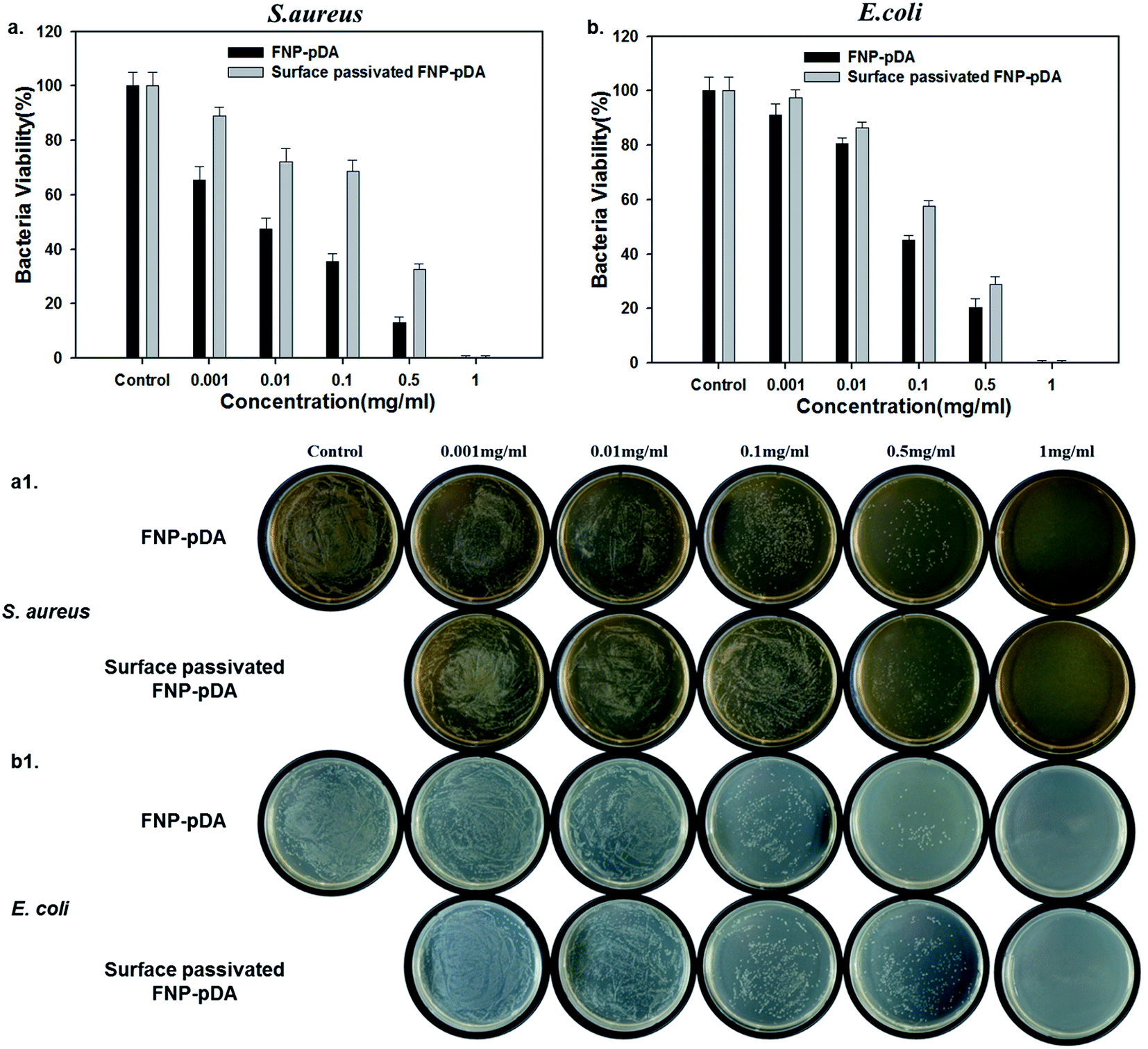 | ||
| Fig. 6 The concentration dependent antibacterial efficiency of FNP-pDA and surface passivated FNP-pDA under 808 nm laser irradiation taken at 5 min post treatment against (a) S. aureus and (b) E. coli. The laser power intensity was 2 W cm−2. | ||
pDA and its derivatives can confer benefits on composite materials when used as independent coating agents. The adhesive properties of dopamine-scaffolds are widely used in the fabrication of a wide variety of coating agents on various substrates.18,23 To examine surface coating efficiency, the carbonized FNP-pDA and surface-passivated FNP-pDA composites were applied to polypropylene (PP), polyethylene terephthalate (PET), polyvinyl chloride (PVC), polystyrene (PS) and silicon (Si) substrates. As can be seen in Fig. 7a, the application of FNP-pDA and surface-passivated FNP-pDA coating on PP, PET, PVC, and PS substrate resulted in decreased hydrophobicity, which was confirmed by the decrease of the water contact angle.24,25 In contrast to those substrates, Si substrates showed increased water contact angles between 36° and 43° arising from the bare Si surface, which were more hydrophilic compare to FNP-pDA and PEG passivated FNP-pDA coating. However, all of the FNP-pDA coated substrates maintained 60–70° static contact, revealing the uniformity of the surface modification with FNP-pDA coating. In addition to contact angle, the fluorescence properties were evaluated from coated surface by using confocal laser scan microscope (CLSM) dependent on excited laser (nm). The fluorescence emission shows blue, green and red color.26 The observed light emission was also noticeable increased from surface-passivated FNP-pDA coating substrate in related with its precursor FNP-pDA coating (Fig. 7b). These results have revelled promising potentiality to draw fluorescence image on versatile substrate.
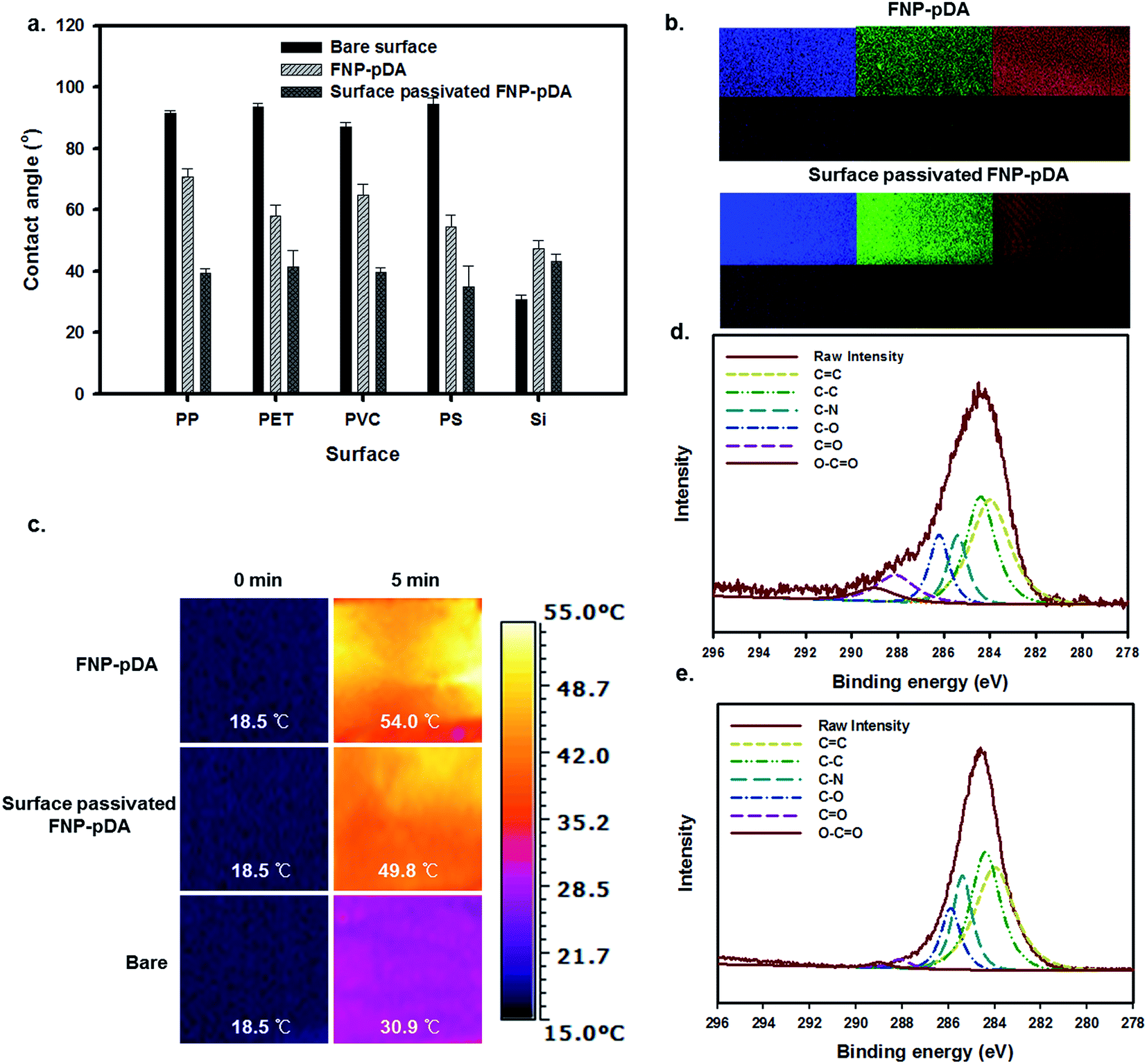 | ||
| Fig. 7 (a) Water contact angle measurements on polypropylene (PP), polyethylene terephthalate (PET), polyvinyl chloride (PVC), polystyrene (PS) and silicon (Si) substrate with application of FNP-pDA and surface passivated FNP-pDA coating material, respectively. (b) The confocal laser scan microscope (CLSM) image from FNP-pDA and surface passivated FNP-pDA coated (10 mg mL−1) polypropylene surface. (c) The near infrared (NIR)-irradiated photothermal response (Δ = Tn − T0) of FNP-pDA and surface passivated FNP-pDA-coated PET substrate. The C1s core level XPS spectra for compositional structural of (d) FNP-pDA and (e) surface passivated FNP-pDA. | ||
Having established that NIR excitation of FNP-pDA results in photothermal conversion, it was then necessary to examine the photothermal sensitivity of this surface.27,28 Irradiation of the FNP-pDA and surface-passivated FNP-pDA surface with the NIR laser demonstrated striking thermal elevation (54 °C) of the coated substrate (PET), which demonstrated the strong photothermal conversion ability of these materials (Fig. 7c).
The XPS C1s spectra of FNP-pDA and surface-passivated FNP-pDA are shown in Fig. 7d and e. The carbon moieties in the spectrum at 283 eV were assigned to CC; the binding energies at 284, 285.5, 286.2, 288 and 289 eV were assigned to C–C, C–N, C–O, CO and O–CO, respectively.12 The intensity of binding energy peaks for FNP-pDA, at about 283 eV for CC and 284 eV for C–C; decreased compared with surface-passivated FNP-pDA. The signals at 288 eV (CO) and 289 eV (O–CO) for surface-passivated FNP-pDA were decreased relative to FNP-pDA. These results demonstrate that both FNP-pDA and surface-passivated FNP-pDA are rich in sp2 and sp3 carbon sites (CC, C–C), which matches the expected constitution of carbon dots (CDs).12,14
Bio-fouling limits the performance of certain materials during biomedical application.29,30 Self-polymerized dopamine has been shown to automatically adhere to a wide range of substrates which could efficiently overcome antifouling properties, however carbonized FNP-pDA had not been examined. Our prepared FNP-pDA coated surface was evaluated by the quantification of nonspecific cell-adhesion and electron microscopic imaging. HeLa cells were cultured according to a previously reported method24 and imaged by optical microscopy. A detailed morphology report of the cell adhesion is presented in Fig. 8. Moderate (60–70%) adhesion of cells onto FNP-pDA coated PP and Si substrates, respectively. Unlike FNP-pDA coating, the surface-passivated FNP-pDA totally prevented cells adhesion owing to natural antifouling properties of PEG. The percentage of cells quantification shows less than 5% present on the PEG passivated FNP-pDA coated surface demonstrated antifouling strategy.
 | ||
| Fig. 8 Surface coated antifouling study (a and b) on bare surface, FNP-pDA coated and surface passivated FNP-pDA coating surface. The cells detachment study by using FNP-pDA and surface passivated FNP-pDA on PP (poly-propylene) and silicon (Si) substrate. | ||
Conclusions
In conclusion, we have synthesized fluorescent nanoparticles from polydopamine (FNP-pDA) that show promise as materials for use in photothermal therapy. The obtained FNP-pDA possess tunable fluorescence emission, small particle size (∼15 nm), and an increased level of sp2 and sp3 carbon atoms. FNP-pDA exhibited striking photothermal conversion of NIR radiation, killing both cancer cell lines and prokaryotic bacteria in vitro, by a specific photothermolytic mechanism. In addition, the surface-passivated FNP-pDA demonstrated remarkable anti-fouling behavior that protected the material surface. Fluorescence profiles combined with measurements of fluorescence lifetimes, demonstrated that surface passivation with polyethylene glycol capped with amine groups produced nanoparticles with a significantly higher quantum yield. Overall, we conclude that precise control over fluorescence nanoparticle synthesis could open exciting opportunities to conduct applied research using our nanomaterial platforms.Acknowledgements
This work was supported by the Grant No. 10048377, 10062079 and R0005237 from the Ministry of Trade, Industry & Energy (MOTIE), and Fusion Research R&D Program from the Korea Research Council for Industrial Science & Technology (No. G02054), Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2014055946).References
- K. Hola, Y. Zhang, Y. Wang, E. P. Giannelis, R. Zboril and A. L. Rogach, Nano Today, 2014, 9, 590 CrossRef CAS.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed.
- C. Ding, A. Zhu and Y. Tian, Acc. Chem. Res., 2013, 47, 20 CrossRef PubMed.
- S. Zhu, J. Zhang, S. Tang, C. Qiao, L. Wang, H. Wang, X. Liu, B. Li, Y. Li, W. Yu, X. Wang, H. Sun and B. Yang, Adv. Funct. Mater., 2012, 22, 4732 CrossRef CAS.
- G. Eda, Y. Y. Lin, C. Mattevi, H. Yamaguchi, H. A. Chen, I. S. Chen, C. W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611 CrossRef CAS.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203 CrossRef CAS PubMed.
- X. Tu, Y. Ma, Y. Cao, J. Huang, M. Zhang and Z. Zhang, J. Mater. Chem. B, 2014, 2, 2184 RSC.
- S. Hong, J. Kim, Y. S. Na, J. Park, S. Kim, K. Singha, G. I. Im, D. K. Han, W. J. Kim and H. Lee, Angew. Chem., Int. Ed., 2013, 52, 9187 CrossRef CAS PubMed.
- R. Liu, S. M. Mahurin, C. Li, R. R. Unocic, J. C. Idrobo, H. Gao, S. J. Pennycook and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 6799 CrossRef CAS PubMed.
- X. Yu, H. Fan, Y. Liu, Z. Shi and Z. Jin, Langmuir, 2014, 30, 5497 CrossRef CAS PubMed.
- S. M. Sharker, S. M. Kim, J. E. Lee, J. H. Jeong, I. In, K. D. Lee, H. Lee and S. Y. Park, Nanoscale, 2015, 7, 5468 RSC.
- D. K. Lim, A. Barhoumi, R. G. Wylie, G. Reznor, R. S. Langer and D. S. Kohane, Nano Lett., 2013, 13, 4075 CrossRef CAS PubMed.
- Y. P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S. Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756 CrossRef CAS PubMed.
- A. T. R. Wiliams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067 RSC.
- S. M. Sharker, C. J. Jeong, S. M. Kim, J. E. Lee, J. H. Jeong, I. In, H. Lee and S. Y. Park, Chem.–Asian J., 2014, 9, 2921 CrossRef CAS PubMed.
- K. Yang, J. Wan, S. Zhang, B. Tian, Y. Zhang and Z. Liu, Biomaterials, 2012, 33, 2206 CrossRef CAS PubMed.
- S. M. Kang, N. S. Hwang, J. Yeom, S. Y. Park, P. B. Messersmith, I. S. Choi, R. Langer, D. G. Anderson and H. Lee, Adv. Funct. Mater., 2012, 22, 2949 CrossRef CAS PubMed.
- M. B. Ahmad, M. Y. Tay, K. Shameli, M. Z. Hussein and J. J. Lim, Int. J. Mol. Sci., 2011, 12, 4872 CrossRef PubMed.
- S. M. Sharker, J. E. Lee, S. H. Kim, J. H. Jeong, I. In, H. Lee and S. Y. Park, Biomaterials, 2015, 61, 229 CrossRef CAS PubMed.
- E. B. Kang, J. E. Lee, J. H. Jeong, G. Lee, I. In and S. Y. Park, J. Ind. Eng. Chem., 2016, 33, 336 CrossRef CAS.
- C. J. Jeong, S. M. Sharker, I. In and S. Y. Park, ACS Appl. Mater. Interfaces, 2015, 7, 9469 Search PubMed.
- Y. J. Oh, C. J. Jeong, S. M. Sharker, S. Y. Lee, I. In and S. Y. Park, Surf. Interface Anal., 2015, 47, 259 CrossRef CAS.
- M. M. Chelsea, S. P. Cooper and A. B. Brennan, Mater. Today, 2010, 13, 36 Search PubMed.
- K. S. Lee, I. In and S. Y. Park, Appl. Surf. Sci., 2014, 313, 532 CrossRef CAS.
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., 2012, 124, 12381 CrossRef.
- S. H. Kim, E. B. Kang, C. J. Jeong, S. M. Sharker, I. In and S. Y. Park, ACS Appl. Mater. Interfaces, 2015, 7, 15600 Search PubMed.
- S. M. Sharker, S. M. Kim, S. H. Kim, I. In, H. Lee and S. Y. Park, J. Mater. Chem. B, 2015, 3, 5833 RSC.
- H. E. Yong, K. Krishnamoorthy, K. T. Hyun and S. J. Kim, J. Ind. Eng. Chem., 2015, 29, 39 CrossRef CAS.
- K. Sun, L. Song, Y. Xie, D. Liu, D. Wang, Z. Wang, W. Ma, J. Zhu and X. Jiang, Langmuir, 2011, 27, 5709 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08196g |
| ‡ Sung Han Kim and Shazid Md. Sharker contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |