
An urchin-like Ni3ZnC0.7–carbon nanotube-porous carbon composite derived from metal–organic gel as a cathode material for rechargeable Li–O2 batteries†
Yanqing Lai,
Wei Chen,
Zhian Zhang*,
Yongqing Gan and
Jie Li
School of Metallurgy and Environment, Central South University, Changsha, Hunan 410083, China. E-mail: zhangzhian@csu.edu.cn; Tel: +86 731 88830649
First published on 4th May 2016
Abstract
An urchin-like Ni3ZnC0.7–carbon nanotube-porous carbon composite was synthesized, for the first time, by one-step direct thermolysis of a metal–organic gel in a conventional horizontal tube furnace without using any additional carrier gas or catalyst. The urchin-like particles of the obtained composite with a particle size of ∼2 μm exhibit a sphere-like core, and CNTs grow outward from the core. And the Ni3ZnC0.7 nano-particles are embedded in the composite, including at the end of the nanotubes. When used as the cathode material of Li–O2 batteries, the composite exhibits excellent electrochemical performances, delivering a good cycle performance and having a discharge capacity of ∼7390 mA h g−1carbon+catalyst at 0.1 mA cm−2.
Introduction
As one of the most promising battery systems, rechargeable Li–O2 batteries have attracted intense interest in recent years because of their high energy density.1–3 However, several challenges, including large polarization, terrible rate capability and serious capacity fading, originating from the sluggish oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) kinetics, are necessary to be overcome before the marketing of Li–O2 batteries.4–6 The carbon materials and the catalyst, which are the main components of the cathode of Li–O2 batteries, affect the battery performance to a large extent.7–9 Therefore, in order to promote the performance of Li–O2 batteries, extensive research efforts have been devoted to exploit these two materials.In recent years, numerous carbon materials have been designed.10,11 As one kind of one-dimensional carbon material with strong mechanical strength and excellent electronic conductivity, carbon nanotubes (CNTs) have been exploited as the electrode materials of Li–O2 batteries for a long time.12–19 On the other hand, depending on the high porosity and the large surface area, which are beneficial for the transport of reactant (oxygen and Li+) and the storage of insoluble discharging product (Li2O2), porous carbon (PC) is also widely used as the cathode material.20–22 There is every reason to believe that the electrochemical performance of Li–O2 batteries will be improved if we can find the appropriate method to combine the two materials together.
In order to further promote the electrochemical performance of Li–O2 batteries, the addition of the catalyst to cathode is required. In recent studies, the metal carbides exhibit excellent catalytic ability.23–26 Kwak et al.24 synthesized Mo2C/CNTs as cathode material for Li–O2 batteries, which presented excellent electrochemical properties. Li et al.23 prepared Fe/Fe3C carbon nanofibers via a facile electrospinning method. And Li's research demonstrates that Fe/Fe3C has excellent OER catalytic activity for rechargeable non-aqueous Li–O2 batteries. More importantly, in addition to the excellent catalytic performance, metal carbides can be obtained during the carbonization process. This characteristic makes it possible to synthesis a catalyst-carbon composite via simple one-step carbonization process. Therefore, it will be an attractive subject to take metal carbides as the catalyst for Li–O2 batteries.
Recently, metal–organic gels (MOGs), as a new kind of coordination complexes, attracts more and more attention. MOGs is a kind of materials constituted by metal irons, organic ligands and part of solvent. Compared to MOFs, the synthesis condition for MOGs is much gentler with low temperature and short reaction time, and the yield is high. More importantly, the solvent encapsulated in MOGs can be seen as high quality carbon sources. This feature makes it an ideal precursor for the preparation of carbon materials. Here, in our work, urchin-like Ni3ZnC0.7–carbon nanotubes-porous carbon (NZC–PCCNTs) was prepared via simply carbonizing MOGs (MOG-NZ). While used as the cathode materials of Li–O2 batteries, it exhibits excellent electrochemical properties.
Experiment
Synthesis of NZC–PCCNTs composite
All chemicals and solvents were purchased from commercial sources and used without further purification. Metal–organic gel, MOG-NZ, was prepared by the following method. 45 mmol of Ni(CH3COO)2·4H2O and 15 mmol of Zn(CH3COO)2·4H2O were dissolved into 500 ml of N,N-dimethylformamide (DMF) to obtain a bright green solution which was marked as reaction solution A. Then, 36 mmol of H2BDC and 8.5 ml of triethylamine (TEA) were added into 400 ml of DMF with ultrasonic treatment, until the clear solution marked as reaction solution B was obtained. After that, the reaction solution B was pour into the reaction solution A under stirring. After reaction for 1 h, the gel, MOG-NZ, was obtained by vacuum filtration. Before carbonization, the gel was washed by DMF for three times.The NZC–PCCNTs composite was synthesized by the simple pyrolysis of the gel, MOG-NZ, without drying. In this route, the gel, MOG-NZ, was transferred into a tube furnace in a ceramic boat and heated to 700 °C at a heating rate of 5 °C min−1 under an argon atmosphere. After 10 h, the sample was cooled down to room temperature in the furnace under inert atmosphere, and NZC–PCCNTs was harvested.
Materials characterizations
X-ray diffraction patterns (XRD) were collected by a Rigaku3014 using graphite-monochromated Cu Kα radiation. Field-emission scanning electron microscopy (SEM) images taken on a Nova NanoSEM 230 and transmission electron microscopy (TEM) images obtained by using a Tecnai G2 20ST were employed to observe the morphology of the samples and/or the electrodes before and after discharge/charge. Raman spectra (Raman) were recorded on a Jobin-Yvon LabRAM HR-800 Raman spectrometer. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) measurements were performed on Baird PS-6. N2 adsorption/desorption measurements were performed at 77 K by using a Quantachrome instrument (Quabrasorb SI-3MP).Electrochemical tests
Electrochemical measurements were carried out in Li–O2 coin cells with 3 × Φ 1.5 mm holes in the centre of the positive pans in an evenly distributed pattern to allow O2 passage. A Li foil was used as the anode for the Li–O2 batteries, which was separated from the O2 electrode by a glass microfiber separator. The O2 electrodes (10 mm in diameter) were prepared by casting a mixture containing the as-prepared NZC–PCCNTs composite (80 wt%), Super P (10 wt%) and PVDF (10 wt%) onto a Ni foam current collector, followed by drying at 50 °C for 12 h in a vacuum oven, and the obtained electrode is denoted here as the NZC–PCCNTs electrode. The mass loading of active materials is about 1 mg cm−2. The same method was used to fabricate the commercial CNTs electrode, and this electrode was named the CNTs electrode. The electrolyte consists of 1 M LiTFSI in tetraethylene glycol dimethyl ether (TEGDME), and the amount of electrolyte in a cell is about 70 μl. The Li–O2 cells were constructed in an argon atmosphere glove box (Universal 2440/750) with oxygen and water contents of less than 1 ppm. The galvanostatic discharge/charge measurement of the Li–O2 battery was performed at a current density of 0.1–0.5 mA cm−2 in the potential range of 2.0–4.4 V under a LAND CT2001A system. It is noted that the specific capacity was calculated based on the total mass of carbon and catalyst. All measurements were undertaken in 1 atm dry oxygen to avoid any complications related to H2O and CO2.Results and discussion
In our research trying to synthesize Ni doped MOF-5 in room temperature via the method proposed by Tranchemontagne et al.,27 we found the addition of Ni2+ has a capability to form gels. The photographs of the as-prepared gel, MOG-NZ, are shown in Fig. 1. MOG-NZ is green and has gel like appearance with encapsulated solvent in it. It can be observed from Fig. 1b that MOG-NZ in the bottle of the beaker is strong enough to hold in the inverted beaker. This phenomenon indicates the formation of the gel. And the photograph shown in Fig. 1a proves the good self-sustaining nature of MOG-NZ, which is another important characteristic that can suggest the formation of the gel. Furthermore, Fig. 1c shows that the as-prepared MOG-NZ can be tailored into any shapes by carefully cutting. This result further confirms the formation of the gel. | ||
| Fig. 1 (a) The photographs of the as-prepared gel, MOG-NZ, (b) snapshot of inverted beaker of MOG-NZ and (c) tailoring the gel to make the shape of CSU. | ||
In order to synthesize the NZC–PCCNTs composite, MOG-NZ was directly carbonized under Ar atmosphere without pre-drying treatment. The morphology of the as-prepared NZC–PCCNTs composite was investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 2a, the obtained NZC–PCCNTs composite with urchin-like morphology has a sphere-like core, from which CNTs grow outward. And its particle size is about 2 μm. TEM images (Fig. 2b–d) further confirm that the urchin-like structure is composed of porous carbon core and CNTs shell. The tentacle-like CNTs can effectively improve the electronic conductivity of the NZC–PCCNTs composite. In addition, there are abundant of duck nano-particles distributing in the composites, including at the end of the nanotubes. Moreover, the hollow inert structures and the tube walls with distinct lattice fringes can be clearly seen in the HRTEM image shown in Fig. 2c. The thickness of the tube walls is about 5–10 nm, and the distance between the two adjacent planes is about 0.34 nm corresponding to (002) plane of carbon. The HRTEM image of the duck particle, which is exhibited in Fig. 2d, shows that the distance between the distinct lattice fringes of the duck particle is 0.21 nm. Combining with the XRD pattern exhibited in Fig. 2e, we can reasonably speculate that the duck particles in the composite are Ni3ZnC0.7, and the lattice fringes in the particle shown in Fig. 2d corresponding to the (111) plane of Ni3ZnC0.7 (JCPDS, no. 28-0713). These results demonstrate that the urchin-like Ni3ZnC0.7–carbon nanotubes-porous carbon composite has been successfully synthesized via the carbonization of MOG-NZ. The weight ratio of the Ni3ZnC0.7 catalyst in the NZC–PCCNTs composite measured by ICP-AES measurements is ∼30%.
 | ||
| Fig. 2 (a) SEM image of NZC–PCCNTs, (b) TEM image of NZC–PCCNTs, HR-TEM image of (c) CNT and (d) Ni3ZnC0.7 particle in NZC–PCCNTs, (e) XRD pattern of NZC–PCCNTs, and (f–i) elemental mapping images of NZC–PCCNTs. | ||
To identify the elements distribution of the NZC–PCCNTs composite material, the EDX analysis for C, N, Ni and Zn were carried out as the TEM test of the composite material was performed. The C, N, Ni and Zn elemental mapping images in Fig. 2f–i demonstrate that the Ni and Zn elemental are overlap with the Ni3ZnC0.7 nanoparticles in the NZC–PCCNTs composite, and the C and N elemental are evenly distributed over the porous carbon and CNTs of the NZC–PCCNTs composite and overlaps with them. From this result, it can be further confirmed the presence of Ni3ZnC0.7 and carbon material in the NZC–PCCNTs composite material and the successful synthesis of the NZC–PCCNTs composite material.
Based on the structure and morphology of the NZC–PCCNTs composite we observed, it can be proposed that the presence of Ni3ZnC0.7 is important for the formation of the NZC–PCCNTs composite. The gel (MOG-N) synthesized in the absence of Zn2+ was carbonized under the same condition. The obtained composite (NC) was tested by the XRD and SEM. The XRD pattern of NC in Fig. S1a† shows that NC composite is composed of carbon and Ni, and it can't be seen the urchin-like morphology and CNTs in the NC composite from the SEM image in Fig. S1b.† These results reveal the important role of Ni3ZnC0.7 in the formation of NZC–PCCNTs. We also carbonized the dry MOG-NZ, in which the solvent was totally removed. The SEM image of the obtained composite (NZCD) in Fig. S2† reveals the important role of the solvent, which supplies abundant carbon source, in the formation of the NZC–PCCNTs composite.
In the XRD pattern of the NZC–PCCNTs composite, we can also observe the diffraction peaks of carbon, which indicate the existence of carbon. For further investigating the detailed structure information of carbons in NZC–PCCNTs, the Raman spectrum measurement was performed. As shown in Fig. 3a, two typical Raman peaks of carbon, namely D-band and G-band, can be seen at 1354 and 1575 cm−1 of the Raman spectrum. The relative intensity of G-bands and D-band (IG/ID), which can verify the crystalline degree of carbon materials, is about 1.22, suggesting the high graphitization degree of the carbon in the as-prepared sample.28 The well graphitized carbon may ensure good electronic conductivity and is beneficial to the enhancement of its performance as cathode material.29 The result of the N2 adsorption–desorption measurements (Fig. 3b) demonstrates that the total specific surface area and pore volume are 276 m2 g−1 and 0.4 cm3 g−1, respectively. These two values are both higher than the normal commercial CNTs (82 m2 g−1 and 0.25 cm3 g−1). Meanwhile, the XPS measurement (Fig. 3c and d) reveals the doping of N into NZC–PCCNTs, and the nitrogen content is about 4.2%. The high-resolution spectrum of N shown in Fig. 3d can be deconvoluted into three peaks centring at 398.50, 399.08 and 400.99 eV, assigned to pyridinic, pyrrolic and graphitic N. As previously reported, all of these N species can promote the ORR activity of N-doped carbon, although graphitic and pyridinic N is more active than their pyrrolic counterpart.30,31
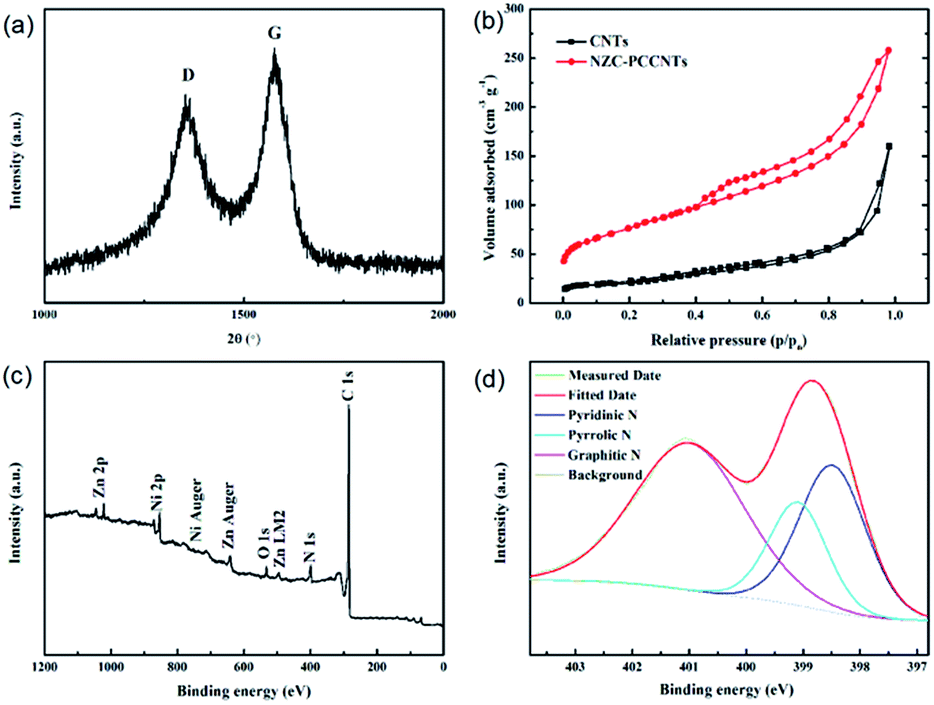 | ||
| Fig. 3 (a) Raman spectrum, (b) the nitrogen adsorption/desorption isotherms of NZC–PCCNTs (the inset is the pore size distribution), (c) survey spectrum and (d) high-resolution N (1s) binding energy spectrum of NZC–PCCNTs. | ||
To gain straight insight into the electrochemical performance of NZC–PCCNTs in Li–O2 battery, the NZC–PCCNTs cathode was assembled and the galvanostatic charge–discharge measurements were performed in O2 atmosphere at the voltage range of 2.0–4.4 V (vs. Li/Li+). The discharge and charge voltage profiles of the Li–O2 battery with the NZC–PCCNTs cathode, which were discharged and recharged at the current density of 0.1 mA cm−2carbon+catalyst, were given in Fig. 4a. The NZC–PCCNTs cathode delivers a discharge capacity of ∼7390 mA h g−1carbon+catalyst and a charge capacity of ∼7160 mA h g−1carbon+catalyst, which is comparable to previous reports.32–34 And these two values are much higher than that of CNTs cathode (a discharge capacity of ∼2200 mA h g−1carbon+catalyst and a charge capacity of ∼2620 mA h g−1carbon+catalyst, respectively). The NZC–PCCNTs cathode also shows a higher discharge voltage plateau of 2.75 V than that of CNTs cathode (∼2.68 V). Meanwhile, the NZC–PCCNTs cathode exhibits a charge voltage at 4.03 V, which is considerably lower than that of CNTs electrode (∼4.22 V). And these two values are also comparable to that of other reported catalysts.32,35–37 As most of reported Li–O2 batteries, the widely used capacity limited protocol was also employed in our work. The cycle performance of the Li–O2 battery using the NZC–PCCNTs cathode was also investigated by confining the discharge/charge capacities to 800 mA h g−1carbon+catalyst at a current density of 0.1 mA cm−2. As shown in the Fig. 4b–d The NZC–PCCNTs electrode shows better cycle performance over 42 cycles with stable reversible capacities than that of CNTs cathode. While increasing the applied current density, the NZC–PCCNTs electrode also shows a better rate performance than that of CNTs cathode (Fig. 4e and f). It is rationally deduced that the better performances of the NZC–PCCNTs electrode may be attributed to the synergistic effect of the good catalytic activity of Ni3ZnC0.7 and N-doped carbon, the existence of the porous carbon core and the high electric conduction of CNTs that grow outward from the core. The results of the electrochemical tests suggest that the NZC–PCCNTs composite is suitable for the cathode material of Li–O2 batteries.
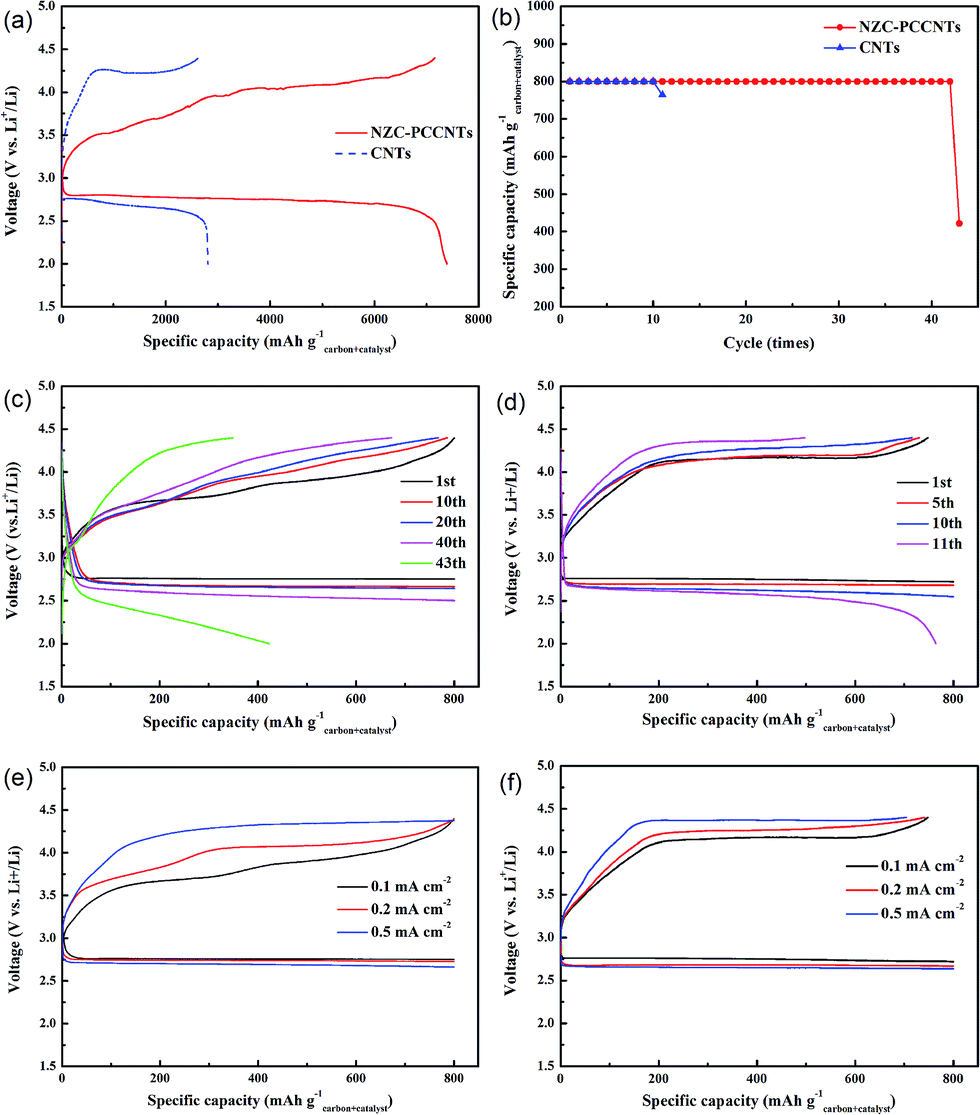 | ||
| Fig. 4 (a) Discharge–charge curves of the NZC–PCCNTs electrode and CNTs electrode between 2.0 and 4.4 V at 0.1 mA cm−2, (b) cycle performances of Li–O2 batteries with the NZC–PCCNTs electrode and CNTs electrode, discharge/charge curves for selected cycles of (c) the NZC–PCCNTs electrode and (d) CNTs electrode with fixed capacity of 800 mA h g−1, rate capability of (e) the NZC–PCCNTs electrode and (f) CNTs electrode at different current densities. | ||
Since many researches demonstrate that the electrolyte is not completely stable in lithium–O2 batteries, and the decomposition of electrolyte may contribute to the capacity of lithium–O2 batteries.38–40 The ex situ XRD measures were carried out here to identify whether the discharge and charge capacity of the batteries with NZC–PCCNTs electrode are originated from the generation and decomposition of Li2O2. As presented in Fig. 5, the XRD patterns of the pristine, discharged, and recharged electrodes were obtained, respectively. According to Fig. 5, the two typical peaks corresponding to the Li2O2 phase were emerged at 33.1 and 34.8°, after the electrode was discharged to 2.0 V at a current density of 0.1 mA cm−2.41,42 When the cell was fully recharged to 4.4 V, these two peaks related to the Li2O2 phase were found fully disappeared, indicating that the Li2O2 phase formed in the discharging process has been reversibly decomposed during the subsequent charging process. This result confirms that the discharge and charge capacity of the Li–O2 batteries with the NZC–PCCNTs electrodes are mainly derived from the reversible formation and decomposition of Li2O2, instead of the decomposition of electrolyte.
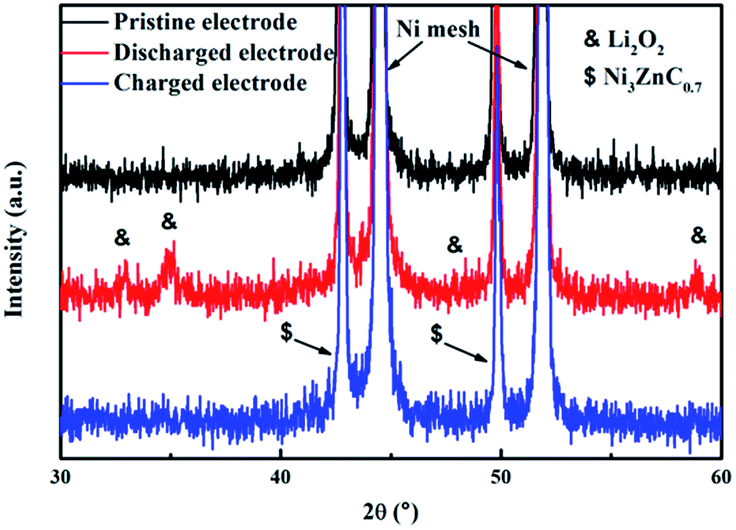 | ||
| Fig. 5 XRD patterns of the pristine, discharged, and recharged NZC–PCCNTs electrode. | ||
Conclusions
In summary, urchin-like Ni3ZnC0.7–carbon nanotubes-porous carbon composite was synthesized via a pyrolysis procedure of metal–organic gel (MOG-NZ). The solvent DMF, Zn2+ and Ni2+ in the MOG-NZ play an important role in the formation of urchin-like morphology of the obtained composite, which possesses a sphere-like core with CNTs growing outward from it and has Ni3ZnC0.7 nano-particles embedded in the carbon material. This method opens a new way for the preparation of CNTs. And this is an unprecedented method to combine CNTs with porous carbon synchronously. While the NZC–PCCNTs composite was used as the cathode material of Li–O2 batteries, it exhibits a lower over-potential, higher discharge capacity (a discharge capacity of ∼7390 mA h g−1carbon+catalyst at 0.1 mA cm−2) and better cycling stability (over 42 cycles) that commercial CNTs. The excellent electrochemical performance of NZC–PCCNTs indicates its potential as a cathode material for Li–O2 batteries.Notes and references
- R. S. Kalubarme, H. S. Jadhav, C.-N. Park, K.-N. Jung, K.-H. Shin and C.-J. Park, J. Mater. Chem. A, 2014, 2, 13024–13032 CAS.
- D. Oh, J. Qi, B. Han, G. Zhang, T. J. Carney, J. Ohmura, Y. Zhang, Y. Shao-Horn and A. M. Belcher, Nano Lett., 2014, 14, 4837–4845 CrossRef CAS PubMed.
- A. Riaz, K. N. Jung, W. Chang, K. H. Shin and J. W. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 17815–17822 CAS.
- C. K. Lee and Y. J. Park, Chem. Commun., 2015, 51, 1210–1213 RSC.
- F. Li, T. Zhang and H. Zhou, Energy Environ. Sci., 2013, 6, 1125–1141 CAS.
- Y. Shao, F. Ding, J. Xiao, J. Zhang, W. Xu, S. Park, J.-G. Zhang, Y. Wang and J. Liu, Adv. Funct. Mater., 2013, 23, 987–1004 CrossRef CAS.
- J. Lu, L. Li, J. B. Park, Y. K. Sun, F. Wu and K. Amine, Chem. Rev., 2014, 114, 5611–5640 CrossRef CAS PubMed.
- M. D. Bhatt, H. Geaney, M. Nolan and C. O'Dwyer, Phys. Chem. Chem. Phys., 2014, 16, 12093–12130 RSC.
- Z. L. Wang, D. Xu, J. J. Xu and X. B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- W. Jun Lee, U. N. Maiti, J. M. Lee, J. Lim, T. H. Han and S. O. Kim, Chem. Commun., 2014, 50, 6818–6830 RSC.
- A. D. Roberts, X. Li and H. Zhang, Chem. Soc. Rev., 2014, 43, 4341–4356 RSC.
- M. L. Thomas, K. Yamanaka, T. Ohtab and H. R. Byon, Chem. Commun., 2015, 51, 3977–3980 RSC.
- S. Liu, Z. Wang, C. Yu, Z. Zhao, X. Fan, Z. Ling and J. Qiu, J. Mater. Chem. A, 2013, 1, 12033 CAS.
- R. Mi, H. Liu, H. Wang, K.-W. Wong, J. Mei, Y. Chen, W.-M. Lau and H. Yan, Carbon, 2014, 67, 744–752 CrossRef CAS.
- G.-L. Tian, Q. Zhang, B. Zhang, Y.-G. Jin, J.-Q. Huang, D. S. Su and F. Wei, Adv. Funct. Mater., 2014, 24, 5956–5961 CrossRef CAS.
- G.-L. Tian, M.-Q. Zhao, D. Yu, X.-Y. Kong, J.-Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 2251–2259 CrossRef CAS PubMed.
- H. Yuan, L. Deng, X. Cai, S. Zhou, Y. Chen and Y. Yuan, RSC Adv., 2015, 5, 56121–56129 RSC.
- J.-H. Kim, A. G. Kannan, H.-S. Woo, D.-G. Jin, W. Kim, K. Ryu and D.-W. Kim, J. Mater. Chem. A, 2015, 3, 18456–18465 CAS.
- J. Song, C. Zhu, S. Fu, Y. Song, D. Du and Y. Lin, J. Mater. Chem. A, 2016, 4, 4864–4870 CAS.
- J. Li, H. Zhang, Y. Zhang, M. Wang, F. Zhanga and H. Nie, Nanoscale, 2013, 5, 4647–4651 RSC.
- J. Li, Y. Zhang, W. Zhou, H. Nie and H. Zhang, J. Power Sources, 2014, 262, 29–35 CrossRef CAS.
- H. Nie, H. Zhang, Y. Zhang, T. Liu, J. Lia and Q. Lai, Nanoscale, 2013, 5, 8484–8487 RSC.
- J. Li, M. Zou, L. Chen, Z. Huang and L. Guan, J. Mater. Chem. A, 2014, 2, 10634–10638 CAS.
- W. J. Kwak, K. C. Lau, C. D. Shin, K. Amine, L. A. Curtiss and Y. K. Sun, ACS Nano, 2015, 9, 4129–4137 CrossRef CAS PubMed.
- D. Kundu, R. Black, B. Adams, K. Harrison, K. R. Zavadil and L. F. Nazar, J. Phys. Chem. Lett., 2015, 6, 2252–2258 CrossRef CAS PubMed.
- Z. Wang, J. Sun, Y. Cheng and C. Niu, J. Phys. Chem. Lett., 2014, 5, 3919–3923 CrossRef CAS PubMed.
- D. J. Tranchemontagne, J. R. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef CAS.
- M. A. Rahman, X. Wang and C. Wen, J. Appl. Electrochem., 2013, 44, 5–22 CrossRef.
- Y. Li, J. Wang, X. Li, D. Geng, M. N. Banis, R. Li and X. Sun, Electrochem. Commun., 2012, 18, 12–15 CrossRef CAS.
- Q. Li, P. Xu, W. Gao, S. Ma, G. Zhang, R. Cao, J. Cho, H. L. Wang and G. Wu, Adv. Mater., 2014, 26, 1378–1386 CrossRef CAS PubMed.
- Y. Hou, T. Huang, Z. Wen, S. Mao, S. Cui and J. Chen, Adv. Energy Mater., 2014, 4, 1400337 Search PubMed.
- J. Zhang, P. Li, Z. Wang, J. Qiao, D. Rooney, W. Sun and K. Sun, J. Mater. Chem. A, 2015, 3, 1504–1510 CAS.
- Y. Cao, M.-S. Zheng, S. Cai, X. Lin, C. Yang, W. Hu and Q.-f. Dong, J. Mater. Chem. A, 2014, 2, 18736–18741 CAS.
- G. Gnanakumar, M. Christy, H. Jang and K. S. Nahm, J. Power Sources, 2015, 288, 451–460 CrossRef CAS.
- K. Zhang, L. Zhang, X. Chen, X. He, X. Wang, S. Dong, L. Gu, Z. Liu, C. Huang and G. Cui, ACS Appl. Mater. Interfaces, 2013, 5, 3677–3682 CAS.
- W.-H. Ryu, T.-H. Yoon, S. H. Song, S. Jeon, Y.-J. Park and I.-D. Kim, Nano Lett., 2013, 13, 4190–4197 CrossRef CAS PubMed.
- S. H. Kang, K. Song, J. Jung, M. R. Jo and Y.-M. Kang, J. Mater. Chem. A, 2014, 2, 19660–19664 CAS.
- B. Sun, X. Huang, S. Chen, J. Zhang and G. Wang, RSC Adv., 2014, 4, 11115–11120 RSC.
- W. Xu, J. Hu, M. H. Engelhard, S. A. Towne, J. S. Hardy, J. Xiao, J. Feng, M. Y. Hu, J. Zhang, F. Ding, M. E. Gross and J. Zhang, J. Power Sources, 2012, 215, 240–247 CrossRef CAS.
- N. Mozhzhukhina, L. P. Méndez, D. Leo and E. J. Calvo, J. Phys. Chem. C, 2013, 117, 18375–18380 CAS.
- B. Sun, J. Zhang, P. Munroe, H.-J. Ahn and G. Wang, Electrochem. Commun., 2013, 31, 88–91 CrossRef CAS.
- W.-H. Ryu, T.-H. Yoon, S. H. Song, S. Jeon, Y.-J. Park and I.-D. Kim, Nano Lett., 2013, 13, 4190–4197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08218a |
| This journal is © The Royal Society of Chemistry 2016 |