
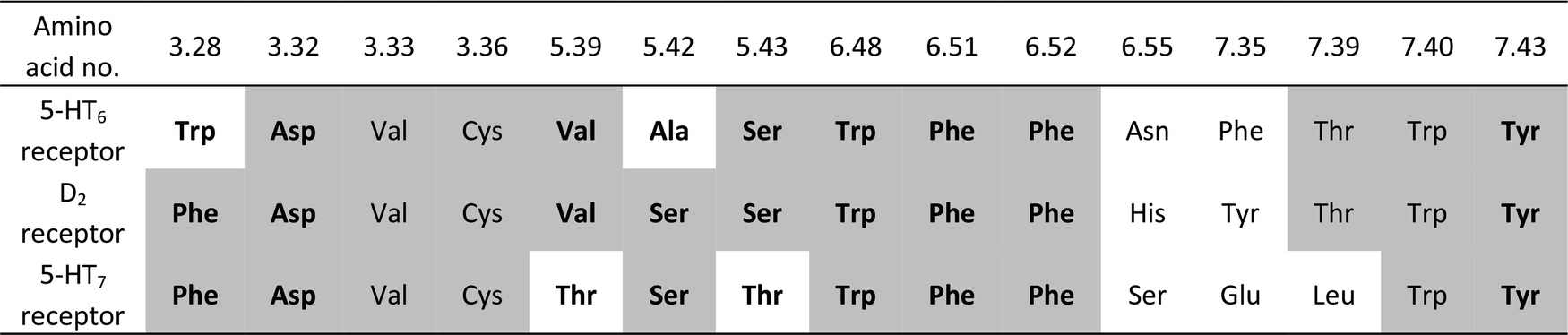
Halogen bonding enhances activity in a series of dual 5-HT6/D2 ligands designed in a hybrid bioisostere generation/virtual screening protocol†
Jakub Staroń,
Dawid Warszycki,
Rafał Kurczab,
Grzegorz Satała,
Ryszard Bugno,
Adam Hogendorf and
Andrzej J. Bojarski*
Department of Medicinal Chemistry, Institute of Pharmacology Polish Academy of Sciences, 12 Smętna Street, 31-343 Kraków, Poland. E-mail: bojarski@if-pan.krakow.pl; Tel: +48 12 6623365
First published on 27th May 2016
Abstract
A novel hybrid bioisostere generation/virtual screening method combined with narrowing of chemical space through similarity to compounds that are active at the second target was successfully applied for the development of structurally new dual 5-HT6/D2 receptor ligands. Consequently, a series of derivatives of the found hit 1d (N-[2-(dimethylamino)ethyl]-N-(2-phenylethyl)aniline) was synthesized. The most active 5-HT6/D2 ligands also showed affinity for 5-HT7R and 5-HT2AR. The para-chloroaniline derivative was identified as a potent dual 5-HT6/5-HT7 receptor antagonist (Ki = 24 nM and Kb = 30 nM, Ki = 4 nM and Kb = 1.4 nM, respectively). In the case of halogen-containing compounds, interesting structure–activity relationships were observed at 5-HT6, D2 and 5-HT7 receptors, and the ligand–receptor complexes were subsequently examined using a molecular modelling approach that combined quantum-polarized ligand docking (QPLD) and Molecular-Mechanics-Generalized-Born/Surface Area (MM/GBSA) free-energy calculation, which permitted the identification of putative halogen binding pockets.
Introduction
New drugs can be designed or searched for in silico using a structure-based or ligand-based approach or both methods together.1 Structure-based approaches require the structure (obtained either through crystallographic or NMR methods) of a biological target (e.g., protein) that is, in certain cases, difficult to obtain. Indeed, to date, the crystal structures of most serotonin and dopamine (except 5-HT1BR, 5-HT2BR and D3) receptors have not been determined. In turn, ligand-based methods utilize structures of known compounds, both endogenous and exogenous, that function at a given biological target. Among the ligand-based methods is bioisosteric replacement, which includes the modification of the chemical structure of a drug in a way that a new compound with similar biological and modified pharmacokinetic properties is acquired.2,3 Bioisosteric replacement is commonly used for the development of a series of analogues in hit-to-lead and/or lead optimization processes.Virtual screening (VS) campaigns are usually aimed at identifying compounds that are active at one selected biological target. It is well known, however, that many drugs interact with more than one target, such as therapeutics that are used for mental disorders with a complex aetiology.4–6 The design of ligands that display a given pattern of activities for CNS receptors is however a challenging task. Because the bioisostere generation/virtual screening method was successfully applied for the development of new 5-HT6R ligands,7 we assumed that a similar approach combined with narrowing of the chemical space through similarity to ligands of the second target may result in the identification of structurally novel compounds that are active at two given receptors. This assumption could provide a starting point for rational design of ligands with a more complex profile of pharmacological activity. Thus, following the therapeutic properties revealed by agents of serotonergic and dopaminergic systems, the 5-HT6 receptor was selected as a primary target,10,11 whereas activity at dopamine D2 receptor was additionally sought.12,13 It should be mentioned, however, that the verification of the methodological approach, rather than focusing on development of drug candidates, was the main objective of the project. Nevertheless such dual 5-HT6/D2 activity profile has recently been reported for hybrid compounds that display antidepressant-like effects in animal models together with pro-cognitive properties.14 Moreover, pro-cognitive efficacy of 5-HT6R antagonists is also suggested as an adjunctive for the primary function of dopamine D2 receptors, in antipsychotic drug action.8,9
Here, we report the design of new ligands with dual 5-HT6R/D2R biological activity through a multi-step in silico screening protocol. The developed group of N-[2-(dimethylamino)ethyl]-N-(2-phenylethyl)aniline derivatives was additionally tested for affinity for 5-HT1A, 5-HT2A and 5-HT7 receptors. Among the group of developed derivatives, halogen-containing compounds showed interesting structure–activity relationships at 5-HT6, D2 and 5-HT7 receptors, and thus their complexes were further investigated using hybrid quantum mechanic/molecular mechanic (QM/MM) methods.
Bioisostere query
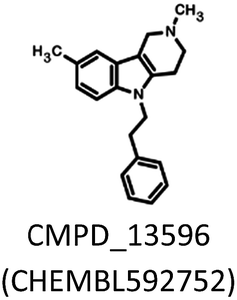
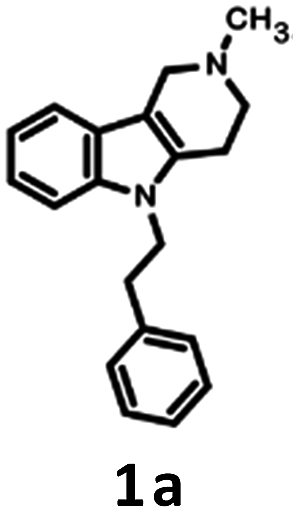
The applied bioisosteric query was designed to identify 5-HT6R ligands that are similar to any D2R ligand and thus potentially exhibit the desired dual 5-HT6R/D2R activity (Fig. 1). Initially, a set of 4806 5-HT6R ligands (Ki < 100 nM) extracted from the ChEMBL database was used for bioisostere generation with the PipelinePilot program.15 Next, the obtained group of 165![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 657 daughter structures was compared to a set of 3396 D2R ligands that were also obtained from the ChEMBL database. Only 853 bioisosteres had a Tanimoto similarity (Tc) higher than 0.9, whereas 53 were identical to the corresponding D2R ligands. These 906 structures that were daughters of the 95 parent structures were subsequently virtually screened for 5-HT6R affinity. The screening protocol consisted of several steps that included Lipinski and Veber rules, physicochemical properties, ADME/Tox, 5-HT6R pharmacophores and docking to 5HT6R homology models.16 As a result, a group of 31 bioisosteres that were daughters of 20 5-HT6R ligands was obtained. Visual inspection of these 20 parent structures revealed that they included only 8 chemical scaffolds (see ESI,† page 2). The specificity of the PipelinePilot program caused that none of the obtained scaffolds was unique because they were mostly simple derivatives of their parent compounds. The PipelinePilot utilizes only bioisosteric replacements that can be found in bioactive molecules (taken from the BIOSTER database17), and thus it has a limited ability to create a new unique chemical scaffold. However, the vBrood program18 provides analyses of the electro- and stereo-topological properties of molecular fragments and substitutes the fragment with another similar fragment according to established program settings. This procedure allows vBrood to produce dozens of thousands of new, unique chemical scaffolds, the majority of which are, however, nonsense. Eight scaffolds obtained in the first round of the VS protocol were therefore processed in vBrood and provided 26791 bioisosteres, which were subsequently evaluated using the same virtual screening protocol against 5-HT6R. One hundred and two bioisosteres, out of these 26791, passed all VS filters, among which four compounds (1d–1g) were selected in terms of their synthetic accessibility and chemical scaffold novelty (Table 1). The chemical scaffolds (1a–1c) of the parents were synthesized rather than the parents themselves because this study was focused mainly on the effect on activity of the applied bioisosteric replacement. However, among all of the chosen bioisosteres, only 1d was active both at 5-HT6R and D2R. Thus, to investigate the structure–activity relationship, a group of its derivatives 4a–4t was obtained.
657 daughter structures was compared to a set of 3396 D2R ligands that were also obtained from the ChEMBL database. Only 853 bioisosteres had a Tanimoto similarity (Tc) higher than 0.9, whereas 53 were identical to the corresponding D2R ligands. These 906 structures that were daughters of the 95 parent structures were subsequently virtually screened for 5-HT6R affinity. The screening protocol consisted of several steps that included Lipinski and Veber rules, physicochemical properties, ADME/Tox, 5-HT6R pharmacophores and docking to 5HT6R homology models.16 As a result, a group of 31 bioisosteres that were daughters of 20 5-HT6R ligands was obtained. Visual inspection of these 20 parent structures revealed that they included only 8 chemical scaffolds (see ESI,† page 2). The specificity of the PipelinePilot program caused that none of the obtained scaffolds was unique because they were mostly simple derivatives of their parent compounds. The PipelinePilot utilizes only bioisosteric replacements that can be found in bioactive molecules (taken from the BIOSTER database17), and thus it has a limited ability to create a new unique chemical scaffold. However, the vBrood program18 provides analyses of the electro- and stereo-topological properties of molecular fragments and substitutes the fragment with another similar fragment according to established program settings. This procedure allows vBrood to produce dozens of thousands of new, unique chemical scaffolds, the majority of which are, however, nonsense. Eight scaffolds obtained in the first round of the VS protocol were therefore processed in vBrood and provided 26791 bioisosteres, which were subsequently evaluated using the same virtual screening protocol against 5-HT6R. One hundred and two bioisosteres, out of these 26791, passed all VS filters, among which four compounds (1d–1g) were selected in terms of their synthetic accessibility and chemical scaffold novelty (Table 1). The chemical scaffolds (1a–1c) of the parents were synthesized rather than the parents themselves because this study was focused mainly on the effect on activity of the applied bioisosteric replacement. However, among all of the chosen bioisosteres, only 1d was active both at 5-HT6R and D2R. Thus, to investigate the structure–activity relationship, a group of its derivatives 4a–4t was obtained.
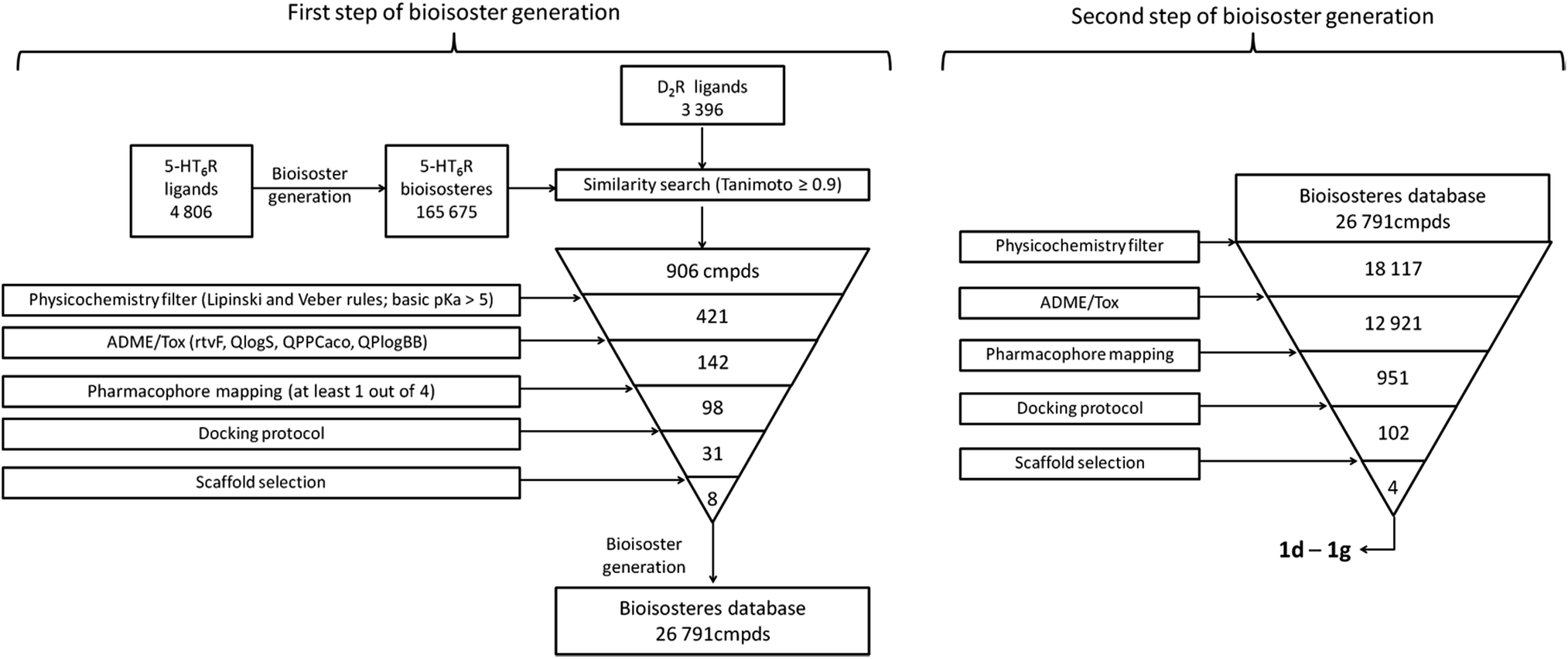 | ||
| Fig. 1 Workflow of the applied protocol for developing a dual ligand that acts at 5-HT6R and D2R. From the structures of known 5-HT6R ligands, a database of bioisosteres was generated (using a PipelinePilot program15), which was subsequently compared with D2R ligands. The obtained structures with a Tanimoto coefficient > 0.9 were evaluated using the 5-HT6R VS protocol. Structures that passed all filters were used in a second bioisostere generation step (using the vBrood program18), and the obtained structures were evaluated again using the same VS filters. The final hits were selected considering the synthetic accessibility and novelty of the chemical scaffolds. | ||
| Parent structure | Chemical scaffold | Daughter structure |
|---|---|---|
 |
 |
 |
 |
 |
 |
 |
||
 |
 |
 |
Synthesis
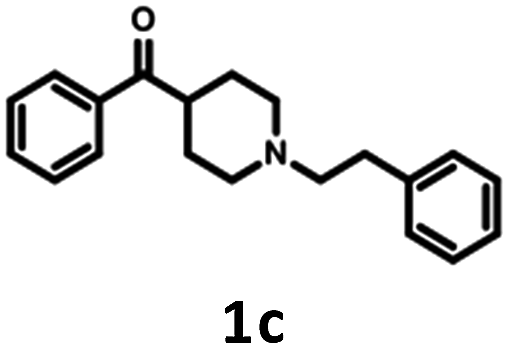
The parent compound 1c was synthesized according to a previously described method in patent US 2010/0120792 (Scheme 1).19,20 The first step of the synthesis utilized the reaction of phenylhydrazine with N-methyl-4-piperidone in sulfuric acid, which produced N-methyl-2,3,4,5-tetrahydro-1H-pyrido[4,3-b]indole (1a). Next, the product was conjugated to phenylacetylene using KOH, (Bu4N)2SO4 in DMSO and, finally, hydrogenated over 10% Pd/C in methanol to afford target product 1c.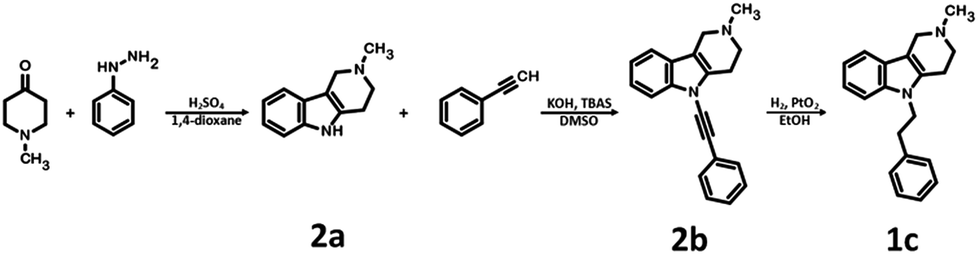 | ||
| Scheme 1 Synthesis of 2-methyl-5-(2-phenylethyl)-1H,2H,3H,4H,5H-pyrido[4,3-b]indole 1c. | ||
The synthesis of bioisosteres 1d, 4a–4t consisted of two steps (Scheme 2). First, reaction of the appropriate anilines with an alkyl chloride (2-chloro-N,N-dimethylethylamine, 2-chloro-N,N-diethylethylamine or 2-chloro-N,N-dimethylpropylamine) in isopropanol with K2CO3 provided intermediates 3a–3u. Second, reaction of the intermediate with phenylacetaldehyde in DCE with triacetoxyborohydride yielded the final products 1d, 4a–4t.
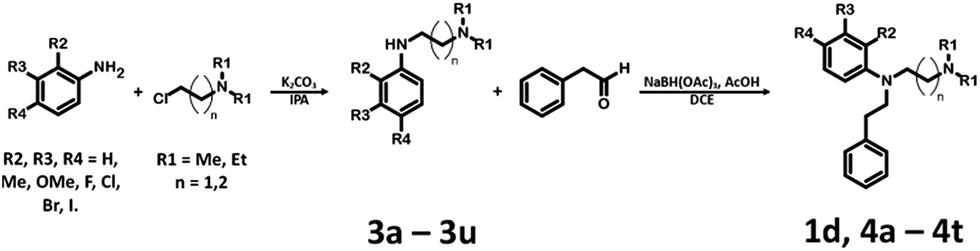 | ||
| Scheme 2 Synthesis of N-[2-(dimethylamine)ethyl]-N-(2-phenylehtyl)aniline derivatives 1d, 4a–4t. | ||
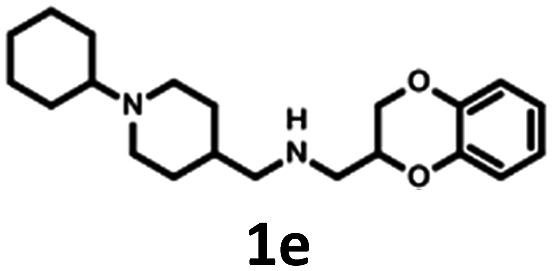
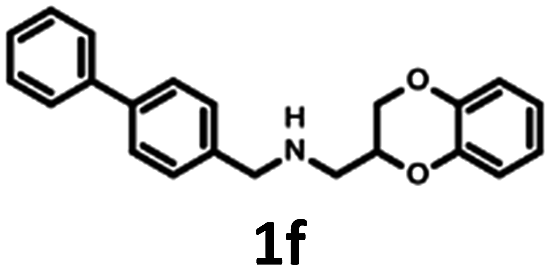
Synthesis of 1b, 1e and 1f was achieved by the conjugation of 5d with 5i, 5j and 6b in acetonitrile with K2CO3 (Schemes 3 and 4). Intermediate 5d was synthesized by, first, bromination of ethyl acrylate, followed by conjugation to catechol, reduction of the ester group with LiAlH4 and subsequent esterification with tosyl chloride. Intermediates 5i and 5j were obtained by the reaction of 2,4-dinitrochlorobenzene with isonicotinamide, followed by substitution with aniline. The obtained compound 5f was hydrogenated under pressure over platinum or palladium to generate 5g and 5h. The products were reduced using LiAlH4 to produce 5i and 5j. Intermediate 6b was synthesized by Suzuki–Miyaura coupling of 4-bromobenzaldehyde with phenylboronic acid, followed by the reaction with hydroxylamine and subsequent reduction with NaBH4. Unfortunately, the attempts to synthesize bioisostere 1g were unsuccessful, and the synthesis of parent 1c was also abandoned.
 | ||
| Scheme 3 Synthesis of (2,3-dihydro-1,4-benzodioxin-2-ylmethyl)[(1-phenyl-piperidin-4-yl)methyl]amine 1b and [(1-cyclohexylpiperidin-4-yl)methyl](2,3-dihydro-1,4-benzodioxin-2-ylmethyl)amine 1e. | ||
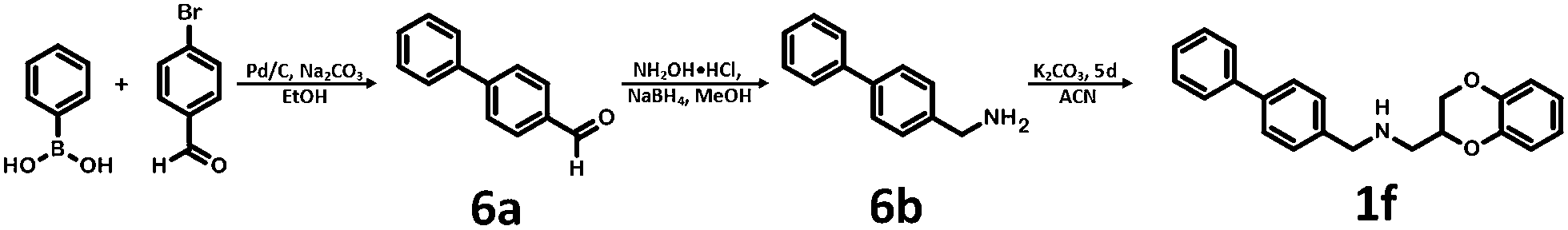 | ||
| Scheme 4 Synthesis of (2,3-dihydro-1,4-benzodioxin-2-ylmethyl)[(4-phenyl-phenyl)methyl]amine 1f. | ||
Pharmacology
Radioligand binding assays were used to determine the affinity and selectivity profiles of the synthesized compounds for human serotonin 5-HT1AR, 5-HT2A, 5-HT6R, 5-HT7bR and D2LR, which were stably expressed in HEK293 cells. This procedure was accomplished via the displacement of [3H]-8-OH-DPAT (187 Ci mmol−1) for 5-HT1AR, [3H]-Ketanserin (47.3 Ci mmol−1) for 5-HT2A, [3H]-LSD (85.2 Ci mmol−1) for 5-HT6R, [3H]-5-carboxyamidotryptamine (5-CT, 39.2 Ci mmol−1) for 5-HT7bR and [3H]-Raclopride (74.4 Ci mmol−1) for D2LR. Each compound was tested in triplicate at 7 to 8 different concentrations (10−11 to 10−4 M). The inhibition constants (Ki) were calculated using the Cheng–Prusoff equation,21 and the results are expressed as the mean of at least two independent experiments (Table 2). The antagonistic properties of 4m on 5-HT6R and 5-HT7R were evaluated based on its ability to inhibit cAMP production induced by the agonist 5-CT (100 nM in the case of 5-HT6R, 10 nM in the case of 5-HT7R) in HEK293 cells overexpressing 5-HT6 or 5-HT7 receptors (Table 3). The experiments were performed in triplicate at 8 concentrations (10−11 to 10−4 M).
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd. | n | Substitution | Ki [nM] | |||||||
| R1 | R2 | R3 | R4 | 5-HT6 | D2 | 5-HT7 | 5-HT1A | 5-HT2A | ||
| a Binding affinity, Ki, expressed as the average of at least two independent experiments; the maximum S.D. did not exceed 34% (see ESI, page 5); n.d. – not determined. | ||||||||||
| 1a | — | — | — | — | — | 22 | 77 | 8 | 217 | 29 |
| 1b | — | — | — | — | — | 2280 | 210 | 430 | 10 | n.d. |
| 1d | 1 | Me | H | H | H | 63 | 476 | 121 | 1682 | 233 |
| 1e | — | — | — | — | — | 2675 | 1609 | 3428 | 22 | n.d. |
| 1f | — | — | — | — | — | 3760 | 110 | 857 | 101 | n.d. |
| 4a | 1 | Et | H | H | H | 51 | 266 | 205 | 7614 | 128 |
| 4b | 2 | Me | H | H | H | 67 | 55 | 623 | 4486 | 228 |
| 4c | 1 | Me | H | H | F | 23 | 37 | 31 | 5707 | 58 |
| 4d | 1 | Me | H | H | Me | 25 | 33 | 10 | 482 | 70 |
| 4e | 1 | Me | H | H | CF3 | 61 | 274 | 30 | 5341 | 809 |
| 4f | 1 | Me | H | H | OMe | 234 | 1147 | 141 | >10000 |
863 |
| 4g | 1 | Me | H | Me | H | 264 | n.d. | 2253 | 605 | 777 |
| 4h | 2 | Me | H | H | F | 38 | 80 | 38 | 8969 | 27 |
| 4i | 1 | Me | H | F | H | 64 | 594 | 748 | >10000 |
312 |
| 4j | 1 | Me | Cl | H | Cl | 597 | 1176 | 387 | >10000 |
472 |
| 4k | 1 | Me | H | OMe | H | 355 | 1738 | 4512 | >10000 |
721 |
| 4l | 2 | Me | H | H | Me | 83 | 108 | 24 | 2140 | 204 |
| 4m | 1 | Me | H | H | Cl | 24 | 153 | 4 | 5960 | 212 |
| 4n | 1 | Me | Me | H | H | 380 | n.d. | 1040 | >10000 |
348 |
| 4o | 1 | Me | Naphthyl | H | 137 | 423 | 2037 | 4749 | 621 | |
| 4p | 1 | Me | H | F | F | 21 | n.d. | 114 | >10000 |
79 |
| 4q | 1 | Me | H | H | Br | 90 | 505 | 19 | n.d. | 295 |
| 4r | 1 | Me | H | H | I | 79 | 691 | 10 | n.d. | 372 |
| 4s | 1 | Me | Me | H | Me | 347 | 1751 | 597 | n.d. | 518 |
| 4t | 1 | Me | H | Cl | H | 150 | 675 | 353 | n.d. | 311 |

Results and discussion
The Tc among 102 structures, obtained through tandem bioisosteric queries and the investigation of 5-HT6R and D2R ligand databases, ranged from 0.43 to 0.88. The obtained bioisostere 1d was active at both 5-HT6 and D2 receptors, validating the effectiveness of the designed bioisosteric query for creating a multi-receptor ligand (Table 2). Consequently, compound 1d was chosen as the lead for a SAR series (4a–4t).Bioisosteres 1e and 1f were found to be inactive at 5-HT6R, but they possessed moderate-to-low affinity for D2R (1e Ki = 1609 nM, 1f Ki = 110 nM) and high-to-moderate affinity for 5-HT1AR (1e Ki = 22 nM, 1f Ki = 101 nM). Interestingly, substitution of the phenyl group with cyclohexane (compounds 1b and 1e) resulted in a loss of activity at D2R and only a 2-fold decrease in affinity for 5-HT1AR. In contrast, substitution of the phenylpiperidine group with a biphenyl group (compounds 1b and 1f) resulted in a 2-fold increase in affinity for D2R and a 10-fold decrease in affinity for 5-HT1AR. This result suggests that aromatic interactions may be more important for ligand–receptor (L–R) interactions in D2R compared with 5-HT1AR.
Compared with the parent compound 1a, the hit 1d exhibited 3- to 15-fold lower affinity for all targets. However, the introduction of a fluorine and methyl at position 4 of the aniline aromatic ring (4c and 4d, respectively) resulted in compounds that possessed affinity comparable to the parent 1d for 5-HT6R (4c Ki = 23 nM, 4d Ki = 25 nM) and were 2-fold more active at D2R (4c Ki = 37 nM, 4d Ki = 33 nM).
An affinity similar to 5-HT6R was also obtained for compounds 4m and 4p (Ki = 24 and Ki = 21, respectively), but they were 2-fold less active at D2R compared with the parent compound. Compound 1a, in addition to its affinity for 5-HT6R and D2R, was also highly active at 5-HT7R and 5-HT2A (Ki = 8 and 29 nM, respectively) and moderately active at 5-HT1A (Ki = 217 nM). Only one compound bearing the 3-(N,N-dimethylamino)propyl and 4-fluorophenyl group (4h), achieved comparable affinity for 5-HT2A (Ki = 27 nM) but was less active at other targets. Compound 4m, in turn, was twice as active towards 5-HT7 receptor (Ki = 4 nM) as the parent 1a, together with a comparable affinity for 5-HT6R (Ki = 24 nM), but it exhibited 2- to 27-fold lower affinity for other targets. In addition, functional activity assays revealed that 4m possessed antagonistic properties at 5-HT6R and 5-HT7R, with the latter being comparable to the reference compound SB-269970 (Table 3). Analysis of structure–activity relationship in a series of 1d derivatives revealed a preference for halogens (chlorine, bromine and iodine), especially in the case of binding to 5-HT7R. It appears that there may be a specific site in the 5-HT7 receptor binding pocket that can form halogen bonds with a ligand. Among the three mentioned halogens, iodine forms the strongest halogen bonds.24 However, the iodine-substituted compound 4r exhibited lower affinity for 5-HT7R (Ki = 10 nM) than the chlorine-substituted compound 4m (Ki = 4 nM), although its affinity was higher than the bromine derivative 4q (Ki = 19 nM). This finding indicates that the putative halogen binding pocket may be large enough to accommodate chlorine but too small to fully accommodate larger halogens. Nevertheless, the halogen-bonding properties of iodine allowed it, in part, to counter this unfavourable steric hindrance and resulted in a higher affinity for 5-HT7R than for bromine. A similar observation was obtained for the binding to 5-HT6R; the chlorine-substituted compound (4m) belongs to derivatives with the highest 5-HT6R affinity, together with fluorine and methyl-substituted compounds (4c, 4d). However, in this case, compounds (4q, 4r) that were substituted with heavier halogens, bromine and iodine, exhibited an approximately 4-fold lower affinity (Ki = 90 nM and Ki = 79 nM, respectively) than compound 4m, suggesting that the putative halogen binding pocket in 5-HT6R is smaller than that in 5-HT7R.
The binding affinity data suggested that a similar halogen binding pocket might also be present in D2R. This hypothesis is supported by the observation that the introduction of a chlorine increases the affinity 3-fold compared with an unsubstituted hit 1d. However, introduction of a bromine and iodine resulted in a decrease in affinity, which might be explained by the potentially small volume of the putative halogen binding cavity. This hypothesis is supported by the observation that compounds containing small groups, such as methyl and fluorine (4d, 4c, respectively), showed the highest affinity for D2R (Ki = 33 nM and 37 nM, respectively) and that compound containing a 4-trifluoromethyl group (3f) displayed a lower affinity (D2R Ki = 274 nM) than those with a 4-methyl group (4d) – with van der Waals volumes equal to 39.8 and 21.6 Å3 for CF3 and CH3, respectively.25
To gain insight into the observed changes in affinity of the obtained series of compounds, they were all re-docked to homology models of 5-HT6, D2 and 5-HT7 receptors. The combination of QPLD from the Schrödinger Suite with MM-Generalized-Born/Surface Area (MM/GBSA) calculations was used to obtain L–R complexes because this approach is able to describe the anisotropy of the electron density around halogen atoms, which is a key feature during halogen bond examination. Only the top scored complexes, according to ΔG, were considered. The binding modes (Fig. 2) were proposed based on the mutual spatial arrangement of particular ligand fragments and 5-HT6, D2 and 5-HT7 receptor binding pockets. All of the obtained L–R complexes exhibited very consistent binding modes (within each receptor type), in line with the findings of Kooistra et al.26 and potentially associated with the similar binding pocket construction of these receptors (Table 4). In addition to the crucial charge-assisted hydrogen bond with Asp3.32, all of the docked ligands formed at least one specific aromatic interaction (CH–π or π–π stacking) with Phe6.51 or Phe6.52 residues. Additionally, the second aromatic ring of a ligand was usually targeted to Phe3.28 (D2R/5-HT7R) and Trp3.28 (5-HT6R).
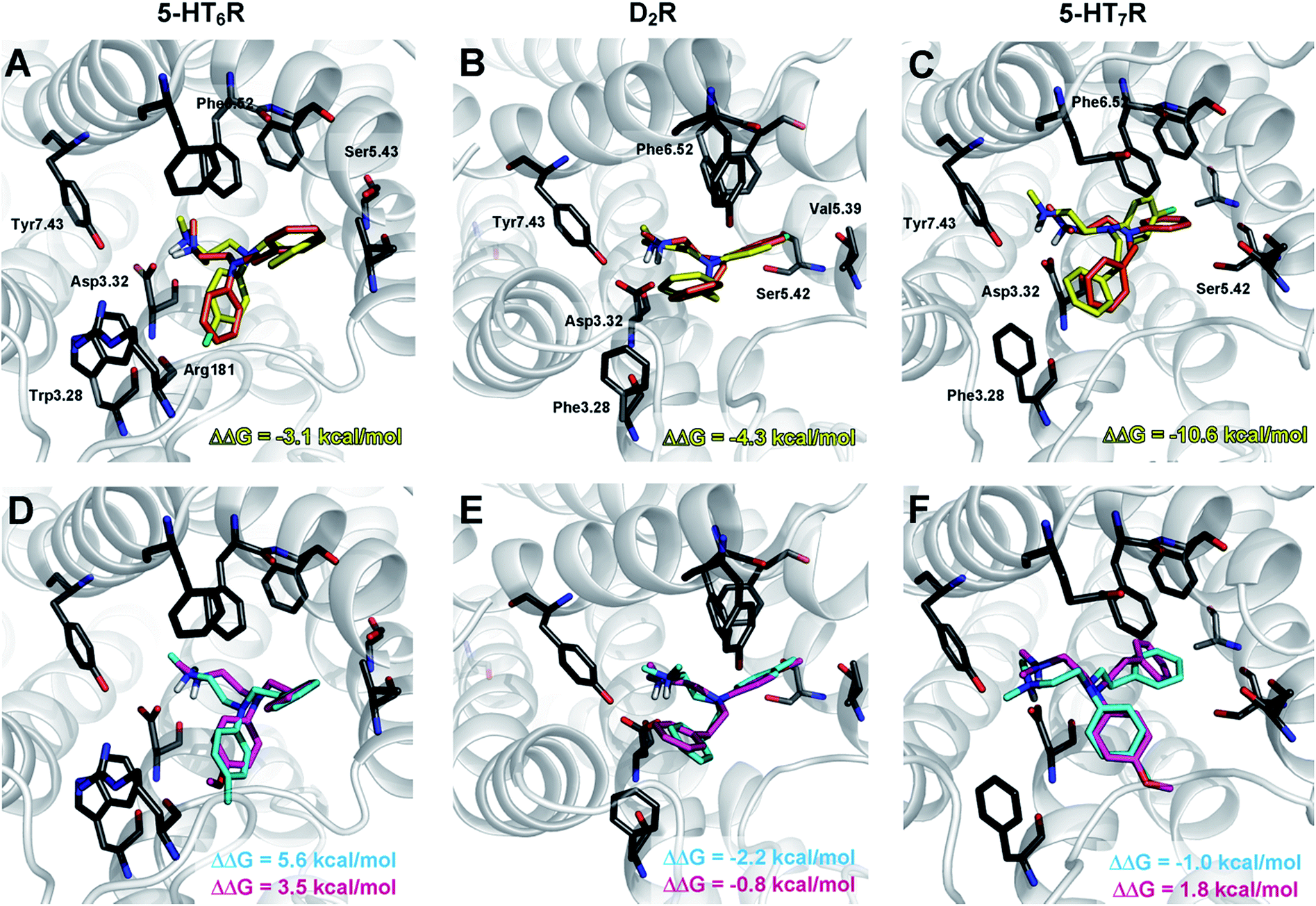 | ||
| Fig. 2 Representative L–R complexes (top scores based on ΔG) of selected ligands with 5-HT6, D2, and 5-HT7 receptors. Figures (A–C) show a comparison of docking poses for compounds 1d (orange) and 4m (yellow), whereas figures (D–F) show a comparison for compounds 4d (cyan) and 4f (magenta). Amino acids that were selected as crucial for the binding of the presented compounds are shown as sticks. A basic nitrogen atom forms a charge-assisted hydrogen bond with aspartic acid Asp3.32, an aniline aromatic ring (in the case of D2R and 5-HT7R complexes) interacts with Phe6.52, whereas in the case of 5-HT6R, the same interaction is formed by a phenylethyl moiety. The ΔΔG [kcal mol−1] value shows the difference between ΔG of complexes of a particular compound (4m, 4d, 4f) and an unsubstituted analogue 1d. In most cases, the ΔΔG values correspond to the binding affinity; the greatest energy gain was obtained by the introduction of a chlorine atom (4m, yellow), regardless of the receptor. | ||
The exchange of a methyl with a methoxy group in the para position resulted in a 9- (for 5-HT6R) to 35-fold (D2R) decrease in affinity. Analysis of the binding modes showed that the methoxy group occupied the same binding cavity as the methyl group (Fig. 2E), which formed many hydrophobic interactions that stabilized the L–R complex (e.g., Val5.39, Ser5.42, Phe6.52, and His6.55 in D2R). It should also be noted that for the methoxy substituent, no hydrogen bonding with side chain amino acids was detected (e.g., Ser5.42 in D2R), and thus it destabilized the L–R complex through unfavourable polar interactions.
The recognized halogen interaction appeared to possess a highly directional nature. To explain the 4- (D2R) to 88-fold (5-HT7R) decrease in activity by shifting the chlorine atom from position 4 to 3 in the phenyl ring, interaction spheres were plotted onto relevant backbone carbonyl oxygens (Fig. 3). In each case, the 4-chloro substituent was positioned within the energetically favourable areas of the sphere, whereas in the meta substitution, the 3-Cl atom pointed outside of the sphere, indicating that halogen bonding did not occur.
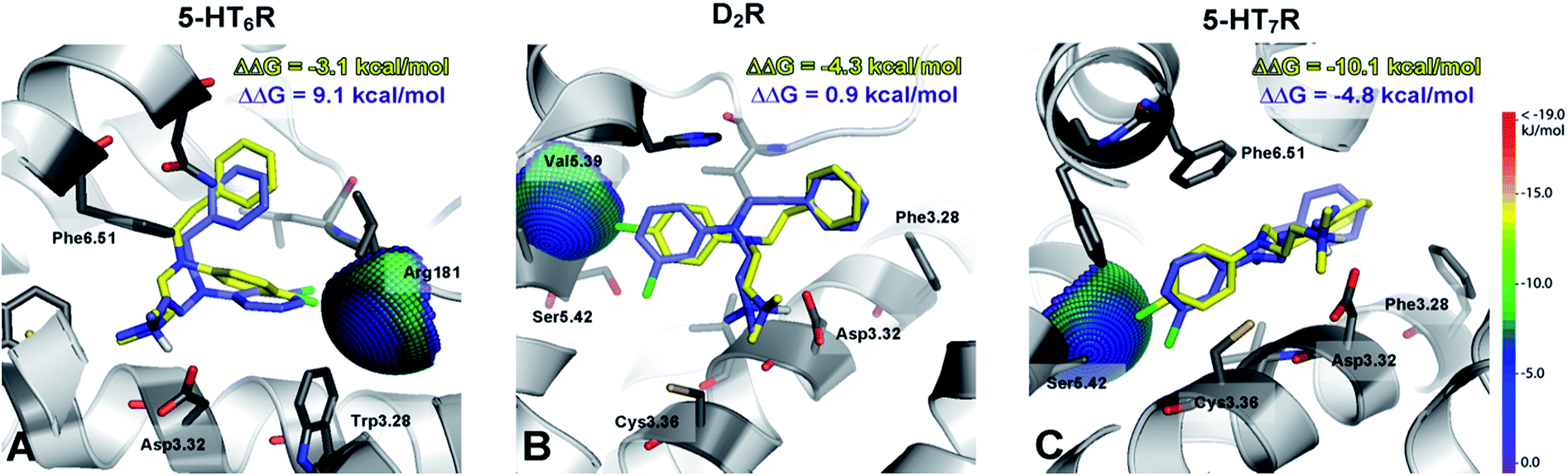 | ||
| Fig. 3 A superposition of the top scored poses of 4-Cl (4m, yellow) and 3-Cl (4t, blue) derivatives against putative halogen binding pocket interaction spheres. The ΔΔG [kcal mol−1] value shows the difference between the ΔG of complexes of a particular compound (4m, 4t) and an unsubstituted analogue 1d. The higher binding energy value for 3-Cl than for 4-Cl derivatives illustrates the highly directional nature of the identified halogen bond interaction. The methodology applied has been described by Wilcken et al.27 Geometrical parameters for the halogen bonds are: 5-HT6R d(Cl⋯O) = 3.7 Å, ∠(C–Cl⋯O) = 141°, D2R d(Cl⋯O) = 3.2 Å, ∠(C–Cl⋯O) = 126°, 5-HT7R d(Cl⋯O) = 2.9 Å, ∠(C–Cl⋯O) = 131°. | ||
Conclusions
An average hit rate for virtual screening to identify new compounds with affinity for a given biological target seldom exceeds 10%. The multi-step protocol applied herein differed from standard virtual screening by the introduction of additional bioisosteric derivatization steps and a similarity filter for the ligand of the second target. These modifications allowed us to obtain new chemical scaffolds that would not be identified in commonly utilized virtual screening techniques for commercially available compound databases. Among 102 structures that fulfilled all of the VS filters, only four were chosen for further evaluation. Among them, one compound (1d) possessed the desired dual 5-HT6R/D2R activity, two (1e, 1f; bioisosteres of the same parent) were inactive at 5-HT6R and the synthesis of one (1g) was unsuccessful. It is probable that the remaining screening hits might also exhibit activity similar to 1d, but their synthesis was determined to be more challenging.The hybrid QM/MM analysis performed for the series of 1d derivatives allowed to identify a putative halogen binding pocket in 5-HT6, D2 and 5-HT7 receptors. It must be stressed that halogen bonds in complexes with serotonin receptors have only recently been proposed in the case of 5-HT6R28 and 5-HT7R29 but with different anchoring points: the carbonyl oxygen of Pro4.60/Thr5.46 and Cys3.36(sulfur)/Thr3.37, respectively. Here, the characteristics of putative halogen binding pockets also differed between the investigated receptors (5-HT6, D2 and 5-HT7) because the affinity gain resulting from the introduction of the halogen atom was not equivalent and depended on the volume of the halogen binding cleft; the most profound gain was observed for the chloro derivative 4m, which resulted in a 30-fold increase in affinity for 5-HT7R (compared with the hit 1d).
Because the geometry of the recognized halogen bonds (see Fig. 3) is not optimal, they should be regarded rather as weak interactions. It has to be stressed, however, that receptor conformation is not optimized during Schrödinger QPLD protocol, thus detailed halogen bonding parameters (distance, sigma hole angle) and contribution of hydrophobic interaction would have required further studies including, e.g., full quantum mechanics optimization of L–R complexes.
Experimental
The 1H NMR spectra were recorded using a Bruker Avance III HD 400 NMR spectrometer. Mass spectra were recorded using a TQD Waters LC/MS spectrometer with electrospray ionization. Substrates and solvents were purchased from Sigma-Aldrich and Apollo Scientific company and used without further purification.Detailed synthetic, biomolecular and molecular modelling procedures are presented in ESI.†
General procedure 1 for the synthesis of N-[2-(dialkylamino)alkyl]anilines (3a–3u)
A 100 ml round bottom flask was charged with aniline (0.054 mol), appropriate (2-chloroalkyl)dialkylamine hydrochloride (0.018 mol), anhydrous potassium carbonate (0.11 mol) and isopropanol (50 ml). The reaction was maintained at reflux for 12 hours. After completion, the reaction mixture was filtered, the residuum was washed with isopropanol and the solvent was evaporated under reduced pressure. The product was purified by column chromatography on silica using ethyl acetate:methanol (9:1).
General procedure 2 for the synthesis of N-[2-(dialkylamino)alkyl]-N-(2-phenylethyl)anilines (1d, 4a–4t)
A 100 ml round bottom flask was charged with appropriate N-[2-(dialkylamino)alkyl]aniline (6 mmol), phenylacetaldehyde (12 mmol), triacetoxyborohydride (9 mmol) and dichloroethane (20 ml). The reaction was maintained at room temperature for 24 hours. After completion, distilled water (100 ml) was added, and the reaction mixture was extracted with dichloromethane (2 × 100 ml). The collected organic extracts were washed with brine, dried over anhydrous magnesium sulfate and evaporated under reduced pressure. The product was purified by column chromatography on silica using ethyl acetate:methanol (9:1).
Acknowledgements
The study was partially supported by the Polish-Norwegian Research Programme operated by the National Centre for Research and Development under the Norwegian Financial Mechanism 2009–2014 in the frame of the Project PLATFORMex (Pol-Nor/198887/73/2013) and by the National Science Center Grant No. DEC-2014/15/D/NZ7/01782.References
- G. Sliwoski, S. Kothiwale, J. Meiler and E. W. Lowe, Pharmacol. Rev., 2014, 66, 334–395 CrossRef PubMed.
- L. M. Lima and E. J. Barreiro, Curr. Med. Chem., 2005, 12, 23–49 CrossRef CAS PubMed.
- S. Schann, K. Pompermayer, J. Feldman, B. Pfeiffer, P. Renard, E. Scalbert, P. Bousquet and J. Ehrhardt, J. Med. Chem., 2001, 44, 1588–1593 CrossRef CAS PubMed.
- R. Morphy and Z. Rankovic, J. Med. Chem., 2005, 48, 6523–6543 CrossRef CAS PubMed.
- D. H. Kim, M. J. Maneen and S. M. Stahl, Neurotherapeutics, 2009, 6, 78–85 CrossRef CAS PubMed.
- J. L. Medina-Franco, M. A. Giulianotti, G. S. Welmaker and R. A. Houghten, Drug Discovery Today, 2013, 18, 495–501 CrossRef PubMed.
- J. Staroń, D. Warszycki, J. Kalinowska-Tłuscik, G. Satała and A. J. Bojarski, RSC Adv., 2015, 5, 25806–25815 RSC.
- A. Nikiforuk, Rev. Neurosci., 2014, 25, 367–382 CrossRef CAS PubMed.
- K. Fijał, P. Popik and A. Nikiforuk, Psychopharmacology, 2014, 231, 269–281 CrossRef PubMed.
- A. Wesołowska and A. Nikiforuk, Neuropharmacology, 2007, 52, 1274–1283 CrossRef PubMed.
- A. Quiedeville, M. Boulouard, V. Da Silva Costa-Aze, F. Dauphin, V. Bouet and T. Freret, Rev. Neurosci., 2014, 25, 417–427 CAS.
- A. Zhang, J. L. Neumeyer and R. J. Baldessarini, Chem. Rev., 2007, 107, 274–302 CrossRef CAS PubMed.
- N. Ye, J. L. Neumeyer, R. J. Baldessarini, X. Zhen and A. Zhang, Chem. Rev., 2013, 113, PR123–PR178 CrossRef PubMed.
- M. Kołaczkowski, M. Marcinkowska, A. Bucki, J. Śniecikowska, M. Pawłowski, G. Kazek, A. Siwek, M. Jastrzębska-Więsek, A. Partyka, A. Wasik, A. Wesołowska, P. Mierzejewski and P. Bienkowski, Eur. J. Med. Chem., 2015, 92, 221–235 CrossRef PubMed.
- W. A. Warr, J. Comput.-Aided Mol. Des., 2012, 26, 801–804 CrossRef CAS PubMed.
- R. Kurczab, M. Nowak, Z. Chilmonczyk, I. Sylte and A. J. Bojarski, Bioorg. Med. Chem. Lett., 2010, 20, 2465–2468 CrossRef CAS PubMed.
- I. Ujváry, Pestic. Sci., 1997, 51, 92–95 CrossRef.
- BROOD Fragment Replacement for Medicinal Chemistry, Version 1.1.2 and 2.0.0, OpenEye Scientific Software, Inc., Santa Fe, NM, USA, 2010 Search PubMed.
- A. A. Ivashchenko, et al., U.S. 0120792, 2010.
- Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions, Marvin 6.2.2, 2014, ChemAxon (http://www.chemaxon.com).
- Y.-C. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS PubMed.
- N. Upton, T. T. Chuang, A. J. Hunter and D. J. Virley, Neurotherapeutics, 2008, 5, 458–469 CrossRef CAS PubMed.
- J. J. Hagan, G. W. Price, P. Je, N. J. Deeks, T. Stean, D. Piper, M. I. Smith, N. Upton, A. D. Medhurst, D. N. Middlemiss, G. J. Riley, P. J. Lovell, S. M. Bromidge and D. R. Thomas, Br. J. Pharmacol., 2000, 130, 539–548 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- F. Leroux, ChemBioChem, 2004, 5, 644–649 CrossRef CAS PubMed.
- A. J. Kooistra, S. Kuhne, I. J. P. de Esch, R. Leurs and C. de Graaf, Br. J. Pharmacol., 2013, 170, 101–126 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, S. Zahn and F. M. Boeckler, J. Comput.-Aided Mol. Des., 2012, 26, 935–945 CrossRef CAS PubMed.
- B. Benhamú, M. Martín-Fontecha, H. Vázquez-Villa, L. Pardo and M. L. López-Rodríguez, J. Med. Chem., 2014, 57, 7160–7181 CrossRef PubMed.
- V. Canale, P. Guzik, R. Kurczab, P. Verdie, G. Satała, B. Kubica, M. Pawłowski, J. Martinez, G. Subra, A. J. Bojarski and P. Zajdel, Eur. J. Med. Chem., 2014, 78, 10–22 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08714k |
| This journal is © The Royal Society of Chemistry 2016 |