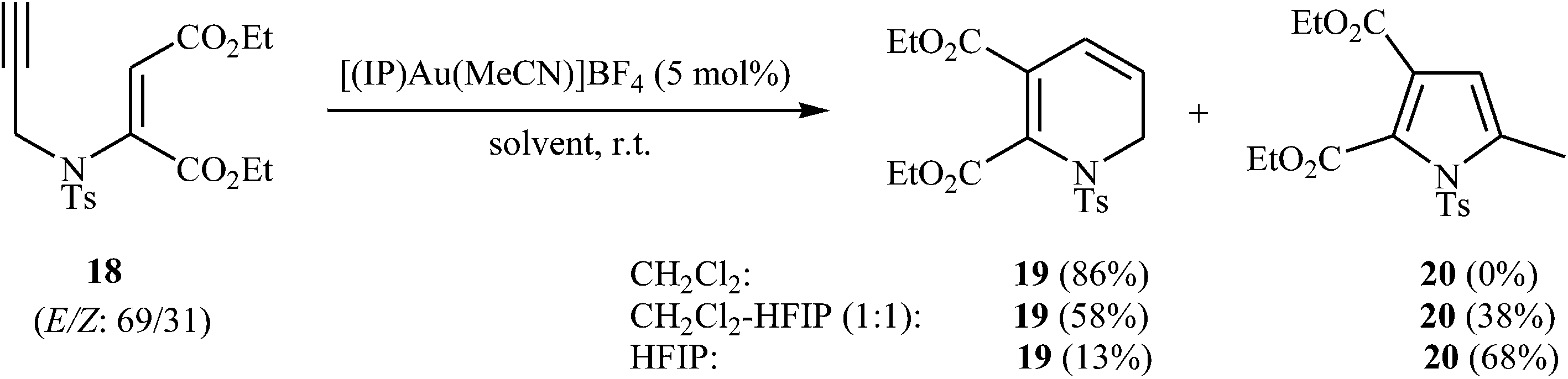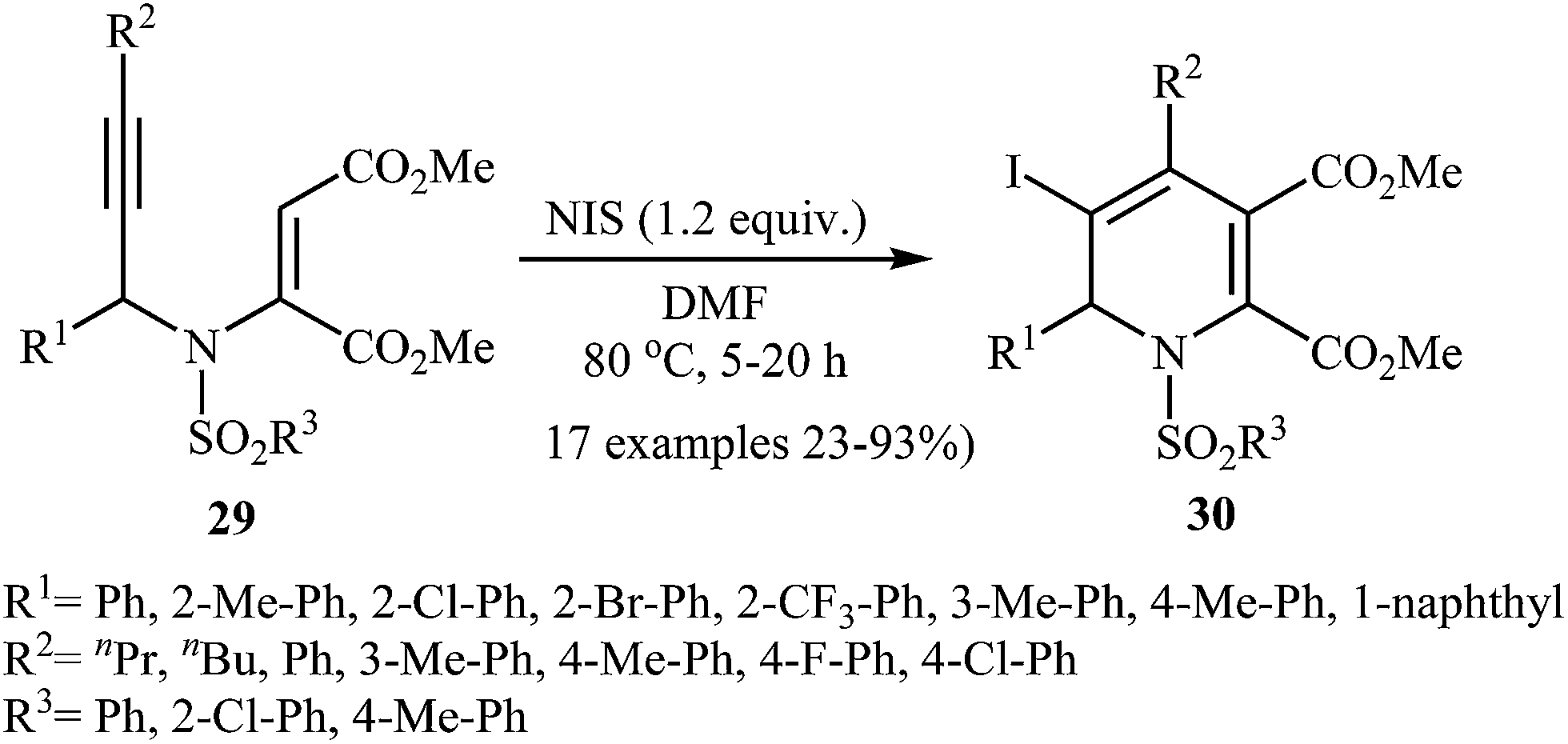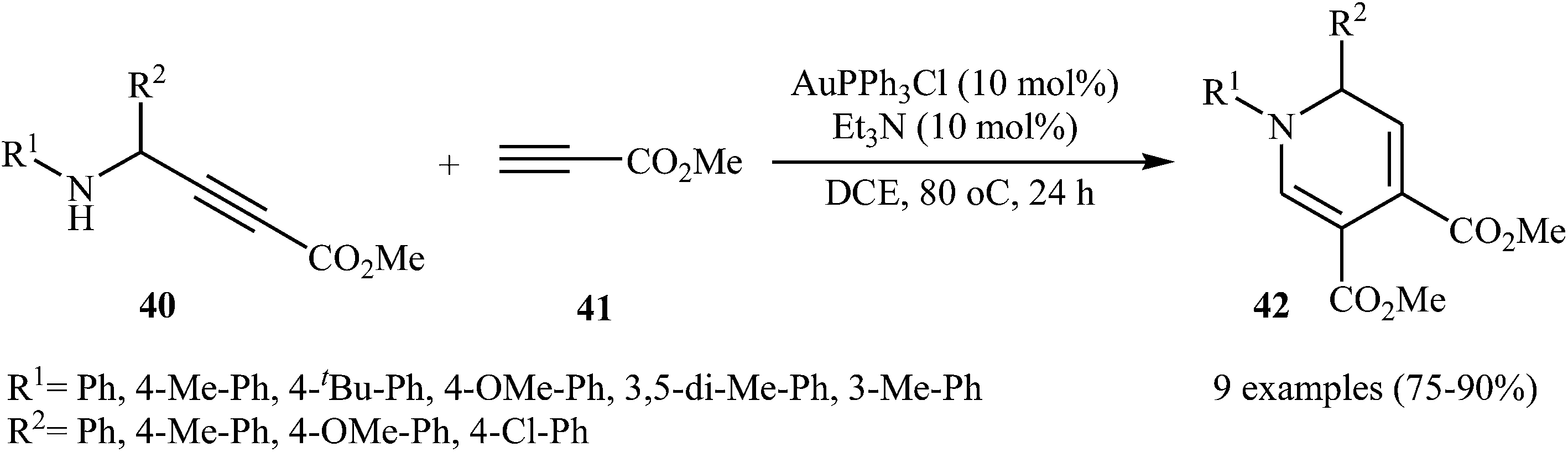DOI:
10.1039/C6RA08720E
(Review Article)
RSC Adv., 2016,
6, 71662-71675
New page to access pyridine derivatives: synthesis from N-propargylamines
Received
5th April 2016
, Accepted 24th June 2016
First published on 18th July 2016
Abstract
The pyridine is one of the simplest, yet most important N-heterocycles. This ring is utilized in more than 100 drugs and this number is increasing by the day. Therefore, synthesis of these compounds using new protocols is always interesting. This review surveys research work on the preparation of pyridines and their ring fused analogues from N-propargylamines. In this review, we have classified these reactions based on the type and the desired products.
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his Ph.D. degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as Associate Professor. His research interests include Theoretical Organic Chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in inorganic chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed his Ph.D. degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as Assistant Professor. Her research interests include inorganic and organic synthesis, new methodologies in nano material synthesis. |
 Ladan Edjlali | Ladan Edjlali was born in Tabriz, Iran, in 1960. She received her B.S. degree in Applied Chemistry from University of Tabriz, Iran, and her M.S. degree in organic chemistry from University of Tabriz, Tabriz, Iran, in 1993 under the supervision of Prof. Y. Mirzaei. She completed his Ph.D. degree in 2000 under the supervision of Prof. Y. Mirzaei and Prof. S. M. Golabi. Now she is working at Islamic Azad University, Tabriz Branch as Associate Professor. Her research interests include organic synthesis and new methodologies in organic synthesis. |
 Ahmadreza Bekhradnia | Ahmadreza Bekhradnia is associated professor at Mazandaran University of Medical Sciences, Iran. His current research interests focus on pharmaceutical intermediates, ranging from synthesis to study of biological and photophysical properties, as well as application of molecular modeling in mechanistic investigation. He received his PhD degree in organic chemistry from Tarbiat Modares University-Tehran, Iran (2005). During his sabbatical period, he worked on the mechanism of transition metal-catalyzed cross-coupling reaction, both from an experimental and computational viewpoint under the supervision of Prof. Per-Ola Norrby in Gothenburg University, Sweden (2012–2013). |
 Mehdi D. Esrafili | Mehdi D. Esrafili, was born in Shabestar, Iran, in 1981. He received his MS & Ph.D (honor) at Tarbiat Modares University. Then, he joined Kyoto University under the supervision of Professor Keiji Morokuma. Dr Esrafili is currently working as Head of Laboratory and working group on computational chemistry at University of Maragheh, Iran. His researches are centered on the intermolecular interactions and reaction mechanism using electronic structure methods. |
1. Introduction
N-Heterocycles are some of the most important building blocks in organic and medicinal chemistry.1 Among these, pyridines continues to play a vital role in the development of human medicines. This ring is utilized in nucleic acid bases and more than 100 drugs and this number is increasing by the day.2 Due to diversity of these analogs in the therapeutic response profile, many researchers have been working to explore synthetic routes for this skeleton. The most well-known methodologies for the synthesis of pyridine cores include the Bohlmann–Rahtz,3 Bönnemann,4 Kröhnke,5 and Hantzsch6 reactions. However, most methods are scarcely used due to the lack of generality or selectivity, the requirement of expensive metal catalysts, the production of harmful waste streams, the harsh reactions conditions involved, or poor yields.7 Therefore, the search for novel straightforward approaches for the synthesis of functionalized pyridines remains a challenge.
N-Propargylamines are one of the most important and versatile intermediates in organic synthesis. They are attractive starting materials for the synthesis of various nitrogen heterocycles.8 Recently, we published two review papers that cover the synthesis of pyrrole9 and quinoline10 derivatives from these compounds. Synthesis of pyridines from N-propargylamines provides a new page to titled compounds that in the most cases, provides milder conditions and simpler procedures than previously reported examples. In this review, we have classified these reactions based on the type (e.g. cyclization of N-propargyl enaminones, condensation of N-propargylamine and carbonyl compound, inter- and intra-molecular [2 + 2 + 2] cyclization of N-propargylamines) and the desired products (e.g. pyridines, hydropyridines, ring fused pyridines).
2. Pyridines
2.1. From N-propargyl β-enaminones
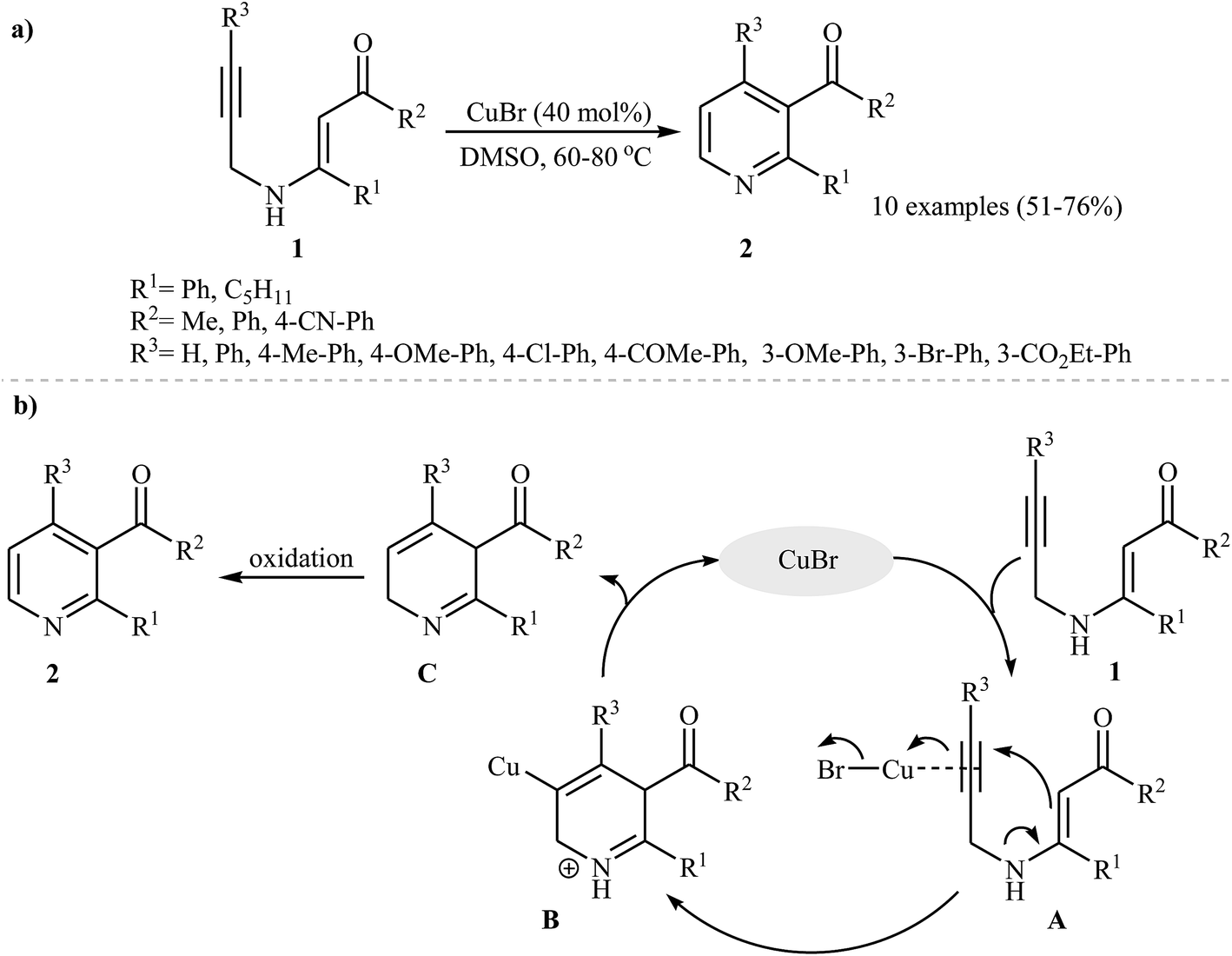
β-Enaminones are one of the most important and versatile intermediates in organic synthesis, because they exhibit a dual behavior as electrophiles and nucleophiles.11 They were successfully transformed into various N-heterocycles via inter and intramolecular reactions.12 The formation of pyridine derivatives can be accomplished by intramolecular cyclization of easily available13 N-propargylic β-enaminones. Pioneering work on this subject was developed by Cacchi and co-workers, those who carried out the Cu(I)-catalyzed intramolecular cyclization of a series of N-propargylic β-enaminones 1 under an argon atmosphere. They tested several catalysts and solvents, and the system CuBr/DMSO was found to be superior. Under optimized conditions, the reaction tolerates both electron-poor and electron-rich aryl rings in the alkyne terminus and gave the products 2 in moderate to good yields (Scheme 1a), whereas pyrroles were formed when using Cs2CO3 as catalyst at room temperature. The mechanism proposed to explain this reaction starts with the generation of the intermediate A via coordination of the alkyne moiety with copper, and then the 6-endo-dig cyclization of A furnishes intermediate B. Subsequently, substitution of the C–H bond for the C–Cu bond of intermediate B gives the intermediate C with concomitant regeneration of CuBr. Finally, the oxidation of C affords the observed products 2 (Scheme 1b).14
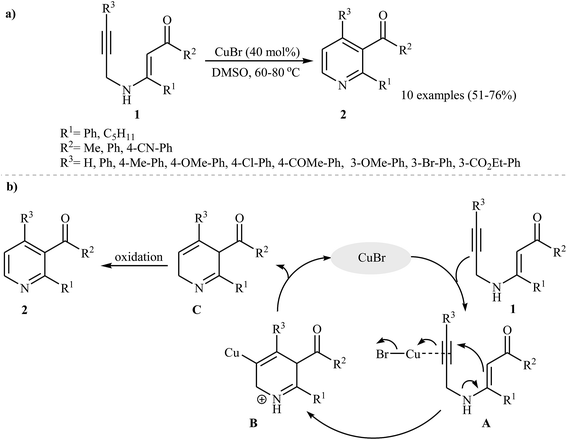 |
| | Scheme 1 (a) Synthesis of pyridine 2 via Cu-catalyzed intramolecular cyclization of N-propargylic β-enaminones 1; (b) proposed mechanism for formation of 2. | |
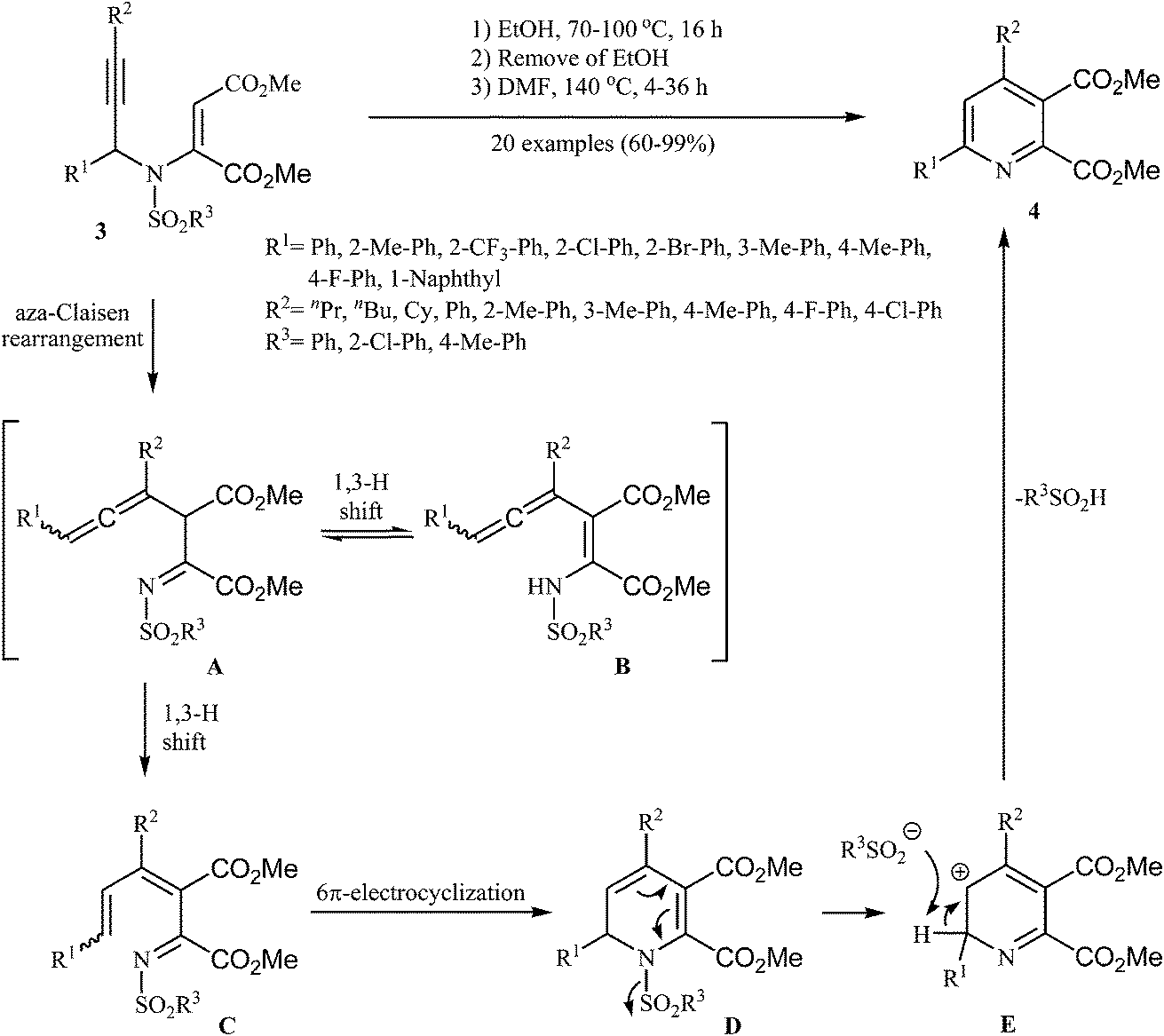
Later, Wan and co-workers extended this chemistry to an aza-Claisen rearrangement/6π-electrocyclization/elimination cascade of N-sulfonyl, N-propargylic β-enaminones 3 to highly substituted pyridines 4 by a one-pot three-steps reaction (Scheme 2). These steps involves: (i) heating of starting N-sulfonyl, N-propargylic β-enaminones 3 in ethanol at 70–100 °C for 16 h under an argon atmosphere; (ii) removing of the ethanol solvent under vacuum; and (iii) adding of DMF and heating of the reaction mixture at 140 °C for 4–36 h under an argon atmosphere. Under optimized conditions, the reaction tolerates electron-neutral, electron-poor and electron-rich aryl rings at R1, R2 and R3 and gave the corresponding pyridines 4 in good yields, but extension of the reaction to substrates with R1 = alkyl was failed. It is noted that the Z-isomer substrates does not work well in this protocol.15
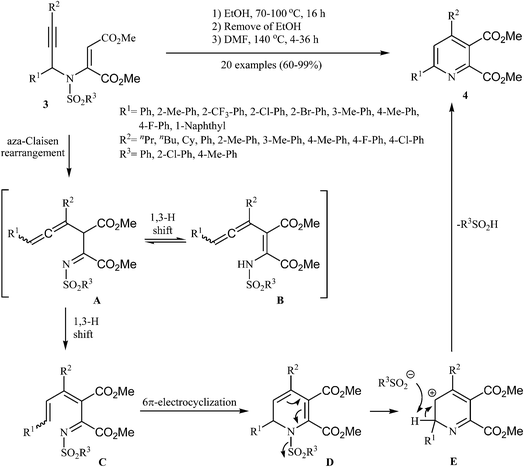 |
| | Scheme 2 Aza-Claisen rearrangement/6π-electrocyclization/elimination sequence to 4. | |
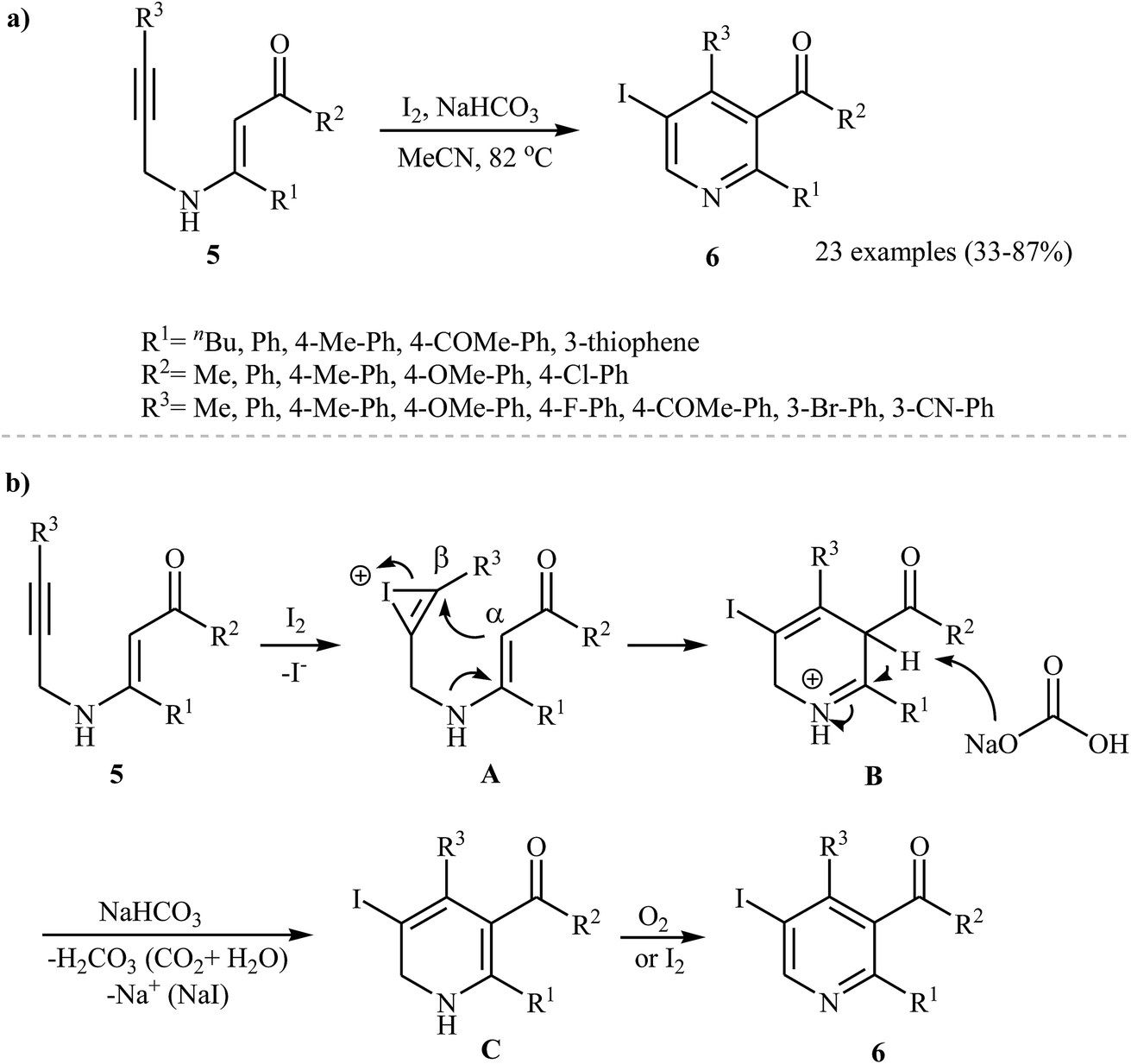
Recently, Karabiyikoglu, Kelgokmen, and Zora described a general and highly efficient synthesis of a wide variety of 5-iodopyridines 6 by the iodine-mediated electrophilic cyclization of corresponding N-propargylic β-enaminones 5 in the presence of sodium bicarbonate as a base. This process was run in refluxing acetonitrile, tolerated various functional groups (including fluoride, bromide, methoxy, cyano, and ketone), and in most cases, provided iodopyridines 6 in good to high yields (Scheme 3a). The mechanistic course of this reaction sequence is shown in Scheme 3b, and involves the initial formation of the iodonium intermediate A from the reaction of the alkyne moiety of starting β-enaminone 5 and iodine. The electrophilic cyclization of this intermediate gives intermediate B, which undergoes deprotonation with base to produce dihydropyridine C. Finally, oxidation of C with air O2 affords the observed pyridines 6.16 The applicability of the above cyclization products to the synthesis of more functionalized pyridines was demonstrated by transition-metal-catalyzed processes, such as Sonogashira and Suzuki-Miyaura cross-coupling reactions by the same research team.17
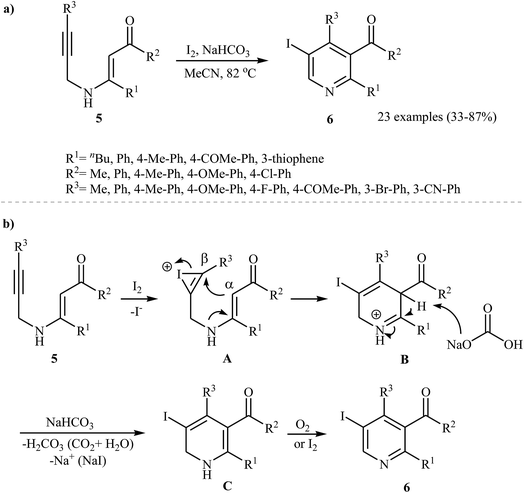 |
| | Scheme 3 (a) Synthesis of 5-iodopyridines 6 by the iodine-mediated electrophilic cyclization of corresponding N-propargylic β-enaminones 5; (b) proposed mechanism for formation of 6. | |
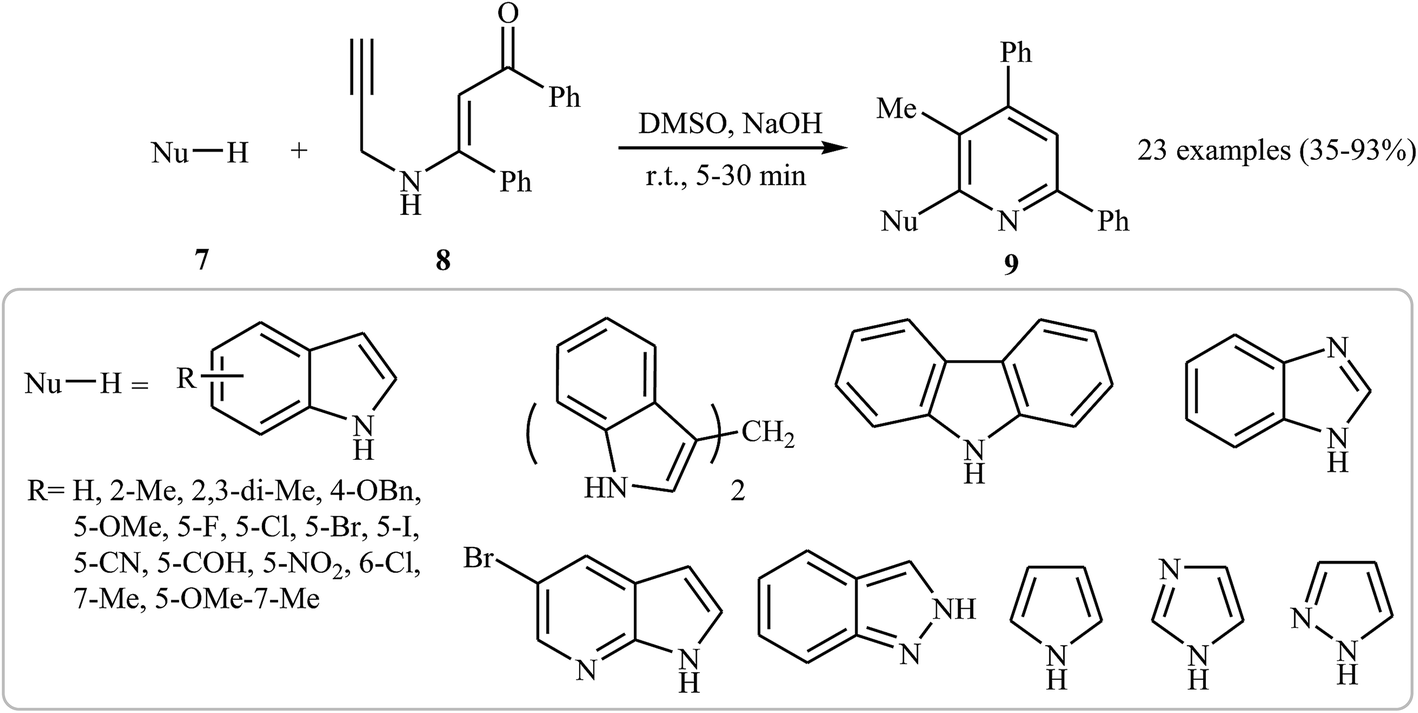
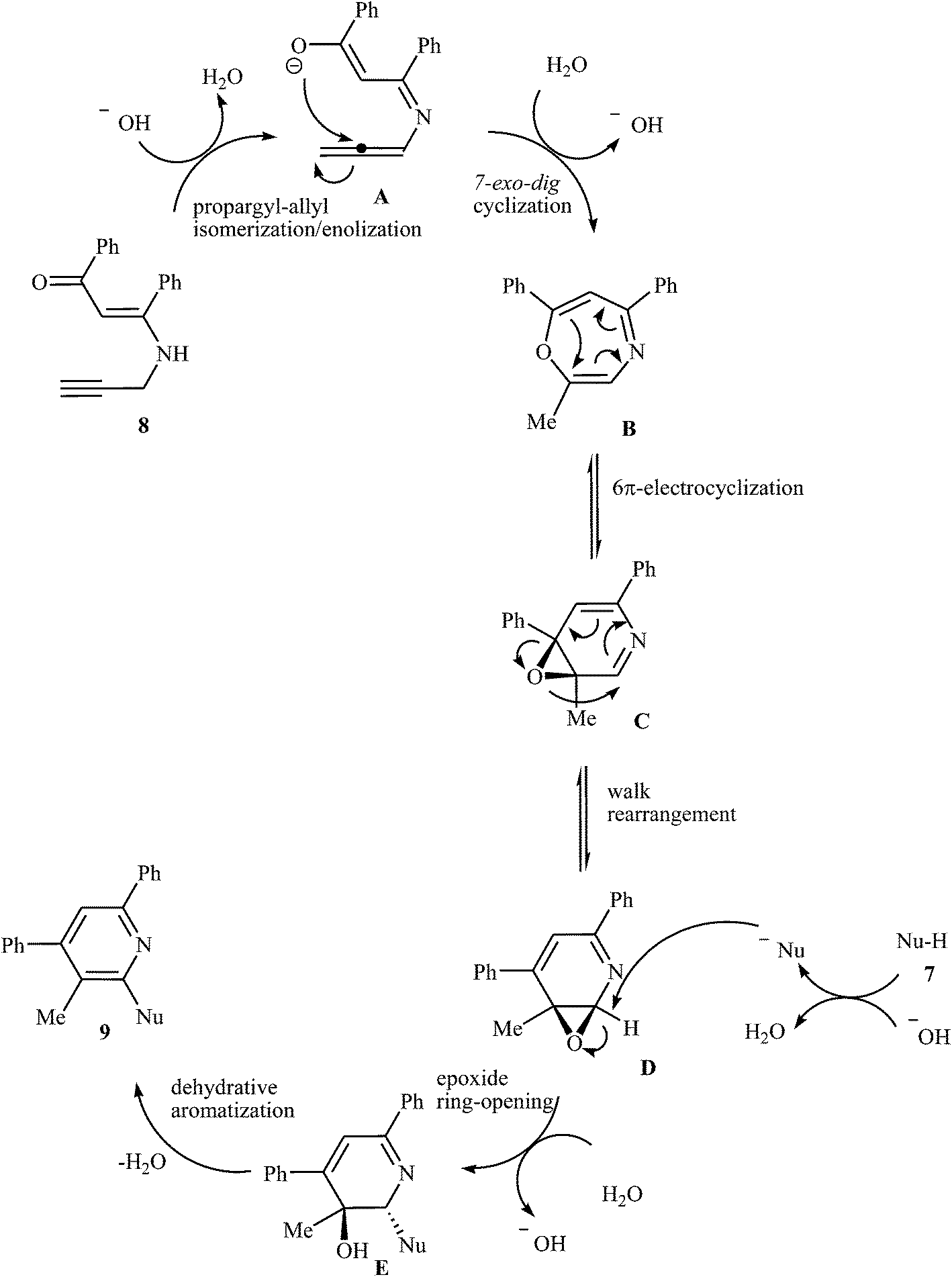
In 2015, an interesting and high efficiency strategy for synthesis of a variety of 2-(1-heteroaryl) pyridines 9 through a base-catalyzed N-pyridylation of N-heteroarenes 7 with N-propargyl β-enaminone 8 was reported by Cheng et al. This process allowed the formation of one pyridine core and one C–N bond in one pot, where water is only theoretical by-product. A diverse set of nitrogen nucleophilic source, such as indole carbazole, pyrrole, imidazole, pyrazole, and benzimidazole were tolerated in this procedure (Scheme 4). However, the reaction does not work with benzylamine, aniline, acetanilide, succinimide, and benzyl thiol. It is noted that the authors examined the scope of N-propargyl β-enaminones and found that alkyl groups cannot be used as substituent in the R1 and R2. The mechanism proposed for this transformation involves the formation of iminoenolate intermediates A by propargyl–allenyl isomerization/enolization cascade reaction of enaminones 8. This intermediate undergoes 7-exo-dig cyclization to form 1,4-oxazepines B. The formation of intermediate D occurs next via a 6π-electrocyclization/walk rearrangement cascade reaction, followed by its ring opening by substitution reaction with N-heteroarene to give a trans-2,3-dihydropyridine intermediate E that, after dehydrative aromatization, transforms to the final product 9 (Fig. 1).18
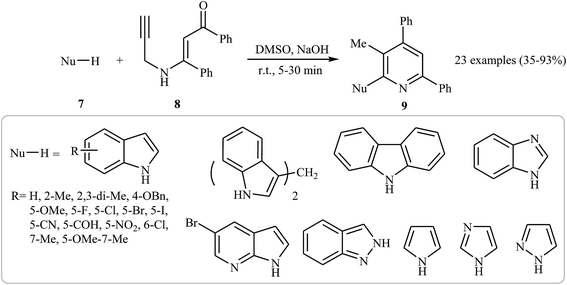 |
| | Scheme 4 One-pot synthesis of highly substituted pyridines 9 via base-catalyzed cascade reaction of N-heteroarenes 7 and N-propargyl β-enaminone 8. | |
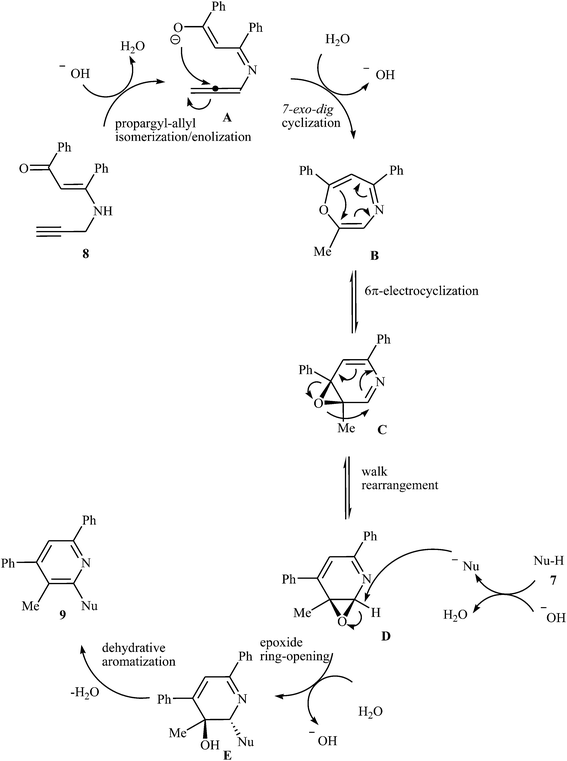 |
| | Fig. 1 Proposed reaction mechanism for synthesis of 9 from 7 and 8. | |
2.2. From N-propargylamine and ketones or aldehydes
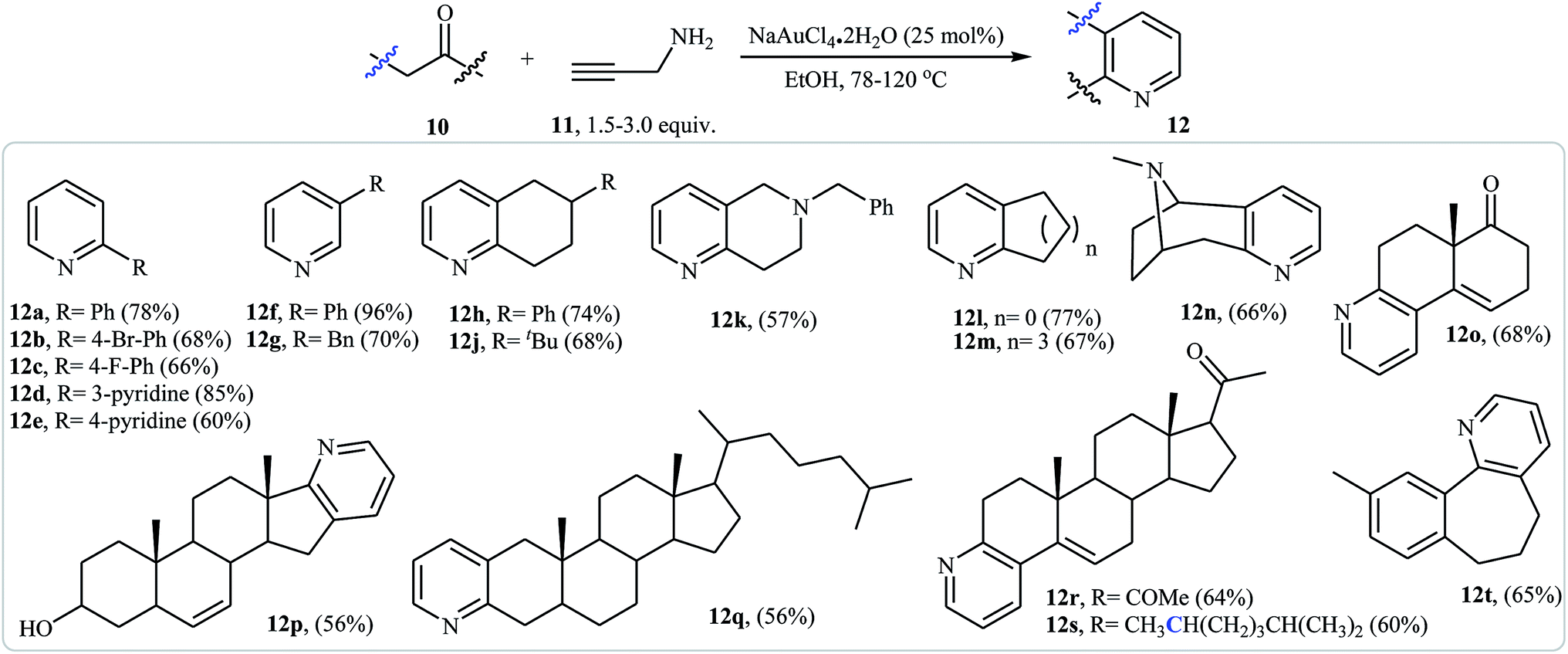
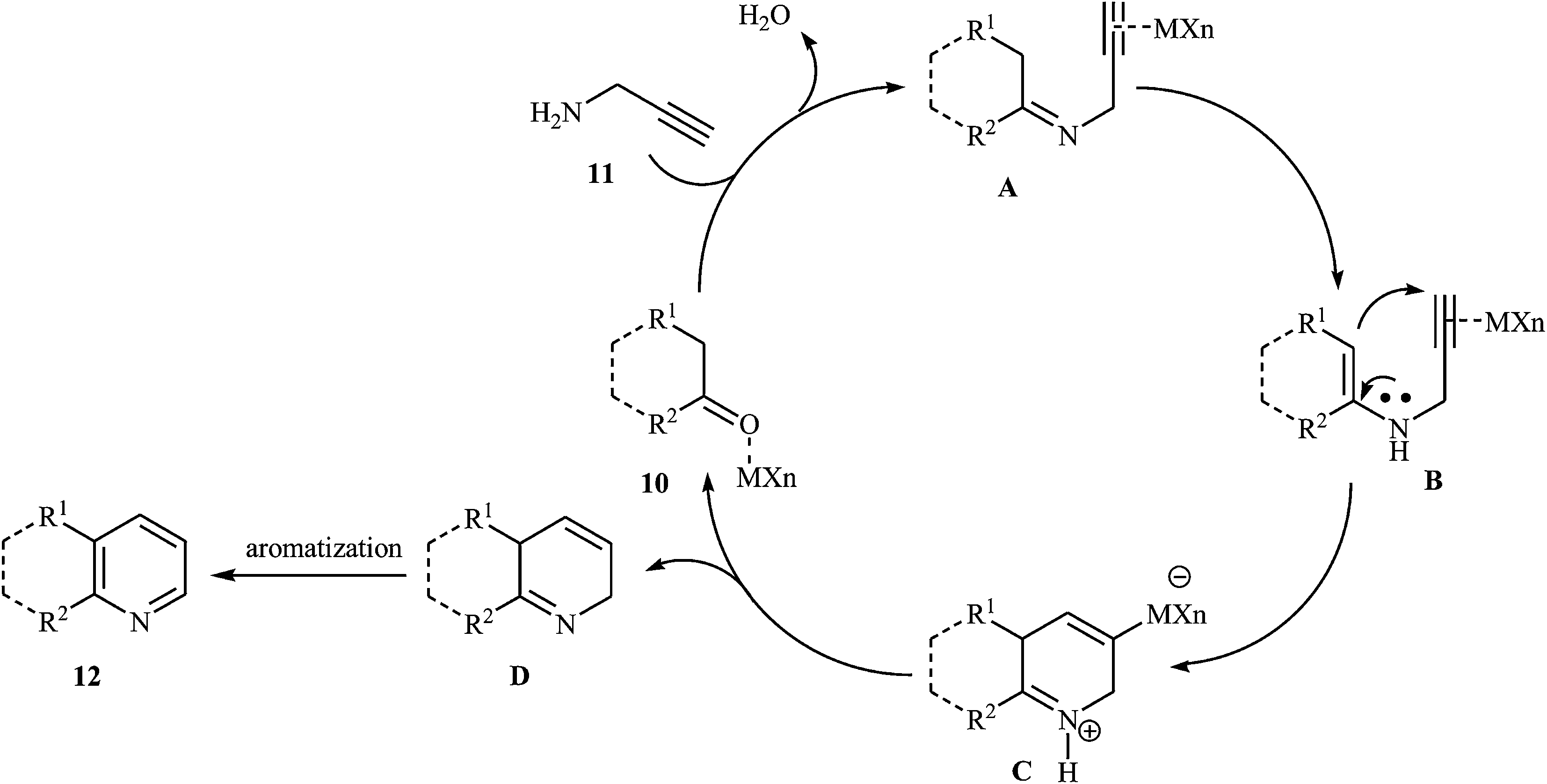
An interesting approach toward the synthesis of pyridine derivatives by treatment of carbonyl compounds with N-propargylamine in the presence of NaAuCl4·2H2O, was developed by Arcadi et al. Thus, a variety of functionalized pyridines 12 were synthesized via the Au-catalyzed amination/annulation/aromatization reaction of ketones or aldehydes bearing α-hydrogens 10 and N-propargylamine 11 in refluxing ethanol (Scheme 5). The mechanism proposed for this transformation involves the formation of the imino intermediate A by Au-catalyzed condensation reaction of the ketone 10 with N-propargylamine 11. This intermediate undergoes imine–enamine isomerization to form intermediate B. The formation of organometallic intermediate C occurs next, followed by its protonolysis to give the dehydropyridine D that transforms to the final product by a dehydrogenation process (Fig. 2).19
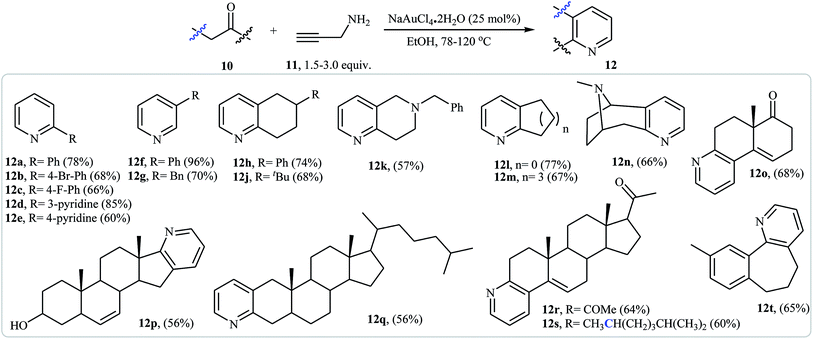 |
| | Scheme 5 Synthesis of disubstituted pyridines 12 from carbonyl compounds 10 and N-propargylamine 11. | |
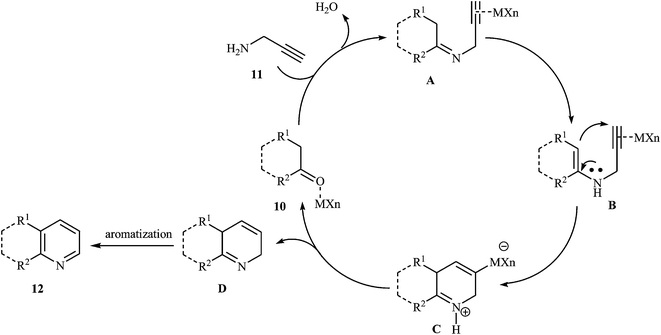 |
| | Fig. 2 Proposed mechanistic pathways for the formation of 12. | |
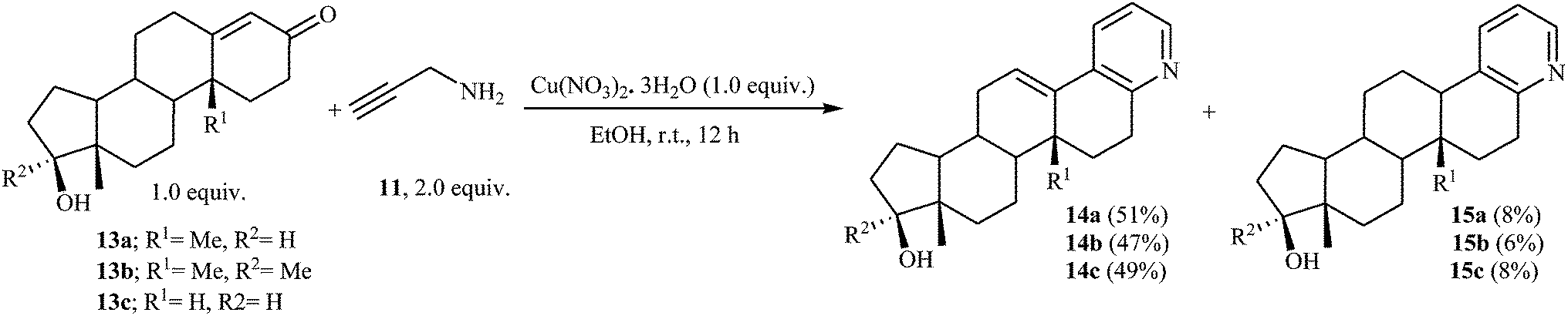
In a closely related investigation, Yan, Li, and Rao also described that the reaction of steroidal carbonyl compounds 13 with N-propargylamine 11 in presence of Cu(NO3)2·3H2O as catalyst, produced corresponding 3,4-fused pyridine compounds 14 in moderate yields (Scheme 6).20
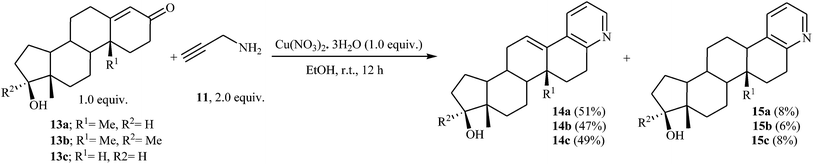 |
| | Scheme 6 Cu-Catalyzed synthesis of A-ring fused steroidal pyridines 14 via treatment of steroidal carbonyl compounds 13 with N-propargylamine 11. | |
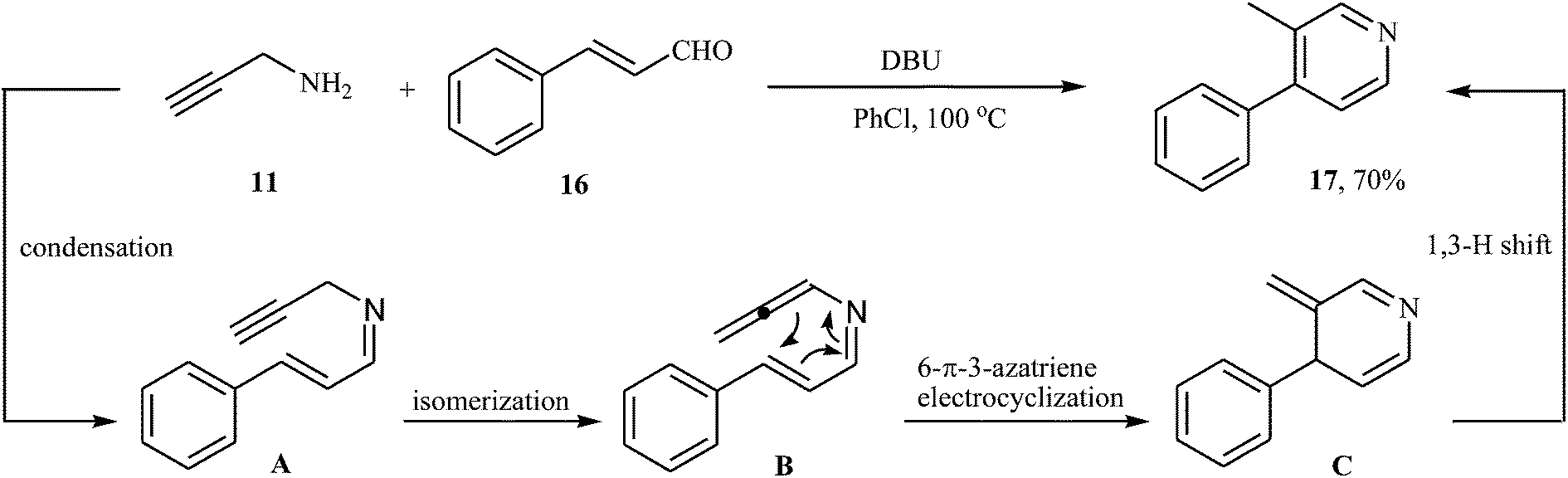
In 2015, H. Zhai and co-workers reported one example of pyridine preparation from direct condensation of N-propargylamine 11 and cinnamaldehyde 16 in the presence of DBU as base in PhCl. As shown in Scheme 7 the target pyridine 17 was obtained in yield of 70% via a condensation/isomerization/6-π-3-azatriene electrocyclization/1,3-H shift sequential process (Scheme 7).21
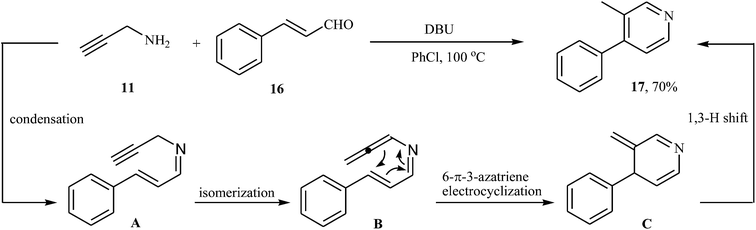 |
| | Scheme 7 Synthesis of pyridine 17 from direct condensation of N-propargylamine 11 and cinnamaldehyde 16. | |
3. Dihydropyridines
3.1. From N-propargyl β-enaminones
The dihydropyridines are an important building class of organic compounds and widely found in many biologically active natural or synthetic products.22 The first examples of transformation of N-propargyl β-enaminones to dihydropyridines were reported by the groups of Saito, in 2010, when they attempted to find the optimal conditions for the preparation of highly substituted pyrroles through intramolecular cyclization of N-propargyl β-enaminone derivatives. They showed that 1,2-dihydropyridine 19 were formed in high yield from enaminoester 18 through 6-endo-dig cyclization in simple process employing [(IP)Au(MeCN)]BF4 as catalyst in CH2Cl2 as solvent and at room temperature. Interestingly, this protocol showed different reaction behaviors depending on the employed solvent. By using HFIP (hexafluoroisopropanol) as co-solvent the yield of 19 is decreased in favor of the pyrrole 20 (Scheme 8).23
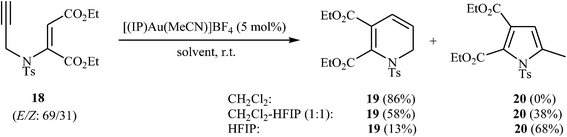 |
| | Scheme 8 Au-Catalyzed conversion of enaminoester 18 to 1,2-dihydropyridine 19 and pyrrole 20. | |
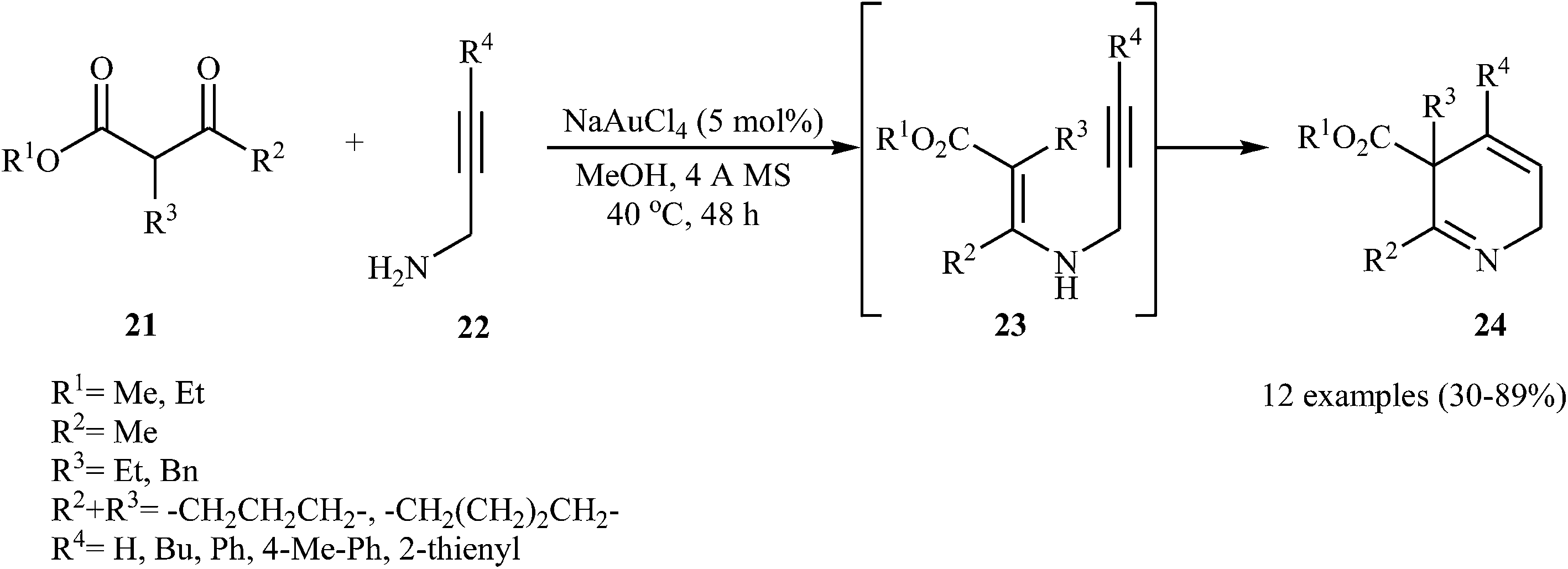
In 2011, Fañanás and co-workers reported an elegant study on the preparation of rare 2,5-dihydropyridines 24 by the reaction of various commercially available β-ketoesters 21 and N-propargylamines 22 in the presence of NaAuCl4 as catalyst in methanol. The authors suggested that the reaction proceeds via the formation of N-propargylic β-enaminoester intermediates 23 through condensation of 21 and 22, which after a 6-endo-dig cyclization process delivers the 2,5-dihydropyridine systems 24 in moderate to high yields (Scheme 9). They showed that the best results were obtained with N-propargylamines unsubstituted at the triple bond or with an aromatic ring at this position.24
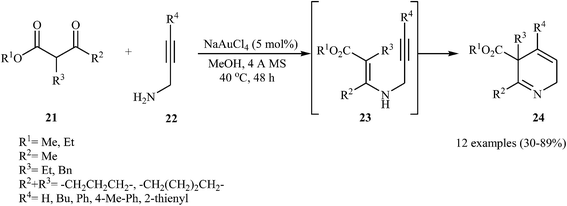 |
| | Scheme 9 Synthesis of 2,5-dihydropyridines 24 via Au-catalyzed reaction between β-ketoesters 21 and N-propargylamines 22. | |
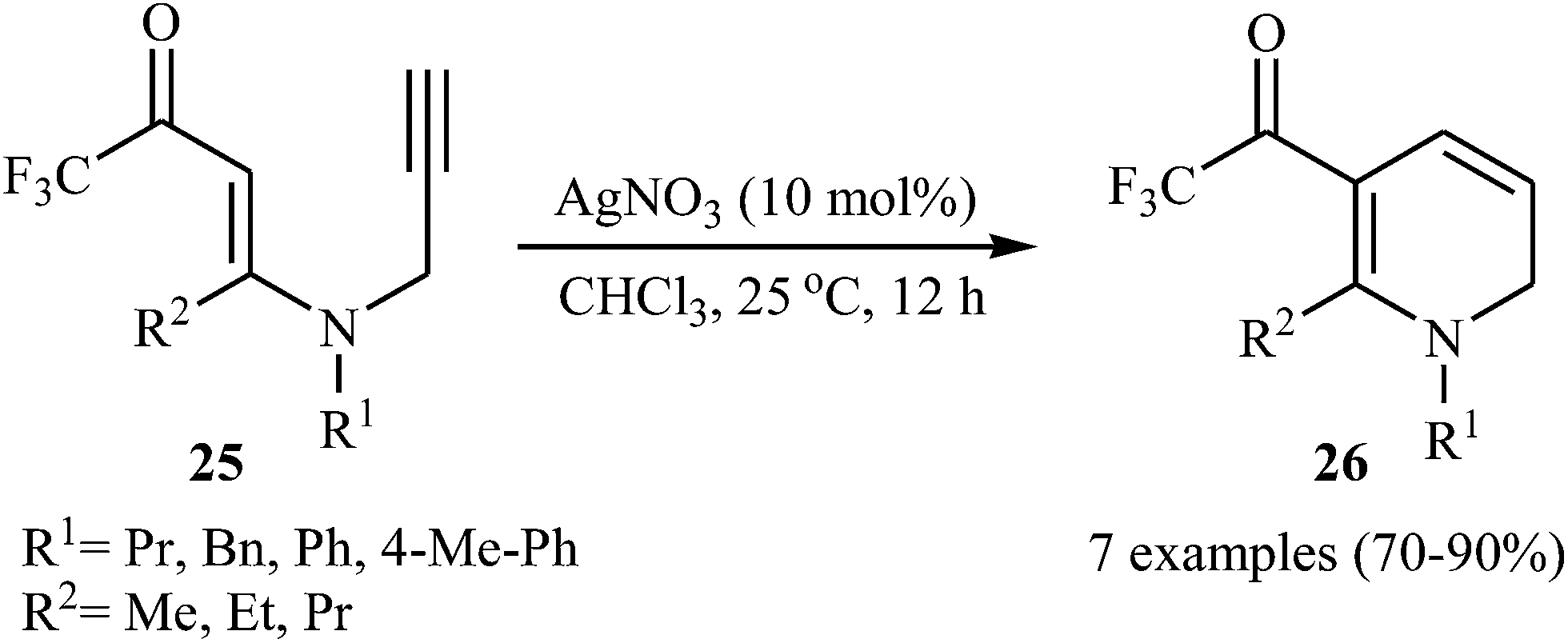
Subsequently, Martins's group studied the Ag-catalyzed cyclization of N-propargylic β-enaminones. Thus, a variety of trifluoromethylated 1,2-dihydropyridine 26 were synthesized in good to high yields from trifluoromethylated N-propargylic β-enaminones 25 through 6-endo-dig cyclization using the AgNO3/CHCl3 system under a nitrogen atmosphere at room temperature (Scheme 10).25
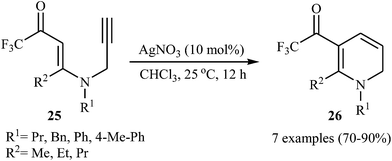 |
| | Scheme 10 Ag-Catalyzed 6-endo-dig cyclization of trifluoromethylated N-propargylic β-enaminones 25. | |
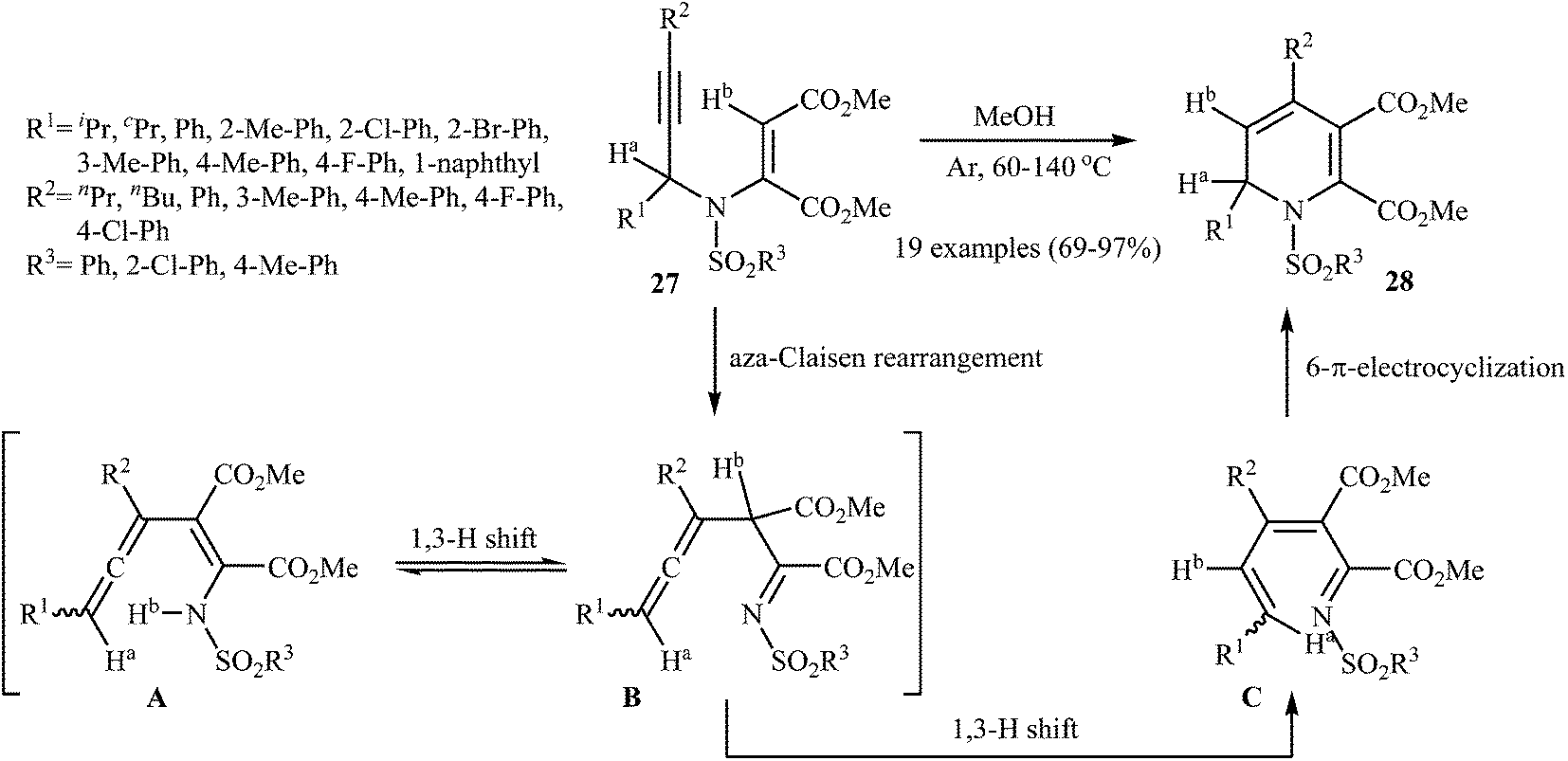
An interesting study about metal-free intramolecular cyclization of N-sulfonylated N-propargylic β-enaminones 27 in the preparation of 1,2-dihydropyridines 28, using methanol as solvent, was published by Wan and coworkers. The reaction tolerated the substrate with R1 and R2 being both aryl and alkyl groups. It was also found that the electronic character of the sulfonyl group (R3) had little effect on the facility of reaction. They suggested that the formation of 28 occurs via an aza-Claisen rearrangement followed by a sequential 1,3-H shift and 6π-electrocyclization by the mechanism described in Fig. 3. It is noted that this process can also be scaled up to provide multigram quantities of the desired product without sacrificing the yield or outcome of the methodology.26
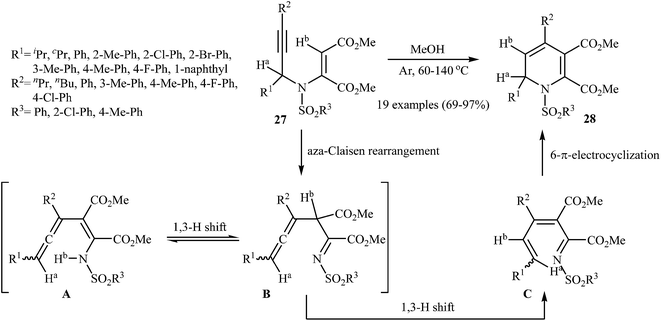 |
| | Fig. 3 Metal-free cyclization of N-sulfonylated N-propargylic β-enaminones 27 to 1,2-dihydropyridines 28. | |
The same authors also developed a new method for synthesis of iodo-substituted 1,2-dihydropyridine derivatives. They showed that N-propargylic β-enamiones 29 in the treatment with N-iodosuccinimide (NIS) in DMF at 80 °C, underwent electrophilic iodocyclization to afford 3-iodo-1,2-dihydropyridines 30 in moderate to high yields (Scheme 11). However, the reaction does not work with substrates having alkyl groups on the carbon bearing the nitrogen atom.26a
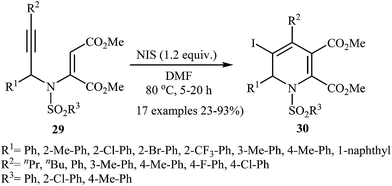 |
| | Scheme 11 Synthesis of fully substituted pyridines 30 via iodo-cyclization of N-propargylic β-enamiones 29. | |
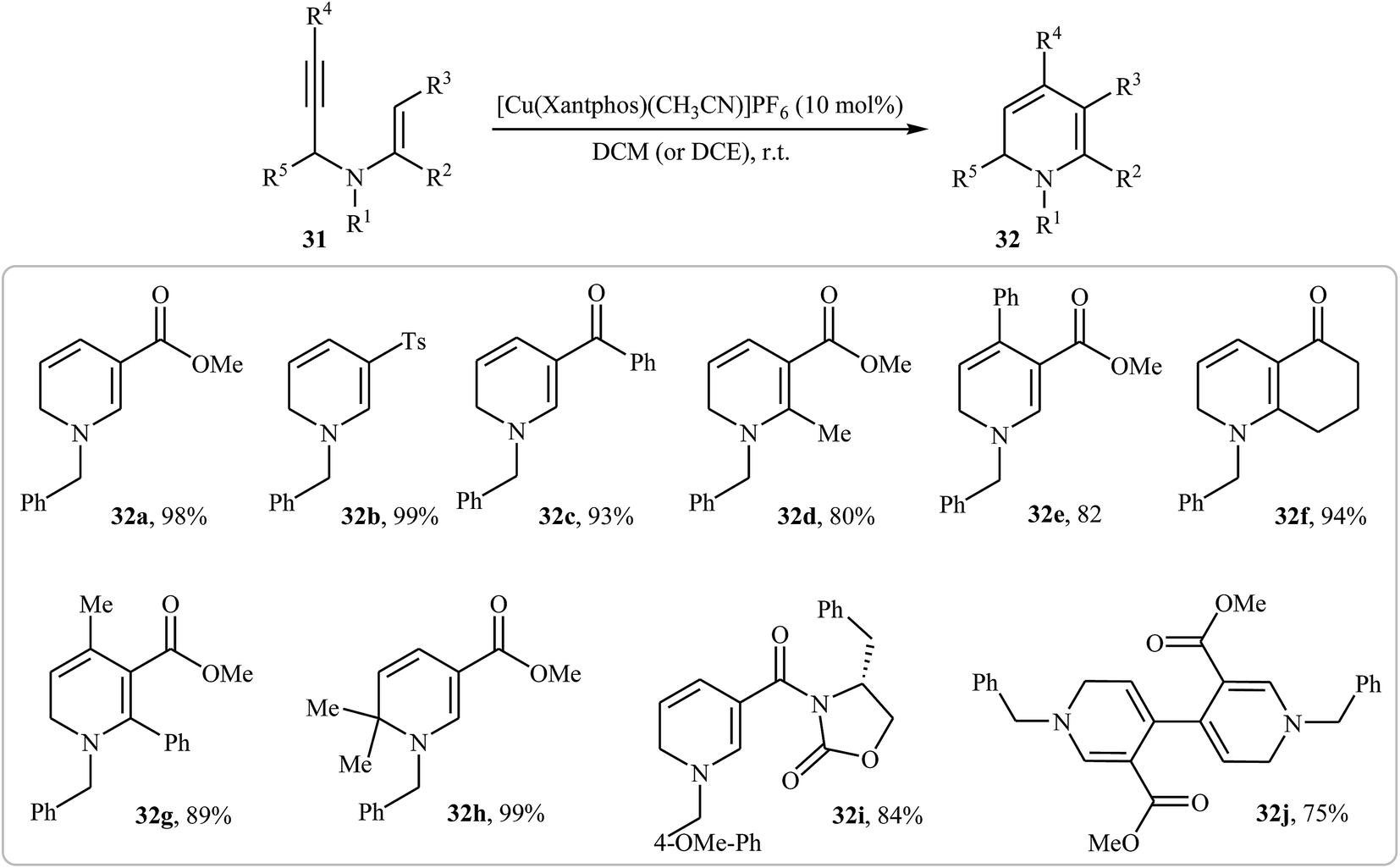
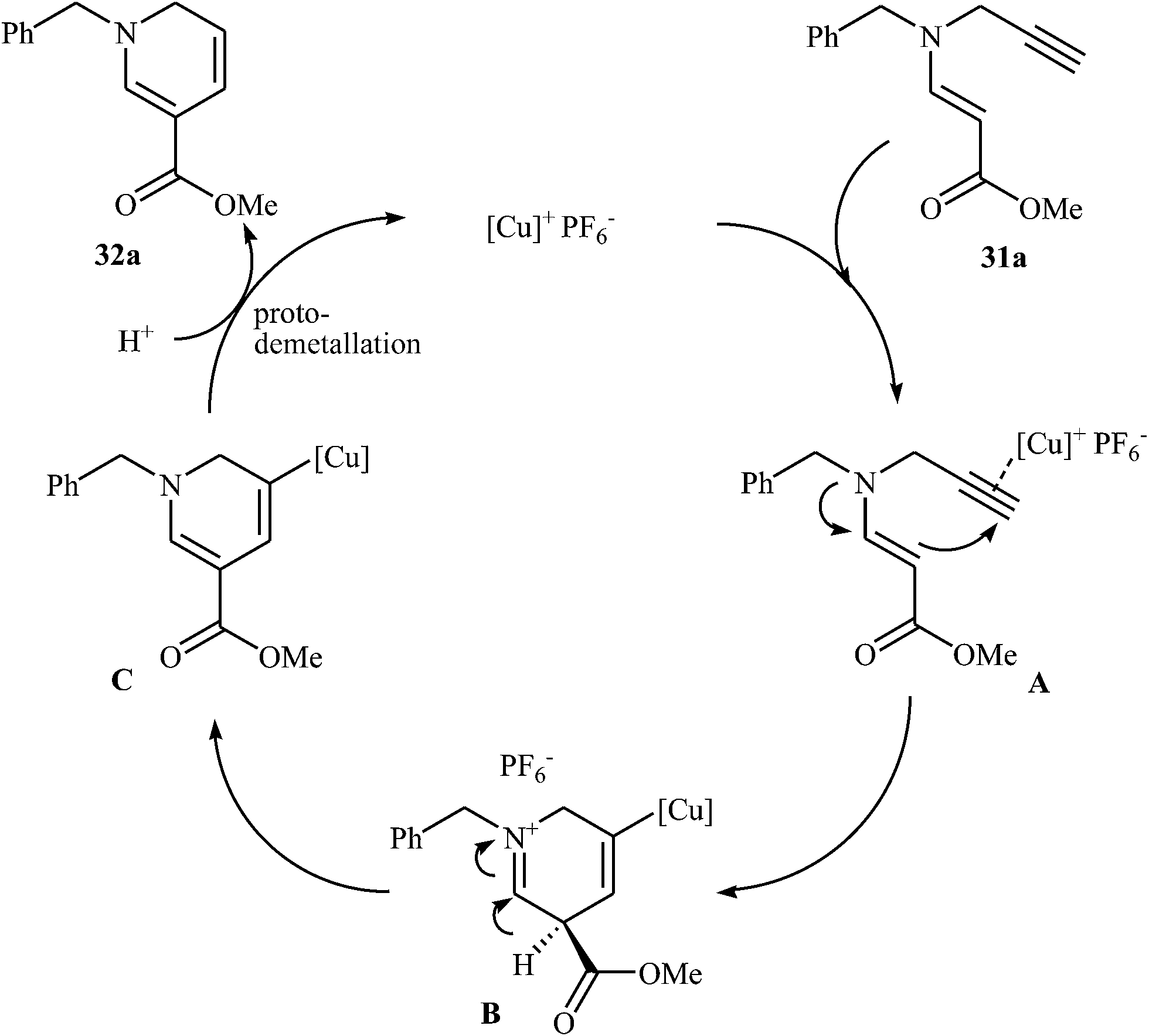
More recently, Oguri and co-workers showed that a series of multiply substituted 1,6-dihydropyridines 32 were formed from N-propargylic β-enaminocarbonyls 31 through 6-endo cyclization employing [Cu(Xantphos)(CH3CN)]PF6 as catalyst, in CH2Cl2 at room temperature (Scheme 12). According to mechanistic studies, it proceeds through the coordination of Cu(I) to the triple bond of 31 to form the electrophilic π-complex A, following the intramolecular nucleophilic attack of pendate enamine onto the activated triple bond to give ionic intermediate B, which undergoes a deprotonation and protodemetallation sequence to produce the observed 1,6-dihydropyridines 32 (Fig. 4).27
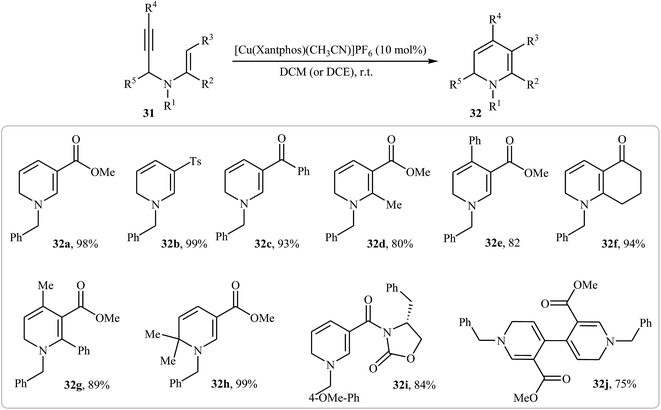 |
| | Scheme 12 Synthesis of multiply substituted 1,6-dihydropyridines 32 through 6-endo cyclization of N-propargylic β-enaminocarbonyls 31. | |
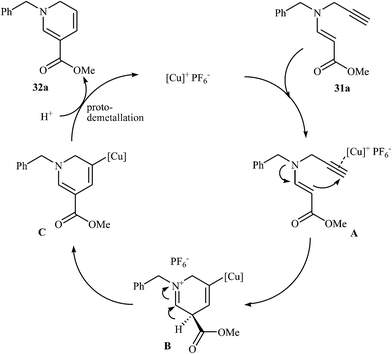 |
| | Fig. 4 Proposed mechanistic pathways for the formation of dihydropyridines 32. | |
3.2. From N-vinylpropargylamines
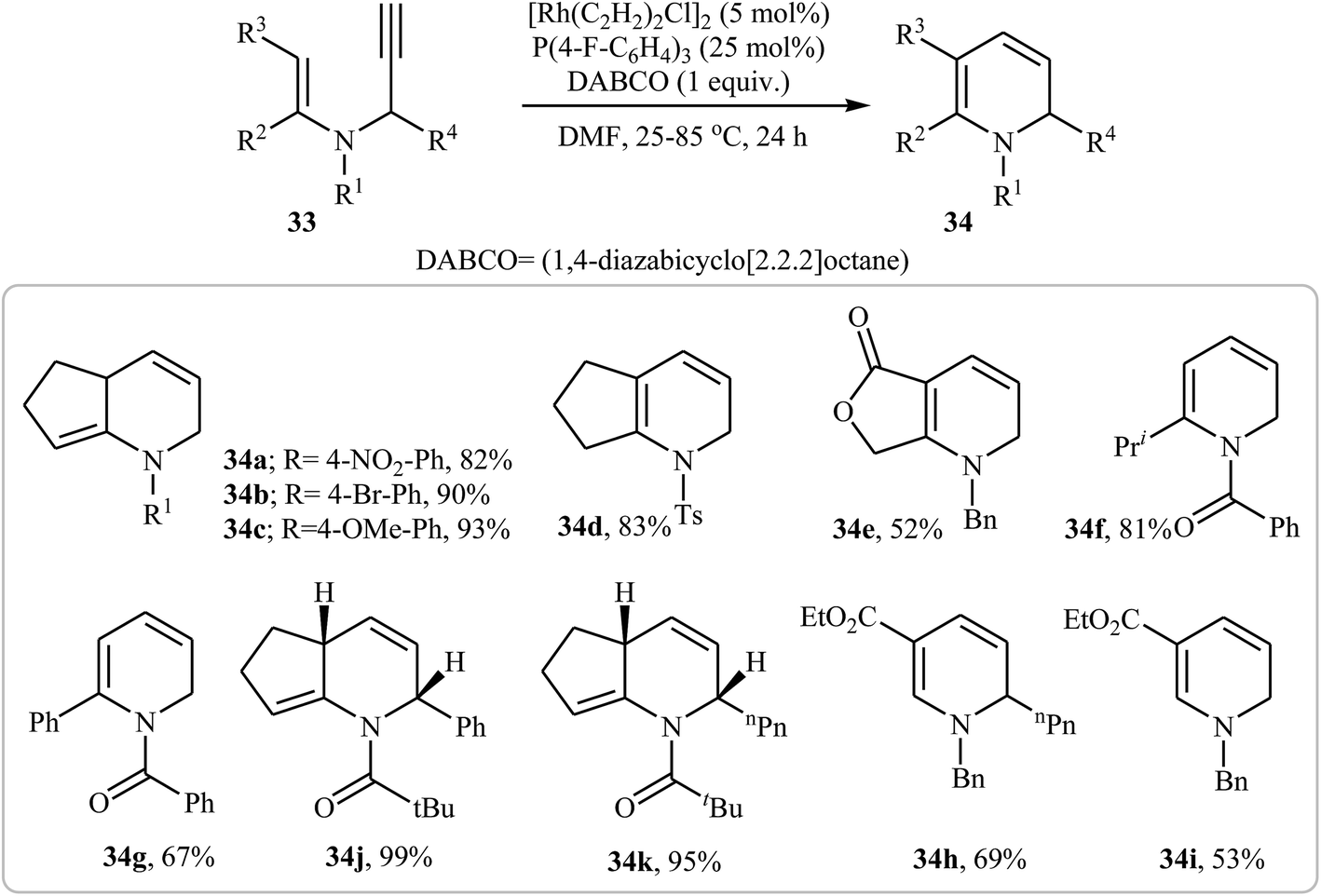
In 2006, Kim and Lee studied the possibility of synthesizing dihydropyridines 34 from N-vinylpropargylamines 33 through an Rh-catalyzed cycloisomerization process. Thus, the careful analysis of the optimized reactions revealed that the optimum condition for this cycloisomerization was the addition of [Rh(C2H2)2Cl]2 (5 mol%), P(4-F-C6H4)3 (25 mol%), and DABCO (100 mol%), at room temperature, to a solution of N-vinylpropargylamines in DMF (0.1 M). The optimized protocol tolerated a variety of functional groups, such as nitro, bromo, ketone, and methoxy, and generally provided corresponding dihydropyridine 34 in good yields (Scheme 13).28
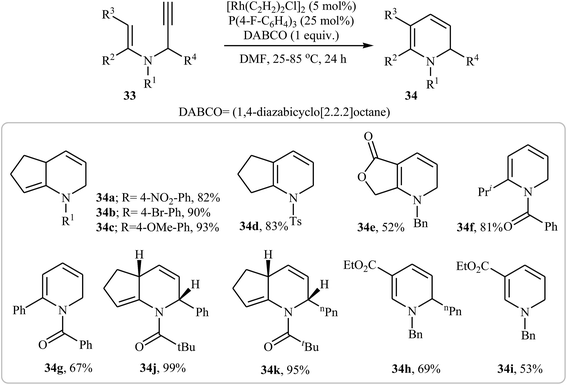 |
| | Scheme 13 Synthase of dihydropyridines 34 from N-vinylpropargylamines 33 through Rh-catalyzed cycloisomerization process. | |
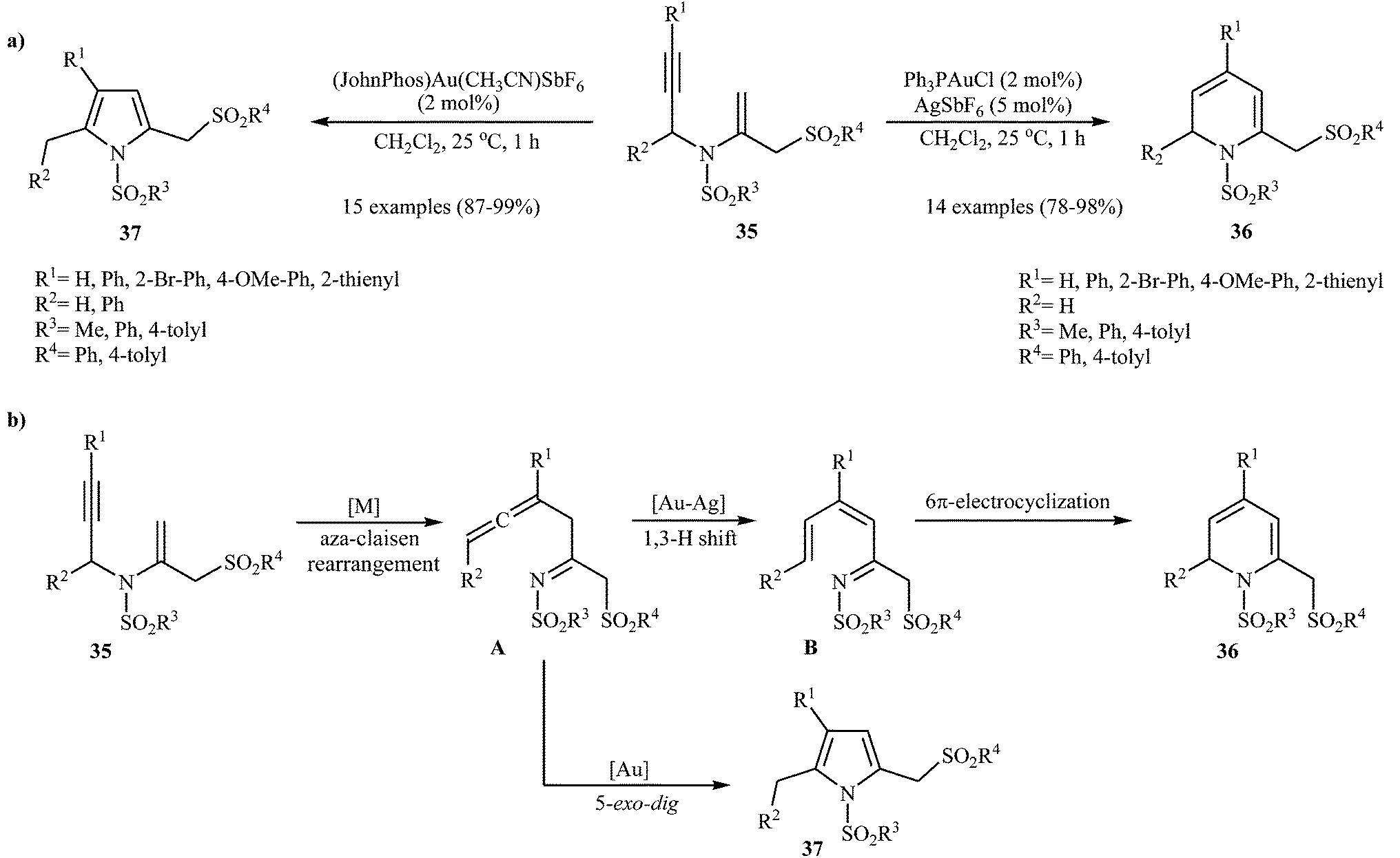
Pyridinium-1-sulfonate was applied to selective oxidation of several versatile compounds.29a,b Recently, Menon et al. reported that N-sulfonylated 2-sulfonylmethyl-1,2-dihydropyridines 36 could be prepared via the Ag–Au-catalyzed cycloisomerization of the corresponding N-vinylpropargylamines 35 (Scheme 14a). However, the authors reported a limitations in their methodology when they attempted to react substrate having a phenyl substituent at the carbon bearing the nitrogen atom. In this case, they only observed the pyrrole product, with no formation of desired product. It should be noted that when the reaction was carried out in the presence of Echavarren's gold(I) catalyst, it led to corresponding pyrroles 37 as sole products. The following reaction pathway has been proposed: the process is initiated by metal-catalyzed aza-Claisen rearrangement of 35 to give β-allenyl imine A (gold-mediated 5-exo-dig cyclisation of A and subsequent aromatisation via hydrogen shift affords the pyrrole derivative 37), which after tautomerization generates aza-triene B. Subsequent 6-π-aza-electrocyclization gives 1,2-dihydropyridines 36 (Scheme 14b).29c
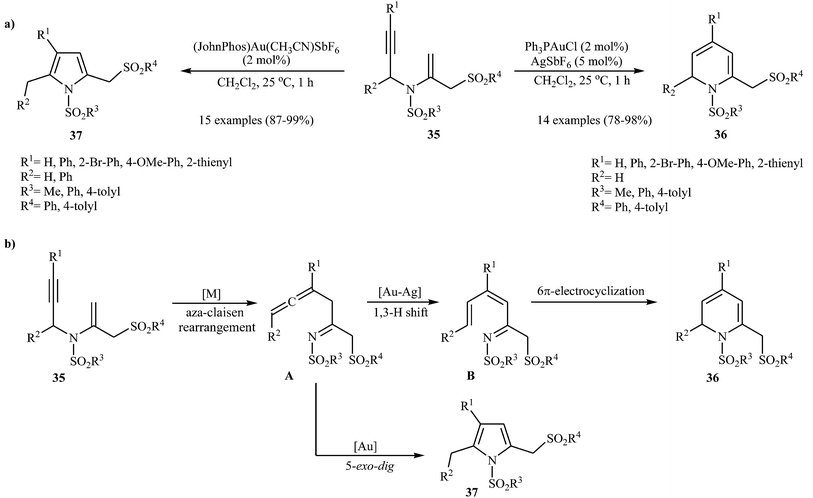 |
| | Scheme 14 (a) Au-Catalysed synthesis of 2-sulfonylmethyl dihydropyridines 36 and pyrroles 37; (b) proposed mechanism for formation of 36 and 37. | |
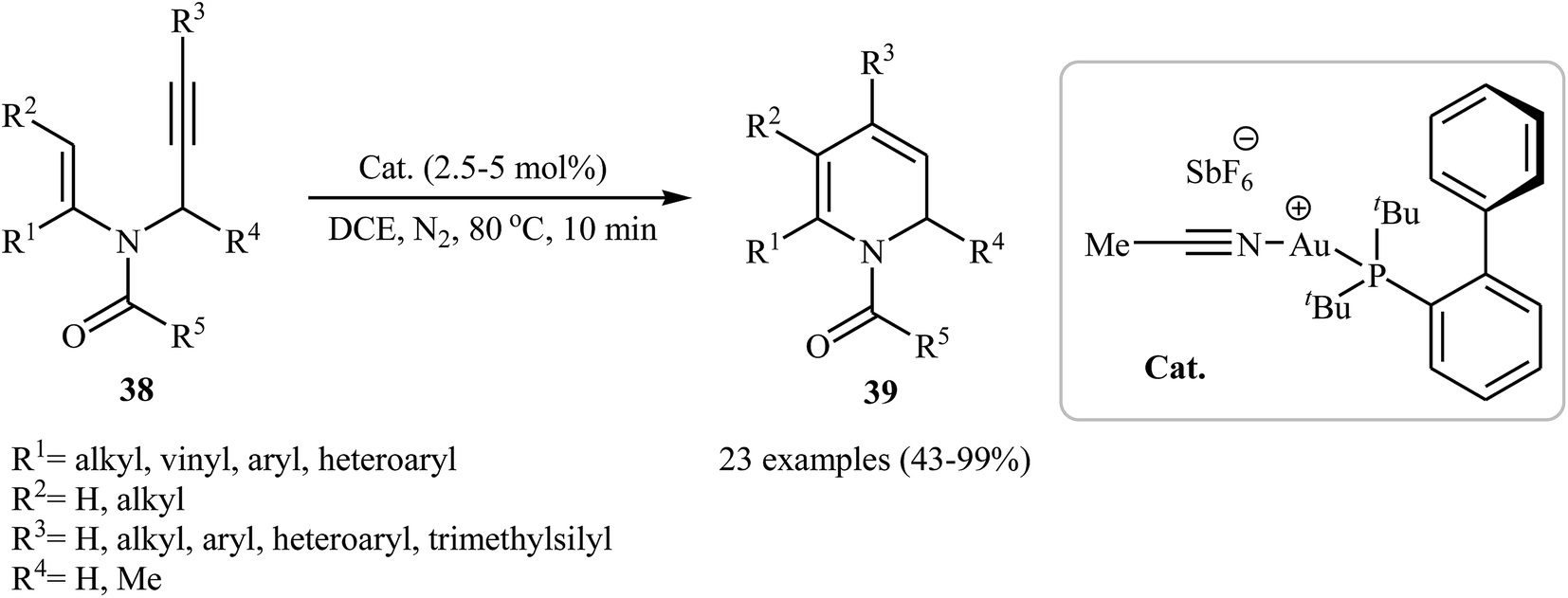
Along this line, Wang and co-workers reported the gold(I)-catalyzed intramolecular cyclization of N-vinylpropargylamines 38 into di- to penta-substituted 1,2-dihydropyridine derivatives 39 (Scheme 15). The mechanism proposed for this transformation is similar to the one described in Scheme 1.30
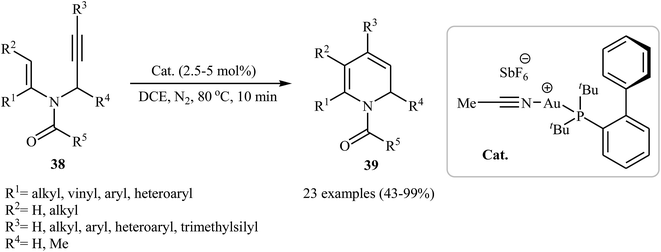 |
| | Scheme 15 Au-Catalyzed intramolecular cyclization of N-vinylpropargylamines 38 into di- to penta-substituted 1,2-dihydropyridines 39. | |
3.3. From N-propargylamines and alkynes
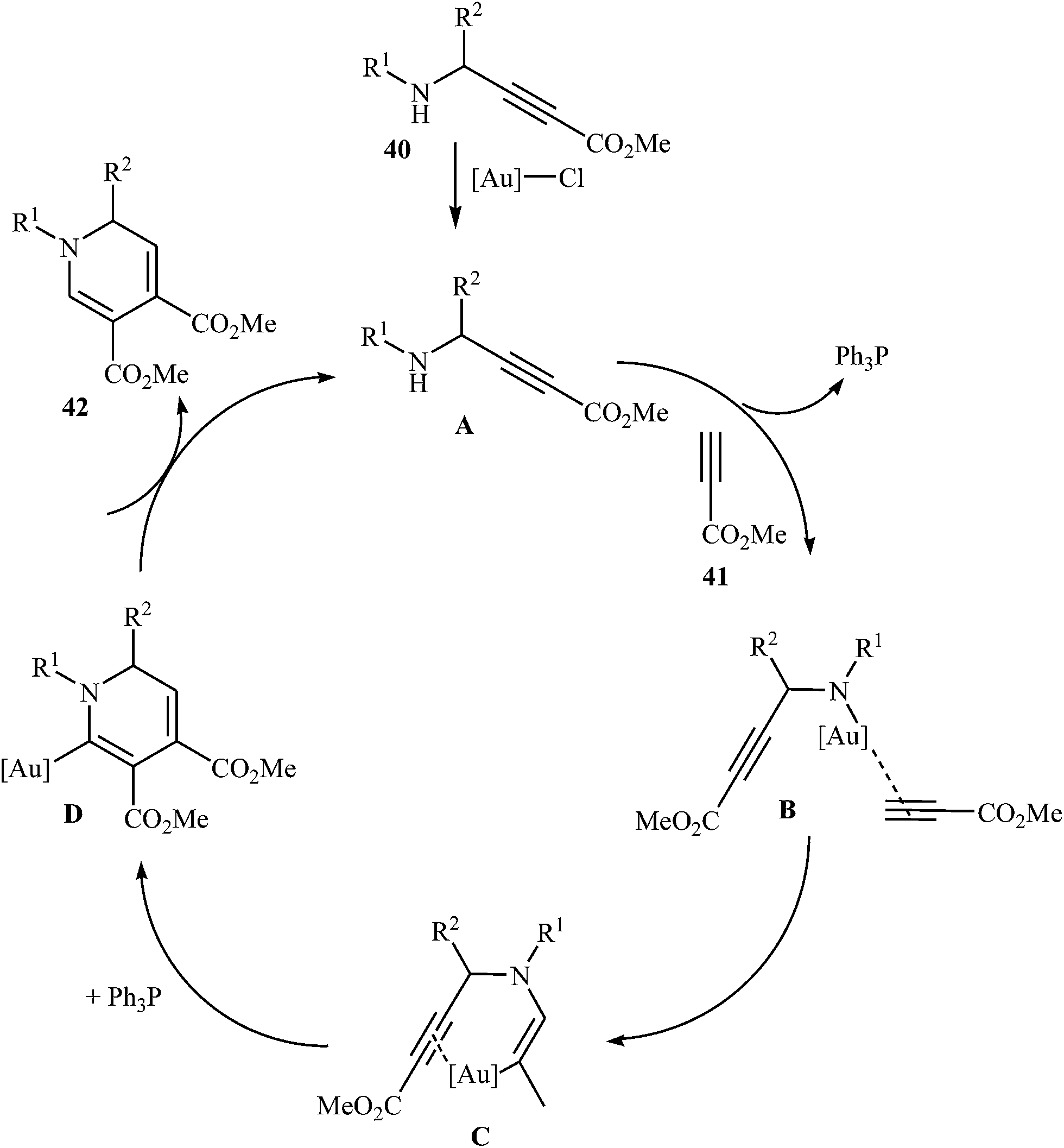
In 2015, Y. Li and co-workers developed the synthesis of multisubstituted dihydropyridines by Au-catalyzed cascade reaction of N-propargylamines and alkynes. Thus, the reaction of N-phenyl N-propargylamines 40 and methyl propiolate 41 furnishes 3,4-carboxylated 1,6-dihydropyridines 42 in moderate to high yields (Scheme 16). The results demonstrated that the rate of cyclization was considerably influenced by electronic characters of the substituents on the aromatic rings of propargylamine. Generally, the substrates bearing electron-donating groups, like Me and MeO in the para- or meta-position, gave good yields, while an electron-withdrawing group, like a Cl group, gave moderate yields. Moreover, strong electron-withdrawing groups like CF3 and NO2 inhibited the cyclization completely. The author proposed mechanism for this cyclization is depicted in Fig. 5.31
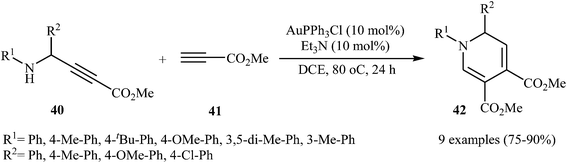 |
| | Scheme 16 Cascade synthesis of 3,4-carboxylated 1,6-dihydropyridines 42 from N-propargylamines 40 and methyl propiolate 41. | |
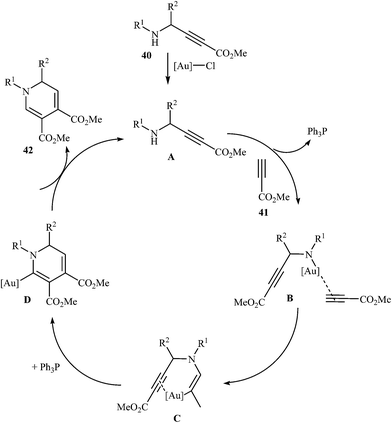 |
| | Fig. 5 Proposed mechanism for formation of dihydropyridines 42. | |
4. Piperidines
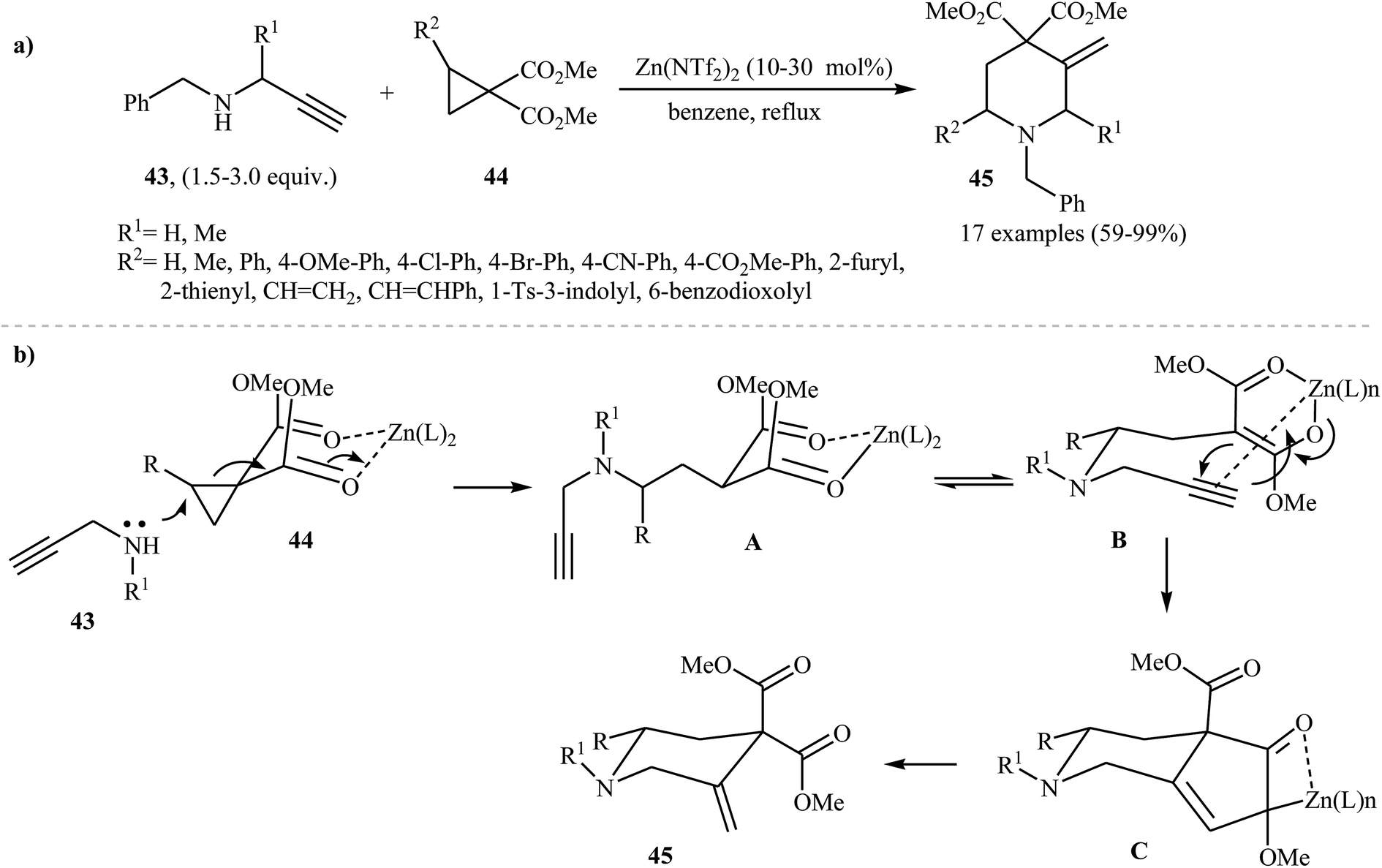
The use of N-propargylamines as starting substrates in the synthesis of piperidines has been scarcely studied; in fact, only two examples of such a reaction were reported in the literature. In 2009, Leblod, Leduc, and Kerr by studying the chemistry and application of the benzyl-protected propargyl amines 43, discovered that they reacted with 1,1-cyclopropane diesters 44 in the presence of Zn(NTf2)2 as catalyst to produce highly substituted piperidines 45 in good to excellent yields (Scheme 17a). According to the proposed mechanism, the key steps of the reaction involves a cyclopropane ring-opening and a Conia-ene cyclization (Scheme 17b).32
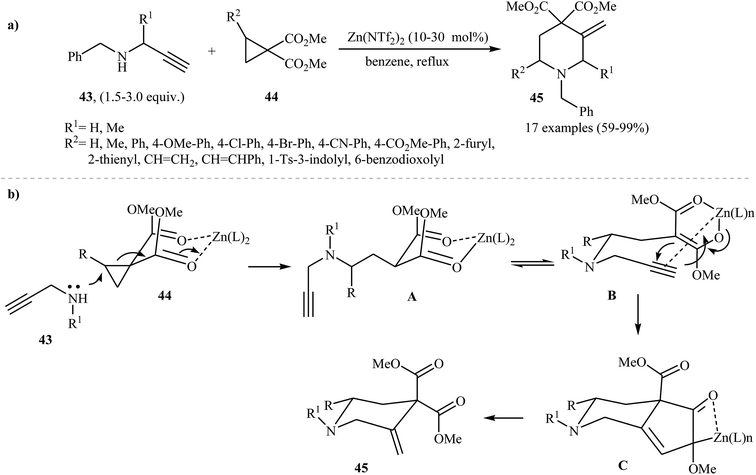 |
| | Scheme 17 (a) Zn(II)-catalyzed synthesis of highly substituted piperidines 45 from propargyl amines 43 and 1,1-cyclopropane diesters 44; (b) proposed mechanistic pathways for the formation of piperidines 45. | |
5. Fused pyridine ring
Synthesis of fused pyridine rings from N-propargylamines are scarcely reported. However, there are dozen papers on synthesis of quinolines (benzene fused pyridine) from titled compounds. We have not discussed about this reactions here, because we summarized this rout of synthesis of guinolines in other there.10 Dihydropyrrolo-pyrridines are another ring fused pyridines that can be synthesizes from N-propargylamines and nitriles through [2 + 2 + 2] cyclization.33 We also have not discussed about this reactions due to they are belonged to the synthesis of dihydropyrroles (not pyridines) from N-propargylamines.
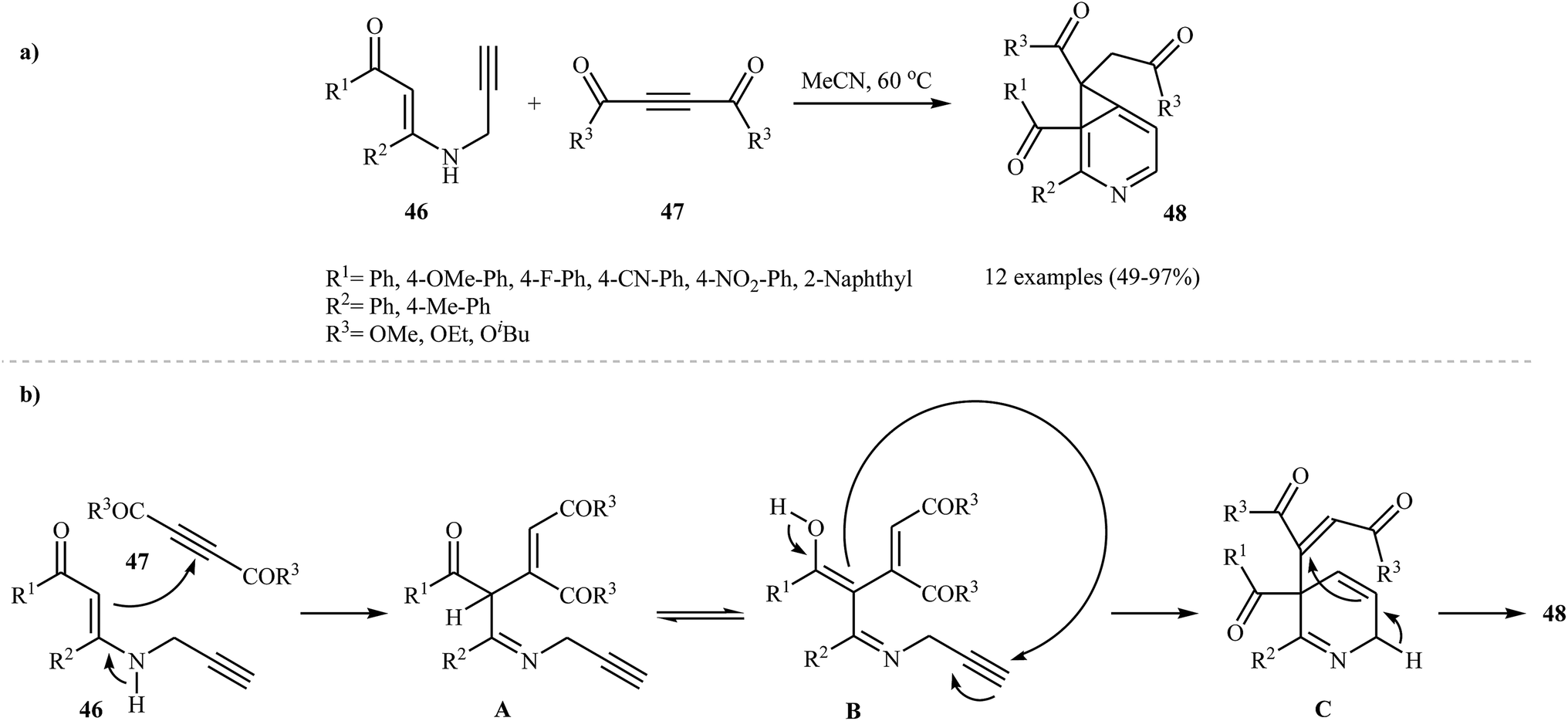
As shown in the Section 2.1. and 3.1., N-propargylic β-enaminones are interesting starting materials to access substituted pyridines. Based on duel role of these compounds, Karunakar and co-workers were able to demonstrate that a series of pyridine systems 48 could be obtained from the reaction of N-propargylic β-enaminones 46 with acetylenedicarboxylates 47 under catalyst and base-free conditions in acetonitrile (Scheme 18a). The results showed that N-propargylamines with electron-donating groups gave higher yields than those with electron-withdrawing groups. For acetylenedicarboxylates 47, the decreasing order of reactivity is diethyl but-2-ynedioate > di-tert-butyl but-2-ynedioate > dimethyl but-2-ynedioate. According to the proposed mechanism, the reaction proceeds in four consecutive steps: (i) formation of an iminic intermediate A through nucleophilic addition of 46 to the electrophiles 47, (ii) tautomerization of A to form intermediate B, (iii) intramolecular cyclization of intermediate B to produce cyclic intermediate C and (iv) cyclopropanation of C to the expected product 48 (Scheme 18b).34
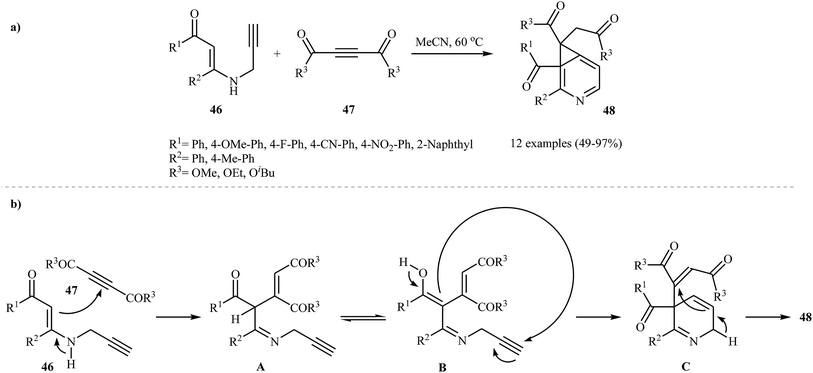 |
| | Scheme 18 (a) Synthesis of fused pyridines 48 from treatment of N-propargylic β-enaminones 46 with acetylenedicarboxylates 47; (b) proposed mechanistic pathways for the formation of 48. | |
6. Conclusion
This review provides concise overview on the synthesis of pyridines and their ring fused analogues from N-propargylamines. This new page to access pyridine derivatives in most cases has many advantages over more conventional methodologies, which can be summarized as follows: (i) shorter synthetic routes; (ii) high atom economy; (iii) environmentally friendly processes; (iv) high yielding, wide in scope and many more. More work is still necessary to expand the N-propargylamines to the synthesis of different types of pyridine derivatives. We conclude this review by hoping that it will stimulate researchers to develop the highly versatile and extremely effective procedures to synthesis of diverse of pyridines from N-propargylamines.
References
-
(a) S. B. Ferreira and C. R. Kaiser, Expert Opin. Ther. Pat., 2012, 22, 1033–1051 CrossRef CAS PubMed;
(b) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC;
(c) A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Saudi Pharm. J., 2013, 21, 1–12 CrossRef PubMed;
(d) C. Liu, K. Kevin, J. Li and S. Sakya, Mini-Rev. Med. Chem., 2004, 4, 1105–1125 CrossRef PubMed;
(e) E. Vessally and M. Abdoli, J. Iran. Chem. Soc., 2016, 13, 1235–1256 CrossRef CAS;
(f) A. Shafiee, M. Z. Kassaee and A. R. Bekhradnia, J. Heterocycl. Chem., 2007, 44, 471–474 CrossRef CAS;
(g) E. Vessally, A. Hassanpour, R. Hosseinzadeh-Khanmiri, M. Babazadeh and J. Abolhasani, Monatsh. Chem., 2016 DOI:10.1007/s00706-016-1762-2;
(h) L. Dinparast, H. Valizadeh, E. Vessally and M. B. Bahadori, J. Mol. Struct., 2016, 1105, 118–127 CrossRef CAS;
(i) Z. Asadi, M. B. Asnaashariisfahani, E. Vessally and M. D. Esrafili, Spectrochim. Acta, Part A, 2015, 140, 585–599 CrossRef CAS PubMed;
(j) E. Vessally, E. Fereyduni, H. Shabrendi and M. D. Esrafili, Spectrochim. Acta, Part A, 2013, 116, 65–73 CrossRef CAS PubMed.
-
(a) A. E. Goetz and N. K. Garg, Nat. Chem., 2013, 5, 54 CrossRef CAS PubMed;
(b) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 CrossRef PubMed;
(c) S. Arshadi, A. R. Bekhradnia and A. Ebrahimnejad, Can. J. Chem., 2011, 89, 1403–1409 CrossRef CAS;
(d) M. Z. Kassaee and A. R. Bekhradnia, J. Biosci. Bioeng., 2003, 95, 526–529 CrossRef CAS PubMed.
- F. Bohlmann and D. Rahtz, Chem. Ber., 1957, 90, 2265–2272 CrossRef CAS.
- H. Bönnemann, Angew. Chem., Int. Ed. Engl., 1985, 24, 248–262 CrossRef.
- F. Kröhnke, W. Zecher, J. Curtze, D. Drechsler, K. Pfleghar, K. Schnalke and W. Weis, Angew. Chem., Int. Ed. Engl., 1962, 1, 626–632 CrossRef.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1881, 14, 1637–1638 CrossRef.
-
(a) M. Zheng, P. Chen, W. Wu and H. Jiang, Chem. Commun., 2016, 52, 84–87 RSC;
(b) S. Kankala, R. Pagadala, S. Maddila, C. S. Vasam and S. B. Jonnalagadda, RSC Adv., 2015, 5, 105446–105452 RSC.
-
(a) K. Goutham, D. Ashok Kumar, S. Suresh, B. Sridhar, R. Narender and G. V. Karunakar, J. Org. Chem., 2015, 80, 11162–11168 CrossRef CAS PubMed;
(b) T. P. Willumstad, P. D. Boudreau and R. L. Danheiser, J. Org. Chem., 2015, 80, 11794–11805 CrossRef CAS PubMed;
(c) K. Komeyama, R. Igawa and K. Takaki, Chem. Commun., 2010, 46, 1748–1750 RSC;
(d) J. Meng, Y.-J. Li, Y.-L. Zhao, X.-B. Bu and Q. Liu, Chem. Commun., 2014, 50, 12490–12492 RSC;
(e) M.-C. P. Yeh, M.-N. Lin, C.-H. Hsu and C.-J. Liang, J. Org. Chem., 2013, 78, 12381–12396 CrossRef CAS PubMed;
(f) Z. Jiang, P. Lu and Y. Wang, Org. Lett., 2012, 14, 6266–6269 CrossRef CAS PubMed;
(g) B. Alcaide, P. Almendros, J. M. Alonso, I. Fernández, G. Gómez-Campillos and M. R. Torres, Chem. Commun., 2014, 50, 4567–4570 RSC;
(h) S. Basceken, S. Kaya and M. Balci, J. Org. Chem., 2015, 80, 12552–12561 CrossRef CAS PubMed.
- E. Vessally, RSC Adv., 2016, 6, 18619–18631 RSC.
- E. Vessally, L. Edjlali, A. Hosseinian, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 49730–49746 RSC.
- A.-Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463–8480 CrossRef CAS.
-
(a) K. R. Reddy, A. S. Reddy, R. Shankar, R. Kant and P. Das, Asian J. Org. Chem., 2015, 4, 573–583 CrossRef CAS;
(b) N. RaoáMangina, Org. Biomol. Chem., 2014, 12, 2869–2873 RSC;
(c) J.-P. Wan, Y. Zhou, Y. Liu and S. Sheng, Green Chem., 2016, 18, 402–405 RSC;
(d) J.-P. Wan, S. Zhong and Y. Liu, Synthesis, 2015, 47, 3611–3617 CrossRef CAS;
(e) C. Zheng, Y. Wang and R. Fan, Org. Lett., 2015, 17, 916–919 CrossRef CAS PubMed;
(f) K. Goutham, D. Ashok Kumar, S. Suresh, B. Sridhar, R. Narender and G. V. Karunakar, J. Org. Chem., 2015, 80, 11162–11168 CrossRef CAS PubMed.
- M. A. Martins, M. Rossatto, L. D. Prola, L. Pizzuti, D. N. Moreira, P. T. Campos, C. P. Frizzo, N. Zanatta and H. G. Bonacorso, Ultrason. Sonochem., 2012, 19, 227–231 CrossRef CAS PubMed.
- S. Cacchi, G. Fabrizi and E. Filisti, Org. Lett., 2008, 10, 2629–2632 CrossRef CAS PubMed.
- X. Xin, D. Wang, X. Li and B. Wan, Tetrahedron, 2013, 69, 10245–10248 CrossRef CAS.
- S. Karabiyikoglu, Y. Kelgokmen and M. Zora, Tetrahedron, 2015, 71, 4324–4333 CrossRef CAS.
-
(a) Y. Kelgokmen and M. Zora, RSC Adv., 2016, 6, 4608–4621 RSC;
(b) E. Karadeniz, M. Zora and N. Z. Kılıçaslan, Tetrahedron, 2015, 71, 8943–8952 CrossRef CAS;
(c) A. Bekhradnia and P.-O. Norrby, Dalton Trans., 2015, 44, 3959–3962 RSC.
- G. Cheng, Y. Weng, X. Yang and X. Cui, Org. Lett., 2015, 17, 3790–3793 CrossRef CAS PubMed.
- G. Abbiati, A. Arcadi, G. Bianchi, S. Di Giuseppe, F. Marinelli and E. Rossi, J. Org. Chem., 2003, 68, 6959–6966 CrossRef CAS PubMed.
- J.-Z. Yan, J. Li and G.-W. Rao, Steroids, 2007, 72, 736–739 CrossRef CAS PubMed.
- H. Wei, Y. Li, K. Xiao, B. Cheng, H. Wang, L. Hu and H. Zhai, Org. Lett., 2015, 17, 5974–5977 CrossRef CAS PubMed.
-
(a) W. Gati, M. M. Rammah, M. B. Rammah, F. o. Couty and G. Evano, J. Am. Chem. Soc., 2012, 134, 9078–9081 CrossRef CAS PubMed;
(b) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed;
(c) P. Jochmann, T. S. Dols, T. P. Spaniol, L. Perrin, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2010, 49, 7795–7798 CrossRef CAS PubMed;
(d) F. Gao, B. Gong, T. A. Christopher, B. L. Lopez, A. Karasawa and X. L. Ma, Br. J. Pharmacol., 2001, 132, 869–878 CrossRef CAS PubMed.
- A. Saito, T. Konishi and Y. Hanzawa, Org. Lett., 2009, 12, 372–374 CrossRef PubMed.
- F. J. Fananás, T. Arto, A. Mendoza and F. Rodriguez, Org. Lett., 2011, 13, 4184–4187 CrossRef PubMed.
- M. A. Martins, M. Rossatto, C. P. Frizzo, E. Scapin, L. Buriol, N. Zanatta and H. G. Bonacorso, Tetrahedron Lett., 2013, 54, 847–849 CrossRef CAS.
-
(a) X. Xin, D. Wang, F. Wu, X. Li and B. Wan, J. Org. Chem., 2013, 78, 4065–4074 CrossRef CAS PubMed;
(b) A. R. Bekhradnia, S. Arshadi and S. A. Siadati, Chem. Pap., 2014, 68, 283–290 CAS.
- H. Mizoguchi, R. Watanabe, S. Minami, H. Oikawa and H. Oguri, Org. Biomol. Chem., 2015, 13, 5955–5963 CAS.
- H. Kim and C. Lee, J. Am. Chem. Soc., 2006, 128, 6336–6337 CrossRef CAS PubMed.
-
(a) A. R. Bekhradnia, F. Zahir and S. Arshadi, Monatsh. Chem., 2008, 139, 521–523 CrossRef CAS;
(b) M. Z. Kassaee and A. R. Bekhradnia, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 2025 CrossRef CAS;
(c) S. Undeela, S. Thadkapally, J. B. Nanubolu, K. K. Singarapu and R. S. Menon, Chem. Commun., 2015, 51, 13748–13751 RSC.
- X. Zhang, X.-M. Xu, L. Zhao, J. You, J. Zhu and M.-X. Wang, Tetrahedron Lett., 2015, 56, 3898–3901 CrossRef CAS.
- Y. Li, Z. Wu, Y. Pan, H. Huang, J. Shi, H. Bu, H. Ma, Y. Liang and B. Huang, Synlett, 2015, 11, 834–838 CrossRef.
- T. P. Lebold, A. B. Leduc and M. A. Kerr, Org. Lett., 2009, 11, 3770–3772 CrossRef CAS PubMed.
-
(a) Y. Zhou, J. A. Porco and J. K. Snyder, Org. Lett., 2007, 9, 393–396 CrossRef CAS PubMed;
(b) D. D. Young and A. Deiters, Angew. Chem., Int. Ed., 2007, 119, 5279–5282 CrossRef;
(c) A. M. Rodriguez, C. Cebrián, P. Prieto, J. I. García, A. de la Hoz and Á. Díaz-Ortiz, Chem.–Eur. J., 2012, 18, 6217–6224 CrossRef CAS PubMed;
(d) S. Medina, G. Domínguez and J. Pérez-Castells, Org. Lett., 2012, 14, 4982–4985 CrossRef CAS PubMed;
(e) K. Kashima, M. Ishii and K. Tanaka, Eur. J. Org. Chem., 2015, 1092–1099 CrossRef CAS;
(f) V. Richard, M. Ipouck, D. S. Mérel, S. Gaillard, R. J. Whitby, B. Witulski and J. L. Renaud, Chem. Commun., 2014, 50, 593–595 RSC;
(g) F. Xu, C. Wang, D. Wang, X. Li and B. Wan, Chem.–Eur. J., 2013, 19, 2252–2255 CrossRef CAS PubMed;
(h) C. Wang, D. Wang, F. Xu, B. Pan and B. Wan, J. Org. Chem., 2013, 78, 3065–3072 CrossRef CAS PubMed;
(i) G. Onodera, Y. Shimizu, J. Kimura, J. Kobayashi, Y. Ebihara, K. Kondo, K. Sakata and R. Takeuchi, J. Am. Chem. Soc., 2012, 134, 10515–10531 CrossRef CAS PubMed.
- K. Goutham, V. Nagaraju, S. Suresh, P. Raghavaiah and G. V. Karunakar, RSC Adv., 2014, 4, 21054–21059 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.