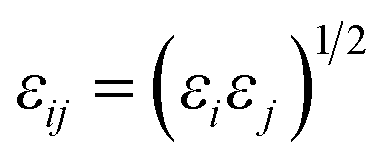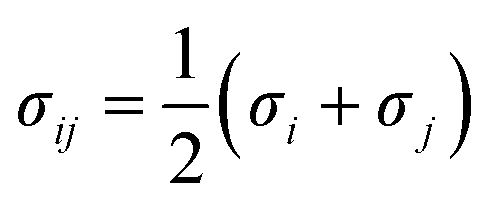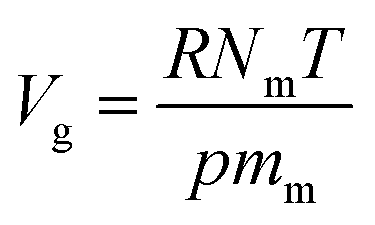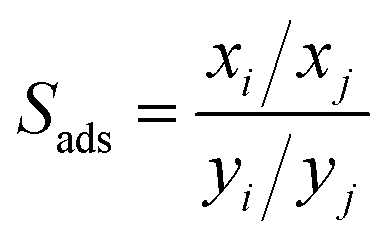DOI:
10.1039/C6RA08778G
(Paper)
RSC Adv., 2016,
6, 91093-91101
Tuning the adsorption and separation properties of noble gases and N2 in CuBTC by ligand functionalization†
Received
5th April 2016
, Accepted 3rd September 2016
First published on 5th September 2016
Abstract
The Grand Canonical Monte Carlo method was used to investigate the adsorption and separation properties of noble gas and N2 mixtures on a MOF material, namely, CuBTC, functionalized with different groups, including amino, hydroxyl and fluorine groups, in order to understand the potential applications of the materials in noble gas separation. Binary equimolar mixtures of Xe/Ar, Xe/Kr and Kr/Ar as well as nonequimolar mixtures of Xe/N2 and Kr/N2 containing 0.01% (molar fraction) noble gas were examined. Amino-functionalized CuBTC displayed an attractive interaction with all the noble gases due to the high polarity of amino-functionalized benzene and its large van der Waals interaction with all the gases, whereas the hydroxyl groups exhibited a lower impact on the adsorption properties of CuBTC. On the other hand, functionalization with fluorine groups largely decreased the adsorption capacity for all the noble gases compared to the parent Cu-BTC. The functional groups also exhibited different effects on the adsorption selectivity to the noble gases over N2. The amino-functionalized CuBTC showed enhanced selectivities towards specific noble gases over N2 or the other noble gases. Hydroxyl and fluorine groups exhibited similar selectivities with the original CuBTC. The difference in the adsorption selectivity is mainly attributed to the enhanced electron delocalization and the polarizability of the aromatic ring due to the presence of amino groups, which comprise the best binding sites for the noble gases and N2. According to the simulation results, it can be concluded that amino-functionalized CuBTC can enhance both the adsorption capacity and the selectivity of specific noble gases over N2 or other noble gases at low pressure from 0 to 100 kPa at 292 K.
Introduction
Noble gases, which occupy only about 0.94% of the air and which have low chemical reactivity, are now widely used in many fields, such as tracer applications,1 lighting2 and imaging.3 For example, Ar, Kr and Xe are used to fill light tubes or windowpanes due to their low thermal conductivity and high density. Xe is also applied as a fuel in satellite launchers and as a narcotic in the medical sector. However, noble gases only possess fairly low concentrations in the air, and their inertia chemical properties make them difficult to separate from the air or from each other. The current extraction methods for the noble gases are cryogenic rectification, catalytic reaction and pressure swing adsorption (PSA). The former two methods are energetically expensive, while PSA is low cost but has difficulty providing a satisfying adsorption and separation performance due to the fully occupied valence shell of the noble gases. Therefore, it is significant but also challenging to develop adsorption materials to enable PSA to effectively purify and separate noble gases from the air.
Nanoporous adsorbents, such as activated carbon,2 zeolites,4 molecular sieves5 and single-walled carbon nanotube bundles,6,7 are usually used for noble gas capture and purification. For example, Foroutan et al. studied the adsorption behaviour of ternary mixtures of noble gases inside single-walled carbon nanotube bundles, and found that heavier gases were more easily adsorbed than lighter ones on the single-walled carbon nanotube bundles.6
In the past few years, metal-organic frameworks (MOFs) have drawn extensive attention due to their promising properties and tuneable structures. Compared to other conventional nanoporous materials, MOFs have the advantages of a high surface area, large pore volume and tuneable structure,8–10 which allow them to be deployed in many fields, such as adsorption and separation,11 catalysis,12 ferromagnetics,13 florescence,14 drug carrier.15 One of the most significant applications of MOFs is gas adsorption and separation. However, so far there are few reports on the adsorption and purification of noble gases by MOFs, although Greathouse et al. reported the adsorption and separation of noble gases in IRMOF-1 using Grand Canonical Monte Carlo (GCMC) simulation and found that the selectivity of Xe to Ar or Kr may correlate with the van der Waals well depth of each gas.16 Thallapally et al. compared the adsorption of Xe on NIDOBDC and activated carbon, and they observed that NIDOBDC was more selective for xenon over krypton compared to over activated carbon.2 Parkes et al. studied the effect of the pore size and framework topology on selective noble gas adsorption in air and discovered that heavier and more polarizable gases were more easily separated by MOFs than lighter and less polarizable gases.17 Wang et al. compared the adsorption and separation properties of Xe in several MOFs and COMs (covalent-organic materials), concluding that Xe uptake on the materials is entirely consistent with their accessible surface area. In addition, the order of selectivities of Xe/N2 on the materials follow the order of their difference in isosteric heats.18 The recent studies imply that MOFs have potential benefits in adsorption and separation compared to conventional adsorbents due to their tuneable structures and compositions.
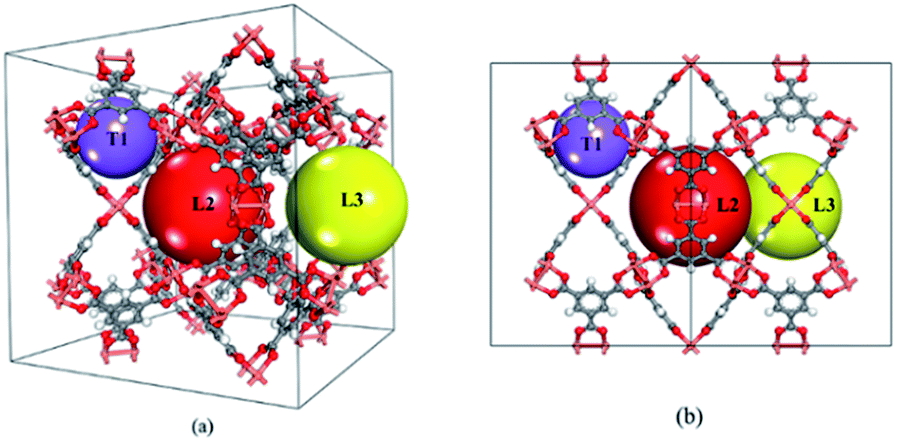
HKUST-1 (also called CuBTC) is one of the most well-studied MOFs, and was first reported by Chui et al. in 1999.19 It consists of three types of cages, namely T1, L2 and L3 cages (see Fig. 1). T1 cages (small tetrahedral-shaped pockets with a diameter of 5–7 Å) are surrounded by two large cages (the L2 and L3 cages) formed by four BTC linkers and that are recognized as apolar in nature. The L3 cages are larger (11–12 Å) and more polar than L2 cages (10–13.5 Å) due to the open copper site located inside the L3 cages. Its gas separation applications have been confirmed during the past few decades.20–22 Besides, CuBTC can be easily synthesized using copper salts and terephthalate under mild conditions.19 Recent reports show that CuBTC has a high adsorption capacity for noble gases, exhibiting an excellent potential for separating noble gases from the air or from other noble gases.18,23–25 One unique feature of MOFs is the tuneable functional groups on their surface, which can lead to a large variety of adsorption properties for gas adsorption. Therefore, functionalized MOFs have drawn significant attention for gas adsorption and separation over the past few years. For example, Couck et al. found that the functionalized group can strengthen the interaction between the framework with CO2, resulting in an enhanced affinity for CO2.26 Cai et al. investigated the impact of alkyl-functionalized BTC on the adsorption and separation properties of copper-based CuBTC and found that functionalized CuBTC was able to suppress water adsorption to one-third of that in CuBTC while maintaining a comparable CO2 and CH4 excess uptake of up to 5 bar. This shows that the proper functionalization of MOFs can promote a strong interaction between the target gas and an open-metal site in MOF and weaken the interaction between water and the open-metal MOF.27 Clearly, functionalization is one of the most effective methods to tune MOFs with more promising properties for gas adsorption and separation. Due to the wide range of available ligands for MOFs, systematic investigation of the relationship between the structure and their adsorption and separation properties using computational methods is highly desired. It is also useful as it is even more challenging to understand the impact of functionalization on MOFs for adsorbing noble gases because the gases are chemically inert substances without a dipole moment or quadrupole.
 |
| | Fig. 1 Schematic of (a) 3D view and (b) left view of the CuBTC model. | |
Therefore, a GCMC simulation study was carried out to investigate the adsorption and separation properties of CuBTCs functionalized with different ligands. Three functionalized-CuBTCs were theoretically constructed by inserting fluorine, hydroxyl, and amino groups onto the aromatic ring of the organic linker. GCMC simulations were then carried out to evaluate the adsorption and separation properties of CuBTC and of functionalized-CuBTCs. DFT calculations were employed to investigate the electrostatic potential of the cluster, the preferred adsorption site and the polarizability of the aromatic rings of the organic linker. The aim of this work was to understand noble gas adsorption on the fluorine-, hydroxyl- and amino-functionalized CuBTCs and to investigate the potential applications of the materials in noble gas adsorption and separation.
Simulation details
Model construction and charge calculation
The crystal structure of CuBTC was constructed from the X-ray diffraction data. According to the previous report,28 the copper nodes and the ben-1,3,5-tricarboxylate (BTC) linkers construct a diametric copper paddle wheel, which is the structural building block for a three-dimensional (3D) face-centred CuBTC unit cell. Generally, the open-metal site is covered with a hydroxyl group or other solvents, which can then be removed to form an activated framework.
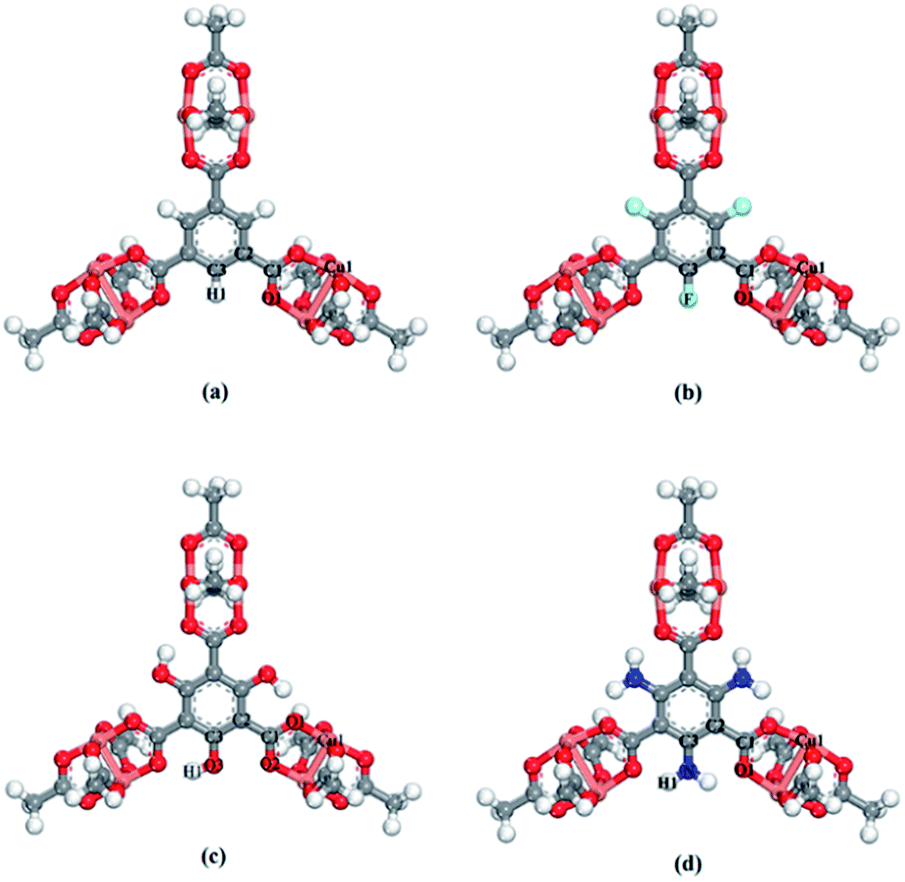
The X-functionalized (X = –F, –OH, –NH2) CuBTC structures were constructed on the basis of the parent CuBTC. The original CuBTC metal cluster was cut down from a CuBTC unit cell, and methyl groups were subsequently inserted into the non-saturated site. The metal cluster of functionalized CuBTC was constructed by substituting the hydrogen atoms of the aromatic ring with the X groups. Finally, the functionalized metal cluster was optimized by the Gaussian 09 (ref. 29) withwb97xd/6-311+g(2df,p) method (the same as in the binding energy calculations) for non-metallic atoms and lan2ldz for copper and Xe atoms while fixing the original structure and replicating the position of the X groups on the CuBTC framework. This constructed model can be properly used to accurately reflect X-functionalized CuBTC due to the unchanged symmetry after insertion of the functionalized groups, which are small in size and have less impact on the original CuBTC. The optimized metal clusters are displayed in Fig. 2. The similar styles of functionalization have been reported by other researchers. For instance, Katharina constructed an amino-functionalized CuBTC and discovered that it had the same topology style with the original CuBTC,30 while Yang et al. synthesized several functionalized CuBTCs by functionalization of the original one with groups including methyl, ethyl, methoxyl, bromo, nitro and acetamide, and demonstrated that the last three functionalized CuBTCs maintained the same topology structure as CuBTC.31 The energy of the clusters and the energy gap (Egap) between the HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) levels are listed in Table 1. The energies of the three functionalized clusters were slightly larger than that of the original one and Egap of them were slightly smaller than that of the original one. These obtained results may demonstrate functionalized structures were less stable than the original one. However, functionalizations enhanced molecular weight and atom numbers of metal clusters, which can increase the energy of the metal cluster. Besides, functionalized metal clusters still remained regular and undistorted after being optimized. Therefore, in consideration of all compared results, it can be safely concluded that it is possible for the three functionalized CuBTCs constructed in this article. Also, the same method was applied to calculate the charges of the atoms. Here, the optimized clusters were fixed when the charges were being calculated. All the charges of the atoms are given in Table S1.†
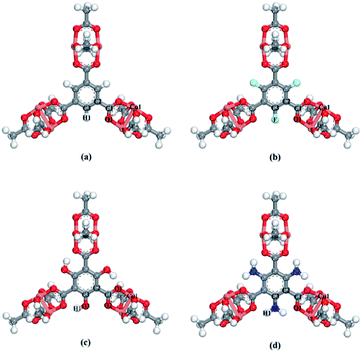 |
| | Fig. 2 The metal clusters for (a) CuBTC, (b) 3F-CuBTC, (c) 3OH-CuBTC and (d) 3NH2-CuBTC, respectively (copper: orange, oxygen: red, carbon: grey, hydrogen: white, nitrogen: dark blue, fluorine: light blue). | |
Table 1 Energies of the four clusters and the energy gap between the HOMO and LUMO levels of the four clusters
| Clusters |
CuBTC |
3F-CuBTC |
3OH-CuBTC |
3NH2-CuBTC |
| Energy (a.u.) |
−4030.78 |
−4327.4 |
−4255.2 |
−4080.1 |
| Egap (eV) |
1.40 |
1.38 |
1.34 |
1.33 |
Force field
Potential parameters for the MOFs were taken from mixed force field including the Universal Force Field (UFF)32 and the all-atom OPLS (OPLS-AA)33 force field, which were found to be in agreement with the experimental results from Allendorf.34 The noble gases model refers to that used by Pellenq and Levitz35 and Loef.36 N2 was defined as a three-site model, including two N atoms and a COM atom, with the van der Waals interactions and atomic charge reported by Siepmann.37 All the interatomic potential parameters are listed in Table S2.† All the LJ cross interaction parameters were calculated by the Lorentz–Berthelot mixing rules:| |
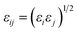 | (1) |
| |
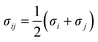 | (2) |
where ε and σ represent the energy well depth and van der Waals radii, respectively.
GCMC simulation
Simulated adsorption isotherms were generated by the GCMC method using the Music code from Snurr.38 The lattice parameters of all the MOFs are listed in Table S3,† and were optimized by the Forcite model in Materials Studio with an ultrafine basis set.39 The simulation was operated on 2 × 2 × 2 unit cells. Simulation was performed at 292 K with a pressure ranging from 1 kPa to 100 kPa. The excess uptake, Nex, was transformed from the absolute uptake, Nabs, to compare it with the experimental data using the following equation (reported Düren et al.38)where ρg and Vg represent the density of the bulk gas calculated from PR EOS and the free pore volume of the MOFs, respectively. Vg was generated by the method of Myers et al.40 from the ideal gas law:| |
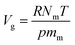 | (4) |
where R is the gas constant, T is the temperature, p is the pressure and Nm is the number of adsorbed probe molecules per molar mass mm of the adsorbents, which is calculated for helium (σ = 2.64 Å, ε/kb = 10.90 K) gas in the MOFs at low temperature and ambient pressure.
The adsorption selectivity (Sads) is often used to indicate the separation ability of nanoporous materials. The adsorption selectivity of component i over component j is defined as
| |
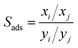 | (5) |
where
x and
y represent the molar fraction of the two components in the adsorbed and bulk phase, respectively.
41
The binding energy (BE) was calculated in order to evaluate the interaction strength of the gas molecules on different binding sites of adsorbents. In the optimization process, the metal clusters were fixed in order to replicate the bulk behaviour, while the gas molecules were nearly relaxed at the possible adsorption sites. The value of BEs can then be given by the difference between the products and reactants, including the energy of Basis Set Superposition Error (EBSSE).42
| | |
BE = EMOF–gas − EMOF − Egas + EBSSE
| (6) |
where
EMOF–gas is the total energy of the adsorbate–adsorbent system in the equilibrium state, while
EMOF and
Egas are the non-adsorbed MOF structure and gas molecule, respectively.
Results and discussion
Adsorption isotherms
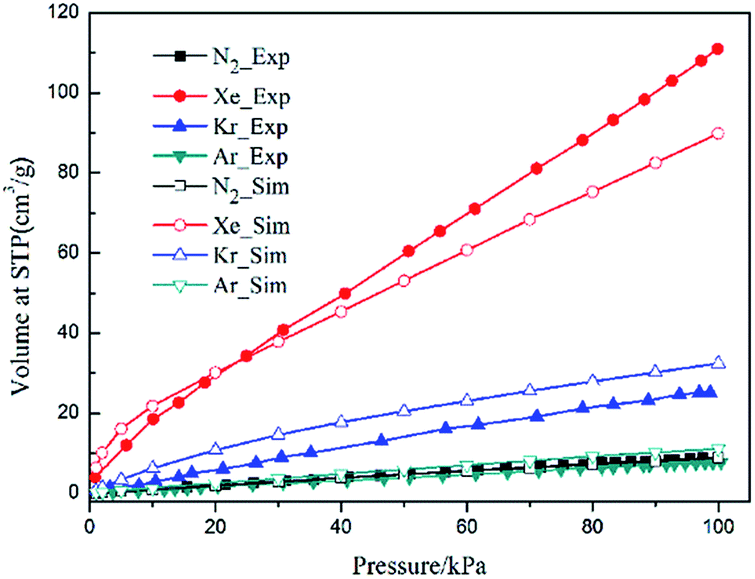
Single component adsorption isotherms of different gas molecules on CuBTC at 292 K were first simulated, and then compared to experimental data (Fig. 3). The experiment data were obtained from the work of Allendorf et al.34 Although a slight difference existed between the simulation and experimental data, the GCMC simulation could still reproduce the experimental results with a certain accuracy, indicating that the force field used in this study was reasonable for the simulating adsorption and separation properties of CuBTC and of the functionalized CuBTCs. Previous work also confirmed that such results can match well with the experimental data.34 Overall, the force field is reasonable for simulating the adsorption and separation properties of CuBTC and of the functionalized CuBTCs.
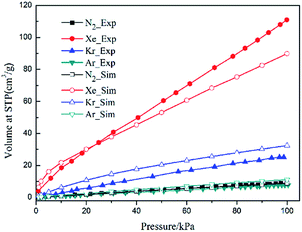 |
| | Fig. 3 Comparison of the experimental and simulation N2 and noble gases adsorption isotherms at 292 K. | |
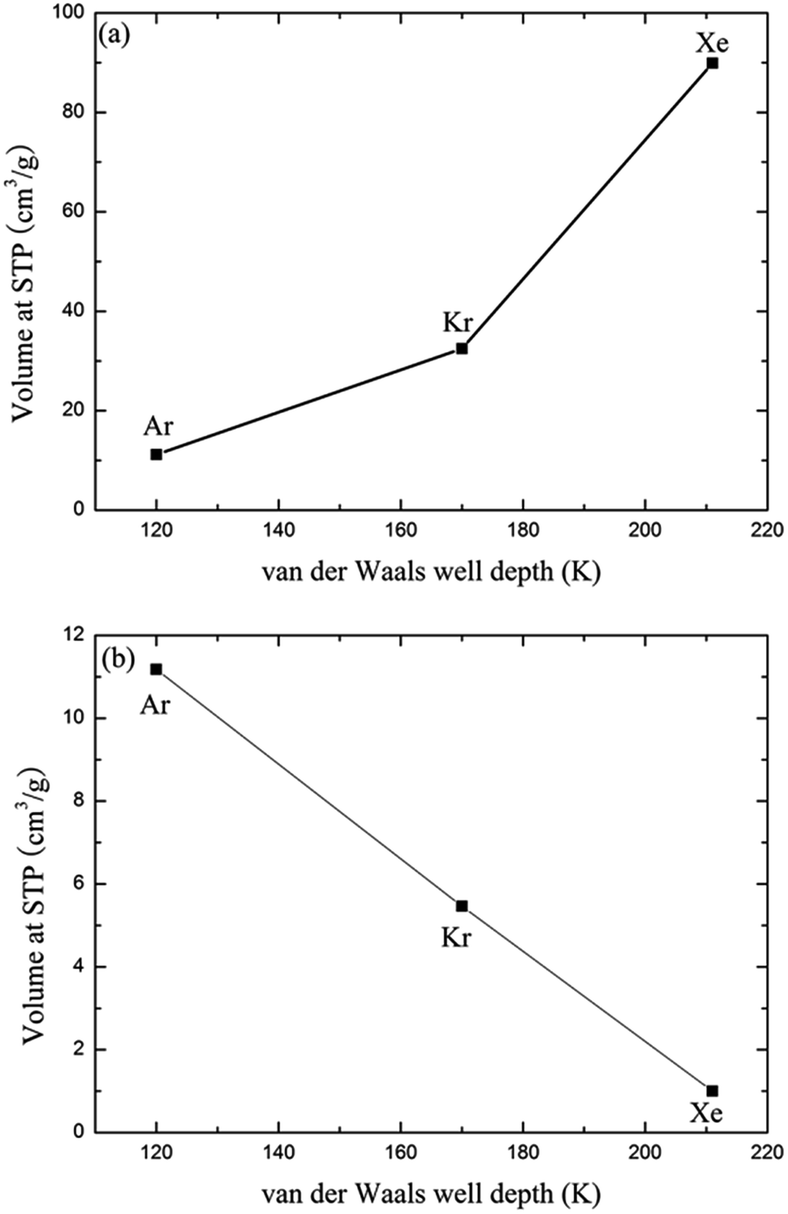
Since the outer shell of noble gas atoms is fully occupied by electrons and the atoms are spherically symmetrical, coulomb interactions, dipole–dipole interactions and hydrogen-bonding interactions have little effect on the binding between noble gases and the MOFs. The charge-induced, dipole-induced and dispersion forces may, however, play roles in the adsorption of noble gases. Both the experimental and simulation results suggest that the adsorption of noble gases on CuBTC favours molecules. This result may be due to the different electronic polarizability, size and van der Waals well depths (ε) of the noble gas atoms. The electronic polarizability of Xe, Kr and Ar, expressed in units of (4πε) Å, have been reported as 4.04, 2.48 and 1.63, respectively.43 Electronic polarizability can result in the displacement of the electron cloud. The larger the displacement is, the more inducible an interaction will happen in the adsorption process. Therefore, Ar and Kr showed a lower interaction with the known binding sites in CuBTC compared to Xe. In addition, the ε of the noble gas atoms may affect their adsorption properties in the framework. Greathouse compared the van der Waals well depths of Xe, Kr and Ar with experimental data, and found out that noble gases with a larger van der Waals well depth tend to be preferentially adsorbed by adsorbents at low pressure.16 Fig. 4 compares the adsorption capacity of CuBTC for Ar and Kr and the adsorption selectivity to Xe in equimolar Xe/Kr and Xe/Ar mixtures at 100 kPa using the van der Waals well depths as a coordinator. The results clearly suggest that the adsorption capacity is correlated to the van der Waals well depth. A similar correlation was also observed for the van der Waals diameter σ (listed in Table S2†). Therefore, we conclude that the adsorption capacity for noble gases on CuBTC closely correlates with the ε and σ of the noble gas atoms. Here, Xe selectivity was larger in the Xe/Ar mixture due to the ratio of εXe/εAr being larger than εXe/εKr. Since N2 (the main composition of air) is a nonpolar gas with a little quadrupole moment, N2 can be treated as an inert gas to a certain degree.
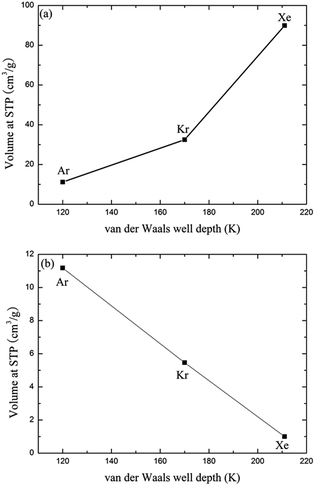 |
| | Fig. 4 Comparison of the adsorption and separation properties of the noble gases by ε. (a) Comparison between ε and the adsorption volume of the noble gases at 100 kPa; (b) comparison between ε and the selectivities of Xe over Kr and Ar at 100 kPa. | |
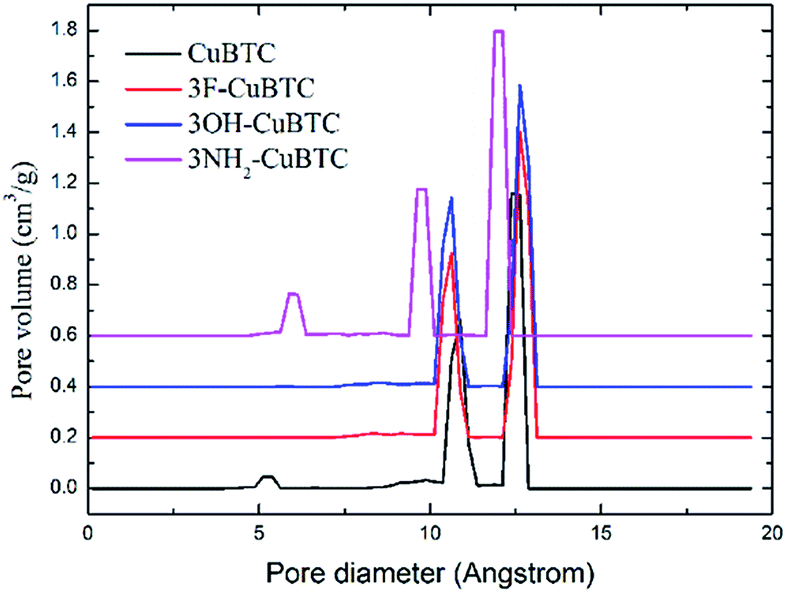
When the functionalized groups were introduced into the original CuBTC metal cluster, it was observed that the angle between the C–X bond (X groups with aromatic carbon atoms) and the aromatic plane was consistent with that between the C–H bond with the aromatic plane in the original CuBTC metal cluster. The structural properties of X-CuBTC and CuBTC, including the accessible surface area (Sacc), pore volume (Vp) and density of MOFs (ρMOF), are displayed in Table 2. The pore size distribution (PSD), listed in Fig. 5, was calculated from the Poreblazer code developed by Sarkisov et al.44 The values of Sacc and Vp decreased with the addition of functional groups into the framework. Noticeably, a larger functional group (–NH2![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) OH
OH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) F) led to smaller values of Sacc and Vp. As shown in the pore size distributions (Fig. 5), the peaks could be assigned as T1, L2 and L3 cage types from left to right, respectively. The T1 cages centred at 5 Å are invisible in 3OH-CuBTC and 3F-CuBTC, indicating that the small cages can be easily blocked by the functional groups. This implies that the presence of inserted groups gives rise to a steric hindrance effect in the small T1 cages. In contrast, the –OH and –F groups did not affect the size distribution of the L2 cages (the peak centred at 11 Å) and the L3 cages (the peak centred at 11 Å), suggesting that there are no functional groups in the L2 cages and L3 cages of these two functionalized CuBTCs. Interestingly, the diameter of the T1 cages in 3NH2-CuBTC increased to 5.9 Å, while the diameter of the L2 and L3 cages decreased to 9.7 Å and 12 Å, respectively. The enlargement of the T1 cages may be attributed to the gas adsorption of the MOF at low pressure, and the reduction in size of the L2 and L3 cages may increase the interaction between the gas and MOF. Commonly, when a gas is adsorbed in the L2 and L3 cages, the MOF may prefer to adsorb the larger gas due to its larger dynamic diameter. The gas would then diffuse into the L2 and L3 cages after the T1 cages were filled. Therefore, the reduction in size of the L2 and L3 cages in 3NH2-CuBTC would help the absorption of the larger gas at high pressure. In addition, the functionalized groups can induce a steric hindrance effect in the adsorption of noble gases and N2.
F) led to smaller values of Sacc and Vp. As shown in the pore size distributions (Fig. 5), the peaks could be assigned as T1, L2 and L3 cage types from left to right, respectively. The T1 cages centred at 5 Å are invisible in 3OH-CuBTC and 3F-CuBTC, indicating that the small cages can be easily blocked by the functional groups. This implies that the presence of inserted groups gives rise to a steric hindrance effect in the small T1 cages. In contrast, the –OH and –F groups did not affect the size distribution of the L2 cages (the peak centred at 11 Å) and the L3 cages (the peak centred at 11 Å), suggesting that there are no functional groups in the L2 cages and L3 cages of these two functionalized CuBTCs. Interestingly, the diameter of the T1 cages in 3NH2-CuBTC increased to 5.9 Å, while the diameter of the L2 and L3 cages decreased to 9.7 Å and 12 Å, respectively. The enlargement of the T1 cages may be attributed to the gas adsorption of the MOF at low pressure, and the reduction in size of the L2 and L3 cages may increase the interaction between the gas and MOF. Commonly, when a gas is adsorbed in the L2 and L3 cages, the MOF may prefer to adsorb the larger gas due to its larger dynamic diameter. The gas would then diffuse into the L2 and L3 cages after the T1 cages were filled. Therefore, the reduction in size of the L2 and L3 cages in 3NH2-CuBTC would help the absorption of the larger gas at high pressure. In addition, the functionalized groups can induce a steric hindrance effect in the adsorption of noble gases and N2.
Table 2 Structural parameters of the CuBTCs
| MOF |
Pore volume (cm3 g−1) |
Sacc (m2 g−1) |
ρcrystal (g cm−3) |
| CuBTC |
0.879 |
1924.49 |
0.805 |
| 3F-CuBTC |
0.708 |
1774.92 |
1.010 |
| 3OH-CuBTC |
0.711 |
1787.92 |
1.014 |
| 3NH2-CuBTC |
0.683 |
1701.00 |
1.034 |
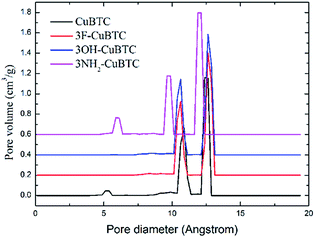 |
| | Fig. 5 Comparison of the pore diameter distribution of the CuBTCs. | |
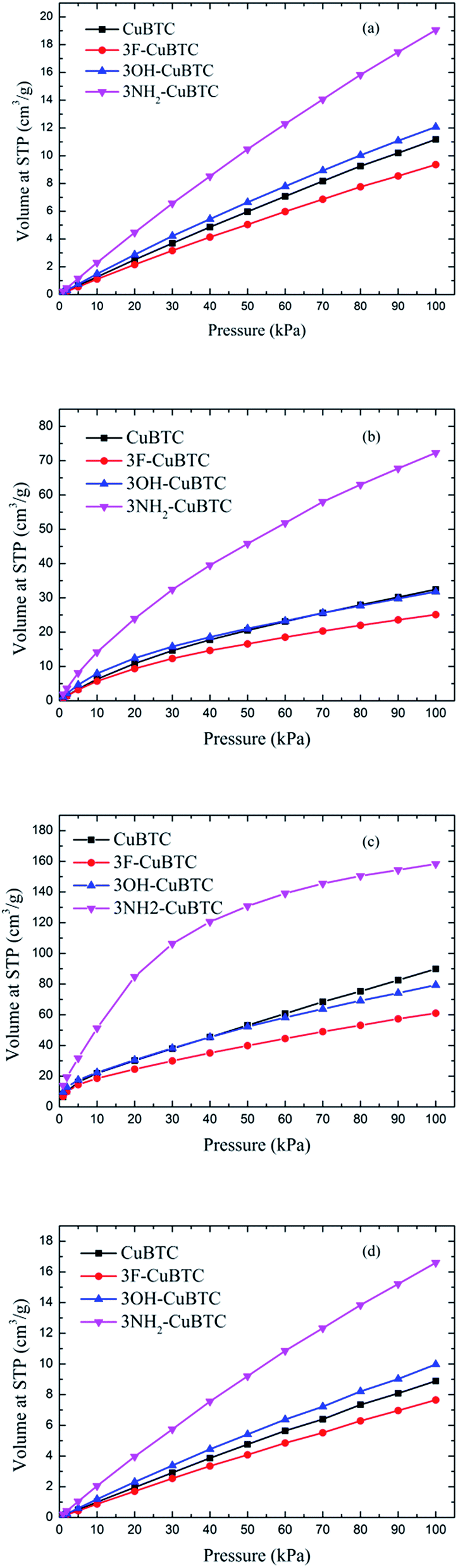
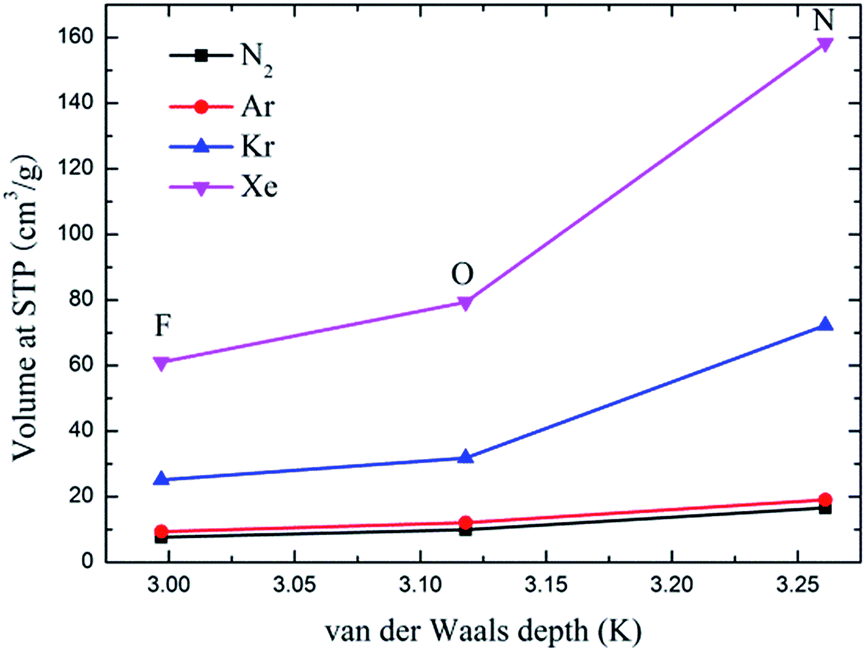
Fig. 6 depicts the adsorption isotherms of noble gases and N2 on CuBTCs with different functional groups at a pressure from 0 to 100 kPa at 292 K. Among the three CuBTC samples, 3NH2-CuBTC exhibited the highest adsorption capacities for all the studied gases, while 3OH-CuBTC showed similar adsorption properties with the parent CuBTC and 3F-CuBTC exhibited a relatively weak adsorption for all the gases. The order of polarity of the functionalized groups is F > OH > NH2. The low adsorption capacity of 3F-CuBTC for noble gases is constructive since the noble gas atoms or the N2 atoms would be favourably adsorbed on the atom or groups with a higher polarizability. This may suggest that noble gases and N2 were not preferred to be adsorbed near the functional groups, particularly at low pressure. In order to understand the adsorption properties of the functionalized CuBTC, the polarizabilities of the functionalized aromatic ring were calculated (listed in Table 3). The increased polarizability follows the order of benzene > 3F-benzene > 3OH-benzene > 3NH2-benzene, which is in agreement with the order of the enhanced value of the adsorption volume of noble gases and N2 at low pressure, except for the 3F-benzene. This indicates that the noble gases and N2 are preferentially adsorbed on the aromatic planes of the CuBTC with different functional groups. The lower capacity for 3F-CuBTC can be attributed to the similar polarizability between benzene and 3F-benzene, rather than the much higher weight of the fluorine atom compared to the hydrogen atom. We also observed that the adsorption capacity of the gases is correlated with the van der Waals well depth of the centre atoms of the functionalized groups. Fig. 7 compares the adsorption volume of gases at 100 kPa in CuBTCs by using the ε of the centre atoms of the functionalized groups and the hydrogen atom. There is a remarkable relationship between the van der Waals well depths of the noble gas and the adsorption volume at 100 kPa. In other words, the inserted group with larger van der Waals well depths contributed more to the adsorption volume of each gas. Although the van der Waals well depth of the F atom is larger than that of the hydrogen atom, the adsorption volume of each noble gas on CuBTC is larger than that on 3F-CuBTC. This was likely to be associated with the 16 times larger molecular weight of the F atom compared to that of the hydrogen atom, which has a larger effect than the van der Waals well depth on the adsorption property.
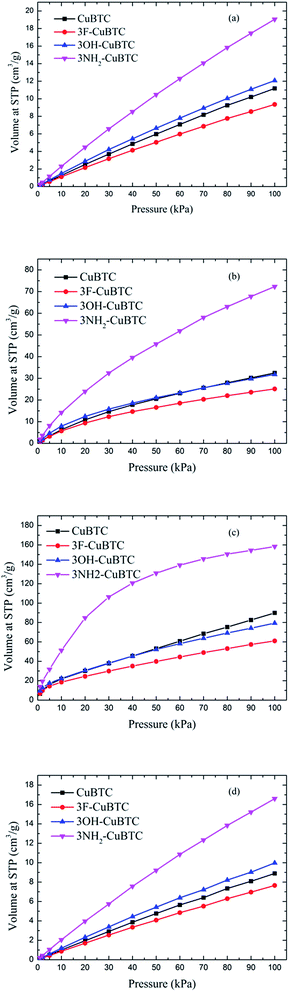 |
| | Fig. 6 Adsorption isotherms of (a) Ar, (b) Kr, (c) Xe and (d) N2 at 292 K. | |
Table 3 Polarizability of the functionalized-benzene derivatives
| Benzenes |
Polarizability (C m2 V−1) |
| Benzene |
70.55 |
| 3F-benzene |
70.82 |
| 3OH-benzene |
88.12 |
| 3NH2-benzene |
103.49 |
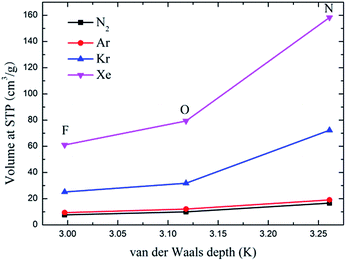 |
| | Fig. 7 Comparison of the adsorption volume of the noble gases and N2 at 100 kPa by the van der Waals well depth of the centre atom of the functionalized groups. | |
Preferential adsorption sites
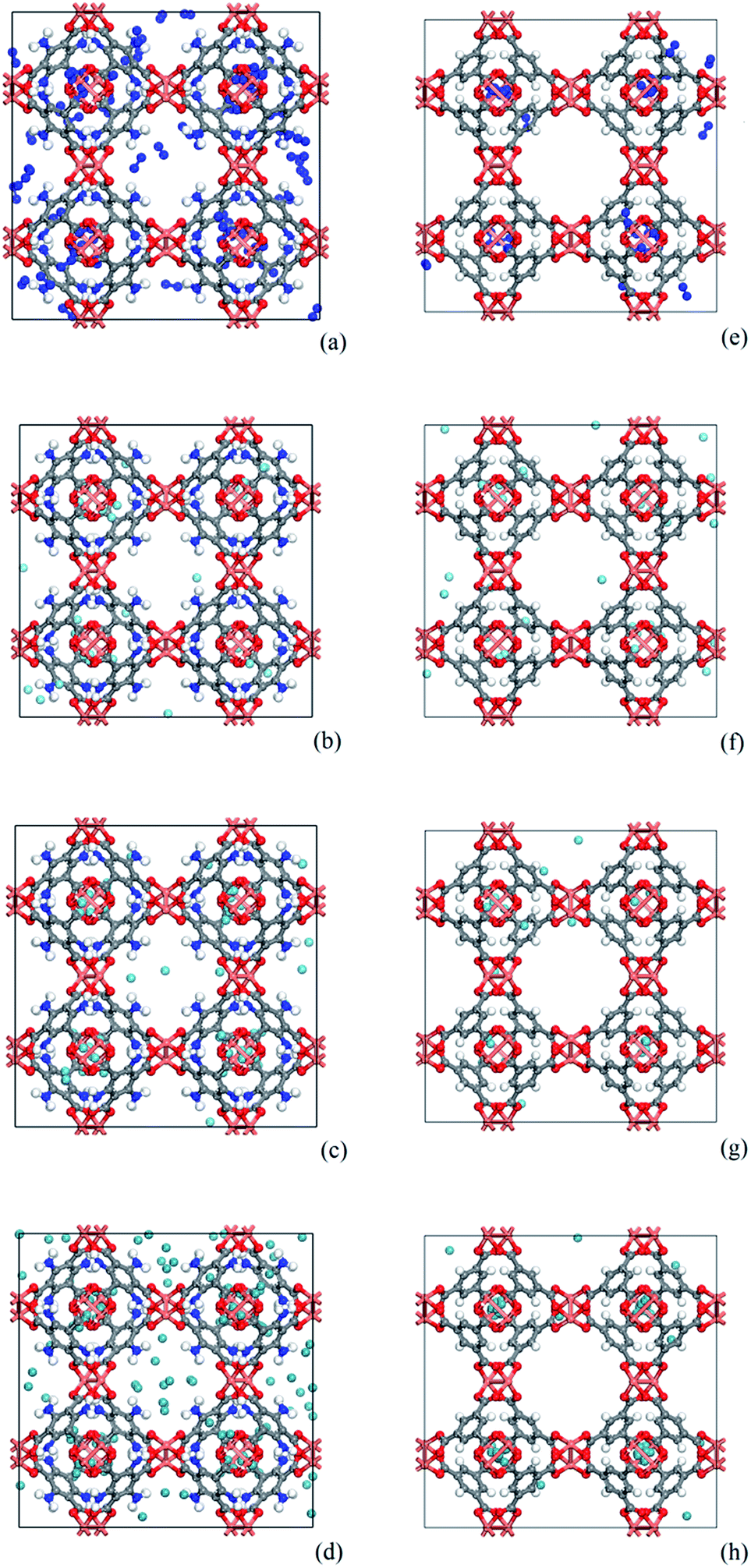
As shown in Fig. 8, snapshots from the production simulation were undertaken to further understand the adsorption mechanism of the noble gases and N2 on the adsorbents with different functionalized groups. Fig. 8(e)–(h) indicate that each gas was preferentially adsorbed in the T1 cages of CuBTC, and then subsequently diffused into the L3 cages after the T1 cages were mostly filled. Some of the gases were adsorbed in L2 cages due to the polarizability regions surrounding the open copper atoms. As we can see, most of the gases were preferentially adsorbed above the centre of the organic linker in CuBTC. A similar phenomenon was found in 3NH2-CuBTC. As shown in Fig. 8(a)–(d), the insertion of amino groups enhanced the adsorption amount of Xe in 3NH2-CuBTC. This phenomenon was attributed to the enhancement in polarizability of the linker and the reduction in size of the L2 and L3 cages. At low pressure, the enhancement in polarizability polarized the adsorbed gases, which consequently enhanced the interaction between the gas atoms with linkers and accelerated the filling of the T1 cages. Also, the reduction in size of the L2 and L3 cages could have decelerated the diffusion of Xe in 3NH2-CuBTC and enhanced the interaction between the gas atoms and the framework. In addition, the noble gases are mainly centred in T1 pockets and L3 pockets, which indicated that noble gases were preferentially bound with aromatic rings or the carbon site connected with the aromatic ring. Similar phenomenon was found on the other functionalized groups.
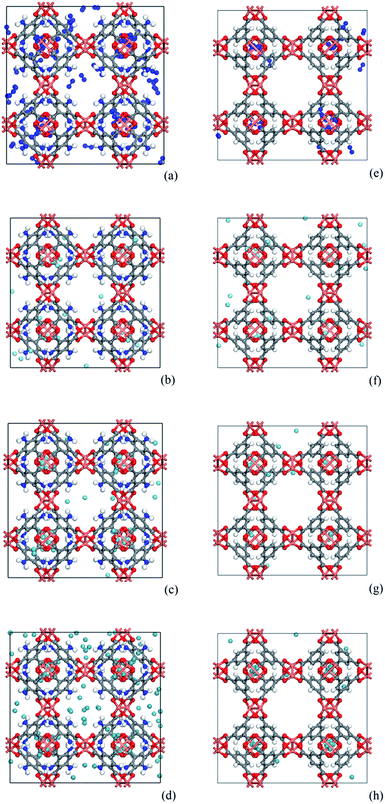 |
| | Fig. 8 Comparison of snapshots of 3NH2-CuBTC and CuBTC. (a)–(d) represent N2 at 100 kPa, Ar at 50 kPa, Kr at 10 kPa and Xe at 5 kPa in 3NH2-CuBTC, respectively; (e)–(h) represent N2 at 100 kPa, Ar at 50 kPa, Kr at 10 kPa and Xe at 5 kPa in CuBTC, respectively (copper: orange, oxygen: red, carbon: grey, hydrogen: white, nitrogen: dark blue, fluorine: light blue, noble gas: light blue, N2: dark blue). | |
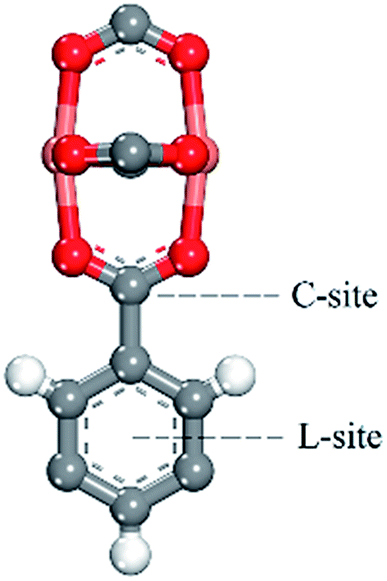
We predicted the possible binding site by comparing the binding energy at the different binding sites. We found there may be two binding sites (in Fig. 9) in the process of adsorbing noble gases. One is the aromatic ring site (L-site) and the other one is the C-site. After the optimization of the adsorption distance, it was found that the noble gas atoms preferred to stay above the aromatic rings and moved away from the C-site. Thus, we calculated the binding energy of Xe with the L-site of the 3NH2-CuBTC and CuBTC clusters. The data showed that E3NH2-CuBTC was −8.319 kJ mol−1, while ECuBTC was −0.035 kJ mol−1. The insertion of amino groups greatly increased the binding energy of Xe with the clusters, which indicates that the amino group can significantly enhance the interaction of gas atoms with the aromatic rings (L-site).
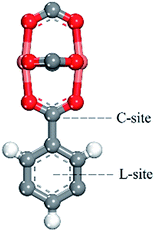 |
| | Fig. 9 Possible binding sites of the noble gases and N2 in CuBTC at low pressure. | |
Adsorptive selectivities
The ideal adsorbed solution theory (IAST) of Myers and Prausnitz45 was applied to calculate the adsorption selectivities of specific noble gases over N2 or the other noble gases. IAST, which assumes that the adsorbed mixture is an ideal solution with a constant temperature and pressure, has been widely used to predict the adsorption selectivities of binary gas mixtures from pure component isotherms.46–48 According to IAST, the chemical potential of the adsorbed solution is considered to be equal to that of the gas phase at equilibrium.45
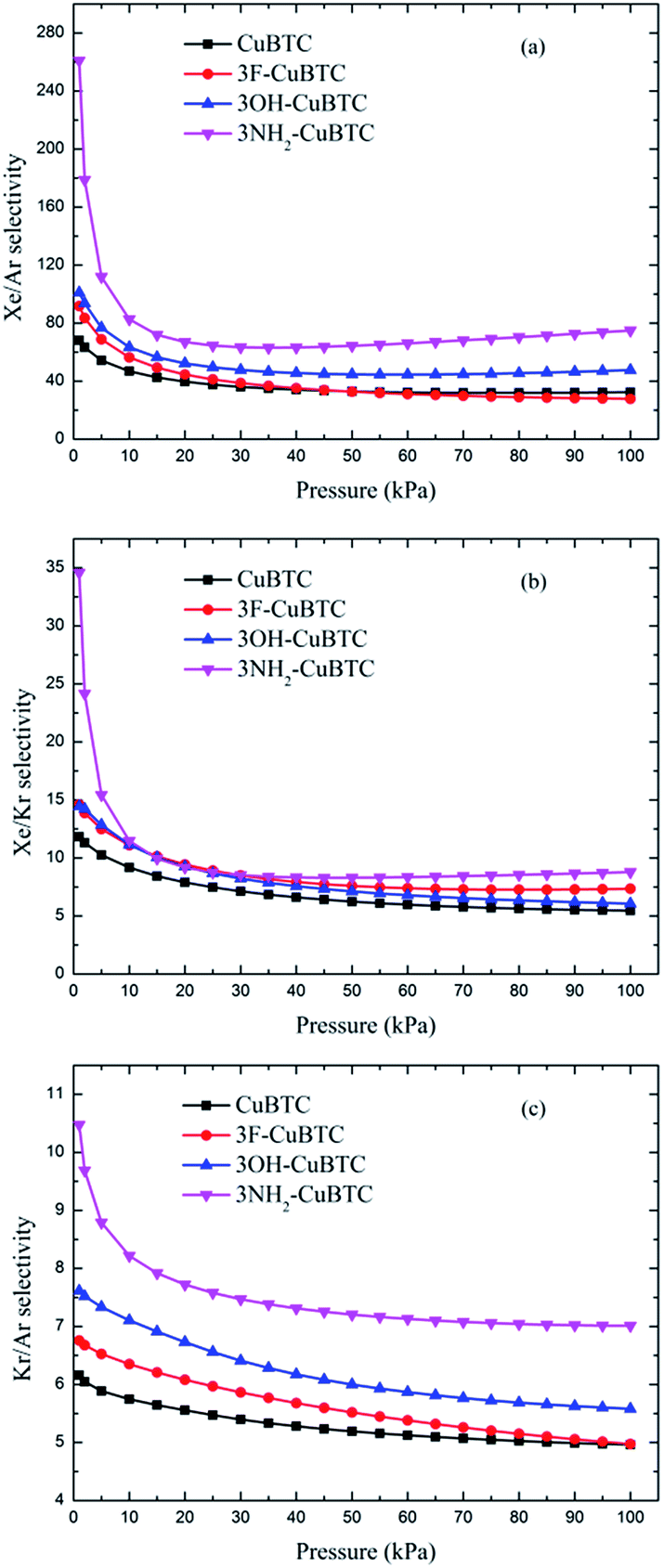
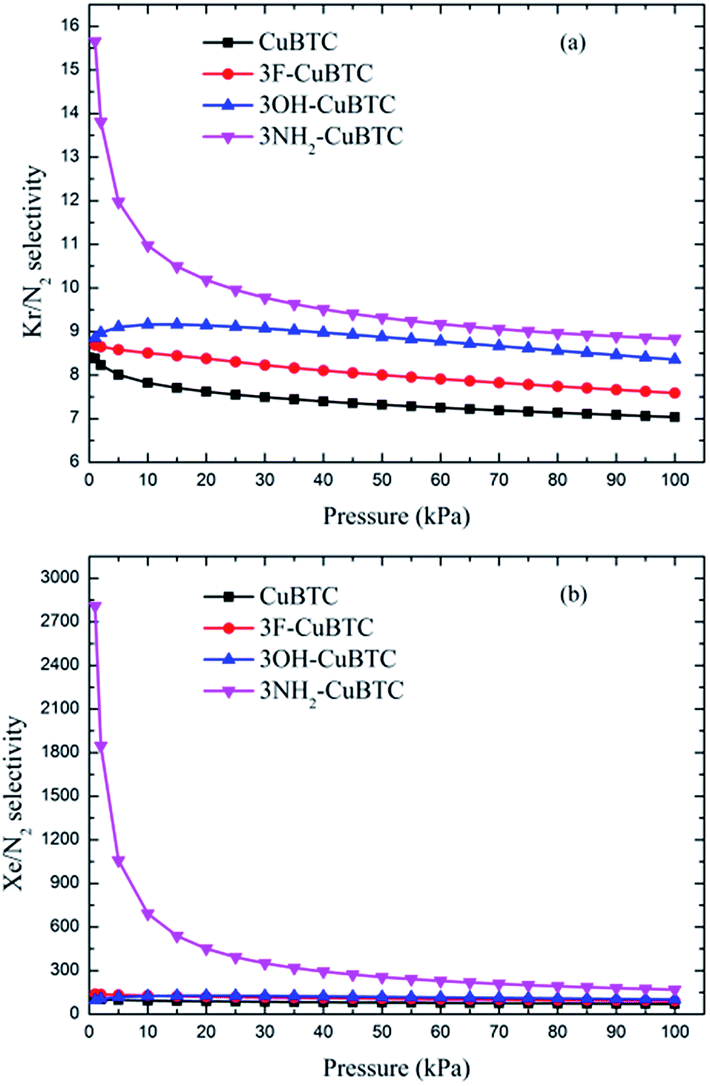
Fig. 10 and 11 display the adsorption selectivities of various noble gases and N2 on the four types of CuBTCs. For the three types of selectivities in Fig. 10, there was a visible enhancement on 3NH2-CuBTC when the pressure was lower than 20 kPa, while the selectivity did not change significantly with increasing the pressure. A similar phenomenon was observed on the other three functionalized CuBTCs. The diameter of the L2 and L3 cages of 3OH-CuBTC was equal to that of 3F-CuBTC, but the polarizability of 3OH-benzene was larger than that of 3F-CuBTC. This larger polarizability would enhance the interaction between the larger gas atoms with the benzene of the L2 and L3 cages at high pressure. Therefore, it can be found that all the selectivities of 3OH-CuBTC were larger than those of 3F-CuBTC. Fig. 11 shows the selectivities for CuBTCs for Xe/N2 and Kr/N2 mixtures, including a 99% molar fraction for N2. For the adsorption selectivity of Xe on N2, 3NH2-CuBTC showed an outstanding performance compared to the three other CuBTCs. Since the molar fraction of N2 is high, a slight change in the adsorption amount of Xe might help in enhancing the selectivity of Xe/N2. The adsorption volume of Xe in 3NH2-CuBTC is nearly twice that in CuBTC. Therefore, 3NH2-CuBTC is the best adsorbent among these three functionalized CuBTCs. The selectivity of Xe/Ar, Xe/Kr, K/Ar and each noble gas over N2 decreased with the increasing pressure, which can be attributed to the differences in the molecular dynamic diameter of each gas (N2: 3.64 Å, Ar: 3.542 Å, Kr: 3.655 Å, Xe: 4.047 Å). With the increasing pressure, the smaller gas atoms have an advantage to be able to flow into the L2 cage or L3 cage easier than the larger gas atoms, resulting in a decrease in the selectivity. In addition, for the selectivity of the other noble gases over N2, there was little interaction between the N2 atoms. Therefore, the competitive adsorption ability of the larger molecules would be minimized.
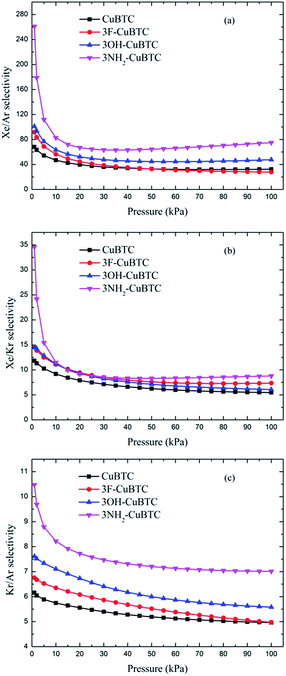 |
| | Fig. 10 Selectivities of CuBTCs for equimolar (a) Xe/Ar, (b) Xe/Kr and (c) Kr/Ar mixtures. | |
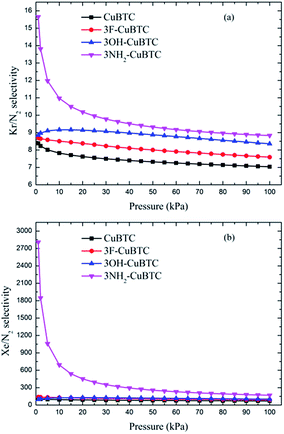 |
| | Fig. 11 Selectivities of CuBTCs for (a) Kr/N2 and (b) Xe/N2 mixtures, including a 99% molar fraction for N2. | |
Herein, we deduced that the polarizability of the functionalized benzenes, the van der Waals well depth and the electron delocalization on the aromatic ring are the main factors that dominate the adsorption property of the functionalized CuBTC. The functionalized groups can influence the electron cloud distribution on the aromatic ring. The three inserted groups can be classified into two types: (i) amino and hydroxyl groups, which are all electron-excluded groups that can average the positive charge from the aromatic ring and move an equal number of electrons to return to the aromatic ring; (ii) fluorine atoms, which represent an electron-attracting group that can minimize the electron density.
Conclusions
The simulation results demonstrate that a mixed force field, including the Universal Force Field and OPLS-AA force field, is suitable to simulate the adsorption and separation of noble gases and N2 on the CuBTC MOF. This force field was used to predict the adsorption and separation properties of noble gases and N2 on CuBTC and on functionalized CuBTC. The simulation results exhibited that amino-functionalized CuBTCs could effectively enhance the adsorption volume and separation property of noble gases over N2 and over the other noble gases at low pressure. The selectivity of Xe over N2 in 3NH2-CuBTC was able to maintain a high value even when the pressure was increased to 100 kPa. The aromatic ring was found to be attractive to noble gases and N2 for its rich electron cloud, which can induce these gases to stay above the ring. The gases with a larger van der Waals well depth can be attractively adsorbed in CuBTCs, while the functionalized groups with a larger van der Waals well depth and smaller size can enhance the adsorption and separation of noble gases over N2 and over the other noble gases. The high polarizability and electron cloud density of the best binding sites can also contribute to the adsorption and separation of the noble gas over N2 and over the other noble gases. More functionalized CuBTCs should be further investigated to explore the adsorption and separation mechanism fully.
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Nos 21436005 and 21576094), the National High Technology Research and Development Program of China (No. 2013AA065005), SRFDP (No. 20130172110012) and Guangdong Natural Science Foundation (S2011030001366).
Notes and references
- Z. T. Lu, P. Schlosser, W. M. Smethie, N. C. Sturchio, T. P. Fischer, B. M. Kennedy, R. Purtschert, J. P. Severinghaus, D. K. Solomon, T. Tanhua and R. Yokochi, Earth-Sci. Rev., 2014, 138, 196–214 CrossRef CAS.
- P. K. Thallapally, J. W. Grate and R. K. Motkuri, Chem. Commun., 2012, 48, 347–349 RSC.
- Z. Liu, T. Araki, Y. Okajima, M. Albert and H. Hatabu, Eur. J. Radiol., 2014, 83, 1282–1291 CrossRef PubMed.
- T. Fujino, M. Furuki, M. Kashitani, K. Onda, J. Kubota, J. N. Kondo, A. Wada, K. Domen, C. Hirose, F. Wakabayashi, M. Ishida, F. Goto and S. S. Kano, J. Chem. Phys., 1996, 105, 279–288 CrossRef CAS.
- A. Ringbom, T. Larson, A. Axelsson, K. Elmgren and C. Johansson, Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment, 2003, 508, 542–543 CrossRef CAS.
- M. Foroutan and A. T. Nasrabadi, Chem. Phys. Lett., 2010, 497, 213–217 CrossRef CAS.
- M. Foroutan and A. T. Nasrabadi, Phys. E, 2011, 43, 851–856 CrossRef CAS.
- N. u. Qadir, S. A. M. Said and H. M. Bahaidarah, Microporous Mesoporous Mater., 2015, 201, 61–90 CrossRef CAS.
- X. Su, Z. Yao, Y. Ye, H. Zeng, G. Xu, L. Wu, X. Ma, Q.-H. Chen, L. Wang, Z. Zhang and S. Xiang, Inorg. Chem., 2016, 55, 983–986 CrossRef CAS PubMed.
- Y. Chen, Z. Li, Q. Liu, Y. Shen, X. Wu, D. Xu, X. Ma, L. Wang, Q.-H. Chen, Z. Zhang and S. Xiang, Cryst. Growth Des., 2015, 15, 3847–3852 CAS.
- R. J. Li, M. Li, X. P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- J. Park, H. Kim and Y. Jung, J. Phys. Chem. Lett., 2013, 4, 2530–2534 CrossRef CAS.
- B. Gole, A. Kumar Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 12137–12139 RSC.
- Q. L. Li, J. P. Wang, W. C. Liu, X. Y. Zhuang, J. Q. Liu, G. L. Fan, B. H. Li, W. N. Lin and J. H. Man, Inorg. Chem. Commun., 2015, 55, 8–10 CrossRef CAS.
- J. A. Greathouse, T. L. Kinnibrugh and M. D. Allendorf, Ind. Eng. Chem. Res., 2009, 48, 3425–3431 CrossRef CAS.
- M. V. Parkes, C. L. Staiger, J. J. Perry, M. D. Allendorf and J. A. Greathouse, Phys. Chem. Chem. Phys., 2013, 15, 9093–9106 RSC.
- Q. Wang, H. Wang, S. Peng, X. Peng and D. Cao, J. Phys. Chem. C, 2014, 118, 10221–10229 CAS.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. Guy Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- J. Liu, J. T. Culp, S. Natesakhawat, B. C. Bockrath, B. Zande, S. G. Sankar, G. Garberoglio and J. Kar Johnson, J. Phys. Chem. C, 2007, 111, 9305–9313 CAS.
- Z. Liang, M. Marshall and A. L. Chaffee, Energy Fuels, 2009, 23, 2785–2789 CrossRef CAS.
- L. Hamon, E. Jolimaître and G. D. Pirngruber, Ind. Eng. Chem. Res., 2010, 49, 7497–9503 CrossRef CAS.
- A. I. Skoulidas, J. Am. Chem. Soc., 2004, 126, 1356–1357 CrossRef CAS PubMed.
- M. V. Parkes, H. Demir, S. L. Teich-McGoldrick, D. S. Sholl, J. A. Greathouse and M. D. Allendorf, Microporous Mesoporous Mater., 2014, 194, 190–199 CrossRef CAS.
- Y. Gurdal and S. Keskin, J. Phys. Chem. C, 2013, 117, 5229–5241 CAS.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- Y. Cai, Y. Zhang, Y. Huang, S. R. Marder and K. S. Walton, Cryst. Growth Des., 2012, 12, 3709–3713 CAS.
- Q. Yang and C. Zhong, J. Phys. Chem. B, 2007, 110, 17776–17783 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, GAUSSIAN 09, Rev. D.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. Fröba, K. Peikert and F. Hoffmann, Chem. Commun., 2012, 48, 11196–11198 RSC.
- Y. Cai, A. R. Kulkarni, Y.-G. Huang, D. S. Sholl and K. S. Walton, Cryst. Growth Des., 2014, 14, 6122–6128 CAS.
- C. J. Casewit, A. K. Rappe, K. S. Colwell, W. A. Goddard III and W. M. Skid, J. Am. Chem. Soc., 1992, 114, 10025–10035 Search PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. T. Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- J. J. Perry, S. L. Teich-McGoldrick, S. T. Meek, J. A. Greathouse, M. Haranczyk and M. D. Allendorf, J. Phys. Chem. C, 2014, 118, 11685–11698 CAS.
- R. J. M. Pellenq and P. E. Levitz, Mol. Phys., 2002, 100, 2059–2077 CrossRef CAS.
- J. J. van Loef, Physica B+C, 1981, 103, 133–157 CrossRef CAS.
- J. J. Potoff and J. Ilja Siepmann, AIChE J., 2001, 47, 1676–1682 CrossRef CAS.
- T. Düren and R. Q. Snurr, J. Phys. Chem. B, 2004, 108, 15703–15708 CrossRef.
- Materials Studio 5.0, Accelrys, San Diego, Ca, 2010 Search PubMed.
- O. Talu and A. L. Myers, AIChE J., 2001, 7, 1160–1168 CrossRef.
- M. Heuchel, R. Q. Snurr and E. Buss, Langmuir, 1997, 13, 6795–6804 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. Schiltz, R. Fourme and T. Prangé, Methods Enzymol., 2003, 374, 83–119 CAS.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248–1257 CrossRef CAS.
- A. L. Myers and J. J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 1–9 Search PubMed.
- Y.-S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 19, 2131–2134 RSC.
- B. Liu, Q. Yang, C. Xue, C. Zhong, B. Chen and B. Smi, J. Phys. Chem. C, 2008, 112, 9854–9860 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08778g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.