
Preparation of multicompartment silica-gelatin nanoparticles with self-decomposability as drug containers for cancer therapy in vitro†
Anhe Wanga,
Yang Yangb,
Xuehai Yana,
Guanghui Maa,
Shuo Bai*a and
Junbai Li*bc
aNational Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: baishuo@ipe.ac.cn
bNational Center for Nanoscience and Technology, Beijing, 100190, China
cKey Lab of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
First published on 18th July 2016
Abstract
We demonstrate multicompartment silica-gelatin nanoparticles (MSGNs), using gelatin doped CaCO3 particles as templates, with self-decomposability in response to body temperature as drug carriers for cancer therapy in vitro. The use of CaCO3 particles provides facile size control of MSGNs in nanoscale without using surfactants. Gelatin involved in the hybrid particles will cover the surface of particles, reducing the toxicity of silica materials caused by Si–OH groups, and moreover plays the role of the cement in the construction to hold the multicompartment structure. Above the gelation temperature, gelatin molecules can escape from particles resulting in the self-decomposition of MSGNs under body conditions. The obtained MSGNs have the potential to be used as an efficient carrier in cancer therapy.
Introduction
In the past decade, silica-based nano/micro- materials have obtained much interest due to their designable porosity and biocompatibility with great potential of applications, such as catalysis, coating and biomedicine.1–6 For instance, silica particles with mesoporous structure have become the most promising candidates in disease diagnosis and therapy.6 Anticancer drugs, proteins and RNA can be loaded within particles through chemical bonding or physical adsorption with high efficiency to meet the demands of therapy.7–12 The hydrophilic surface of the silica particles, which is favourable for cellular internalization, can be modified by various functional groups endowing them with unique chemical, physical and biological properties.13–21 However, silica based materials as drug carriers still suffer from the low rate of decomposition in vitro and in vivo due to their stable network structures and have the risk of accumulation in human body, which may be harmful and limit their further application in diagnosis and therapy field. Several strategies have been developed and partially solve the problems.22–26 For example, Zhang and co-workers have incorporated various molecules (methylene blue doxorubicin, oxazine 725, and LDS751) into silica nanoparticles during the TEOS (tetraethyl orthosilicate) hydrolysis process creating a concentration gradient of drugs within the particles, which could achieve the controllable drug release and trigger the carrier decomposition simultaneously.22 Hao and co-workers reported synthesis of silica/hydroxyapatite nanocomposites as carriers for drug release after decomposition of hybrid particles through the dissolution of HAP in responsive to pH.23 As an ideal candidate for drug delivery and therapy, how to fabricate uniform silica particles with controlled size, large porous structure, self-decomposability, responsivity of body temperature and tumor microenvironment in a facile manner is still in a big challenge.As a major inorganic substances produced in nature, CaCO3 materials have great applications in toothpastes, cosmetics, paper industry, water treatment, catalysis and bio-nanotechnologies due to their properties, such as biocompatibility, porous structure, large surface area, easy production and removal under mild conditions.27,28 Especially the size control of CaCO3 particles from nanometer to micrometer have been intensively investigated and utilized as templates, containers or reactors. Encouraged by the success of using CaCO3 particles as templates to fabricate silica particles with the advantages of involving no surfactants and easily template removal,29–31,35 we aimed to construct sponge-like multicompartment silica nanoparticles (MSN) with self-decomposability in response to body temperature, which may carry multiple payloads in a single carrier and release them in separated spatially manner satisfying the key demands for some critical applications.6,32–34
Here, we demonstrate the use of gelatin doped CaCO3 particles as templates to fabricate multicompartment silica-gelatin nanoparticles (MSGN) with fast self-decomposability to overcome this drawback. In detail, gelatin can cover the surface of MSGN reducing the toxicity of silica materials caused by Si–OH groups, moreover, trigger the decomposition of MSGN. Above the gelation temperature (35–40 °C),36–38 gelatin molecules could escape from MSGN resulting in the self-decomposition of MSGN in body conditions. The obtained MSGN here was certified to be an efficient carrier for anticancer drugs and further showed a good efficacy for cancer therapy in vitro.
Experimental
Materials
Tetraethyl orthosilicate (TEOS), ethylene glycol (EG), gelatin, Na2CO3, CaCl2·2H2O, ethylene diamine tetraacetic acid sodium (EDTA), ammonium hydroxide (30%, wt%) and doxorubicin (DOX) were purchased from Sigma-Aldrich. Cell counting Kit-8 (CCK-8) was from Dojindo Molecular Technologies, Inc. Alexa Fluor 488 WGA, Hoechst 33342 cell marker and FM-4-64 were obtained from Molecular Probes Inc. Other reagents were obtained from Beijing Chemical Reagent Co., Ltd, Beijing, China. The water used throughout the experiment was purified with a Milli-Q integral A10 system from Millipore Co., Billerica, MA, USA.Gelatin doped CaCO3 particle
CaCO3 particles with mean size of 450 ± 80 to 230 ± 30 nm (mean value ± SD, n = 100) doped by gelatin were synthesized as described by our previous work.39 Firstly, gelatin (0.5 g) was added to 10 mL water and heated to 50 °C. Then, the gelatin aqueous solution was added to 200 mL hot EG solution with vigorous stirring. After that, CaCl2 (0.14702, 0.58808 g) was added to the above solution. Finally, 30 mL Na2CO3 aqueous solution (0.10599, 0.42396 g) was injected into CaCl2 and gelatin water/EG solution under vigorous stirring at room temperature, and kept stirring for 2 h. The obtained particles were carefully washed by cool water for one times, and then dried in vacuum. CaCO3 particles without gelatin (about 500 nm) were synthesized by mixing CaCl2 and Na2CO3 water/EG solutions according to the literature procedures.40 In a typical experiment, 100 mL Na2CO3 (0.02 M) water/EG solution (17%, v/v%) was poured in to a solution of 100 mL CaCl2 (0.02 M) water/EG solution (17%, v/v%), and the mixture was intensely agitated on a magnetic stirrer with stirring times of 2 h. The prepared particles were washed with deionized water thoroughly for three times and then dried under vacuum.Preparation of MSGN
A 50 mg amount of CaCO3 particles with or without gelatin were incubated in 2 mL TEOS ethanol solution (75%, v/v%). After the adsorption of TEOS, the particles were separated from the TEOS ethanol solution, and moved to a flask with a mixture solution (ammonium hydroxide 2.5 mL, 30 wt%; water 2.5 mL; ethanol 6 mL). The reaction system was kept under vigorous stirring overnight at room temperature. The final products were washed with water for at least three times, and incubated in EDTA solution (0.1 M, pH 7.4) for 1 h to remove the templates. This step was repeated for at least three times to remove the templates completely. At last, the obtained products were washed with water and dried under vacuum for the following experiments.For other methods, please refer to the ESI† for further details.
Results and discussion
The fabrication of MSGN
Scheme 1 shows the procedure of fabrication of MSGN as a self-decomposable carrier. As templates, gelatin doped CaCO3 particles with different diameters were prepared in water/ethylene glycol (EG) mixture solution under the assistance of gelatin molecules according to our previous work.39 By adjusting the concentration from 0.02 to 0.005 M, the diameter of CaCO3 varied from 450 ± 80 to 230 ± 30 nm (mean value ± SD, n = 100) (Fig. 1a and b). The mechanism of how the gelatin molecules controlling the size of CaCO3 particles have been discussed in our previous work, which could be ascribed to the protection of gelatin molecules during the formation of CaCO3 particles.39 Magnified SEM images show that CaCO3 particles are accumulated with particulate nanoparticles with a size of 10–70 nm, which endows the CaCO3 particles with porous structure and high surface area (Fig. 1a and b, right). It is estimated that the CaCO3 particles (230 ± 30 nm, mean value ± SD, n = 100) possess a large surface area about 204 m2 g−1,39 which provide an ideal templates to construct multicompartment architecture in the following experiment.27,28 Thermogravimetric analysis (TGA) certifies that the particles is composited of gelatin and CaCO3 (see the ESI, Fig. S1†). The mass loss at 250–350 °C is attributed to the combustion of gelatin with the amount of 6.8% in weight.41 After incubated in TEOS ethanol solution overnight at room temperature, the pores of CaCO3 templates were saturated with TEOS molecules. Upon that being incubated in hydrolysis medium (2.5 mL ammonium hydroxide, 30 wt%; water 2.5 mL; ethanol 6 mL), the surface of templates was covered with SiO2 by the hydrolysis of TEOS (Fig. 2). Meanwhile the templates kept integrate morphology during the hydrolysis process, accounting for the formation of multicompartment structure of MSGN. After removal of the templates at mild condition (treated by ethylenediaminetetraacetic acid sodium, EDTA, 0.1 M, pH 7.4), MSGN was obtained with a similar size (480 ± 80 and 250 ± 50 nm, mean value ± SD, n = 100) and surface morphology to CaCO3 particles (Fig. 3). The selected area electron diffraction (SAED) pattern verified that the crystal form of MSGN was amorphous (Fig. 3a, inset). For comparison, MSN with similar structure and size (around 500 nm) was also fabricated templated on CaCO3 without gelatin, which will be taken as control in the following self-decomposable experiment (see the ESI, Fig. S2†). | ||
| Scheme 1 Schematic illustration of the preparation of gelatin doped multicompartment silica nanoparticles (MSGN) and their loading and release of drugs. | ||
 | ||
| Fig. 1 SEM images (right: magnified images) of gelatin doped CaCO3 particles with size of (a) 450 ± 80 nm and (b) 230 ± 30 nm (mean value ± SD, n = 100). | ||
 | ||
| Fig. 2 TEM images (right: magnified images) of surface morphology of CaCO3 particles (230 ± 30 nm, mean value ± SD, n = 100) before (a) and after (b) TEOS hydrolysis treatment. | ||
 | ||
| Fig. 3 TEM and SEM images (right: magnified images) of the MSGN templated on different size of CaCO3 particles. (a) 450 ± 80 nm; (b) 230 ± 30 nm (mean value ± SD, n = 100); inset: SAED patterns of MSGN. | ||
It was clearly observed that the MSGN were comprised of nanoshells with a size between 10 and 80 nm and consistent with the size of building units of CaCO3 (Fig. 3a and b). Moreover, the cross section of MSGN gives the view of multicompartment architecture inside the MSGN, which derived from the removal of template by EDTA treatment (see the ESI, Fig. S3†). It is expected the special structure of MSGN benefits the drugs loading with high efficiency and release in separated spatially manner.
The composition of MSGN
The energy dispersive X-ray spectroscopy (EDX) was employed to determine the composition of MSGN (Fig. 4a). The contents of carbon, nitrogen, oxygen, silicon were about 18.15%, 14.87%, 58.22% and 8.75%, respectively (at%, atomic ratio). In contrast, no peak ascribed to calcium was observed indicating the CaCO3 templates were removed completely. It is reasonable to believe that the nitrogen comes from the residual gelatin after template removal, which suggests the MSGN is comprised of SiO2 and gelatin. Meanwhile the nitrogen element mapping showed that the gelatin distributes over the whole MSGN homogeneously (Fig. 4b). X-ray photoelectron spectroscopy (XPS) results (Fig. 4c), which was consistent with the data of EDX also showed that the surface of MSGN was composed of silicon, carbon, nitrogen and oxygen elements without residual CaCO3. High resolution Si 2p peak analysis showed that there were some differences of chemical environment of silicon between MSGN and MSN (Fig. 4d). In detail, the peak of Si 2p appeared at 103.9 eV, which represented the contribution of silicon atoms of MSN.42 However, the absorption of Si 2p obtained in the MSGN appeared as two peaks. One was at 103.3 eV (zone I) ascribed to the contribution of silicon atoms from MSN. Another peak located at 99.7 eV (zone II) with lower energy indicated that partial silicon atoms in MSGN were occupied by newly formed bonds. At this stage, maybe some nitrogen or carbon atoms in gelatin chains served as electron donors for the formation of the new bonds, resulting in the decrease of silicon binding energy on the surface of MSGN.42 Based on those data, it is believed that the surface of MSGN was covered by gelatin, which could reduce the toxicity of MSGN caused by Si–OH groups and have no use for further modification of surface for biocompatibility.43,44 | ||
| Fig. 4 (a) EDX spectrum of MSGN (250 ± 50 nm, mean value ± SD, n = 100); (b) the corresponding element mapping of MSGN (c) XPS spectrum of MSGN; (d) Si 2p analysis for MSN and MSGN, respectively. | ||
The self-decomposability of MSGN
Biodegradability is critical requirement for non-natural materials used as drug carriers, which means the drug carrier should be decomposed and removed from the body after successful completion of cargo release to the lesion location. To test the biodegradability, MSGNs with different size (480 ± 80 and 250 ± 50 nm, mean value ± SD, n = 100, 0.1 mg mL−1) were incubated in PBS (0.1 M, pH 7.4) or acetate (0.1 M, pH 5.0) buffer solution at 25 or 37 °C to investigate the effect of the pH and temperature on the decomposability of MSGN. The corresponding morphological evolution of the MSGN was recorded by TEM at predetermined intervals, meanwhile MSN was applied as a control (templated on CaCO3 and fabricated in EG/water solution without assistance of gelatin, the details were given in ESI†). As shown in Fig. 5a, most of the MSGN (480 ± 80 nm, mean value ± SD, n = 100) remained after incubation in PBS solution for 24 h at 37 °C, but became hollow and broken gradually after 48 h and left a spherical thin shell after 96 h. Finally, all of the MSGN were damaged and completely collapsed into pieces of fragments after 288 h incubation. However, the same particles incubated in PBS buffer at 25 °C (see the ESI, Fig. S4a†) still kept their integral structure after 144 h incubation and appeared only partial decomposition after 288 h incubation, which could be attributed to the enhanced solubility of silica in PBS buffer solution.45,46 It strongly indicates the significant effect of the gelatin molecules on the decomposition of MSGN. The mechanism may be because the gelatin molecule with random coil state could diffuse out from MSGN up to the gelation temperature (35–40 °C), resulting in the structure damage of MSGN. While the gelatin molecules tent to hold interrupted helical form, preventing their escaping from the MSGN at room temperature.36–38 To exclude the case that the decomposition of MSGN was caused by the increased solubility of SiO2 at high temperature, a sample of MSN was incubated in PBS buffer solution at 37 °C, the TEM data obviously shows there is no morphology change of the particles during the experimental intervals (see the ESI, Fig. S4c†). Moreover, the pH value of environment also affected the decomposition of MSGN, as shown in Fig. S4b (see the ESI†). The morphology of MSGN appeared no visible change after incubation in acetate buffer solution (0.1 M, pH 5.0) at 37 °C during the incubation time, which could be ascribed to the low solubility of gelatin at pH 5.0 near to its pI (4.7–5.2). The results demonstrate that the incorporation of gelatin into MSGN endows the silica particles with self-decomposability in response to the body temperature, which could benefit the interesting cargo loading and release depending on the difference environmental temperature. | ||
| Fig. 5 Typical TEM images of MSGN with size (a) 480 ± 80 and (b) 250 ± 50 nm (mean value ± SD, n = 100) after being incubated in PBS buffer solution (0.1 mg mL−1) at 37 °C. | ||
When decreasing the size of MSGN from 480 ± 80 to 250 ± 50 nm (mean value ± SD, n = 100), the small one had a similar decomposition behavior with that of the big one, but possessed faster decomposition rate, which could be decomposed completely in 144 h (Fig. 5b). It is believed that the larger surface area of small particles gives more contact area with solvent and faster diffusion rate. In addition, a quantitative measurement of Si element in PBS buffer solution (0.1 mg mL−1) incubating MSGN (250 ± 50 nm, mean value ± SD, n = 100) at 37 °C was performed by inductively coupled plasma optical emission spectroscopy (ICP-OES). As Fig. 6a shown, the concentration of Si increased along with the incubation time and reached a balance (14 μg mL−1) at 160 h, which is consistent with the result of TEM (Fig. 5b). Meanwhile it can estimate that about 91% of MSGN incubated in PBS at 37 °C have been decomposed combing the data of EDX which indicated the weigh percentage of Si element in MSGN is about 15.33% (Fig. 4a).
 | ||
| Fig. 6 (a) The concentration of Si element along with the incubation time in PBS buffer solution (MSGN with size about 250 ± 50 nm, mean value ± SD, n = 100); (b) CLSM images of MSGN (labelled by TRITC) incubated with MCF-7 cells for 1 and 9 days, respectively; (c) TEM images of MSGN extracted from MCF-7 cells after 9 days incubation. The MSGN not incubated with cells as control. | ||
The decomposition of MSGN (250 ± 50 nm, mean value ± SD, n = 100) labelled by TRITC was inspected in vitro. After being co-incubated with MSGN for 24 h, MCF-7 cells were investigated by Confocal Laser Scanning Microscopy (CLSM). As indicated in Fig. 6b, the internalization of MSGN by MCF-7 was confirmed by the TRITC fluorescence signal in the cells. The concentrated TRITC fluorescence signal observed in a randomly spotted pattern in the cells (Fig. 6b and S5a in the ESI†) suggested that most of MSGN kept their structure integrity after 1 day incubation. After 9 days incubation with cells, most of the concentrated fluorescent spots in MSGN disappeared, while a rather diffusive fluorescent fragment of spots was observed (Fig. 6b and S5b in the ESI†). It is believed that gelatin molecules escaped from the MSGN or were digested by enzyme inside the cells, resulting in the damage of MSGN. Furthermore, MSGN extracted from MCF-7 cells at 9 d after the uptake showed obvious decomposition in consistent with the data of CLSM (Fig. 6c).
Biocompatibility and application in drug loading and release of MSGN
To assess the safety and biocompatibility of MSGN (480 ± 80 nm, mean value ± SD, n = 100) in vitro, different amount of MSGN was incubated with MCF-7 cells for 24 h to analyze the viability of cells by CCK-8 method (Fig. 7a). No significant cytotoxicity in vitro was observed among the experimental concentration from 0.1 to 1.2 mg mL−1, indicating the excellent biocompatibility of MSGN. It is maybe due to the gelatin covered surface of MSGN shielding the Si–OH groups and reducing their cytotoxicity.43,44 After labelled by FITC, the interaction between MSGN (480 ± 80 nm, mean value ± SD, n = 100) and MCF-7 cells was also investigated by CLSM, which confirmed that the MSGN was internalized by MCF-7 and located in the cytoplasm (Fig. 7b) with high efficiency and without causing obvious damages to the cell, agreeing with the result of viability experiment. It indicates that the MSGN has excellent biocompatibility and can be used as a safe drug carrier.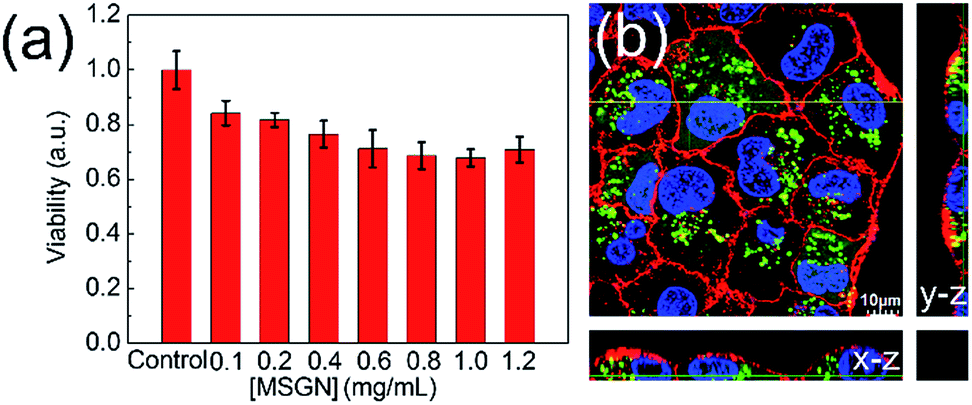 | ||
| Fig. 7 (a) In vitro viability of MCF-7 cells in the presence of MSGN with meaning size 480 ± 80 nm (mean value ± SD, n = 100); (b) 3D CLSM image, x–z (bottom) and y–z (right) views along the two yellow lines, which represent the position where the stack is cut to form the x–z and y–z sections, respectively. The MSGN, nuclei and membrane of MCF-7 were stained by FITC (green fluorescent domains), Hoechst 33342 (blue one) and FM-4-64 (red one), respectively. | ||
Doxorubicin (DOX), a positively charged and widely applied anticancer drug,47 was employed to test the drug encapsulation and release of MSGN (480 ± 80 nm, mean value ± SD, n = 100) in vitro. As shown in TEM images (Fig. 8a), the DOX molecules spontaneously accumulated and formed black spots inside the MSGN due to the electrostatic interaction between MSGN and DOX molecules (denoted as MSGN–DOX). It is estimated that the maximum dosage is about 0.105 mg per mg MSGN. The zeta potential of MSGN after adsorption of DOX is −5.2 mV (MSGN, −28.8 mV), which can only be ascribed to surface adsorption of DOX. Meanwhile, the CLSM also certified that almost all DOX molecules were encapsulated inside the MSGN without obviously leakage of drugs from the MSGN (see the ESI, Fig. S6,† characteristic fluorescence of DOX excited by 559 nm laser).
 | ||
| Fig. 8 (a) TEM images of MSGN (480 ± 80 nm, mean value ± SD, n = 100) loaded with DOX. Insert: magnified TEM images; (b) the release profile of encapsulated DOX from MSGN at different pH; (c) CLSM image of MCF-7 cells incubated with MSGN–DOX for 24 h (fluorescence from DOX excited by 559 nm laser) (d) cytotoxicity of different dosage of MSGN–DOX or free DOX (green one, with the same amount of DOX loaded by MSGN, n = 3, *p < 0.01) to MCF-7 cells, the cells treated without MSGN–DOX or free DOX as control. | ||
It is well known that there are pH differences among the microenvironments of tumor tissues (pH 6.5–6.8), cell endosomes (pH 5.5–6.5) and lysosomes (pH 4.5–5.5) and that of blood stream and normal tissues (pH 7.4).48,49 The samples of MSGN–DOX were incubated in pH 5.0 and 7.4 buffer solutions to assess the drug release in different pH environment. A typical time-dependent release curves was obtained as displayed in Fig. 8b. In the case of pH 7.4, DOX was released slowly from the MSGN. Only about 8.1% of adsorbed DOX was released after 9 h incubation. But about 72% of adsorbed DOX was released during the same time period in pH 5.0 buffer solution. It is demonstrated that the MSGNs could keep the payload in normal physiological environment (pH 7.4) and release them in acidic microenvironments of tumor tissues quickly, which would reduce the side effect of DOX in vivo. We speculates that the surface charge of MSGN becomes neutral at pH 5.0 (zeta potential −0.5 mV), resulting in a weak interaction between DOX and MSGN, which was supported by the fact that the release rata of DOX from MSGN was faster in 0.05 M NaCl than that in 0 and 0.01 M NaCl aqueous solution as depictured in Fig. S7.† Meanwhile DOX has a high solubility in low pH aqueous solution, all of which may contribute to the fast DOX release at pH 5.0.50,51 The release of DOX from the MSGN in vitro was investigated by CLSM. After being incubated with MSGN–DOX for 4 h, the intracellular distribution of the MSGN and the DOX was examined by means of DOX fluorescence signal (excited by 559 nm laser). As shown in Fig. S8 (see the ESI†), concentrated DOX fluorescence signal as a randomly spotted pattern was observed in the cells, suggesting that most DOX molecules were confined inside the MSGN. With the incubation time moving on (24 h), most of the concentrated fluorescent spots disappeared, while a rather diffusive fluorescent background was visualized (Fig. 8c) indicating that the DOX was released from the MSGN and eventually reached the cytoplasma of cells. In addition, a cytotoxicity assay of MSGN–DOX was conducted with MCF-7 cells in vitro using CCK-8 method. The cell viability under various dosages of MSGN–DOX and free DOX was shown in Fig. 8d. In details, cell viability decreased sharply after incubation with different dosages of MSGN–DOX (0.02, 0.04, 0.08 mg mL−1). For example, the viability reduced to 22.7% as the cells were incubated with 0.02 mg mL−1 MSGN–DOX, indicating better therapy efficacy compared with that of free DOX (viability 53.7%, *P < 0.01), which can be explained by that MSGN–DOX can enter cancer cells more easily than free DOX probably through endocytosis mechanism. Once internalized by MCF-7 cells, the MSGN–DOX experience a low pH (endosomes, pH 5.5–6.5 and lysosomes, pH 4.5–5.5) to trigger the release of DOX from the MSGN.52 Therefore, it demonstrated that MSGN delivers drugs in response to the change of pH value, which possibly protects fragile drugs from hard conditions and releases them when reaching the lesion location with more effective therapy.
Conclusions
In conclusion, we demonstrate using gelatin doped CaCO3 particles as templates to fabricate multicompartment silica-gelatin nanoparticles as drug carriers for cancer therapy in vitro. The use of CaCO3 particles provides a facile size control of MSGN in nanoscale without using surfactants. By introducing gelatin molecules through the templates, MSGN shows an excellent biocompatibility and self-decomposes into harmless pieces at body conditions of 37 °C and pH 7.4 in 12 days, which can be ascribed to the collapse of gelatin within MSGN above gelation temperature. As an efficient carrier for loading and transferring anticancer drug, cell experiments demonstrated that the MSGN with DOX had an excellent efficacy to kill cancers compared with that of free DOX in vitro. The methodology presented here offers a facile and economical way to construct self-decomposable drug carrier and facilitates encapsulation and compartmentalization with applications in drug delivery and release in response to pH value and body temperature. There are opportunities to expand the design of multicompartment structure of carriers to include both hydrophilic and hydrophobic drugs and functional nanoparticles for stepwise target drug delivery and release in complicated tumor microenvironment.Acknowledgements
Anhe Wang and Yang Yang contributed equally to this work. This work is financially supported by the Talent Fund of the Recruitment Program of Global Youth Experts, the National Nature Science Foundation of China (51303038, 21273055), National Basic Research Program of China (2013CB932800) and National key foundation for exploring scientific instrument (2013YQ16055108).References
- S. S. Cao, L. Fang, Z. Y. Zhao, Y. Ge, S. Piletsky and A. P. F. Turner, Adv. Funct. Mater., 2013, 23, 2162 CrossRef CAS.
- C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876 RSC.
- X. Du and J. H. He, Nanoscale, 2012, 4, 852 RSC.
- D. S. Moon and J. K. Lee, Langmuir, 2012, 28, 12341 CrossRef CAS PubMed.
- X. Du, X. Y. Li and J. H. He, ACS Appl. Mater. Interfaces, 2010, 2, 2365 CAS.
- Y. Zhao and L. Jiang, Adv. Mater., 2009, 21, 1 Search PubMed.
- S. R. Bhattarai, E. Muthuswamy, A. Wani, M. Brichacek, A. L. Castañeda, S. L. Brock and D. Oupicky, Pharm. Res., 2010, 27, 2556 CrossRef CAS PubMed.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435 CrossRef CAS.
- M. Hartmann and X. Kostrov, Chem. Soc. Rev., 2013, 42, 6277 RSC.
- Y. Yang and J. B. Li, Adv. Colloid Interface Sci., 2014, 207, 155 CrossRef CAS PubMed.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203 RSC.
- T. Suma, K. Miyata, Y. Anraku, S. Watanabe, R. J. Christie, H. Takemoto, M. Shioyama, N. Gouda, T. Ishii, N. Nishiyama and K. Kataoka, ACS Nano, 2012, 6, 6693 CrossRef CAS PubMed.
- D. Tarn, C. E Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792 CrossRef CAS PubMed.
- S. H. Wu, C. Y. Mou and H. P. Lin, Chem. Soc. Rev., 2013, 42, 3862 RSC.
- K. Zhang, L. L. Xu, J. G. Jiang, N. Calin, K. F. Lam, S. J. Zhang, H. H. Wu, G. D. Wu, B. Albela, L. Bonneviot and P. Wu, J. Am. Chem. Soc., 2013, 135, 2427 CrossRef CAS PubMed.
- Y. Chen, H. R. Chen and J. L. Shi, Adv. Mater., 2013, 25, 3144 CrossRef CAS PubMed.
- W. Li, Q. Yue, Y. H. Deng and D. Y. Zhao, Adv. Mater., 2013, 25, 5129 CrossRef CAS PubMed.
- W. Z. Zhou, G. S. Tong, D. L. Wang, B. S. Zhu, Y. Ren, M. Butler, E. Pelan, D. Y. Yan, X. Y. Zhu and S. D. Stoyanov, Small, 2016, 12, 1797 CrossRef CAS PubMed.
- J. H. Xu, F. P. Gao, L. L. Li, H. L. Ma, Y. S. Fan, W. Liu, S. S. Guo, X. Z. Zhao and H. Wang, Microporous Mesoporous Mater., 2013, 182, 165 CrossRef CAS.
- L. Rao, L. L. Bu, J. H. Xu, B. Cai, G. T. Yu, X. L. Yu, Z. B. He, Q. Q. Huang, A. Li, S. S. Guo, W. F. Zhang, W. Liu, Z. J. Sun, H. Wang, T. H. Wang and X. Z. Zhao, Small, 2015, 11, 6225 CrossRef CAS PubMed.
- L. Rao, L. L. Bu, B. Cai, J. H. Xu, A. Li, W. F. Zhang, Z. J. Sun, S. S. Guo, W. Liu, T. H. Wang and X. Z. Zhao, Adv. Mater., 2016, 28, 3460 CrossRef CAS PubMed.
- S. L. Zhang, Z. Q. Chu, C. Yin, C. Y. Zhang, G. Lin and Q. Li, J. Am. Chem. Soc., 2013, 135, 5709 CrossRef CAS PubMed.
- X. H. Hao, X. X. Hu, C. M. Zhang, S. Z. Chen, Z. H. Li, X. J. Yang, H. F. Liu, G. Jia, D. D. Liu, K. Ge, X. J. Liang and J. C. Zhang, ACS Nano, 2015, 9, 9614 CrossRef CAS PubMed.
- D. K. Shen, J. P. Yang, X. M. Li, L. Zhou, R. Y. Zhang, W. Li, L. Chen, R. Wang, F. Zhang and D. Y. Zhao, Nano Lett., 2014, 14, 923 CrossRef CAS PubMed.
- L. Fan, Y. S. Zhang, F. L. Wang, Q. Yang, J. L. Tan, G. Renata, H. Wu, C. J. Song and B. Q. Jin, Biomaterials, 2016, 76, 399 CrossRef CAS PubMed.
- S. S. Zhao, S. L. Zhang, J. Ma, L. Fan, C. Yin, G. Lin and Q. Li, Nanoscale, 2015, 7, 16389 RSC.
- D. Volodkin, Adv. Colloid Interface Sci., 2014, 207, 306 CrossRef CAS PubMed.
- Y. Boyjoo, V. K. Pareek and J. Liu, J. Mater. Chem. A, 2014, 2, 14270 CAS.
- M. Fuji, T. Shin, H. Watanabe and T. Takei, Adv. Powder Technol., 2012, 23, 562 CrossRef CAS.
- Z. Z. Li, L. X. Wen, L. Shao and J. F. Chen, J. Controlled Release, 2004, 98, 245 CrossRef CAS PubMed.
- W. Z. Mao, L. Hu, Q. H. Zhao and C. Y. Gao, Chem. Res. Chin. Univ., 2012, 28, 546 Search PubMed.
- T. Suteewong, H. Sai, R. Hovden, D. Muller, M. S. Bradbury, S. M. Gruner and U. Wiesner, Science, 2013, 340, 337 CrossRef CAS PubMed.
- R. Chandrawati, M. P. van Koeverden, H. Lomas and F. Caruso, J. Phys. Chem. Lett., 2011, 2, 2639 CrossRef CAS.
- K. Maeda, H. Onoe, M. Takinoue and S. Takeuchi, Adv. Mater., 2012, 24, 1340 CrossRef CAS PubMed.
- A. H. Wang, Y. Yang, Y. F. Qi, W. Qi, J. B. Fei, H. C. Ma, J. Zhao, W. Cui and J. B. Li, ACS Appl. Mater. Interfaces, 2016, 8, 8900 CAS.
- S. M. Tosh and A. G. Marangoni, Appl. Phys. Lett., 2004, 84, 4242 CrossRef CAS.
- A. Hayashi and S. Oh, Agric. Biol. Chem., 1983, 47, 1711 CAS.
- M. Zandi, H. Mirzadeh and C. Mayer, Eur. Polym. J., 2007, 43, 1480 CrossRef CAS.
- A. H. Wang, Y. Yang, X. M. Zhang, X. C. Liu, W. Cui and J. B. Li, ChemPlusChem, 2016, 81, 194 CrossRef CAS.
- B. V. Parakhonskiy, A. Haase and R. Antolini, Angew. Chem., Int. Ed., 2012, 51, 1195 CrossRef CAS.
- W. Wei, G. H. Ma, G. Hu, D. Yu, T. Mcleish, Z. G. Su and Z. Y. Shen, J. Am. Chem. Soc., 2008, 130, 15808 CrossRef CAS PubMed.
- M. C. Porté-Durrieu, C. Labrugére, F. Villars, F. Lefebvre, S. Dutoya, A. Guette, L. Bordenave and C. Baquey, J. Biomed. Mater. Res., 1999, 46, 368 CrossRef.
- Y. S. Lin and C. L. Haynes, J. Am. Chem. Soc., 2010, 132, 4834 CrossRef CAS PubMed.
- H. Y. Zhang, D. R. Dunphy, X. M. Jiang, H. Meng, B. B. Sun, D. Tarn, M. Xue, X. Wang, S. J. Lin, Z. X. Ji, R. B. Li, F. L. Garcia, J. Yang, M. L. Kirk, T. Xia, J. I. Zink, A. Nel and C. J. Brinker, J. Am. Chem. Soc., 2012, 134, 15790 CrossRef CAS PubMed.
- X. Y. Huang, N. P. Young and H. E. Townley, Nanomater. Nanotechnol., 2014, 4 DOI:10.5772/58290.
- H. Yamada, C. Urata, Y. Aoyama, S. Osada, Y. Yamauchi and K. Kuroda, Chem. Mater., 2012, 24, 1462 CrossRef CAS.
- Y. Lvov, W. C. Wang, L. Q. Zhang and R. Fakhrullin, Adv. Mater., 2016, 28, 1227 CrossRef CAS PubMed.
- P. Shi, K. G. Qu, J. S. Wang, M. Li, J. S. Ren and X. G. Qu, Chem. Commun., 2012, 48, 7640 RSC.
- X. M. Zhang, Y. X. Lin and R. J. J. Gillies, Nucl. Med., 2010, 51, 1167 CrossRef CAS PubMed.
- P. DeMuth, M. Hurley, C. W. Wu, S. Galanie, M. R. Zachariah and P. DeShong, Microporous Mesoporous Mater., 2011, 141, 128 CrossRef CAS.
- X. Chen, X. Y. Cheng, A. H. Soeriyadi, S. M. Sagnella, X. Lu, J. A. Scott, S. B. Lowe, M. Kavallaris and J. J. Gooding, Biomater. Sci., 2014, 2, 121 RSC.
- W. J. Fang, J. Yang, J. W. Gong and N. F. Zheng, Adv. Funct. Mater., 2012, 22, 842 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10743e |
| This journal is © The Royal Society of Chemistry 2016 |